
Out for a RiPP: challenges and advances in genome mining of ribosomal peptides from fungi
Simon C.
Kessler
 and
Yit-Heng
Chooi
*
and
Yit-Heng
Chooi
*
School of Molecular Sciences, The University of Western Australia, Perth, WA 6009, Australia. E-mail: yitheng.chooi@uwa.edu.au
First published on 28th September 2021
Abstract
Covering up to June 2021
Ribosomally synthesized and post-translationally modified peptides (RiPPs) from fungi are an underexplored class of natural products, despite their propensity for diverse bioactivities and unique structural features. Surveys of fungal genomes for biosynthetic gene clusters encoding RiPPs have been limited in their scope due to our incomplete understanding of fungal RiPP biosynthesis. Through recent discoveries, along with earlier research, a clearer picture has been emerging of the biosynthetic principles that underpin fungal RiPP pathways. In this Highlight, we trace the approaches that have been used for discovering currently known fungal RiPPs and show that all of them can be assigned to one of three distinct families based on hallmarks of their biosynthesis, which are in turn imprinted on their corresponding gene clusters. We hope that our systematic exposition of fungal RiPP structural and gene cluster features will facilitate more comprehensive approaches to genome mining efforts in the future.
 Simon C. Kessler | Simon Kessler graduated from the Technical University of Munich with BS and MS degrees in Molecular Biotechnology. Currently, he is a PhD student at the University of Western Australia under the supervision of Dr Yit-Heng Chooi, investigating the biosynthesis of ribosomally synthesized and post-translationally modified peptides in filamentous fungi. |
 Yit-Heng Chooi | Yit-Heng Chooi received his PhD degree in RMIT University in Melbourne, Australia. He moved to the USA for a postdoctoral position at the University of California, Los Angeles with Prof. Yi Tang in 2009 and then to Australian National University for an Australian Research Council (ARC) Discovery Early Career Researcher Award fellowship in 2013. He joined the School of Chemistry & Biochemistry (now Molecular Sciences) at UWA in 2015 and was awarded an ARC Future Fellowship in 2016. His research interests are in genome-based natural product discovery and elucidating secondary metabolites biosynthesis and their ecological roles. |
1 Introduction
Among the major classes of natural products, ribosomally synthesized and post-translationally modified peptides (RiPPs) have enjoyed a steadily increasing share of research attention within the last decades.1,2 However, this general trend has not been extended to fungi, where the first RiPP was discovered in 2007. Since then, only a handful of fungal RiPPs have been characterized, which stands in contrast to the plethora of known plant and bacterial RiPPs, despite the fact that fungi are prolific producers of natural products. Yet even these few compounds exhibit an impressive array of bioactivities, hinting at the buried biosynthetic potential of the numerous RiPP encoding gene clusters in fungal genomes. Due to their peptide backbone sequences being encoded genetically, RiPPs are highly “evolvable” and are thus attractive engineering targets for generating novel bioactive peptides. Furthermore, biosynthetic enzymes unique to RiPP biosynthesis pathways in fungi are expanding the avenues for introducing novel chemical functionalities in peptide compounds.The developments in fungal RiPPs research have been reviewed in 20193 and as part of a comprehensive review covering RiPPs from bacteria, fungi and plants in 2020.2 With the more recent developments, this perspective seeks to classify the currently known fungal RiPPs into three subfamilies based on their biosynthesis and unique structural hallmarks, and provides an overview on the approaches and tools used in previous and more recent fungal RiPP discovery efforts. Finally, we discuss how the uncovered features of fungal RiPPs can be used to facilitate genome mining in the future.
2 Known fungal RiPPs and their hallmarks
The biosynthetic pathway of RiPPs consists of two main constituents, usually encoded in close proximity on the genome as a biosynthetic gene cluster: the precursor peptide and associated biosynthesis proteins. The core peptide, a sequence of amino acids that makes up the structural backbone of the RiPP is embedded within the precursor peptide. In RiPP biosynthesis, the core peptide is excised from the precursor peptide and endowed with posttranslational modifications to yield the mature peptide. Fungi were not known to produce RiPPs before 2007, when the amatoxins and phallotoxins were shown to be produced from a precursor peptide. This initial discovery was followed by the demonstration of the ribosomal origin of the ustiloxins in 2013, epichloëcyclins in 2015, phomopsins and asperipin-2a in 2016, borosins in 2017 and victorin in 2020. The fungal RiPPs that have been discovered to date are either processed by prolyl oligopeptidases or kexin proteases to excise the core peptides from their precursor peptides. Although the amatoxins and phallotoxins, the founding members of the RiPP family of cycloamanides, and the borosins both are processed by a prolyl oligopeptidase, their biosynthetic pathway is sufficiently distinct to form separate fungal RiPP families (see Sections 2.1 and 2.2). The remaining five groups of RiPPs (ustiloxins, epichloëcyclins, phomopsins, asperipin-2a and victorin) are all processed by a kexin protease (along with serine endopeptidases in some cases4,5), as well as sharing additional features that warrants grouping them into a single RiPP family (see Section 2.3). Like other authors,3 we advocate using “dikaritins” as name for this third family of fungal RiPPs, a previously more narrowly defined term that was originally introduced by Ding et al.6 Accordingly, here we classify fungal RiPPs into three distinct families: the cycloamanides, the borosins and the dikaritins. Below, we describe previous efforts to establish and expand these families, what unique hallmarks of each family these efforts have revealed, and how these features facilitate mining fungal genomes for RiPPs.2.1 Cycloamanides (MSDIN family)
The MSDIN family of peptides, named after the conserved N-terminal sequence of amino acids in their precursor peptides, originally included only the amatoxins, phallotoxins and the virotoxins. A uniting biosynthetic feature of this family of RiPPs is the prolyl oligopeptidase B (POPB), which excises the core peptide and catalyses its head-to-tail macrocyclisation in two steps.7 A small group of related cyclic peptides discovered in the 1960s, cycloamanides A–D,8 as well as antamanide9 and the newly discovered cycloamanides E and F,10 have recently found to be POPB-processed in the same manner as MSDIN peptides and were added to this RiPP family (Fig. 1).10 Subsequently, the name “cycloamanides” has been proposed to supersede “MSDIN peptides” as the new name for this family of fungal RiPPs, in part due to the discovery that the MSDIN sequence is not conserved in all members.11,12 In this perspective we use the term cycloamanides for the RiPP family, with the names cycloamanide A–F referring to six specific family members.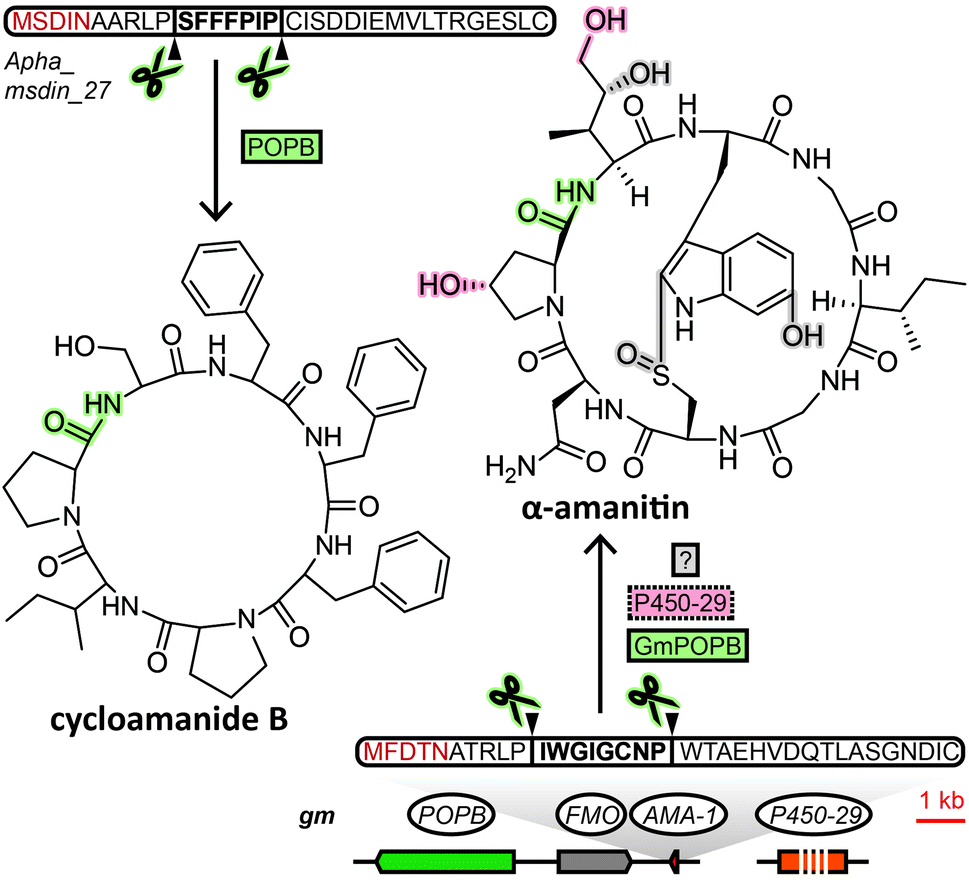 | ||
| Fig. 1 Biosynthesis of two representative cycloamanides. Gene or protein names in solid line boxes/circles indicate a confirmed role in biosynthesis, dotted line boxes/circles indicate a proposed role in biosynthesis. Proteins and the corresponding structural features/bonds they are responsible for are shaded with matching colour. Gm: Galerina marginata, Apha: Amanita phalloides, POPB: prolyl oligopeptidase, FMO: flavin-containing monooxygenase, AMA-1: precursor peptide, P450-29: P450 monooxygenase. | ||
The most notorious members of the cycloamanides are the amatoxins, the poisonous agent responsible for the deadliness of some mushrooms of the genus Amanita that inhibit RNA polymerase II in the liver.13Amanita mushrooms that produce amatoxins also produce the structurally closely related phallotoxins,14 which exhibit toxicity by stabilizing actin,15 but – unlike amatoxins – are poorly absorbed orally.11 While their structures had been known for decades, they were previously assumed to be non-ribosomal peptides, like all other fungal cyclic peptides characterized prior to the discovery of fungal RiPPs. Yet when the genome of a well-known producer of amatoxins and phallotoxins, Amanita bisporigera, was sequenced in 2007, no nonribosomal peptide synthetase (NRPS) genes were found.16 Consequently, the genome was queried with amino acid sequences corresponding to the unmodified amino acid backbone of the peptides. This query returned the precursor peptide genes AMA1 and PHA1 for the amatoxin α-amanitin and the phallotoxin phallacidin, respectively. Using the conserved N- and C-terminal amino acid sequence encoded in those two genes, 13 additional genes were found in the genome of A. bisporigera. Degenerate PCR primers targeting that same conserved region were used to detect four more cycloamanide genes in other Amanita species, while DNA hybridisation experiments linked the presence of cycloamanide genes to toxin production in different Amanita species.16 Together, these were the founding members of the cycloamanides.
Following the initial discovery of the cycloamanide genes in A. bisporigera, subsequent studies have mostly used their conserved features to mine for additional members, either by homology query of sequenced genomes or with degenerate primers for unsequenced strains.
Using homology-based searches Luo et al. identified AMA1 homologs GmAMA1-1/2 (Fig. 1) in Galerina marginata.17 More examples of genome mining for cycloamanide genes with this approach are the discoveries of nine cycloamanide genes in Amanita exitialis,18 18 and 29 in A. subjunquillea and A. pallidorosea, respectively19 and lastly, 110 cycloamanide genes in genomes of seven Amanita and Lepiota species.20 Instances of obtaining cycloamanide-encoding genes by PCR using degenerate primers include the discovery of 24 cycloamanide genes in six species of Amanita21 and a total of 70 in different Amanita, Galerina and Lepiota species.20
In one study,10 after about 30 unique cycloamanide genes each in A. bisporigera and A. phalloides genomes were identified through homology search, tandem mass spectrometry was used to confirm the presence of the corresponding compounds. In A. phalloides extracts several known cycloamanides were detected, as well as novel cycloamanides E and F,10 related to the immunosuppressive22 peptides cycloamanides A–D that were structurally elucidated in the 1970s8 and are now part of the cycloamanide family of RiPPs.
In an effort to adapt genome mining tools developed for bacterial RiPPs to mine fungal genomes, Vignolle et al. discovered numerous putative cycloamanide-like precursor peptide genes in several species of Trichoderma,26 a surprising result if confirmed, since cycloamanide production had previously only been found in species from genera Amanita, Galerina and Lepiota (with the exception of a single species from genus Conocybe23).
Overall, genome mining for cycloamanides has relied mostly on characteristics that were described upon the discovery of their biosynthetic origin in 2007, although the name-giving MSDIN motif turned out not to be perfectly conserved even within Amanita, with more variations in other genera.10,19,20 Nevertheless, the degree of conservation of the precursor peptide is sufficient for homology-based searches in sequenced genomes or transcriptomes and they have prevailed as the most reliable approach for detecting cycloamanide genes, while genome annotation pipelines10 and degenerate PCR methods25 have been shown to miss cycloamanide genes.
Regarding cycloamanide-associated enzymes, the prolyl oligopeptidase POPB performs core peptide excision and head-to-tail cyclization for cycloamanide peptides. The POPB gene appears to be exclusive to genomes of cycloamanide-producing species,19 yet POPB and cycloamanide precursor peptide genes co-localize within the genome only in some cases.10,24,27,28 Therefore, the POPB gene can only be used as a general predictor of the presence of cycloamanide genes in the genome. Different members of the peptide family show varying degrees of post-translational modifications beyond macrocyclization,10 installed by as-of-yet unidentified enzymes, except for a predicted flavin-containing monooxygenase and a cytochrome P450 in G. marginata.11,12 Since POPB appears to be the only processing enzyme universal to all cycloamanide biosynthesis pathways, genome mining for cycloamanide genes cannot rely on gene clustering of signature decorating enzymes with the precursor peptide like in other RiPP families.2
2.2 Borosins (omphalotins)
Borosins are a family of head-to-tail cyclized, heavily backbone N-methylated peptides with strong nematicidal activity,29,30 known as omphalotins prior to the discovery of their ribosomal origin. Their structures were elucidated in 199729 and the genome of their producer, the mushroom Omphalotus olearius, was sequenced in 2012.31 With their ribosomal origin obfuscated by their structural similarity to non-ribosomal peptide cyclosporine and the fact that backbone N-methylation had only been observed in non-ribosomal peptides, it was not until 2017 that two groups independently discovered omphalotins to be RiPPs.32,33 Both groups found the precursor peptide gene ophA/ophMA by using the omphalotin linearized amino acid backbone sequence to query the O. olearius genome. Borosin precursor peptides were found to be fused to the methyltransferase responsible for their signature backbone N-methylations. Methylation has been shown to be carried out in trans between two precursor peptides interlocking as a homodimer.34,35 Currently, catalytic activity is still unique to the precursor peptides of the borosin family among all fungal RiPPs. Clustered with ophA/ophMA was prolyl oligopeptidase gene ophP, which, expressed heterologously alongside ophA/ophMA, yielded omphalotin A, the least post-translationally decorated member of the borosin family.33 Additional genes located next to ophA/ophMA are likely involved in the biosynthesis of other omphalotins (Fig. 2). This is supported by the finding of a homologous gene cluster in Dendrothele bispora, which contains a ophA/ophMA homologue as well as homologues to six predicted genes in immediate genomic vicinity of ophA/ophMA.32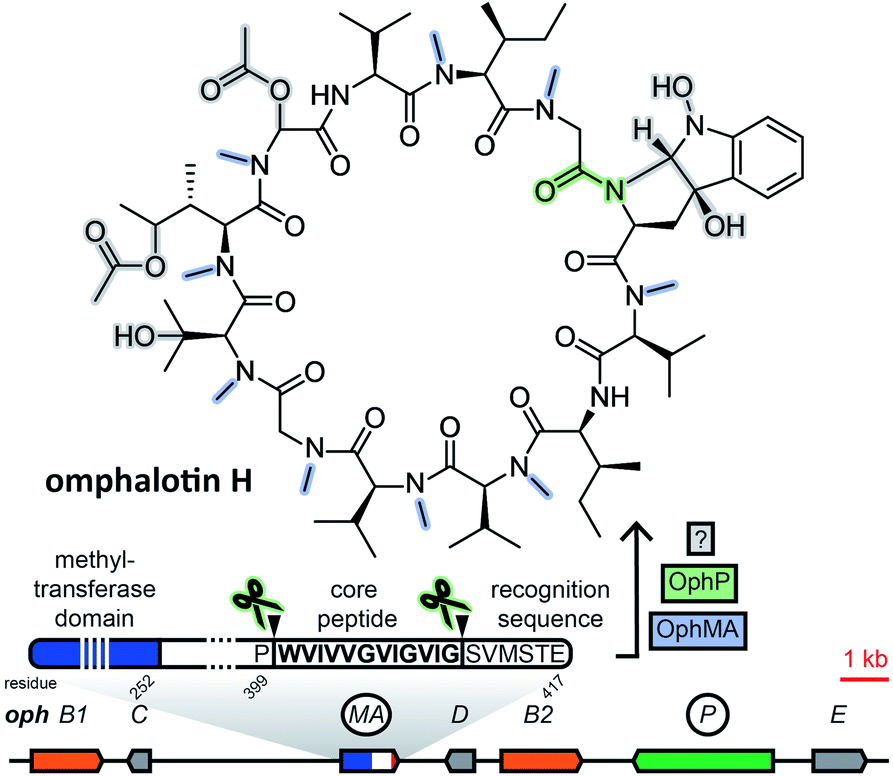 | ||
| Fig. 2 Biosynthesis of a representative borosin. Gene or protein names in solid line boxes/circles indicate a confirmed role in biosynthesis. Proteins and the corresponding structural features/bonds they are responsible for are shaded with matching colour. Oph: Omphalotus olearius; B1, B2: P450 monooxygenase; C: NTF2-like; MA: methyltransferase fused to precursor peptide; D: acyltransferase; P: prolyl oligopeptidase; E: F-box/RNI-like. | ||
The borosin-associated methyltransferases have been shown to form a distinct group within their protein family.35 This feature was exploited by Quijano et al. to mine available fungal genomes for more omphalotin precursor peptide genes and their corresponding gene clusters by using the methyltransferase domain of ophMA as query.36 After checking hits against available transcriptome data and manual curation, the authors ended up with 42 and 12 ophMA homologues from basidiomycetes and ascomycetes, respectively. Analysis of genes clustered with these homologues revealed very little conservation of predicted gene functions.
As demonstrated by Quijano et al.,36 mining for borosin precursor peptide genes is facilitated by their N-terminal fusion to a highly conserved, borosin-specific tetrapyrrole methylase domain.35
2.3 Dikaritins
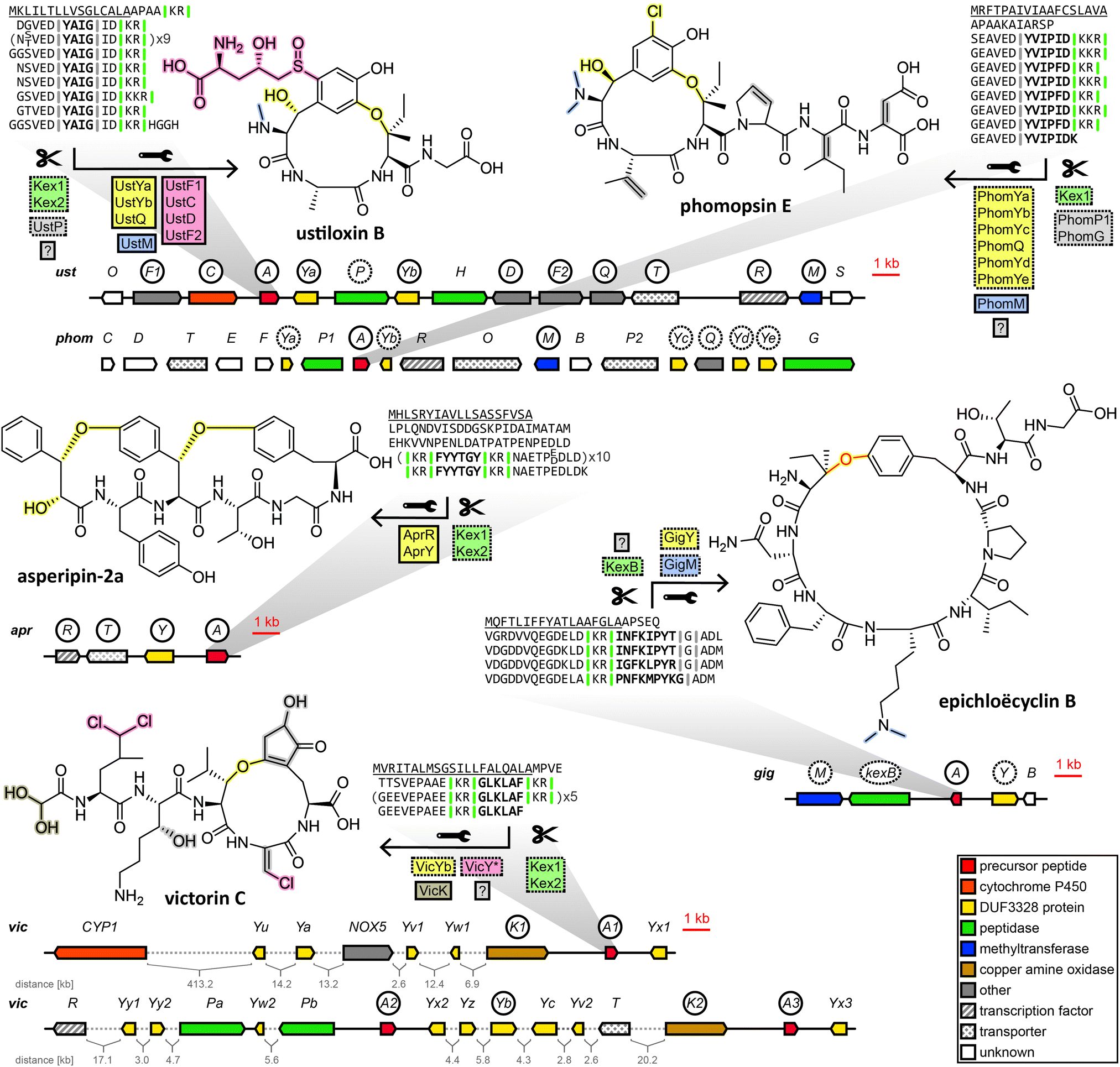 | ||
| Fig. 3 Biosynthesis of representative dikaritins. In precursor peptide sequences, the signal peptide is underlined, the core peptide is highlighted in bold and cutsites are indicated as vertical lines. Gene or protein names in solid line boxes/circles indicate a confirmed role in biosynthesis, dotted line boxes/circles indicate a proposed role in biosynthesis. Posttranslational modification proteins are listed under/over the wrench symbol and corresponding features in the structure are highlighted with matching colours. Peptidases (green boxes) are listed under/over the scissors symbol and match corresponding cutsites (vertical lines in precursor peptide sequences) of the same color. Genes are colored according to predicted functions listed in the bottom right corner. The phomopsin gene cluster is not publicly available, the figure is based on published coding sequences. The epichloëcyclin ether bond (red) is not verified experimentally. In victorin biosynthesis, VicY* refers to an unknown combination of DUF3328 proteins encoded in the gene cluster. | ||
Subsequently, the ustiloxin gene cluster was confirmed by gene disruption experiments39 and heterologous pathway reconstruction.40 Importantly, it was shown that the expression of two DUF3328 protein genes ustYa/Yb along with a tyrosinase and the precursor peptide gene from A. flavus in A. oryzae was necessary and sufficient to produce an ustiloxin intermediate devoid of post-translational modifications except for a single hydroxylation and the macrocycle formed via ether bond between the first and the third amino acid of the core peptide backbone.40
Drawing on two distinguishing features of the ustiloxins gene cluster, the prevalence of ustY-homologues and the presence of multiple repetitions of the core peptide within the precursor peptide, Nagano et al. developed a pipeline to mine genomes of Aspergillus species for ustiloxin-like gene clusters.41ustY-homologues were detected by BLAST search, and hits discarded if they did not feature the conserved double HXXHC-motif suspected to be part of the DUF3328 active site. To mine for ustiloxin-like precursor peptide genes, candidates were assessed based on three criteria: the presence of a signal peptide detected by SignalP4.1, the presence of a proposed Kex2 endopeptidase recognition site consisting of two consecutive basic amino acids, and the presence of repeats of at least six amino acids to account for conserved leader or follower peptide sequences joined to the core peptide. Candidates passing a threshold score based on these criteria were then checked to be within 10 kb of an ustY-homologue. This way, an additional 94 ustiloxin-like precursor peptide genes were detected. One of them, identified in the genome of A. flavus, was linked to its corresponding RiPP compound by gene deletion and named asperipin-2a.41
In a 2016 study Ding et al. revealed phomopsins to be fungal RiPPs. Phomopsins are a group of compounds closely related to ustiloxins in regards to bioactivity and structure, but with a backbone of six amino acids instead of four and featuring a chlorination on an activated carbon.42 After Ding et al. sequenced the genome of their producer Phomopsis leptostromiformis and were unable to identify an NRPS genes suitable to produce phomopsins in the genome, a query with the unmodified phomopsin backbone amino acid sequence returned the phomopsin precursor peptide gene phomA. phomA and two of the genes clustered with it, methyltransferase phomM and tyrosinase phomQ were shown to be expressed together and deletion of phomQ resulted in the loss of production of a compound that was subsequently isolated and identified as a phomopsin. Ding et al. then went on to mine fungal genomes for phomopsin-like gene clusters by homology to phomA, phomQ or phomM. Of the gene clusters that were identified that way, only those containing a PhomQ-like tyrosinase, multiple DUF3328-containing proteins and a methyltransferase, among others, were considered phomopsin-like. The authors named the products of these gene clusters “dikaritins” due to their prevalence in the fungal subkingdom of Dikarya.6
We endorse these additions to the same fungal RiPP family as the ustiloxins and phomopsins, and further include victorin into the same family, for which we have also used the name dikaritins in this perspective for continuity and clarity reasons. Although the dikaritins are united by a set of common features, recent discoveries have demonstrated that defining the boundaries of this RiPP family is not straightforward. One hallmark of dikaritins is the architecture of their precursor peptides. All dikaritin precursor peptides possess an N-terminal signal sequence, followed by multiple perfect or imperfect repeats that each contain a core peptide, sequence stretches of unknown function and – since dikaritins are kexin protease-processed in contrast to the prolyl oligopeptidase-processed borosins and cycloamanides – at least one kexin protease recognition site. This recognition site is a motif of two basic residues, either KK, KR or RR,50 at one or both ends of the core peptide. The fact the kexin cut site is not always immediately adjacent to the core peptide at both ends indicates the involvement of additional proteases in the maturation process of some dikaritins.
However, recently it has been shown that dikaritin precursor peptides share their architecture not only between themselves but also with a much larger group of peptides called Kex2-processed repeat proteins (KEPs), most of which are not RiPPs.51,52 Consequently, genome mining for dikaritins based solely on the characteristics of the precursor peptide will yield both dikaritins and KEPs,52 which leads to the question of what biosynthetic features separate the two. A recent survey of 1461 fungal strains for KEPs suggested a potential evolutionary relationship between larger linear KEP-derived peptides with various biological functions (including yeast α-mating factor) and KEP-derived RiPPs.52 It seems plausible that dikaritins could have evolved from repurposing KEPs by recruiting additional posttranslational modification enzymes, increasing structural complexity and obtaining secondary metabolite functions. Therefore, genome mining for dikaritins can be facilitated by incorporating both the precursor peptide features and characteristic posttranslational modification enzymes. One reliable structural hallmark of dikaritins among fungal small cyclic peptides has been the ether bond-containing macrocycle.53 Much evidence points to the DUF3328 protein to catalyse its formation. DUF3328 protein genes have been found in close proximity to over 20% of KEP genes in the most recent surveying of fungal genomes for KEPs,52 and they are found in the gene clusters of ustiloxins, phomopsins, asperipin-2a, epichloëcyclins and victorin. However, many unanswered questions remain. For one, DUF3328 protein genes are not exclusive to dikaritin gene clusters. They have been found in a borosin-type gene cluster,36 in the gene clusters of non-ribosomal peptides like cyclochlorotines54,55 and astins,56 in the gene cluster of the atpenin A4-producing polyketide synthase–nonribosomal peptide synthetase57 or in rare cases, clustered with genes encoding putative fungal KEP-derived pheromones.52 Furthermore, their biosynthetic function remains enigmatic. While DUF3328 proteins have been established to be involved in the formation of the ether bond in ustiloxins,40 their role in other RiPP pathways remains elusive. In the heterologous reconstruction of the asperipin-2a pathway, no step of the biosynthetic pathway was conclusively linked to any one protein, as expressing different incomplete sets of genes from the gene cluster still resulted in the detection of trace amounts of the finished compound, and no pathway intermediates were shown.58
In a very recent breakthrough, the individual biosynthetic functions of three DUF3328 proteins encoded in the gene cluster of the non-ribosomal peptide cyclochlorotine have been elucidated.55 While one DUF3328 protein catalyses an unusual intramolecular transacylation that is unlikely to be prevalent in dikaritins, the other two catalyse a hydroxylation and a chlorination on an unactivated carbon, respectively, both reactions highly relevant to dikaritin biosynthesis. Ustiloxins, phomopsins, asperipin-2a and victorin are all hydroxylated, and, more importantly, both victorin and phomopsin are chlorinated, both lacking any predicted halogenase in their gene clusters. Instead the phomopsin gene cluster features five DUF3328 genes, three more than the gene cluster of ustiloxin, which is structurally very similar to phomopsin but is not chlorinated. Victorin is chlorinated on unactivated carbons multiple times, without any genes previously known to catalyse such a reaction encoded in the genome of its producer, but with numerous DUF3328 proteins encoded in the genomic vicinity of the victorin precursor peptide genes.46 Victorin, ustiloxin and phomopsin also feature β-hydroxy amino acids, in which the enzymes responsible for their formation are yet to be unambiguously determined, but evidence points towards DUF3328 proteins as well. This indicates that these DUF3328 proteins fulfil a diverse set of biosynthetic functions, which is in line with the number of DUF3328 genes in the dikaritin gene clusters that often exceeds the number presumably needed for ether bond formation in dikaritins. However, a similarity network analysis of DUF3328 protein sequences (not including victorin-associated DUF3328 protein sequences) by Jiang et al.55 did not show any clustering of dikaritin-associated DUF3328 proteins with the newly elucidated halogenating DUF3328 proteins, suggesting DUF3328 protein sequences are highly variable and functional prediction remains difficult at this stage.
Lastly, we have noted that NCBI CD search appears to be unreliable at predicting DUF3328 domains from query sequences,46 possibly obscuring the prevalence of these genes in previous genome surveys. Instead, checking for the presence of two “HXXHC” motifs41 appears to be a reliable approach to detecting DUF3328 proteins.
Overall, the biosynthetic functions of DUF3328 proteins appear to be highly diverse yet are still poorly understood. In their 2020 review, Montalbán-López et al. proposed the DUF3328 protein as the class-defining enzyme of dikaritins,2 but until those that catalyse ether bond formation can be bioinformatically distinguished from DUF3328 proteins with other functions, they remain an unreliable predictor of gene clusters encoding ether bridge-cyclized dikaritins. Yet for the purposes of genome mining a very strict classification is probably not necessary. Overall, KEP-derived peptides, cyclic or not, if endowed with post-translational modifications such as those afforded by DUF3328 proteins, appear to be an attractive target in the search for small, bioactive peptides. Genome mining for these peptides can simply rely on the architecture of the precursor peptide gene, with the use of associated posttranslational modification genes as a rough indicator of the chemical complexity of the mature peptide.
3 Tools and strategies for mining of fungal RiPPs
Genome mining strategies for RiPPs in general have been covered in previous reviews.59,60 Here, we will focus on fungal RiPPs specifically. Discovery of the founding members (except for ustiloxins) of the fungal RiPP families has so far been confined to the identification of their corresponding biosynthetic gene cluster working backwards from the chemical structure in a top-down approach. Subsequent bottom-up genome mining efforts to expand these families of RiPPs were mostly homology-based. For cycloamanides, homology to the highly conserved leader sequence of known precursor peptides has been shown to be a reliable indicator, whereas for borosins homology to methyltransferase OphM has been successfully used to discover new family members. Genome mining for dikaritin gene clusters can utilize the occurrence of repeated stretches of amino acids containing kexin cleavage sites in the precursor peptide, in combination with a predicted DUF3328 protein encoded in close genomic proximity. Our lab has recently implemented a similar approach in a dikaritin gene search tool named fRIPPA, accessible online.† Beyond these approaches, few resources are available that facilitate the discovery of novel fungal RiPPs.The discovery of the ustiloxin gene cluster with MIDDAS-M38 remains the only example of the discovery of a novel RiPP family through bottom-up genome mining. However, MIDDAS-M is not a RiPP-specific discovery pipeline, rather it identifies a gene cluster merely based on co-regulation of its genes. Therefore, MIDDAS-M is independent of the actual gene sequences and the class of natural product encoded in the gene cluster, known or unknown. However, to be detected by the tool, a gene cluster needs to exhibit markedly different expression levels between two conditions, captured by transcriptomics data. Additionally, once a gene cluster is identified, further bioinformatic or manual curation is needed to determine its biosynthetic nature. Despite these caveats motif-independent approaches in general have great potential to discover new families of fungal RiPPs, as well as other novel secondary metabolite gene clusters.
Vignolle et al. have developed a pipeline which adapted the bacterial RiPP mining tool RiPPMiner for fungal RiPP discovery26 and relies on extensive exclusion steps to reduce a large pool of candidate genes to a manageable list of genes potentially encoding RiPP precursor peptides. The authors used the pipeline to identify potential cycloamanide-encoding genes in Trichoderma genomes, albeit none were verified experimentally to encode RiPPs.
AntiSMASH is a popular secondary metabolite gene cluster prediction tool for bacterial and fungal genomes that supports the identification of fungal RiPP gene clusters, but is very narrowly focused on a highly specific set of rules. Classification of a gene cluster is based on the predicted functions of its genes and the recognition of a fungal RiPP cluster is triggered by the presence of a peptidase combined with either a tetrapyrrole methylase gene, a cytochrome P450 gene or a DUF3328 protein gene.‡ Requiring a peptidase to be encoded in the fungal RiPP gene cluster limits the ability of antiSMASH to detect them, since in many cases the peptidase processing the RiPP also fulfils a more general role in the metabolism of the host and is therefore encoded elsewhere in the genome. This has been shown for borosins, where a survey of borosin gene clusters has found that some do not feature any peptidase gene.36 Likewise, dikaritin gene clusters do not always contain a peptidase gene, as is the case for the asperipin-2a gene cluster58 and the ustiloxin gene cluster in Ustilaginoidea virens.61 Not much is known about the enzymes responsible for the post-translational modifications of cycloamanides and their corresponding genes, but at least in the case of G. marginata the precursor peptide gene is located in the genomic vicinity of a cytochrome P450 gene and a prolyl oligopeptidase gene,11,12 which matches one detection criterion for fungal RiPPs of the antiSMASH algorithm. However, it is not known if this gene pairing is a common feature of cycloamanide biosynthesis.
Overall, current bioinformatic tools lack both the specificity and sensitivity to mine for anything but narrowly defined groups of fungal RiPP gene clusters instead of the full repertoire of RiPP compounds encoded in fungal genomes. A fundamental dilemma for the bioinformatics tools and pipelines used for mining fungal RiPP, such as antiSMASH and fRIPPA, is that to a degree their recognition is based on prior knowledge of structure and function of RiPP genes and gene clusters, and is thus less likely to recognize unrelated and potentially novel RiPPs. However, tools that are more agnostic to biosynthetic classes, such as MIDDAS-M38 or the clusterfinder tool62 embedded in antiSMASH, are not specific to RiPPs and can have high rates of false positives (e.g. detecting gene clusters not responsible for secondary metabolite biosynthesis). This highlights the complimentary nature of traditional natural product isolation and the importance of continued linking of fungal peptide compounds to gene clusters, as exemplified by the discovery of the genetic basis for amanitin, phomopsin, omphalotin and victorin biosynthesis. Establishing relationships between new classes of RiPPs and their gene clusters will propel genome mining of a wider range of RiPPs.
4 Conclusions
Small cyclic peptides generally display desirable properties as pharmaceutical drug leads, and the limited number of discovered fungal RiPPs exhibit diverse bioactivities, ranging from RNA polymerase inhibition (amanitin), inhibition of actin depolymerisation (phalloidin), immunosuppression (cycloamanides A–D), nematicidal activity (borosins), modulation of microtubular function (phomopsins and ustiloxins) to triggering plant cell death in susceptible host via binding to thioredoxin (victorin). Additionally, fungal RiPP biosynthesis pathways feature enzymatic transformations that are very rare or even unique among RiPPs – like chlorination of unactivated carbon centres, sulfinylation, oxidative deamination or peptide backbone N-methylation – and therefore may offer access to exotic chemical structures not obtainable otherwise. The inherent malleability of RiPP biosynthesis makes these enzymes promising tools for rational peptide engineering efforts, though this is still in its infancy for fungal RiPPs. This also highlights the importance of continuing the biosynthetic investigation of RiPP pathways.Genome surveys have hinted at the ubiquitousness of RiPP gene clusters in fungal genomes that has barely been tapped into. Future bioinformatic tools should incorporate the hallmarks of fungal RiPPs outlined in this paper to allow for more comprehensive detection of RiPP gene clusters in fungal genomes. Continuing advances in genome sequencing, improved bioinformatic gene cluster predictions based on a better understanding of fungal RiPP biosynthesis and approaches to connect gene clusters to compounds in a high-throughput manner should make this biosynthetic wealth more accessible than ever.
5 Conflicts of interest
There are no conflicts to declare.6 Acknowledgements
This work was supported by Australian Research Council (ARC) Discovery Projects (DP200101880) and Cooperative Research Centres Project (CRCPFIVE00119). YHC was supported by an ARC Future Fellowship (FT160100233). SCK was supported by a University of Western Australia scholarship.7 Notes and references
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martinez, S. K. Nair, Y. Nicolet, S. Rebuffat, H. G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J. K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2020, 38, 130–239 RSC.
- E. Vogt and M. Künzler, Appl. Microbiol. Biotechnol., 2019, 103, 5567–5581 CrossRef CAS.
- N. Nagano, M. Umemura, M. Izumikawa, J. Kawano, T. Ishii, M. Kikuchi, K. Tomii, T. Kumagai, A. Yoshimi, M. Machida, K. Abe, K. Shin-Ya and K. Asai, Fungal Genet. Biol., 2016, 86, 58–70 CrossRef CAS PubMed.
- M. Umemura, N. Nagano, H. Koike, J. Kawano, T. Ishii, Y. Miyamura, M. Kikuchi, K. Tamano, J. Yu, K. Shin-ya and M. Machida, Fungal Genet. Biol., 2014, 68, 23–30 CrossRef CAS PubMed.
- W. Ding, W. Q. Liu, Y. Jia, Y. Li, W. A. van der Donk and Q. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3521–3526 CrossRef CAS PubMed.
- R. M. Sgambelluri, M. O. Smith and J. D. Walton, ACS Synth. Biol., 2018, 7, 145–152 CrossRef CAS.
- A. Gauhe and T. Wieland, Liebigs Ann. Chem., 1977, 1977, 859–868 CrossRef.
- T. Wieland, Angew. Chem., Int. Ed. Engl., 1968, 7, 204–208 CrossRef CAS.
- J. A. Pulman, K. L. Childs, R. M. Sgambelluri and J. D. Walton, BMC Genomics, 2016, 17, 1038 CrossRef PubMed.
- J. Walton, The Cyclic Peptide Toxins of Amanita and Other Poisonous Mushrooms, Springer, 2018 Search PubMed.
- R. M. Sgambelluri, PhD Thesis, Michigan State University, 2017.
- D. A. Bushnell, P. Cramer and R. D. Kornberg, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1218–1222 CrossRef CAS.
- J. D. Walton, H. E. Hallen-Adams and H. Luo, Biopolymers, 2010, 94, 659–664 CrossRef CAS.
- J. E. Estes, L. A. Selden and L. C. Gershman, Biochemistry, 1981, 20, 708–712 CrossRef CAS.
- H. E. Hallen, H. Luo, J. S. Scott-Craig and J. D. Walton, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19097–19101 CrossRef CAS.
- H. Luo, H. E. Hallen-Adams, J. S. Scott-Craig and J. D. Walton, Fungal Genet. Biol., 2012, 49, 123–129 CrossRef CAS.
- P. Li, W. Q. Deng, T. H. Li, B. Song and Y. H. Shen, Gene, 2013, 532, 63–71 CrossRef CAS.
- H. Luo, Q. Cai, Y. Luli, X. Li, R. Sinha, H. E. Hallen-Adams and Z. L. Yang, IMA Fungus, 2018, 9, 225–242 CrossRef.
- Z. He, P. Long, F. Fang, S. Li, P. Zhang and Z. Chen, BMC Genomics, 2020, 21, 440 CrossRef CAS PubMed.
- P. Li, W. Deng and T. Li, Toxicon, 2014, 83, 59–68 CrossRef CAS.
- Z. Wieczorek, I. Z. Siemion, M. Zimecki, E. Bolewska-Pedyczak and T. Wieland, Peptides, 1993, 14, 1–5 CrossRef CAS.
- J. H. Diaz, Wilderness Environ. Med., 2018, 29, 111–118 CrossRef.
- Y. Luli, Q. Cai, Z. H. Chen, H. Sun, X. T. Zhu, X. Li, Z. L. Yang and H. Luo, BMC Genomics, 2019, 20, 198 CrossRef PubMed.
- Y. He, C. H. Zhang, W. Q. Deng, X. Y. Zhou, T. H. Li and C. H. Li, Toxicon, 2020, 183, 61–68 CrossRef CAS.
- G. A. Vignolle, R. L. Mach, A. R. Mach-Aigner and C. Derntl, BMC Genomics, 2020, 21, 258 CrossRef CAS.
- H. Luo, S. Y. Hong, R. M. Sgambelluri, E. Angelos, X. Li and J. D. Walton, Chem. Biol., 2014, 21, 1610–1617 CrossRef CAS.
- H. Luo, H. E. Hallen-Adams, J. S. Scott-Craig and J. D. Walton, Eukaryotic Cell, 2010, 9, 1891–1900 CrossRef CAS.
- O. Sterner, W. Etzel, A. Mayer and H. Anke, Nat. Prod. Lett., 1997, 10, 33–38 CrossRef CAS.
- J. C. Liermann, T. Opatz, H. Kolshorn, L. Antelo, C. Hof and H. Anke, Eur. J. Org. Chem., 2009, 2009, 1256–1262 CrossRef.
- G. T. Wawrzyn, M. B. Quin, S. Choudhary, F. Lopez-Gallego and C. Schmidt-Dannert, Chem. Biol., 2012, 19, 772–783 CrossRef CAS.
- N. S. van der Velden, N. Kalin, M. J. Helf, J. Piel, M. F. Freeman and M. Künzler, Nat. Chem. Biol., 2017, 13, 833–835 CrossRef CAS.
- S. Ramm, B. Krawczyk, A. Mühlenweg, A. Poch, E. Mösker and R. D. Süssmuth, Angew. Chem., Int. Ed. Engl., 2017, 56, 9994–9997 CrossRef CAS PubMed.
- H. Song, N. S. van der Velden, S. L. Shiran, P. Bleiziffer, C. Zach, R. Sieber, A. S. Imani, F. Krausbeck, M. Aebi, M. F. Freeman, S. Riniker, M. Künzler and J. H. Naismith, Sci. Adv., 2018, 4, eaat2720 CrossRef CAS.
- C. Ongpipattanakul and S. K. Nair, ACS Chem. Biol., 2018, 13, 2989–2999 CrossRef CAS PubMed.
- M. R. Quijano, C. Zach, F. S. Miller, A. R. Lee, A. S. Imani, M. Künzler and M. F. Freeman, J. Am. Chem. Soc., 2019, 141, 9637–9644 CrossRef CAS.
- Y. Koiso, Y. Li, S. Iwasaki, K. Hanaoka, T. Kobayashi, R. Sonoda, Y. Fujita, H. Yaegashi and Z. Sato, J. Antibiot., 1994, 47, 765–773 CrossRef CAS.
- M. Umemura, H. Koike, N. Nagano, T. Ishii, J. Kawano, N. Yamane, I. Kozone, K. Horimoto, K. Shin-ya, K. Asai, J. Yu, J. W. Bennett and M. Machida, PLoS One, 2013, 8, e84028 CrossRef.
- M. Umemura, N. Nagano, H. Koike, J. Kawano, T. Ishii, Y. Miyamura, M. Kikuchi, K. Tamano, J. Yu, K. Shin-ya and M. Machida, Fungal Genet. Biol., 2014, 68, 23–30 CrossRef CAS PubMed.
- Y. Ye, A. Minami, Y. Igarashi, M. Izumikawa, M. Umemura, N. Nagano, M. Machida, T. Kawahara, K. Shin-Ya, K. Gomi and H. Oikawa, Angew. Chem., Int. Ed. Engl., 2016, 55, 8072–8075 CrossRef CAS PubMed.
- N. Nagano, M. Umemura, M. Izumikawa, J. Kawano, T. Ishii, M. Kikuchi, K. Tomii, T. Kumagai, A. Yoshimi, M. Machida, K. Abe, K. Shin-Ya and K. Asai, Fungal Genet. Biol., 2016, 86, 58–70 CrossRef CAS.
- C. C. J. Culvenor, J. A. Edgar, M. F. Mackay, C. P. Gorst-Allman, W. F. O. Marasas, P. S. Steyn, R. Vleggaar and P. L. Wessels, Tetrahedron, 1989, 45, 2351–2372 CrossRef CAS.
- R. D. Johnson, G. A. Lane, A. Koulman, M. Cao, K. Fraser, D. J. Fleetwood, C. R. Voisey, J. M. Dyer, J. Pratt, M. Christensen, W. R. Simpson, G. T. Bryan and L. J. Johnson, Fungal Genet. Biol., 2015, 85, 14–24 CrossRef CAS PubMed.
- C. J. Eaton, M. P. Cox, B. Ambrose, M. Becker, U. Hesse, C. L. Schardl and B. Scott, Plant Physiol., 2010, 153, 1780–1794 CrossRef CAS PubMed.
- K. A. Green, D. Berry, K. Feussner, C. J. Eaton, A. Ram, C. H. Mesarich, P. Solomon, I. Feussner and B. Scott, New Phytol., 2020, 227, 559–571 CrossRef CAS PubMed.
- S. C. Kessler, X. Zhang, M. C. McDonald, C. L. M. Gilchrist, Z. Lin, A. Rightmyer, P. S. Solomon, B. G. Turgeon and Y. H. Chooi, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 24243–24250 CrossRef CAS PubMed.
- J. Lorang, T. Kidarsa, C. S. Bradford, B. Gilbert, M. Curtis, S. C. Tzeng, C. S. Maier and T. J. Wolpert, Science, 2012, 338, 659–662 CrossRef CAS PubMed.
- J. M. Zhang, X. M. Li, X. Han, R. J. Liu and J. G. Fang, Trends Pharmacol. Sci., 2017, 38, 794–808 CrossRef CAS.
- S. Luo and S. H. Dong, Molecules, 2019, 24, 1541 CrossRef CAS.
- N. C. Rockwell, D. J. Krysan, T. Komiyama and R. S. Fuller, Chem. Rev., 2002, 102, 4525–4548 CrossRef CAS PubMed.
- M. Le Marquer, H. San Clemente, C. Roux, B. Savelli and N. Frei Dit Frey, BMC Genomics, 2019, 20, 64 CrossRef PubMed.
- M. Umemura, Fungal Biol. Biotechnol., 2020, 7, 11 CrossRef.
- X. Wang, M. Lin, D. Xu, D. Lai and L. Zhou, Molecules, 2017, 22, 2069 CrossRef.
- T. Schafhauser, N. Kirchner, A. Kulik, M. M. Huijbers, L. Flor, T. Caradec, D. P. Fewer, H. Gross, P. Jacques, L. Jahn, J. Jokela, V. Leclere, J. Ludwig-Muller, K. Sivonen, W. J. van Berkel, T. Weber, W. Wohlleben and K. H. van Pee, Environ. Microbiol., 2016, 18, 3728–3741 CrossRef CAS PubMed.
- Y. Jiang, T. Ozaki, C. Liu, Y. Igarashi, Y. Ye, S. Tang, T. Ye, J. I. Maruyama, A. Minami and H. Oikawa, Org. Lett., 2021, 23, 2616–2620 CrossRef.
- T. Schafhauser, L. Jahn, N. Kirchner, A. Kulik, L. Flor, A. Lang, T. Caradec, D. P. Fewer, K. Sivonen, W. J. H. van Berkel, P. Jacques, T. Weber, H. Gross, K. H. van Pée, W. Wohlleben and J. Ludwig-Müller, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26909–26917 CrossRef CAS PubMed.
- U. Bat-Erdene, D. Kanayama, D. Tan, W. C. Turner, K. N. Houk, M. Ohashi and Y. Tang, J. Am. Chem. Soc., 2020, 142, 8550–8554 CrossRef CAS.
- Y. Ye, T. Ozaki, M. Umemura, C. Liu, A. Minami and H. Oikawa, Org. Biomol. Chem., 2018, 17, 39–43 RSC.
- A. H. Russell and A. W. Truman, Comput. Struct. Biotechnol. J., 2020, 18, 1838–1851 CrossRef CAS.
- Z. Zhong, B. He, J. Li and Y. X. Li, Synth. Syst. Biotechnol., 2020, 5, 155–172 CrossRef PubMed.
- T. Tsukui, N. Nagano, M. Umemura, T. Kumagai, G. Terai, M. Machida and K. Asai, Bioinformatics, 2015, 31, 981–985 CrossRef CAS.
- P. Cimermancic, M. H. Medema, J. Claesen, K. Kurita, L. C. Wieland Brown, K. Mavrommatis, A. Pati, P. A. Godfrey, M. Koehrsen, J. Clardy, B. W. Birren, E. Takano, A. Sali, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 412–421 CrossRef CAS PubMed.
Footnotes |
| † https://github.com/gamcil/frippa |
| ‡ https://github.com/antismash/antismash/tree/master/antismash/detection/hmm_detection/cluster_rules |
| This journal is © The Royal Society of Chemistry 2022 |