
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Squalamine and trodusquemine: two natural products for neurodegenerative diseases, from physical chemistry to the clinic
Ryan
Limbocker
 a,
Silvia
Errico
bc,
Denise
Barbut
d,
Tuomas P. J.
Knowles
ce,
Michele
Vendruscolo
*c,
Fabrizio
Chiti
*b and
Michael
Zasloff
*df
a,
Silvia
Errico
bc,
Denise
Barbut
d,
Tuomas P. J.
Knowles
ce,
Michele
Vendruscolo
*c,
Fabrizio
Chiti
*b and
Michael
Zasloff
*df
aDepartment of Chemistry and Life Science, United States Military Academy, West Point, New York 10996, USA
bDepartment of Experimental and Clinical Biomedical Sciences, University of Florence, Florence 50134, Italy. E-mail: fabrizio.chiti@unifi.it
cCentre for Misfolding Diseases, Yusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK. E-mail: mv245@cam.ac.uk
dEnterin Inc., 3624 Market Street, Philadelphia, Pennsylvania 19104, USA
eCavendish Laboratory, Department of Physics, University of Cambridge, Cambridge CB3 0HE, UK
fMedStar-Georgetown Transplant Institute, Georgetown University School of Medicine, Washington, DC 20010, USA. E-mail: maz5@georgetown.edu
First published on 26th October 2021
Abstract
Covering: 1993 to 2021 (mainly 2017–2021)
Alzheimer's and Parkinson's diseases are neurodegenerative conditions affecting over 50 million people worldwide. Since these disorders are still largely intractable pharmacologically, discovering effective treatments is of great urgency and importance. These conditions are characteristically associated with the aberrant deposition of proteinaceous aggregates in the brain, and with the formation of metastable intermediates known as protein misfolded oligomers that play a central role in their aetiology. In this Highlight article, we review the evidence at the physicochemical, cellular, animal model and clinical levels on how the natural products squalamine and trodusquemine offer promising opportunities for chronic treatments for these progressive conditions by preventing both the formation of neurotoxic oligomers and their interaction with cell membranes.
 Ryan Limbocker | Ryan Limbocker is an Assistant Professor in the Department of Chemistry and Life Science at the United States Military Academy at West Point. He received his B.Sc. in Chemistry from the University of Kansas. He matriculated at the University of Cambridge as a Gates Cambridge and St. John's College Benefactor's Scholar and graduated with a Ph.D. in Chemistry under the supervision of Professor Sir Christopher Dobson and Professor Michele Vendruscolo. At West Point, Ryan teaches general and upper-level chemistry courses, is a passionate academic mentor, and leads a research group that investigates protein misfolding diseases and chemical and biological threat agents. |
 Silvia Errico | Silvia Errico is a PhD student in Biomedical Sciences at the Department of Experimental and Clinical Biomedical Sciences, Section of Biochemistry, at the University of Florence, Italy. She received her B.Sc. in Medical-Diagnostic Biotechnology in 2014 and Master's degree in Medical and Pharmaceutical Biotechnology in 2018 from the University of Florence. She worked as a Research Assistant at the Department of Chemistry, Centre for Misfolding Diseases, at the University of Cambridge, UK, since 2018, and she started her PhD at the University of Florence in 2020. Her scientific interests include protein misfolding diseases and compounds for their potential treatment, with a particular focus on their interaction with lipid membrane models. |
 Denise Barbut | Denise Barbut is a Professor of Neurology & Former Chief of the Neurovascular Division and of the Stroke research Program at Weill-Cornell Medical College. Her scientific interests have centered on neuroprotection and neurodegenerative disorders. She trained as a neurologist in the UK and the USA, receiving her M.D. from University College, London. Her post-graduate training included Imperial College, the Brompton, the National Hospital for Neurology and Neurosurgery and Cornell University. She is a Fellow of the Royal College of Physicians of the UK. Subsequently she became Professor of Neurology at Cornell University. She holds close to 200 patents and has published over 70 peer-reviewed articles. Her current research interests are focused on the role of aminosterols in the prevention and treatment of neurodegenerative diseases. She co-founded Enterin with Michael Zasloff in 2016, and currently serves as President, CMO, and Board Member. |
 Tuomas P. J. Knowles | Tuomas Knowles studied Biology at the University of Geneva, and Physics at ETH Zurich and obtained his PhD working at the Cavendish Laboratory and the Nanoscience Centre in Cambridge. In 2008 he was elected to a Research Fellowship at St John's College, Cambridge, and was then appointed to a University Lectureship in Physical Chemistry in 2010, joining the faculty at the Department of Chemistry at Cambridge University. He then successively held a University Readership between 2013 and 2015 and a Professorship since 2015 in the Department of Chemistry. Since 2016 he is Professor of Physical Chemistry and Biophysics in the Department of Chemistry and at the Cavendish Laboratory, and is co-director of the Cambridge Centre for Misfolding Diseases. He has published over 300 papers and has been named Highly Cited Researcher by the Web of Science. He is the recipient of multiple international prizes, including the Sackler Prize for Biophysics and the Corday-Morgan Prize from the Royal Society of Chemistry. |
 Michele Vendruscolo | Michele Vendruscolo is Professor of Biophysics, Director of the Centre for Misfolding Diseases and Director of the Chemistry of Health Laboratory at the Department of Chemistry of the University of Cambridge where he moved 20 years ago. His work is aimed at establishing the fundamental principles of protein aggregation, and at exploiting these principles to develop methods for drug discovery in neurodegenerative diseases. He has published over 450 scientific papers and 20 patents, and given over 500 invited lectures at international meetings. He is the founder, director and chief scientific officer of Wren Therapeutics, a drug discovery company that targets protein misfolding diseases. |
 Fabrizio Chiti | Fabrizio Chiti is a Full Professor at the Department of Experimental and Clinical Biomedical Sciences, Section of Biochemistry, at the University of Florence, Italy. He studied Biological Sciences at the University of Florence, and attained a PhD degree (D.Phil) in Chemistry in 2000 from the University of Oxford, under the supervision of Professor Sir Christopher Dobson. His post-doc work, carried out in the field of protein aggregation and amyloid formation, was carried out at the University of Florence for 2 years and at the University of Cambridge for 1 year. In November 2002 he became Associate Professor of Biochemistry at the University of Florence and is now, starting from December 2010, Full Professor of Biochemistry at the same University. His scientific interests concern the elucidation of protein aggregation processes, the identification of the molecular determinants of the toxicity of protein aggregates, the study of the effect of chaperones on these processes and the mechanism of cellular toxicity. |
 Michael Zasloff | Michael Zasloff is a Professor of Pediatrics and Genetics at Georgetown University and the former Dean of Research and Translational Science at Georgetown University. Dr Zasloff's scientific interests have centered on the innate immune systems of animals and the role of aminosterols in health and disease, and their application to the prevention and treatment of neurodegenerative diseases. Dr Zasloff received his M.D.-Ph.D. from New York University. He was Chief of Human Genetics at the NIH, subsequently Professor of Pediatrics and Genetics at the University of Pennsylvania and Director of the Division of Human Genetics of the Children's Hospital of Philadelphia prior to becoming the Dean of Research and Translational Science at Georgetown University, He is also the founder and Scientific Director of the MedStar-Georgetown Transplant Institute. He has published over 200 peer-reviewed articles and holds over 100 issued US patents. He co-founded Enterin in 2016 with Denise Barbut and currently serves as CSO and Board Member. |
1 Introduction
Neurodegenerative diseases are among the most prevalent afflictions of old age in our society.1 These conditions are characterized by a progressive loss of neurons in the central nervous system and are still largely incurable. The most common and widespread of these pathologies is Alzheimer's disease (AD), which accounts for about two thirds of the cases of dementia and affects approximately 30–40 million people worldwide.2,3 Another neurodegenerative disorder affecting both the central and peripheral nervous systems is Parkinson's disease (PD), which affects more than 10 million people worldwide.4 Overall, over 35 neurodegenerative diseases have been characterized to date. While some of them are extremely rare, some others are quite common. In the United States of America alone, neurodegenerative diseases were responsible for 272 644 deaths and 3 011 484 disability adjusted life years in 2016.5One of the most common histopathological traits associated with neurodegenerative diseases is the presence of large protein aggregates that can accumulate in the extracellular space of the brain or within neurons.1,6,7 These aggregates are formed by well-defined constituent proteins, and are known as amyloid plaques formed extracellularly by the amyloid-β (Aβ) peptide and neurofibrillary tangles formed intracellularly by the tau protein in AD and intracellular Lewy bodies by α-synuclein in PD.1,6–8 Genetic and circumstantial evidence indicates that the conversion of the initially native and soluble protein into protein aggregates, or their precursor oligomers, is one of the main drivers of neurodegeneration in such diseases.6,7
At present there is an urgent need for effective treatments that significantly slow down the progression of neurodegenerative diseases. After diagnosis, AD and PD patients are typically treated with symptomatic drugs that do not have the ability to interrupt or reverse neurodegeneration,9–11 and the situation is not any different in other neurodegenerative disorders. In June 2021, the FDA has approved using the accelerated approval pathway aducanumab (branded as Aduhelm), a monoclonal antibody targeting Aβ aggregates, therein providing the first disease-modifying drug against AD,12 although the treatment is very costly and is also considered controversial by a few experts. A new hope is offered by a category of natural compounds initially discovered and identified in the gastrointestinal tract of dogfish sharks (Squalus acanthias). These compounds have demonstrated over the years therapeutic properties on a number of diseases and, in the last four years, have also shown to be promising small molecules for the treatment of PD and AD.
2 Discovery of squalamine and trodusquemine in sharks and their effects in human pathologies
It was demonstrated in the 1980's that vertebrates express peptides with antimicrobial activity, which were shown to be a vital arm of the innate immune system.13–15 A search for antimicrobial compounds across vertebrate evolution led to the discovery, in 1993, that stomach extracts of the dogfish shark (Squalus acanthias) also contained a molecule with antimicrobial activity.16 This molecule was isolated and investigation of its chemical structure using mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy led to the identification of a novel compound that had not been known until then (Fig. 1): a bile salt with a steroid ring of the type of cholestane, with hydroxyl and sulfate groups at C-7 and C-24, respectively, coupled to a spermidine moiety at C-3 (3beta,5alpha,7alpha,24S).16,17 This natural product exhibited a broad spectrum of anti-infective activity against Gram-positive and Gram-negative bacteria, fungi, and certain protists, and was named squalamine, after the Squalus genus from which it was extracted and its characteristic polyamine group.16 The minimum inhibitory concentration of squalamine as an antimicrobial agent against E. aerogenes, S. cerevisiae, C. albicans, S. aureus, E. faecalis, E. hirae, E. coli, and P. aeruginosa was shown to range from approximately 2 to 25 μg mL−1.18–22 In subsequent studies, both in vitro and in vivo, squalamine was shown to inhibit pathological angiogenesis associated with retinopathy and cancer,23–25 exhibit antiviral activity against both RNA- and DNA-enveloped human viruses,26 and restore aged-related decreases in colonic motility and jejunal vagal firing rates in mice.27 Clinical studies in both cancer and vascular retinopathy were carried out for squalamine.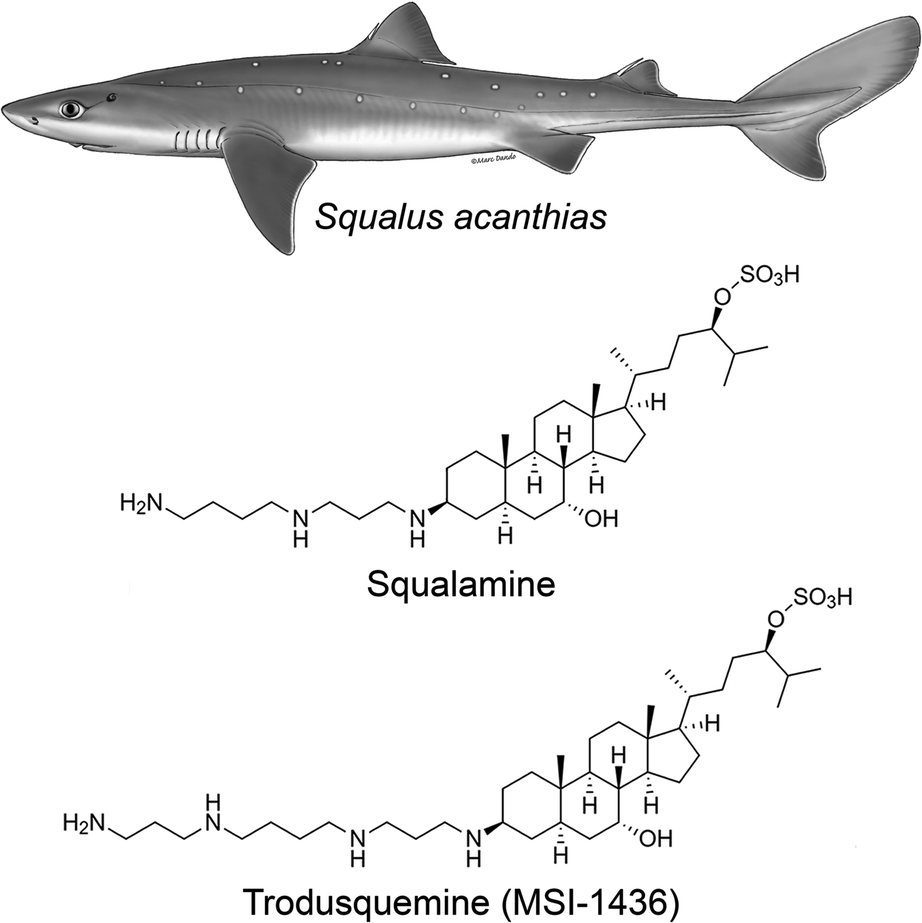 | ||
| Fig. 1 Chemical structures of squalamine and trodusquemine. They were originally isolated from the dogfish shark Squalus acanthias and are now chemically synthesized in the laboratory, enabling systematic chemical and clinical investigations. They differ for the polyamine moiety linked to C-3, which is a spermidine and spermine for squalamine and trodusquemine, respectively. The Squalus acanthias illustration was adapted from the South Andean Huemul website, under the Convention on Migratory Species of Wild Animals (CMS), as authored by Marc Dando. | ||
At physiological pH, squalamine exists as a positively charged zwitterionic molecule, soluble in both water and organic solvents.16 In aqueous solution, squalamine was shown to bind most avidly to membranes containing phospholipids with negatively charged headgroups.28 Due to its net cationic charge, squalamine displaces positively charged proteins bound electrostatically to membranes.26,29 This biophysical property is believed to be the basis of many of the pharmacological properties exhibited by squalamine, such as its antiviral activity.26
Following this discovery, it was found that squalamine was not the only potentially interesting natural product that the shark entrails had in store for us. In 2001, a procedure of extraction of squalamine from larger amounts of dogfish liver, the richest source of squalamine in this animal, led to the discovery of at least seven other aminosterols, present in considerably lower concentrations than squalamine and structurally similar to this molecule, with variations involving mainly the cholestane-like side chain, from C-20 to C-27.18 One of these compounds shared the same steroid cholestane-like moiety as squalamine, but was conjugated at C-3 to spermine, rather than spermidine (Fig. 1). This new natural product was found to have a wide spectrum of antimicrobial activity, similar to squalamine.18 It was initially referred to as MSI-1436 (ref. 18) and was later named trodusquemine by a biopharmaceutical company, a name that became soon popular among scholars.19 The minimum inhibitory concentration of MSI-1436 was found to be approximately four times lower against E. coli, P. aeruginosa, and C. albicans related to squalamine.22
Trodusquemine has been studied in both animals and in humans. It was shown to cross the blood–brain-barrier, suppress appetite and increase insulin sensitivity in rodents and dogs,30,31 most likely by inhibiting protein tyrosine phosphatase 1B (PTP1B).32 PTP1B is an endosomal phosphatase that downregulates many physiologically important signaling pathways, including that of insulin, leptin, as well as many growth factors and cytokines.32 With the recognition that trodusquemine could effectively inhibit PTP1B in vivo, numerous studies were conducted. These investigations revealed that trodusquemine could ameliorate metabolic syndrome in a mouse model of hypothalamic insulin resistance,33,34 reverse atherosclerosis in low-density lipoprotein receptor knockout mice,35 inhibit the growth of malignancy,36 regenerate tail-fin and heart muscle in zebrafish,37 induce the regenerative repair of myocardial infarction and traumatic limb muscle injury by mobilization of stem cells in adult mice,37 relieve stress-induced anxiety in LMO4-deficient mice by targeting the mGluR5 receptors of the limbic system,38 reverse memory impairment, normalize behavior and reduce neuronal loss in a mouse model of AD via a neuronal PTP1B dependent mechanism,39 and ameliorate schizophrenia-like behaviors in LMO4-deficient mice.40 Phase-1 human clinical trials of intravenously administered trodusquemine in obese subjects demonstrated improvement in insulin sensitivity.41
3 Chemical, molecular and cellular effects of squalamine and trodusquemine on protein aggregation
In spite of the many beneficial effects demonstrated by these two aminosterols, it was not until 2017 that a report appeared illustrating that squalamine may have therapeutic potential against neurodegenerative diseases based on a mechanism that either targeted the kinetics of the protein aggregation process or the aggregates of misfolded proteins.42 In fact, this molecule was reported to inhibit α-synuclein aggregation, protect neuronal cells from the action of toxic oligomers formed by this protein and ameliorate deficits of motility and paralysis in a Caenorhabditis elegans model of PD expressing human α-synuclein.42 Within the dopaminergic neurons of the substantia nigra pars compacta of patients with PD, individual α-synuclein molecules aggregate, transitioning from a soluble, intrinsically disordered monomeric state into insoluble β-sheet rich amyloid fibrils with a width of 7–13 nm, which accumulate within neurons as inclusions.1,43 The initiation of the aggregation process through primary nucleation is slow in vitro in a homogenous solution of α-synuclein,44 but can be induced by lipids membranes.45In vitro, squalamine has been demonstrated to compete with monomeric α-synuclein binding to lipid membranes with the result that the membrane-catalysed primary nucleation of α-synuclein starting from the monomeric state was strongly suppressed.42 Moreover, the toxicity of kinetically trapped oligomers of α-synuclein towards human neuroblastoma cells was abolished when cells were exposed to squalamine as a result of the ability of this aminosterol to displace these toxic species from the cell membranes, thus preventing their deleterious activity towards living cells.42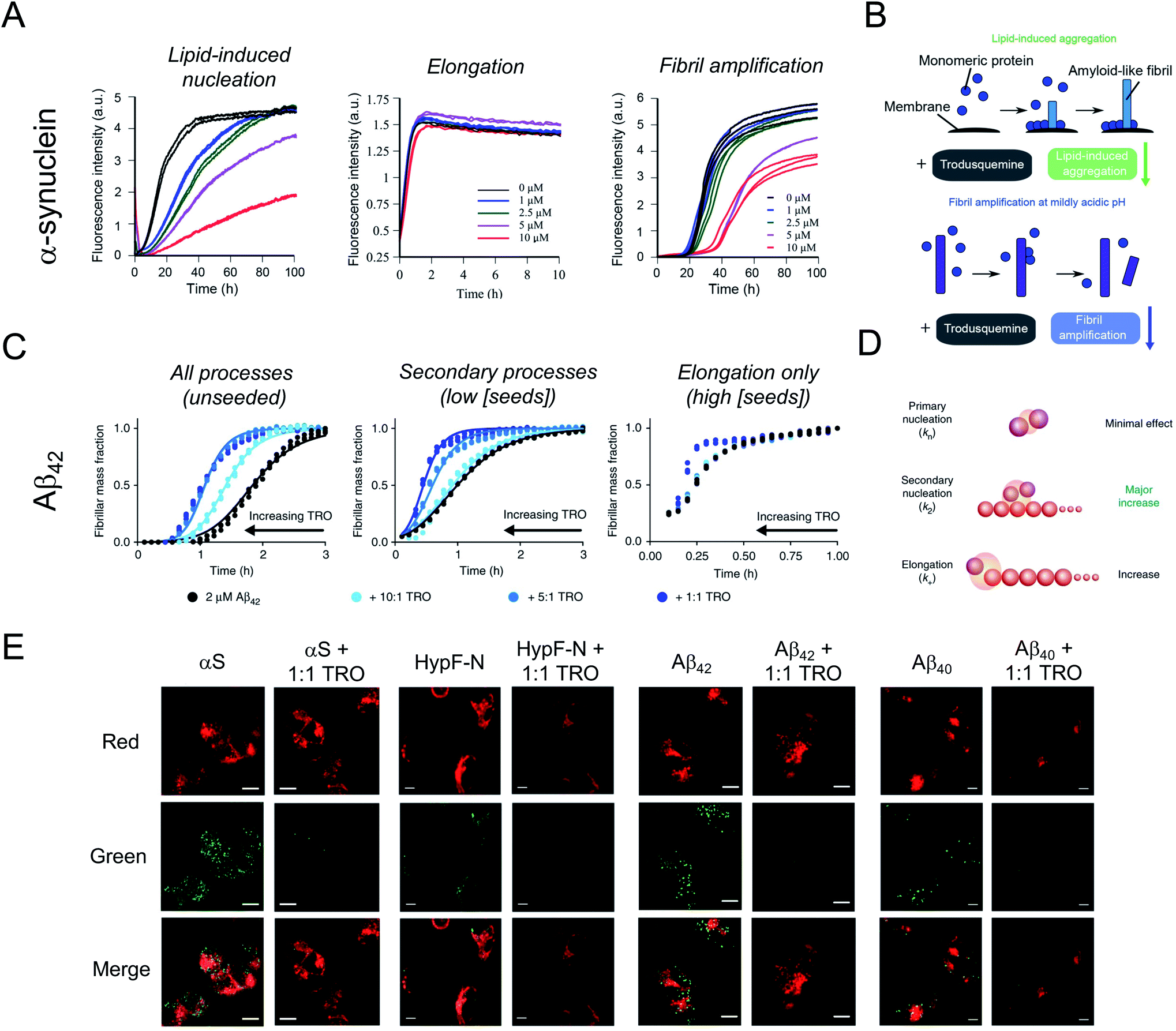 | ||
| Fig. 2 Impact of trodusquemine on α-synuclein and Aβ42 aggregation. (A) Trodusquemine (TRO) inhibits the lipid-induced nucleation and fibril amplification reactions in α-synuclein aggregation, corresponding to primary and secondary nucleation events, respectively, without impacting fibril elongation. (B) Schematic representation of the effect of trodusquemine on the microscopic steps governing α-synuclein aggregation under these conditions. (C) Trodusquemine accelerates the unseeded and seeded aggregation reactions of Aβ42 to varying degrees. (D) Schematic representation of the effect of trodusquemine on the microscopic steps governing Aβ42 aggregation under these conditions. (E) Confocal microscopy analysis of the extent of α-synuclein, HypF-N, Aβ42, or Zn2+-stabilized Aβ40 oligomer binding to the membranes of human neuroblastoma cells in the absence (left panels) or presence (right panels) of an equimolar concentration of trodusquemine shows that aminosterols induce the displacement of oligomers from cell membranes. Red and green channels indicate cell membranes and oligomers, respectively. Scale bars, 10 μm. Panels (A) and (B) and part of panel (E) were adapted with permission from M. Perni, P. Flagmeier, R. Limbocker, R. Cascella, F. A. Aprile, C. Galvagnion, G. T. Heller, G. Meisl, S. W. Chen, J. R. Kumita, P. K. Challa, J. B. Kirkegaard, S. I. A. Cohen, B. Mannini, D. Barbut, E. A. A. Nollen, C. Cecchi, N. Cremades, T. P. J. Knowles, F. Chiti, M. Zasloff, M. Vendruscolo and C. M. Dobson, ACS Chem. Biol., 2018, 13, 2308–2319. Copyright 2018 American Chemical Society. Panels (C) and (D) and part of panel (E) were adapted from R. Limbocker, S. Chia, F. S. Ruggeri, M. Perni, R. Cascella, G. T. Heller, G. Meisl, B. Mannini, J. Habchi, T. C. T. Michaels, P. K. Challa, M. Ahn, S. T. Casford, N. Fernando, C. K. Xu, N. D. Kloss, S. I. A. Cohen, J. R. Kumita, C. Cecchi, M. Zasloff, S. Linse, T. P. J. Knowles, F. Chiti, M. Vendruscolo and C. M. Dobson, Nat. Commun., 2019, 10, 225 published by Springer Nature. Part of panel (E) was adapted from R. Limbocker, B. Mannini, F. S. Ruggeri, R. Cascella, C. K. Xu, M. Perni, S. Chia, S. W. Chen, J. Habchi, A. Bigi, R. P. Kreiser, A. K. Wright, J. A. Albright, T. Kartanas, J. R. Kumita, N. Cremades, M. Zasloff, C. Cecchi, T. P. J. Knowles, F. Chiti, M. Vendruscolo and C. M. Dobson, Commun. Biol., 2020, 3, 1–10 published by Springer Nature. | ||
Shortly thereafter, similarly to squalamine, trodusquemine was shown to inhibit α-synuclein aggregation, displacing both monomers and oligomers of the protein from cell membranes.46 Unlike the previous report on squalamine, which only investigated lipid-induced nucleation, trodusquemine was shown to inhibit both the lipid-induced primary nucleation of α-synuclein as well as a key secondary process in α-synuclein aggregation, namely secondary nucleation (Fig. 2A and B).46 Indeed, by direct binding to the surface of α-synuclein fibrils, trodusquemine also prevented the autocatalytic amplification of α-synuclein fibrils through secondary nucleation,46 a process that is primarily responsible for the proliferation of aggregates, and for the production of toxic oligomeric species once a low but critical concentration of amyloid fibrils have formed.44 Trodusquemine was also shown to displace pre-formed oligomers of α-synuclein from cell membranes of neuroblastoma cells (Fig. 2E). Paradoxically, and importantly, the low concentrations of trodusquemine that were sufficient to displace α-synuclein oligomers from cell membranes were insufficient to cause the displacement of monomeric α-synuclein from liposomes.46 Since monomeric membrane-bound α-synuclein plays a key role in the innate immune system of mammals, selective targeting of the undesired aggregates by the aminosterol would be advantageous.47
It was next found that trodusquemine had an impact on the aggregation of the 42-residue form of the amyloid-β peptide (Aβ42), which plays an important role in the pathogenesis of AD.48 Trodusquemine was found to accelerate, rather than inhibit, the rate of Aβ42 aggregation, predominantly by stimulating the secondary nucleation microscopic step and, to a much lesser extent, fibril elongation (Fig. 2C and D), in contrast to its effects on the aggregation of α-synuclein.46 Trodusquemine appeared, therefore, to shift the Aβ42 oligomer population towards the fibrillar form, therein depleting the pool of oligomers that cause cellular dysfunction.48 Trodusquemine also promoted the displacement of cytotoxic aggregates from cell membranes, similarly to α-synuclein oligomers, thus neutralizing their toxic activity.48,49 The displacement mechanism was studied using Aβ42 ADDL oligomers, zinc-stabilized Aβ40 oligomers and cytotoxic oligomers of the N-terminal domain of the E. coli HypF protein (Fig. 2E). Although the oligomers were displaced from the lipid membranes at therapeutically meaningful concentrations of trodusquemine, neither their size nor their hydrophobicity, two important determinants of oligomer toxicity, were overtly affected.49 Super-stoichiometric concentrations of trodusquemine could instead modulate these properties by increasing both the size and solvent exposed hydrophobicity of oligomers,50,51 but these effects offset each other as they decrease and increase oligomer toxicity, respectively.52,53 Overall, these observations suggested that trodusquemine might be altering the affinity of lipid membranes for these oligomeric species (discussed in greater detail below).
4 Effects of squalamine and trodusquemine on protein aggregation in animal models
The effects of aminosterols on the levels and toxicity of protein aggregates associated with AD and PD have been most extensively studied in C. elegans models of the two diseases.42,46,48 These 1 mm long, transparent nematodes were genetically engineered to express Aβ42 (for the temperature-inducible GMC101 strain) or α-synuclein (for the OW40 strain with α-synuclein fused to yellow fluorescent protein, YFP) in the muscle cells of their body wall, respectively. With ageing, these proteins accumulate, leading to a phenotypic dysfunction in the ability of the worms to thrash and swim, culminating in their paralysis and death. Such phenotypic behaviors were monitored over the worm lifespan for thousands of animals using a recently developed tracking platform54 after the administration of aminosterols to worms by their addition atop nematode growth medium.PD worms were first exposed to various concentrations of squalamine early in their lifespan at the L4 stage of development before microscopically visible α-synuclein accumulated in their tissues.42 As the animals aged, a dose-dependent decrease in the number of α-synuclein-YFP inclusions could be observed for PD worms treated with squalamine relative to untreated PD worms, coincident with an increase in the fitness of the animals, as manifested by the swimming speed, motility (measured in bends per minute), and extent of paralysis.42 These observations paralleled the in vitro studies, in which squalamine inhibited both lipid-induced primary nucleation and the attachment of the resulting aggregates to susceptible cultured cells, therein attenuating the resulting cytotoxicity through two distinct mechanisms. Squalamine also improved the health of worms expressing A30P and A53T mutants of α-synuclein associated with familial forms of PD, with a more robust recovery effect observed for the more aggressive A53T variant than the A30P one.55
Trodusquemine exhibited a more robust effect than squalamine in the PD worms, protecting the animals from developing a full PD phenotype when administered both at a pre-disease onset stage as well when administered several days after phenotype onset (Fig. 3A and B).46 The superior effect of trodusquemine correlates with its ability to inhibit both primary and secondary processes in vitro and to inhibit the binding of the pre-formed oligomers to the membrane of cultured cells at lower doses.46 The overall expression of α-synuclein was not affected by either treatment, as shown by the fluorescence of YFP as a reporter of gene expression.46
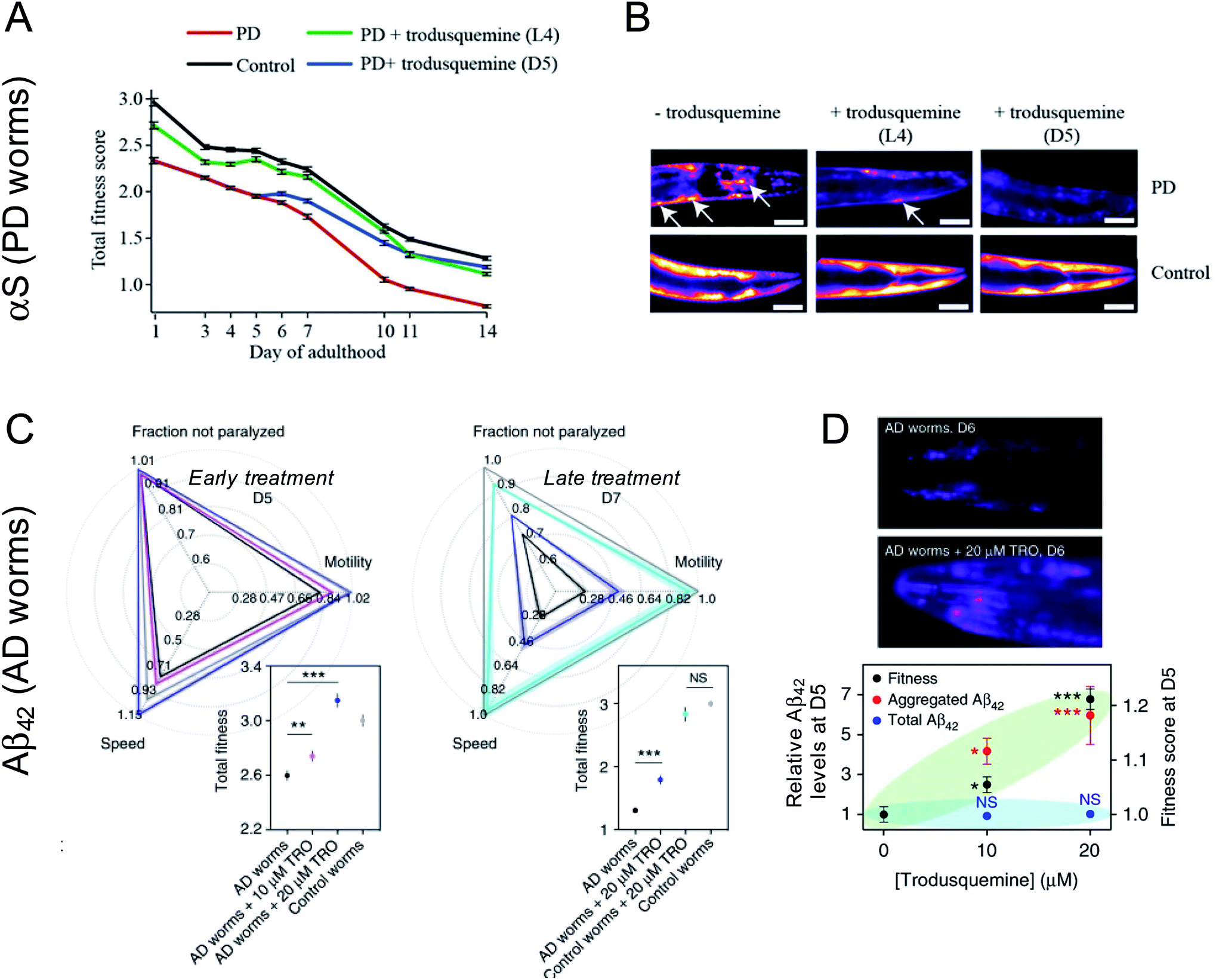 | ||
| Fig. 3 Impact of trodusquemine on AD and PD model worms. (A) Trodusquemine significantly improves the health of PD worms overexpressing α-synuclein-YFP in their muscle cells, when administered before (L4 stage of development) or after (day 5 of adulthood, D5) the onset of toxicity in these animals. (B) Trodusquemine decreases the number of α-synuclein-YFP inclusions in PD worms, when administered before (L4) or after (D5) the onset of toxicity in these animals, a finding that is consistent with its in vitro mechanism of action. Scale bars, 80 μm. (C) Trodusquemine also improves the health of AD model worm overexpressing Aβ42 in their muscle cells, when administered before (L4) or after (D5) the onset of toxicity in the animals. (D) Trodusquemine stimulates the aggregation of Aβ42 in AD worms, which may shift the reactive flux away from the oligomeric state towards the less toxic fibrillar form. Panels (A) and (B) were adapted with permission from M. Perni, P. Flagmeier, R. Limbocker, R. Cascella, F. A. Aprile, C. Galvagnion, G. T. Heller, G. Meisl, S. W. Chen, J. R. Kumita, P. K. Challa, J. B. Kirkegaard, S. I. A. Cohen, B. Mannini, D. Barbut, E. A. A. Nollen, C. Cecchi, N. Cremades, T. P. J. Knowles, F. Chiti, M. Zasloff, M. Vendruscolo and C. M. Dobson, ACS Chem. Biol., 2018, 13, 2308–2319. Copyright 2018 American Chemical Society. Panels C and D were adapted from R. Limbocker, S. Chia, F. S. Ruggeri, M. Perni, R. Cascella, G. T. Heller, G. Meisl, B. Mannini, J. Habchi, T. C. T. Michaels, P. K. Challa, M. Ahn, S. T. Casford, N. Fernando, C. K. Xu, N. D. Kloss, S. I. A. Cohen, J. R. Kumita, C. Cecchi, M. Zasloff, S. Linse, T. P. J. Knowles, F. Chiti, M. Vendruscolo and C. M. Dobson, Nat. Commun., 2019, 10, 225 published by Springer Nature. | ||
Treatment of AD worms with trodusquemine prior to the onset of the disease phenotype increased the accumulation of Aβ42 aggregates in a dose-dependent manner, above that observed in untreated animals.48 This finding is again consistent with observations in vitro, in which trodusquemine was found to potentiate monomer-dependent secondary nucleation. Trodusquemine also increased AD model worm health, as quantified using the phenotypes of speed of swimming, motility and extent of paralysis, when administered before or after the onset of a phenotype without changing the total levels of Aβ42 in these animals, despite causing the deposition of a greater extent of fibrillar aggregates positive for the amyloid-specific dye NIAD-4 when administered early in the worm lifespan (Fig. 3C and D).48 The quantified increase in AD worm health is likely a result of both the displacement of toxic oligomers from cell membranes and their accelerated conversion to the less toxic fibrillar form, consistent with the in vitro studies.48
Trodusquemine treatment extended the lifespan of both wild-type and PD worms.46 Surprisingly, treatment of the PD worms extended their lifespan significantly beyond that observed for untreated wild-type animals and even beyond that for trodusquemine-treated wild-type animals.46 While trodusquemine is known to stimulate growth factor signaling pathways (insulin and leptin) via PTP1B inhibition,32 and to potentiate tissue regeneration following injury in the tissues of various organisms,37 the precise mechanism by which longevity is extended by certain aminosterols in nematode models, and the corresponding significance of such findings to human health, are not well resolved at this time.
In a recent report, intraperitoneal injections of trodusquemine in the hAPP-J20 AD mouse model were shown to inhibit PTP1B and consequently to attenuate hippocampal degeneration (neuronal loss) and recover spatial memory in these mice.39 This study is particularly important because it shows an additional protective mechanism mediated by aminosterols against neurodegeneration, a mechanism resulting from the non-competitive inhibition of PTP1B by trodusquemine and the consequent improvement of glucose metabolism through the disinhibition of glycogen synthase kinase 3β (GSK3β).39
5 Squalamine and trodusquemine act on the physicochemical properties of biological membranes
As described in Sections 3 and 4, in vitro and in vivo studies suggest that the displacement of the toxic oligomers from cell membranes is one of the key steps by which aminosterols reduce aggregate associated toxicity. In principle, the displacement effect may be due to (i) the binding of the aminosterols to the misfolded protein oligomers preventing them from binding to cell membranes, or (ii) the binding of the aminosterols to cell membranes preventing them from binding the oligomers.Neither squalamine nor trodusquemine appear to alter the structural or morphological characteristics of the cytotoxic aggregates from three different proteins at concentrations of the aminosterols that both displace the aggregates from the cell membrane and protect the cells from damage.49–51 These observations do not support the first mechanism outlined above and rather point to a mechanism by which the aminosterols interact with the cellular membrane therein altering its affinity for the aggregates.
In agreement with this idea, recent studies with confocal microscopy, FRET and NMR have demonstrated that water solubilized trodusquemine is able to insert readily into model neuronal membranes (large unilamellar vesicles, LUVs) and the plasma membrane of human neuroblastoma cells (Fig. 4A and B).56 Applying fluorescence quenching and anisotropy experiments with probes that localized in different portions of the membrane, quartz crystal microbalance (QCM) analysis and molecular dynamics (MD) simulations, the location and orientation of trodusquemine within the bilayer could be determined. The positively charged spermine moiety of trodusquemine was positioned on the surface of the external hydrophilic face, whereas the steroidal portion was found to extend downward and penetrate the external hydrophilic layer down to the interface between the hydrophilic and hydrophobic layers. This occurred with a well-defined oblique angle (about 55°) for the major axis of the molecule with respect to the normal to the bilayer plane (Fig. 4C).56
 | ||
Fig. 4 Effects of squalamine and trodusquemine on the physicochemical properties of biological membranes. (A) Confocal microscopy image showing colocalization of trodusquemine-Alexa Fluor® 594 (red) and CHOL-BODIPY-FL LUVs (green). (B) Magnified image of a SH-SY5Y cell incubated for 15 min with 4 μM Alexa Fluor® 594-labelled trodusquemine (probe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trodusquemine of 1:10). (C) Snapshot of MD simulations showing trodusquemine within the membrane (N, C, H, O, S atoms in blue, cyan, white, red and yellow, respectively, lipids in transparent violet, water in cyan sticks). (D) ζ-Potential as a function of temperature for LUVs (filled squares) and trodusquemine-containing LUVs (empty squares), showing the decrease of the total negative charge and the increase of the transition temperature induced by trodusquemine. (E) FRET efficiencies (E) determined for the indicated lipid–lipid pairs in the absence (light grey) and presence (dark grey) of 5 μM trodusquemine. (F) FRET efficiencies (E) determined for the indicated lipid–trodusquemine pairs. (G) Breakthrough force map obtained with AFM from supported lipid bilayers in the presence of 5 μM trodusquemine. (H) Schematic representation of the three major perturbations induced by trodusquemine in a lipid bilayer. All the panels were reproduced from ref. 56 with permission from the Royal Society of Chemistry. :trodusquemine of 1:10). (C) Snapshot of MD simulations showing trodusquemine within the membrane (N, C, H, O, S atoms in blue, cyan, white, red and yellow, respectively, lipids in transparent violet, water in cyan sticks). (D) ζ-Potential as a function of temperature for LUVs (filled squares) and trodusquemine-containing LUVs (empty squares), showing the decrease of the total negative charge and the increase of the transition temperature induced by trodusquemine. (E) FRET efficiencies (E) determined for the indicated lipid–lipid pairs in the absence (light grey) and presence (dark grey) of 5 μM trodusquemine. (F) FRET efficiencies (E) determined for the indicated lipid–trodusquemine pairs. (G) Breakthrough force map obtained with AFM from supported lipid bilayers in the presence of 5 μM trodusquemine. (H) Schematic representation of the three major perturbations induced by trodusquemine in a lipid bilayer. All the panels were reproduced from ref. 56 with permission from the Royal Society of Chemistry. | ||
The incorporation of trodusquemine into the lipid membrane was shown to have significant effects on the physiochemical properties of the membrane.56 First, trodusquemine decreased the total negative charge of the lipid bilayer, contributed by the anionic headgroup of monosialotetrahexosylganglioside 1 (GM1), as determined with ζ-potential measurements (Fig. 4D). Second, it changed the spatial distribution of the constituent lipids of the model neuronal membrane, inducing a separation of GM1 from cholesterol (CHOL) molecules, and the formation of clusters of CHOL molecules and of clusters of GM1 molecules, probably caused by a preferential binding of this aminosterol nearby these two lipids, as observed by lipid–lipid and lipid–trodusquemine FRET experiments, respectively (Fig. 4E and F). Third, it caused an increment of the mechanical resistance of the lipid membrane to indentation, when probed with atomic force microscopy (AFM) applying a force perpendicular to the bilayer plane, and an increase of the membrane phase transition temperature, reflecting a more stable packing of the lipids (Fig. 4G). All these perturbations, schematised in Fig. 4H, contribute significantly to improve the resistance of biological membranes to the toxic action of misfolded protein oligomers, explaining from a physicochemical perspective how these natural products reinforce the membrane making it resistant to the deleterious action of these aberrant species.56
These results suggest the intriguing possibility that aminosterol natural products may have regulatory functions within the lipid homeostasis system, which is the ensemble of cellular processes that regulates the behaviour of lipids in terms of their synthesis, trafficking, localizations, concentrations, interactions, and degradation.57,58 Aminosterols like squalamine and trodusquemine have the ability to modulate the physical state of lipid membranes, a type of activity that has been reported for proteins known to act also as conventional molecular chaperones, including Hsp70, Hsp60, Hsp90, small Hsps,59 as well as lipid-specific proteins, such as fatty acid binding proteins.60
6 Potential of aminosterols as AD and PD drugs and future perspectives
Supported by the studies described above, squalamine has entered human clinical trials for the treatment of PD in 2018, as an orally administered phosphate salt named ENT-01. Although PD is diagnosed based on motor symptoms, resulting from damage to the dopamine-rich substantia nigra pars compacta and caused by the accumulation of intraneuronal α-synuclein aggregates known as Lewy bodies or precursor oligomers,43 ENT-01 is being developed to target α-synuclein aggregates within the enteric nervous system (ENS). In fact, early in the lives of PD patients, in some cases many years before motor symptoms appear, intraneuronal α-synuclein aggregates are present in biopsies of their upper gastrointestinal (GI) tract,61–64 indicating that the disease begins in the GI tract. α-Synuclein is known to serve an immune function starting from childhood, induced during GI infections and attracting macrophages, neutrophils, and activating dendritic cells.65 The extensive surface area of the GI tract and its continued exposure to ingested pathogens, as well as the presence of its own commensal microbes, positions the gut as the first organ to accumulate α-synuclein aggregates within its ENS. Clinically, patients experience constipation due to diminished peristalsis caused by the reduced excitability of the neurons of the myenteric plexus as a result of accumulation of α-synuclein aggregates.66,67 Infections in other areas of the body, or alerted by α-synuclein induced in the GI tract, gradually leads to accumulation of α-synuclein in different divisions of the nervous system, including the brain.68,69 In addition, human epidemiologic and animal model studies suggest that α-synuclein can also migrate from the ENS via the vagus nerve to the brain.65,70 Once induced, α-synuclein outlasts the infection by several months, and in individuals who do not successfully confine the infection, α-synuclein production continues inexorably, leading to the accumulation of neurotoxic aggregates over years and decades. PD occurs when the synthesis of α-synuclein in neurons exceeds clearance, and toxic aggregates are formed faster than they can be cleared, resulting in neuronal damage.After squalamine entered human clinical trials in 2018 as ENT-01, it was reported that the oral administration of this compound to transgenic mice expressing A53T α-synuclein can restore both bowel motility and normal neuronal function.71 Since disruption of bowel function is only observed in mice expressing the A53T mutant, but not wild-type α-synuclein, it has been assumed that bowel dysfunction arises as a consequence of the formation of membrane disruptive oligomers, supporting the hypothesis that squalamine restores electrical activity and bowel motility in the A53T mice through displacement of oligomeric species.71 Restoration of electrical activity of the enteric neurons results in increased flow of electrical signals via the vagus nerve to the brain, with potential positive impact on the normal functioning of the hypothalamus, hippocampus, brain stem and other centers of the brain.
The results of the first phase 1/2a clinical trial using squalamine as ENT-01 in an open label study involving 50 patients (RASMET, identifier NCT03047629) have been published.72 ENT-01 restored bowel motility in over 80% of PD patients with severe constipation. This observation indicates that despite bowel dysfunction in some cases for almost 60 years, the functioning of ENS in PD patients can be restored, and is not, as commonly believed, irreversibly damaged.72 In addition, benefits have been observed in other neurological symptoms, such as memory, hallucinations, motor functioning and circadian rhythm, all of which are currently being further evaluated in an ongoing randomized controlled phase 2b clinical study involving up to 150 patients (KARMET, identifier NCT 03781791).72 Encouraged by these results, randomized, controlled studies involving large numbers of patients with PD dementia or PD psychosis are currently under way.
Following the encouraging results in C. elegans described above, trodusquemine has been recently studied in an aggressive transgenic model of AD and shown to improve memory and prevent neuronal loss.39 In addition, trodusquemine crosses the blood–brain-barrier following parenteral administration.31 For these reasons, aminosterols structurally related to trodusquemine are being developed for the treatment of AD.
7 Author contributions
Conceptualization: R. L., S. E., D. B., T. P. J. K., M. V., F. C., and M. Z.; writing – original draft: R. L., S. E., F. C. and M. Z.; writing – reviewing and editing: R. L., S. E., D. B., T. P. J. K., M. V., F. C., and M. Z.; visualisation: R. L. and S. E.; supervision: F. C. and M. Z.8 Conflicts of interest
M. Z. and D. B. are inventors in patents for the use of squalamine and trodusquemine in the treatment of Parkinson's disease and are co-founders and stockholders in Enterin, Inc. M. V. and T. P. J. K. are founders of Wren Therapeutics Ltd, which is independently pursuing the development of inhibitors of protein aggregation. The remaining authors declare no competing interests. The views expressed herein are those of the authors and do not reflect the position of the United States Military Academy, the Department of the Army, or the Department of Defense.9 Acknowledgements
R. L. acknowledges support from DTRA Service Academy Research Initiative grants and Combat Capabilities Development Command Army Research Laboratory grants. F. C. and S. E. acknowledge the Regione Toscana (FAS-Salute 2014, project SUPREMAL; FAS-Salute 2018, project PRAMA) and the University of Florence (Fondi di Ateneo, RICATEN21). T. P. J. K. acknowledges support from the Frances and Augustus Newman Foundation.10 References
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- Alzheimer's Association, Alzheimer's and Dementia, https://alz.org/alzheimer_s_dementia, accessed June 22, 2021 Search PubMed.
- Alzheimer's Assoication, Dementia vs. Alzheimer's Disease, https://alz.org/alzheimers-dementia/difference-between-dementia-and-alzheimer-s, accessed June 22, 2021 Search PubMed.
- American Parkinson Disease Association, What is Parkinson's Disease, https://www.apdaparkinson.org/what-is-parkinsons/, accessed June 12, 2021 Search PubMed.
- K. E. Thorpe, R. W. Woodruff and A. I. Levey, U.S. Burden of neurodegenerative disease, 2021 Search PubMed.
- N. Bengoa-Vergniory, R. F. Roberts, R. Wade-Martins and J. Alegre-Abarrategui, Acta Neuropathol., 2017, 134, 819–838 CrossRef CAS.
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed.
- M. G. Spillantini and M. Goedert, Lancet Neurol., 2013, 12, 609–622 CrossRef CAS.
- W. V. Graham, A. Bonito-Oliva and T. P. Sakmar, Annu. Rev. Med., 2017, 68, 413–430 CrossRef.
- W. Poewe and P. Mahlknecht, Neurol. Clin., 2020, 38, 255–267 CrossRef.
- J. Cummings, G. Lee, K. Zhong, J. Fonseca and K. Taghva, Alzheimer's Dement.: Transl. Res. Clin. Interv., 2021, 7, e12179 Search PubMed.
- P. Cavazzoni, U.S. Food and Drug Administration, FDA’s Decision to Approve New Treatment for Alzheimer’s Disease, https://www.fda.gov/drugs/news-events-human-drugs/fdas-decision-approve-new-treatment-alzheimers-disease, accessed June 12, 2021 CAS.
- M. Zasloff, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 5449–5453 CrossRef CAS PubMed.
- R. I. Lehrer, M. E. Selsted, D. Szklarek and J. Fleischmann, Infect. Immun., 1983, 42, 10–14 CrossRef CAS PubMed.
- H. Steiner, D. Hultmark, Å. Engström, H. Bennich and H. G. Boman, Nature, 1981, 292, 246–248 CrossRef CAS PubMed.
- K. S. Moore, S. Wehrli, H. Roder, M. Rogers, J. N. Forrest, D. McCrimmon and M. Zasloff, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 1354–1358 CrossRef CAS PubMed.
- S. L. Wehrli, K. S. Moore, H. Roder, S. Durell and M. Zasloff, Steroids, 1993, 58, 370–378 CrossRef CAS PubMed.
- M. Rao, A. Shinnar, L. Noecker, T. Chao, B. Feibush, B. Snyder, I. Sharkansky, A. Sarkahian, X. Zhang, S. Jones, W. Kinney and M. Zasloff, J. Nat. Prod., 2000, 63, 631–635 CrossRef CAS PubMed.
- C. Salmi, C. Loncle, N. Vidal, M. Laget, Y. Letourneux and J. M. Brunel, J. Enzyme Inhib. Med. Chem., 2008, 23, 860–865 CrossRef CAS PubMed.
- K. Alhanout, S. Malesinki, N. Vidal, V. Peyrot, J. M. Rolain and J. M. Brunel, J. Antimicrob. Chemother., 2010, 65, 1688–1693 CrossRef CAS PubMed.
- S. Hraiech, F. Brégeon, J.-M. Brunel, J.-M. Rolain, H. Lepidi, V. Andrieu, D. Raoult, L. Papazian and A. Roch, J. Antimicrob. Chemother., 2012, 67, 2452–2458 CrossRef CAS.
- Y. Shu, S. R. Jones, W. A. Kinney and B. S. Selinsky, Steroids, 2002, 67, 291–304 CrossRef CAS PubMed.
- A. K. Sills, J. I. Williams, B. M. Tyler, D. S. Epstein, E. P. Sipos, J. D. Davis, M. P. McLane, S. Pitchford, K. Cheshire, F. H. Gannon, W. A. Kinney, T. L. Chao, M. Donowitz, J. Laterra, M. Zasloff and H. Brem, Cancer Res., 1998, 58, 2784–2792 CAS.
- R. D. Higgins, R. J. Sanders, Y. Yan, M. Zasloff and J. I. Williams, Invest. Ophthalmol. Vis. Sci., 2000, 41, 1507–1512 CAS.
- J. I. Williams, S. Weitman, C. M. Gonzalez, C. H. Jundt, J. Marty, S. D. Stringer, K. J. Holroyd, M. P. McLane, Q. Chen, M. Zasloff and D. D. V. Hoff, Clin. Cancer Res., 2001, 7, 724–733 CAS.
- M. Zasloff, A. P. Adams, B. Beckerman, A. Campbell, Z. Han, E. Luijten, I. Meza, J. Julander, A. Mishra, W. Qu, J. M. Taylor, S. C. Weaver and G. C. L. Wong, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15978–15983 CrossRef CAS.
- C. L. West, J. Y. Amin, S. Farhin, A. M. Stanisz, Y.-K. Mao and W. A. Kunze, Front. Neurosci., 2019, 13, 955 CrossRef PubMed.
- B. S. Selinsky, Z. Zhou, K. G. Fojtik, S. R. Jones, N. R. Dollahon and A. E. Shinnar, Biochim. Biophys. Acta, 1998, 1370, 218–234 CrossRef CAS.
- R. T. Alexander, V. Jaumouillé, T. Yeung, W. Furuya, I. Peltekova, A. Boucher, M. Zasloff, J. Orlowski and S. Grinstein, EMBO J., 2011, 30, 679–691 CrossRef CAS.
- M. Zasloff, J. I. Williams, Q. Chen, M. Anderson, T. Maeder, K. Holroyd, S. Jones, W. Kinney, K. Cheshire and M. McLane, Int. J. Obes. Relat. Metab. Disord., 2001, 25, 689–697 CrossRef CAS PubMed.
- R. S. Ahima, H. R. Patel, N. Takahashi, Y. Qi, S. M. Hileman and M. A. Zasloff, Diabetes, 2002, 51, 2099–2104 CrossRef CAS.
- K. A. Lantz, S. G. E. Hart, S. L. Planey, M. F. Roitman, I. A. Ruiz-White, H. R. Wolfe and M. P. McLane, Obesity, 2010, 18, 1516–1523 CrossRef CAS PubMed.
- N. R. Pandey, X. Zhou, Z. Qin, T. Zaman, M. Gomez-Smith, K. Keyhanian, H. Anisman, J. M. Brunel, A. F. R. Stewart and H.-H. Chen, J. Neurosci., 2013, 33, 12647–12655 CrossRef CAS PubMed.
- N. R. Pandey, X. Zhou, T. Zaman, S. A. Cruz, Z. Qin, M. Lu, K. Keyhanian, J. M. Brunel, A. F. R. Stewart and H.-H. Chen, Biochem. Biophys. Res. Commun., 2014, 450, 666–672 CrossRef CAS PubMed.
- D. Thompson, N. Morrice, L. Grant, S. Le Sommer, E. K. Lees, N. Mody, H. M. Wilson and M. Delibegovic, Clin. Sci., 2017, 131(20), 2489–2501 CrossRef CAS.
- N. Krishnan, D. Koveal, D. H. Miller, B. Xue, S. D. Akshinthala, J. Kragelj, M. R. Jensen, C.-M. Gauss, R. Page, M. Blackledge, S. K. Muthuswamy, W. Peti and N. K. Tonks, Nat. Chem. Biol., 2014, 10, 558–566 CrossRef CAS.
- A. M. Smith, K. K. Maguire-Nguyen, T. A. Rando, M. A. Zasloff, K. B. Strange and V. P. Yin, npj Regener. Med., 2017, 2, 4 CrossRef.
- Z. Qin, X. Zhou, N. R. Pandey, H. A. Vecchiarelli, C. A. Stewart, X. Zhang, D. C. Lagace, J. M. Brunel, J.-C. Béïque, A. F. R. Stewart, M. N. Hill and H.-H. Chen, Neuron, 2015, 85, 1319–1331 CrossRef CAS.
- K. M. Ricke, S. A. Cruz, Z. Qin, K. Farrokhi, F. Sharmin, L. Zhang, M. A. Zasloff, A. F. R. Stewart and H.-H. Chen, J. Neurosci., 2020, 40, 1581–1593 CrossRef CAS.
- Z. Qin, L. Zhang, S. A. Cruz, A. F. R. Stewart and H.-H. Chen, Neuropsychopharmacology, 2020, 45, 1884–1895 CrossRef CAS PubMed.
- J. Ellis, Multiple Doses of Trodusquemine Improve Glucose Tolerance in Type 2 Diabetic Subjects. 2071-PO (Abs), Vitam. Horm., 69th Scientific Sessions American Diabetes Association, 2009 Search PubMed.
- M. Perni, C. Galvagnion, A. Maltsev, G. Meisl, M. B. D. Müller, P. K. Challa, J. B. Kirkegaard, P. Flagmeier, S. I. A. Cohen, R. Cascella, S. W. Chen, R. Limbocker, P. Sormanni, G. T. Heller, F. A. Aprile, N. Cremades, C. Cecchi, F. Chiti, E. A. A. Nollen, T. P. J. Knowles, M. Vendruscolo, A. Bax, M. Zasloff and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E1009–E1017 CrossRef CAS.
- J. Q. Trojanowski and V. M. Lee, Arch. Neurol., 1998, 55, 151–152 CrossRef CAS.
- A. K. Buell, C. Galvagnion, R. Gaspar, E. Sparr, M. Vendruscolo, T. P. J. Knowles, S. Linse and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7671–7676 CrossRef CAS.
- C. Galvagnion, A. K. Buell, G. Meisl, T. C. T. Michaels, M. Vendruscolo, T. P. J. Knowles and C. M. Dobson, Nat. Chem. Biol., 2015, 11, 229–234 CrossRef CAS.
- M. Perni, P. Flagmeier, R. Limbocker, R. Cascella, F. A. Aprile, C. Galvagnion, G. T. Heller, G. Meisl, S. W. Chen, J. R. Kumita, P. K. Challa, J. B. Kirkegaard, S. I. A. Cohen, B. Mannini, D. Barbut, E. A. A. Nollen, C. Cecchi, N. Cremades, T. P. J. Knowles, F. Chiti, M. Zasloff, M. Vendruscolo and C. M. Dobson, ACS Chem. Biol., 2018, 13, 2308–2319 CrossRef CAS PubMed.
- E. Stolzenberg, D. Berry, D. Yang, E. Y. Lee, A. Kroemer, S. Kaufman, G. C. L. Wong, J. J. Oppenheim, S. Sen, T. Fishbein, A. Bax, B. Harris, D. Barbut and M. A. Zasloff, J. Innate Immun., 2017, 9, 456–463 CrossRef CAS PubMed.
- R. Limbocker, S. Chia, F. S. Ruggeri, M. Perni, R. Cascella, G. T. Heller, G. Meisl, B. Mannini, J. Habchi, T. C. T. Michaels, P. K. Challa, M. Ahn, S. T. Casford, N. Fernando, C. K. Xu, N. D. Kloss, S. I. A. Cohen, J. R. Kumita, C. Cecchi, M. Zasloff, S. Linse, T. P. J. Knowles, F. Chiti, M. Vendruscolo and C. M. Dobson, Nat. Commun., 2019, 10, 225 CrossRef CAS PubMed.
- R. Limbocker, B. Mannini, F. S. Ruggeri, R. Cascella, C. K. Xu, M. Perni, S. Chia, S. W. Chen, J. Habchi, A. Bigi, R. P. Kreiser, A. K. Wright, J. A. Albright, T. Kartanas, J. R. Kumita, N. Cremades, M. Zasloff, C. Cecchi, T. P. J. Knowles, F. Chiti, M. Vendruscolo and C. M. Dobson, Commun. Biol., 2020, 3, 1–10 CrossRef PubMed.
- R. Limbocker, B. Mannini, R. Cataldi, S. Chhangur, A. K. Wright, R. P. Kreiser, J. A. Albright, S. Chia, J. Habchi, P. Sormanni, J. R. Kumita, F. S. Ruggeri, C. M. Dobson, F. Chiti, F. A. Aprile and M. Vendruscolo, Int. J. Mol. Sci., 2020, 21, 4542 CrossRef CAS.
- R. Limbocker, R. Staats, S. Chia, F. S. Ruggeri, B. Mannini, C. K. Xu, M. Perni, R. Cascella, A. Bigi, L. R. Sasser, N. R. Block, A. K. Wright, R. P. Kreiser, E. T. Custy, G. Meisl, S. Errico, J. Habchi, P. Flagmeier, T. Kartanas, J. E. Hollows, L. T. Nguyen, K. LeForte, D. Barbut, J. R. Kumita, C. Cecchi, M. Zasloff, T. P. J. Knowles, C. M. Dobson, F. Chiti and M. Vendruscolo, Front. Neurosci., 2021, 15, 680026 CrossRef PubMed.
- R. P. Kreiser, A. K. Wright, N. R. Block, J. E. Hollows, L. T. Nguyen, K. LeForte, B. Mannini, M. Vendruscolo and R. Limbocker, Int. J. Mol. Sci., 2020, 21, 8651 CrossRef CAS.
- B. Mannini, E. Mulvihill, C. Sgromo, R. Cascella, R. Khodarahmi, M. Ramazzotti, C. M. Dobson, C. Cecchi and F. Chiti, ACS Chem. Biol., 2014, 9, 2309–2317 CrossRef CAS PubMed.
- M. Perni, P. K. Challa, J. B. Kirkegaard, R. Limbocker, M. Koopman, M. C. Hardenberg, P. Sormanni, T. Müller, K. L. Saar, L. W. Y. Roode, J. Habchi, G. Vecchi, N. Fernando, S. Casford, E. A. A. Nollen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, J. Neurosci. Methods, 2018, 306, 57–67 CrossRef.
- M. Perni, A. van der Goot, R. Limbocker, T. J. van Ham, F. A. Aprile, C. K. Xu, P. Flagmeier, K. Thijssen, P. Sormanni, G. Fusco, S. W. Chen, P. K. Challa, J. B. Kirkegaard, R. F. Laine, K. Y. Ma, M. B. D. Müller, T. Sinnige, J. R. Kumita, S. I. A. Cohen, R. Seinstra, G. S. Kaminski Schierle, C. F. Kaminski, D. Barbut, A. De Simone, T. P. J. Knowles, M. Zasloff, E. A. A. Nollen, M. Vendruscolo and C. M. Dobson, Front. Cell Dev. Biol., 2021, 9, 492 Search PubMed.
- S. Errico, G. Lucchesi, D. Odino, S. Muscat, C. Capitini, C. Bugelli, C. Canale, R. Ferrando, G. Grasso, D. Barbut, M. Calamai, A. Danani, M. Zasloff, A. Relini, G. Caminati, M. Vendruscolo and F. Chiti, Nanoscale, 2020, 12, 22596–22614 RSC.
- E. Agmon and B. R. Stockwell, Curr. Opin. Chem. Biol., 2017, 39, 83–89 CrossRef CAS.
- R. H. Unger, G. O. Clark, P. E. Scherer and L. Orci, Biochim. Biophys. Acta, 2010, 1801, 209–214 CrossRef CAS PubMed.
- Z. Balogi, G. Multhoff, T. K. Jensen, E. Lloyd-Evans, T. Yamashima, M. Jäättelä, J. L. Harwood and L. Vígh, Prog. Lipid Res., 2019, 74, 18–30 CrossRef CAS.
- A. D'Anneo, C. C. Bavisotto, A. M. Gammazza, L. Paladino, D. Carlisi, F. Cappello, E. C. de Macario, A. J. L. Macario and M. Lauricella, Cell Stress Chaperones, 2020, 25, 805–820 CrossRef PubMed.
- B. A. Killinger, Z. Madaj, J. W. Sikora, N. Rey, A. J. Haas, Y. Vepa, D. Lindqvist, H. Chen, P. M. Thomas, P. Brundin, L. Brundin and V. Labrie, Sci. Transl. Med., 2018, 10(465), eaar5280 CrossRef PubMed.
- L. L. Edwards, E. M. Quigley and R. F. Pfeiffer, Neurology, 1992, 42, 726–732 CrossRef CAS PubMed.
- H. Braak, R. A. I. de Vos, J. Bohl and K. Del Tredici, Neurosci. Lett., 2006, 396, 67–72 CrossRef CAS PubMed.
- T. Lebouvier, T. Chaumette, P. Damier, E. Coron, Y. Touchefeu, S. Vrignaud, P. Naveilhan, J.-P. Galmiche, S. Bruley des Varannes, P. Derkinderen and M. Neunlist, Gut, 2008, 57, 1741–1743 CrossRef CAS PubMed.
- D. Barbut, E. Stolzenberg and M. Zasloff, J. Parkinson's Dis., 2019, 9, S313–S322 CAS.
- C.-H. Lin, J.-W. Lin, Y.-C. Liu, C.-H. Chang and R.-M. Wu, Parkinsonism Relat. Disord., 2014, 20, 1371–1375 CrossRef PubMed.
- C. Pellegrini, L. Antonioli, R. Colucci, V. Ballabeni, E. Barocelli, N. Bernardini, C. Blandizzi and M. Fornai, Parkinsonism Relat. Disord., 2015, 21, 1407–1414 CrossRef.
- E. L. Beatman, A. Massey, K. D. Shives, K. S. Burrack, M. Chamanian, T. E. Morrison and J. D. Beckham, J. Virol., 2015, 90, 2767–2782 CrossRef.
- J. J. Tomlinson, B. Shutinoski, L. Dong, F. Meng, D. Elleithy, N. A. Lengacher, A. P. Nguyen, G. O. Cron, Q. Jiang, E. D. Roberson, R. L. Nussbaum, N. K. Majbour, O. M. El-Agnaf, S. A. Bennett, D. C. Lagace, J. M. Woulfe, S. Sad, E. G. Brown and M. G. Schlossmacher, J. Neural Transm., 2017, 124, 721–738 CrossRef CAS.
- S. Holmqvist, O. Chutna, L. Bousset, P. Aldrin-Kirk, W. Li, T. Björklund, Z.-Y. Wang, L. Roybon, R. Melki and J.-Y. Li, Acta Neuropathol., 2014, 128, 805–820 CrossRef.
- C. L. West, Y.-K. Mao, T. Delungahawatta, J. Y. Amin, S. Farhin, R. M. McQuade, S. Diwakarla, R. Pustovit, A. M. Stanisz, J. Bienenstock, D. Barbut, M. Zasloff, J. B. Furness and W. A. Kunze, J. Parkinson's Dis., 2020, 10, 1477–1491 CAS.
- R. A. Hauser, D. Sutherland, J. A. Madrid, M. A. Rol, S. Frucht, S. Isaacson, F. Pagan, B. N. Maddux, G. Li, W. Tse, B. L. Walter, R. Kumar, D. Kremens, M. F. Lew, A. Ellenbogen, O. Oguh, A. Vasquez, W. Kinney, M. Lowery, M. Resnick, N. Huff, J. Posner, K. V. Ballman, B. E. Harvey, M. Camilleri, M. Zasloff and D. Barbut, Clin. Park. Relat. Disord., 2019, 1, 2–7 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |