
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthesis of quinolizidine alkaloids in lupins: mechanistic considerations and prospects for pathway elucidation
Davide
Mancinotti
 ,
Karen Michiko
Frick
and
Fernando
Geu-Flores
*
,
Karen Michiko
Frick
and
Fernando
Geu-Flores
*
Section for Plant Biochemistry and Copenhagen Plant Science Centre, Department of Plant and Environmental Sciences, Faculty of Science, University of Copenhagen, Denmark. E-mail: feg@plen.ku.dk; Tel: +45-60571982
First published on 18th March 2022
Abstract
Covering: up to 2022
Quinolizidine alkaloids (QAs) are a class of alkaloids that accumulate in a variety of leguminous plants and have applications in the agricultural, pharmaceutical and chemical industries. QAs are notoriously present in cultivated lupins (Lupinus spp.) where they complicate the use of the valuable, high-protein beans due to their toxic properties and bitter taste. Compared to many other alkaloid classes, the biosynthesis of QAs is poorly understood, with only the two first pathway enzymes having been discovered so far. In this article, we review the different biosynthetic hypotheses that have been put forth in the literature (1988–2009) and highlight one particular hypothesis (1988) that agrees with the often ignored precursor feeding studies (1964–1994). Our focus is on the biosynthesis of the simple tetracyclic QA (−)-sparteine, from which many of the QAs found in lupins derive. We examine every pathway step on the way to (−)-sparteine and discuss plausible mechanisms, altogether proposing the involvement of 6–9 enzymes. Together with the new resources for gene discovery developed for lupins in the past few years, this review will contribute to the full elucidation of the QA pathway, including the identification and characterization of the missing pathway enzymes.
 Davide Mancinotti | Davide Mancinotti obtained his MSc in Chemistry and Molecular Biology from the University of Roskilde, Denmark, with a thesis on the elucidation of the quinolizidine alkaloid pathway in lupins. As a PhD student in the Geu-Flores laboratory at the University of Copenhagen, Denmark, he continues investigating the synthesis of toxic alkaloids in legumes with a focus on discovery and characterization of biosynthetic enzymes and pathway reconstruction in heterologous hosts. |
 Karen Michiko Frick | Karen Michiko Frick received her PhD in 2018 from the University of Western Australia, based at the CSIRO, for her work on the biosynthesis of the lupin alkaloids. She now works in the Geu-Flores laboratory at the University of Copenhagen as a Marie-Curie Postdoctoral Fellow focusing on the long-distance transport of alkaloids in lupin. Her research aim is to facilitate the development of improved lupin crops to promote their use as a sustainable protein source, both in Europe and worldwide. |
 Fernando Geu-Flores | Fernando Geu-Flores obtained his BSc & Licenciatura degrees from the Pontifical Catholic University of Peru (PI: Eric Cosio) and his PhD degree from the University of Copenhagen (PI: Barbara Halkier). After carrying out postdoctoral work at MIT and the John Innes Centre (PI: Sara O'Connor), he established an independent laboratory at the University of Copenhagen. His group investigates the biosynthesis and transport of plant-derived alkaloids with focus on medicinal and anti-nutritional alkaloids. |
1. Introduction
Quinolizidine alkaloids (QAs) are a class of L-lysine-derived alkaloids with over 170 chemical structures occurring predominantly within the legume family (Leguminosae/Fabaceae).1,2 Their presence is especially well-documented among the lupin species (Lupinus spp.),3 some of which are promising protein crops (L. angustifolius, L. albus, L. luteus and L. mutabilis).4 For this reason, QAs are often referred to as “lupin alkaloids”. QAs are also notable in the genera Baptisia, Thermopsis, Genista, Cytisus, Laburnum, and Sophora,1,5 with several Sophora species used as traditional Chinese medicine.6In the cultivated lupins, QAs complicate the end-use of the valuable high-protein grain, as they are unpalatable and can cause acute anticholinergic poisoning in humans and animals.7,8 Accordingly, one of the major breeding aims for lupins is to reduce seed QAs to consistently low levels. This aim has only been met with some success,4 as the available low-QA cultivars can still unpredictably exceed the industry threshold for utilization as food or feed (0.01% and 0.02% dry weight, respectively).9 In addition, low-QA cultivars display a higher susceptibility to herbivores,10,11 which is consistent with a proposed role in plant defense.12
While QAs are a source of concern among lupin farmers and breeders, some QAs exhibit pharmacological activities of interest for the medical field. For example, (−)-sparteine [(−)-8] has both antiarrhythmic13 and anticonvulsant properties,14 and (+)-matrine (6) has proven activity against several types of cancer, including breast15 and ovarian cancers.16 In addition, (−)-cytisine (12) is effective in aiding smoking cessation and has been successfully commercialized for that purpose.17,18 Apart from its pharmaceutical applications, (−)-sparteine [(−)-8] and its (+) version [(+)-8] have found use in the field of chemical synthesis, where they are highly valued as chiral ligands in asymmetric synthesis protocols.19,20
Despite the importance of QAs in agriculture and their potential applications in medicine and chemistry, very little is known about how QAs are biosynthesized. Several biosynthetic pathway hypotheses have been put forth during the last few decades; however, many of them are not in accordance with the precursor feeding experiments carried out in the 1970s and 80s using isotopically labelled compounds. Here, we review these foundational feeding experiments and subsequently describe one biosynthetic hypothesis that fits these often-ignored constraints. Our focus is on the biosynthesis of the tetracyclic QA core represented by (−)-sparteine [(−)-8] from which many QAs are thought to be derived. In our description of the likely QA pathway, we highlight both stereochemical and mechanistic considerations. Used in combination with the advanced genomic and transcriptomic resources recently developed for lupins, this review will contribute to the full elucidation of the QA pathway, including the identification and characterization of the missing pathway enzymes.
2. Occurrence, structural diversity, and biosynthetic origin
Chemically speaking, QAs are alkaloids containing a quinolizidine core (1), i.e. a 1-azabicyclo[4.4.0]decane moiety (Fig. 1A). Within the legume family, QAs are a trademark of the wider genistoid clade (genistoids sensu lato)21,22 and have been used as chemotaxonomic markers for this clade.23 QAs resembling those that accumulate in legumes have also been found sporadically in individual genera within the distantly related families Chenopodiaceae (Anabis), Ranunculaceae (Cimicifuga), Rubiaceae (Readea), and Berberidaceae (Leontice, Caulophyllum). In addition, QAs that are structurally divergent from the legume QAs have been reported in the Lythraceae (Heimia, Decodon), Nympheaceae (Nuphar) and Lycopodiaceae (Lycopodium, Huperzia) families.1,24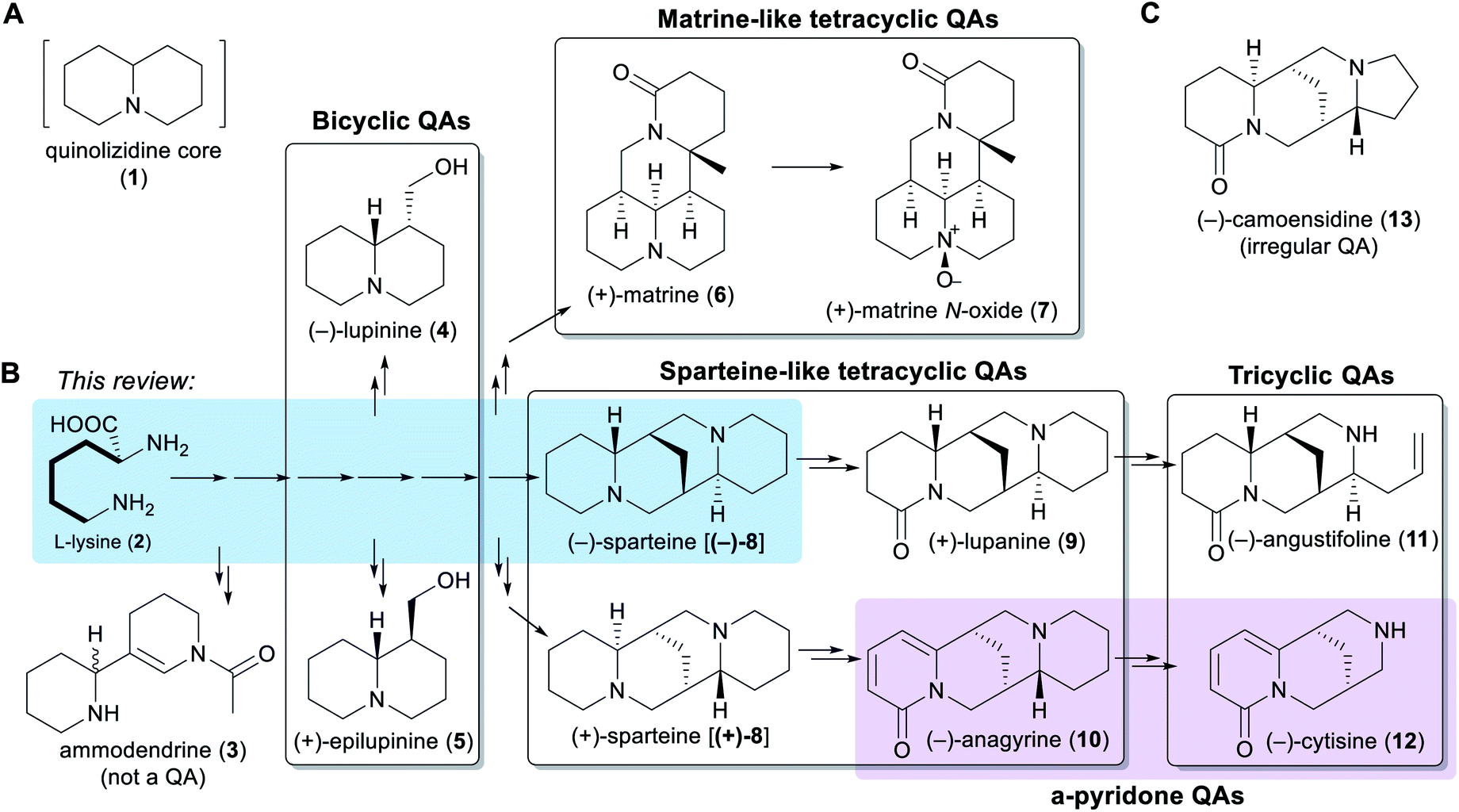 | ||
| Fig. 1 Overview of the different QA sub-classes presented in a biosynthetic context. (A) Structure of the quinolizidine moiety (1) that defines QAs. (B) Overall QA pathway with examples of QAs from a wide range of genistoid legumes. The main QA sub-classes are shown as transparent boxes. The distinction of α-pyridone QAs as a separate sub-class is highlighted in purple. Biosynthetic relationships, including a common origin from L-lysine (2), are indicated via arrows. A single arrow represents one biosynthetic step, while two or more arrows represent an unknown number of biosynthetic steps. The conversion from L-lysine (2) to (−)-sparteine [(−)-8] is highlighted in blue. The bold bonds on the side chain of L-lysine (2) represent the C5 unit that the amino acid donates unabridged for QA biosynthesis. The structure of ammodendrine (3), a probable early by-product of the QA pathway, is also shown. (C) Structure of an irregular QA, (−)-camoensidine (13). | ||
The simplest classification of QAs relates to the number of joined 6-membered rings present in their structures. In this classification, QAs can be bicyclic [e.g. (−)-lupinine (4) and (+)-epilupinine (5)], tricyclic [e.g. (−)-angustifoline (11) and (−)-cytisine (12)], or tetracyclic [e.g. (−)-sparteine [(−)-8] and (+)-matrine (6)]. The tetracyclic QAs can be further divided into two main classes: the sparteine-like QAs [e.g. (−)-sparteine [(−)-8] and (+)-lupanine (9)] and the matrine-like QAs [e.g. (+)-matrine (6) and (+)-matrine N-oxide (7)] (Fig. 1B). Although most QAs are aliphatic, some tri- and tetracyclic QAs contain a pyridone ring [e.g. (−)-anagyrine (10) and (−)-cytisine (12)], and these are sometimes classified separately (purple box in Fig. 1B). In addition, there are a number of irregular QAs with divergent structural features, such as (−)-camoensidine (13) (Fig. 1C).
With the exception of some irregular QAs, the backbones of all legume QAs are exclusively derived from the amino acid L-lysine (2), which donates two C5 units in the case of the bicyclic QAs and three C5 units in the case of the tetracyclic QAs.25,26 The tricyclic QAs are derived from the sparteine-like tetracyclic QAs by oxidative ring-cleavage reactions (Fig. 1B).27,28 Notably, the bipiperidine alkaloid ammodendrine (3) co-occurs with QAs in a variety of QA-containing species21 and is therefore likely to be an early by-product of the biosynthesis of QAs (Fig. 1B).
Typically, genistoid legumes accumulate mixtures of QAs belonging to several different sub-classes. For example, lupins accumulate bicyclic, tricyclic and sparteine-like QAs, while Sophora species accumulate tricyclic, sparteine-like, and matrine-like QAs.1 The accumulation of QAs has been most extensively studied in lupins. Within these, each species features a characteristic QA profile,3 with the exact composition varying according to tissue type,3,29 accession,30 developmental stage,31 growth conditions,30 and time of the day.29 The most common QAs in lupins are those derived from (−)-sparteine [(−)-8] such as (+)-lupanine (9) and (−)-angustifoline (11) (Fig. 1B). This review focuses on the biosynthetic steps that are likely to be required for converting the amino acid precursor L-lysine (2) into (−)-sparteine [(−)-8] (blue box in Fig. 1B).
3. Stereochemical considerations
Like many bioactive natural products, QAs feature a number of stereocenters that are presumably crucial for bioactivity. In particular, the backbone of the sparteine-like QAs features four stereocenters at C6, C7, C9, and C11 (Fig. 2A and B). Considering that each of them can adopt an R or S configuration, 16 different stereoisomers would seem possible. However, the existence of a methylene bridge between C7 and C9 restricts their relative configuration to cis configurations [(7S,9S) or (7R,9R)], thus decreasing the number of possible stereoisomers to 8. In addition, for an unsubstituted sparteine backbone, only 6 of the 8 structures are distinct chemical entities. This is illustrated in Fig. 2C, where the equivalence of the two (6R,11S) forms to their respective (6S,11R) forms can be visualized in terms of a 180° rotation around the z-axis. The 6 chemically distinct sparteine molecules are referred to as (−)-sparteine [(−)-8], (−)-α-isosparteine (14), (−)-β-isosparteine (15), and their respective (+) mirror images (Fig. 2C).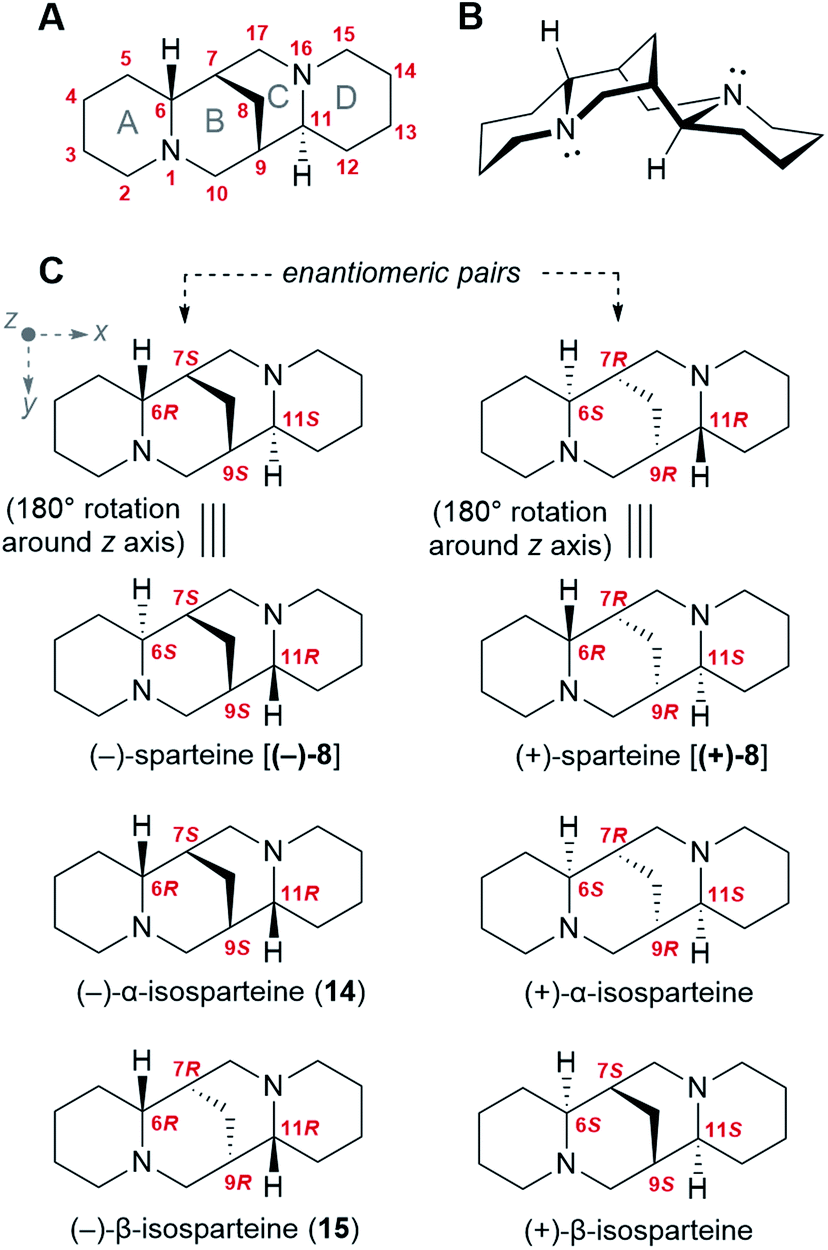 | ||
| Fig. 2 Stereochemistry of the sparteine-like QA backbone. (A) Numbering of the sparteine-like QA backbone as exemplified for (−)-sparteine [(−)-8]. The naming of each joint ring is specified (grey letters A–D). (B) Three-dimensional representation of the most stable conformation of (−)-sparteine [(−)-8]. Rings A, B, and D are in the chair conformation, while ring C is in the boat conformation. (C) Six possible backbone structures varying in the configuration of their four stereocenters. Each structure on the left is the enantiomeric pair of the adjacent structure on the right. The structures of (−)-sparteine [(−)-8] and (+)-sparteine [(+)-8] can each be represented in two different ways that are interconvertible via a 180° rotation around the z axis. The remaining four isomers are all C2 symmetrical around the z axis. Cartesian axes (x, y, z) are defined in grey. | ||
Despite the number of possible stereoisomers, most QA-containing species accumulate only one or two stereoisomeric forms of any given QA, with one of the forms being predominant. In addition, the predominant forms of different QAs in a given species tend to share a particular backbone, suggesting that they belong to a biosynthetic series. This is particularly well documented among lupins, where the most commonly encountered tetracyclic QAs belong to the (−)-sparteine series (6R,7S,9S,11S backbone).3 Exceptions include varieties of L. argenteus, where the (−)-α-isosparteine backbone (6R,7S,9S,11R) predominates,3,32 and varieties of L. sericeus and L. pusillus, where the (−)-β-isosparteine backbone (6R,7R,9R,11R) predominates.3,33,34 The enantiomeric purity of a given QA tends to be high but varies between species. For example, in L. angustifolius and L. polyphyllus, only (+)-lupanine (9) has been reported,35–37 whereas both (+) and (−) enantiomers have been reported in L. albus, albeit with an excess of the (+) form.35
The fact that specific QA backbones predominate in individual plant species suggests that QA backbone formation occurs under stereoselective control. Thus, any proposed biosynthetic hypothesis must account for the selective formation and subsequent preservation of the four aforementioned stereocenters.
4. Evidence from precursor feeding studies
Precursor feeding studies play a vital role in the elucidation of biosynthetic pathways. By administering suitably labelled, putative precursors to biosynthetic tissues and monitoring the incorporation of the label into the metabolites of interest, biochemists can formulate a consistent sequence of biosynthetic steps and thereby make predictions on the nature of the enzymes involved. Extensive work has been done on probing the biosynthesis of sparteine-like QAs by these means. In this section, we will review these foundational feeding studies as a prelude to our presentation of the different biosynthetic hypotheses.Radioactive tracer studies carried out in the 1960s using L. luteus and L. angustifolius provided the first evidence that the carbon skeleton and nitrogen atoms of the sparteine-like QAs are derived exclusively from L-lysine (2).25,38 These early results were confirmed and refined in the 1980s using stably labelled (non-radioactive) precursors and nuclear magnetic resonance (NMR) spectroscopy.39 The exact mode of incorporation of L-lysine (2) is well understood. Two key experiments were the feeding of DL-[2-14C]lysine† to L. angustifolius25 and the feeding of DL-(6-13C)lysine to the same species.26 In these experiments, the single label from either of the two precursors became distributed over the same six carbon atoms of (+)-lupanine (9): C2, C6, C10, C17, C11 and C15 (Fig. 3A). This observation implies that C2 and C6 of L-lysine (2) must become equivalent at some point during biosynthesis. Furthermore, it suggests that (+)-lupanine (9) is made from three discreet five-carbon (C5) units derived from L-lysine (2): two C5 units that constitute the outer rings (rings A and D) and a third C5 unit that accounts for the carbon atoms in between these rings (including the methylene bridge between rings B and C) (Fig. 3B). Taken together, these studies suggest that the skeleton of the sparteine-like QAs originates from three L-lysine (2) molecules via a symmetrical C5 intermediate (i.e. with C2v symmetry).
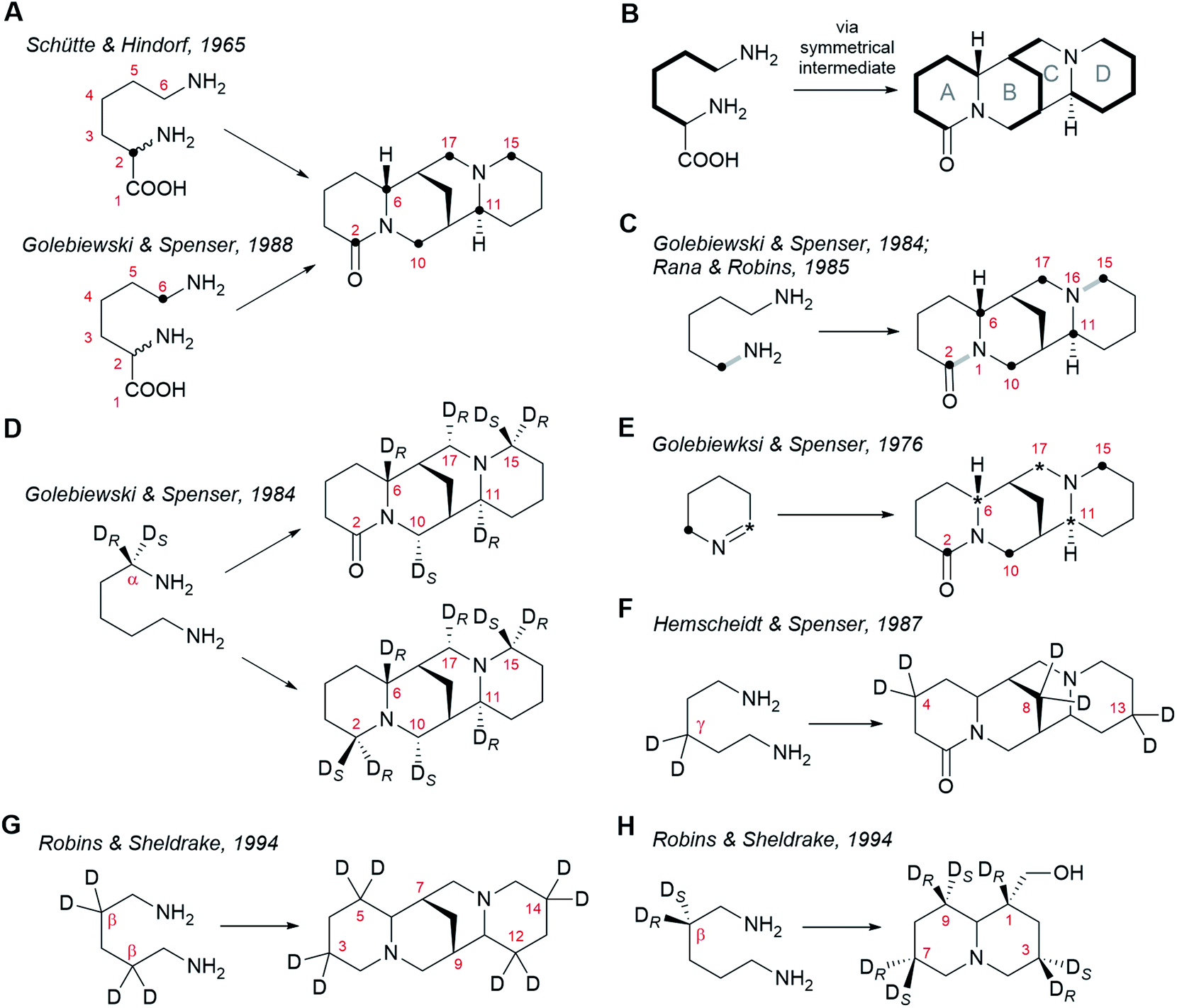 | ||
| Fig. 3 Patterns of incorporation of isotopically labelled precursors into sparteine-like QAs and into the bicyclic QA lupinine (4). (A) Incorporation of DL-[2-14C]lysine25 and DL-[6-14C]lysine26 into (+)-lupanine (9) in L. angustifolius. (B) Model specifying the incorporation of three C5 units derived from L-lysine (2) into (+)-lupanine (9) (based on feeding experiments shown in (A)). (C) Incorporation of (2-15N, 1-13C)cadaverine into (+)-lupanine (9) in L. angustifolius.27,39 The grey bonds in bold indicate how the original 13C–15N bond from labelled cadaverine (16) is incorporated intact into labelled (+)-lupanine (9). (D) Incorporation of (R)-(1-2H)cadaverine and (S)-(1-2H)cadaverine (depicted as a single molecule) into (+)-lupanine (9) in L. angustifolius and into (−)-sparteine [(−)-8] in L. luteus.39 (E) Incorporation of [2-14C]Δ1-piperideine and [6-14C]Δ1-piperideine (depicted as a single molecule) into (+)-lupanine (9) in L. angustifolius.42 (F) Incorporation of (3,3-2H2)cadaverine into (+)-lupanine (9) in L. angustifolius.43 (G) Incorporation of (2,2,4,4-2H4)cadaverine† into (−)-sparteine [(−)-8] in L. luteus.44 (H) Incorporation of (R)-(2-2H)cadaverine and (S)-(2-2H)cadaverine (depicted as a single molecule) into (−)-lupinine (4) in L. luteus.44 | ||
The most obvious candidate for this C5 symmetric intermediate is cadaverine (16), a diamine that can be formed directly from L-lysine (2) by decarboxylation. Indeed, feeding of (1-15N, 1-13C)cadaverine to L. angustifolius caused the labelling of (+)-lupanine (9) at the same six carbons as when feeding DL-[2-14C]lysine or DL-(6-13C)lysine (Fig. 3C).27,39 This experiment also showed that both nitrogen atoms in the tetracyclic QAs derive from cadaverine (16) and therefore, ultimately, from L-lysine (2). The three L-lysine/cadaverine units that make up the tetracyclic QAs possess a total of six nitrogen atoms. Since only two of these are retained in the final QAs, four deamination events must occur. Important clues about these deamination events emerged when observing that the 13C–15N bond of labelled cadaverine was incorporated intact into (+)-lupanine (9) only at positions C2–N1 and C15–N16 (grey bonds in bold in Fig. 3C).27,39 This implies that the cadaverine unit that gives rise to ring A is deaminated at a position that later becomes C6 in (+)-lupanine (9). Likewise, the cadaverine unit giving rise to ring D must be deaminated at a position that later becomes C11. Finally, it also implies that the middle cadaverine unit must lose both of its nitrogen atoms by undergoing deamination at the positions that later become C10 and C17.
The deamination of terminal, linear amines such as cadaverine (16) is typically oxidative, converting the amine into the corresponding aldehyde with concomitant loss of one of the two hydrogens at the alpha carbon (Cα).40 The stereochemistry of hydrogen loss in the four deaminations in QA biosynthesis has been revealed by studying the incorporation of monodeuterated cadaverine precursors labelled at Cα.28,39,41 It was consistently observed in L. angustifolius and L. luteus that the deuterium from (R)-(1-2H)cadaverine enters the sparteine-like QA backbone at C6, C11 and C17, but not at C10 (Fig. 3D).39 This implies that the pro-S hydrogen is the one that is specifically lost from the Cα atoms later to become C6, C11, and C17. In addition, the deuterium from (S)-(1-2H)cadaverine was shown to be incorporated at position C10 (Fig. 3D).39 This confirms that the fourth deamination, which affects only the central C5 unit, proceeds through a different route involving the loss of the pro-R hydrogen from the Cα later to become C10.
The fact that C6, C11, and C17 derive from three different cadaverine molecules that are deaminated with the same stereoselective mechanism suggests a direct deamination of cadaverine (16) into 5-aminopentanal (26) as the sole mode of entry of cadaverine (16) into the pathway. 5-Aminopentanal (26) cyclizes spontaneously via intramolecular Schiff-base formation to yield Δ1-piperideine (17). When L. angustifolius was fed with cyclic Δ1-piperideine labelled at either of the two different carbon atoms adjacent to the nitrogen atom, the label was incorporated into all three C5 units of the sparteine-like QAs (Fig. 3E).42 Specifically, the aldimine carbon (deaminated carbon; asterisk in Fig. 3E) became C6, C11, and C17, and the amine carbon (bold dot in Fig. 3E) became C2, C10 and C15, as expected (compare with Fig. 3D). This regiospecific incorporation also implies that Δ1-piperideine (17) cannot be converted into any compound with C2v symmetry later during biosynthesis.
The experiments described above suggest that the sparteine-like QAs are derived from three units of Δ1-piperideine (17) (or a close, equally asymmetric derivative). One interesting insight into the coupling of these C5 units emerged from observing the relative extent of labelling of the different units in the final QAs. Consistently, the incorporation of label from [2-14C]Δ1-piperideine, [6-14C]Δ1-piperideine, and DL-[6-14C]lysine was more pronounced in the C5 unit corresponding to ring D than in the other two C5 units.26 This indicates that this C5 unit is subject to a lower degree of endogenous dilution before being incorporated in the QA backbone, which suggests that it is added last during biosynthesis. In these experiments, the incorporation of label was equal for the other two C5 units, indicating that the dimerization of Δ1-piperideine (17) is a plausible first step in the coupling of C5 units. The non-enzymatic dimerization of Δ1-piperideine (17) occurs readily at slightly basic pH values (discussed in detail in Section 6).
Finally, a few extra constraints have been revealed by experiments using deuterated cadaverine (16) molecules labelled at positions different than Cα. When cadaverine (16) was doubly deuterated at the γ position and was fed to L. angustifolius, both deuterium atoms were found at positions C4, C8, and C13 of (+)-lupanine (9), as expected (Fig. 3F).43 This means that no direct chemical modifications occur at the carbons that will become C4, C8, and C13 during the entire biosynthesis. By contrast, when cadaverine (16) was fully deuterated at the β positions and was fed to L. luteus, the deuterium atoms were found at C3, C5, C12, and C14 of (−)-sparteine [(−)-8], but not at the bridgehead carbons C7 or C9 (Fig. 3G).44 This implies that both hydrogen atoms at the positions that will become the bridgehead carbons must be lost during biosynthesis. The implications of this on the proposed biosynthetic hypotheses will be discussed in Section 6. One last experiment with cadaverine molecules labelled at the β position is worth mentioning. In this experiment, (R)- and (S)-(2-2H)cadaverine were fed separately to L. luteus, and the incorporation into the bicyclic QA (−)-lupinine (4) was assessed (Fig. 3H). Interestingly, only the deuterium from (R)-(2-2H)cadaverine was retained at C1 of (−)-lupinine (4) (Fig. 3H).44 Assuming that lupinine is a side product of the pathway towards the sparteine-like QAs and that these pathways diverge after the formation of the quinolizidine core (1), the selective loss of the pro-S hydrogen at the position later to become the bridgehead carbon C7 in e.g. (−)-sparteine [(−)-8] [equivalent to C1 in (−)-lupinine (4)] must occur before the pathways diverge. Mechanistic implications of this will be discussed in Section 6.
5. Overview of the different biosynthetic hypotheses
Five different hypotheses/models have been put forth to explain the biosynthetic origin of the sparteine-like QAs (Fig. 4). Common to all is the proposition that the first committed step of the pathway is the decarboxylation of L-lysine (2) to give cadaverine (16). The different hypotheses then vary in the nature of the pathway steps downstream of cadaverine (16). Several of these hypotheses, however, are not consistent with the results of the feeding studies reviewed in the previous section. For example, an early model proposed the direct, single-enzyme conversion of cadaverine (16) into 17-oxosparteine (21) (Fig. 4, Wink & Hartmann, 1979). The model was based on in vitro activities from plant extracts and postulated that 17-oxosparteine (21) would serve as common intermediate towards all other sparteine-like QAs, including (−)-sparteine [(−)-8] and (+)-lupanine (9).45 However, feeding studies showed five years later that deuterium from labelled cadaverine is retained at C17 of both (−)-sparteine [(−)-8] and (+)-lupanine (9) (Fig. 3D).39 This ruled out 17-oxosparteine (21) as possible common intermediate, since it does not possess any hydrogen atoms at C17.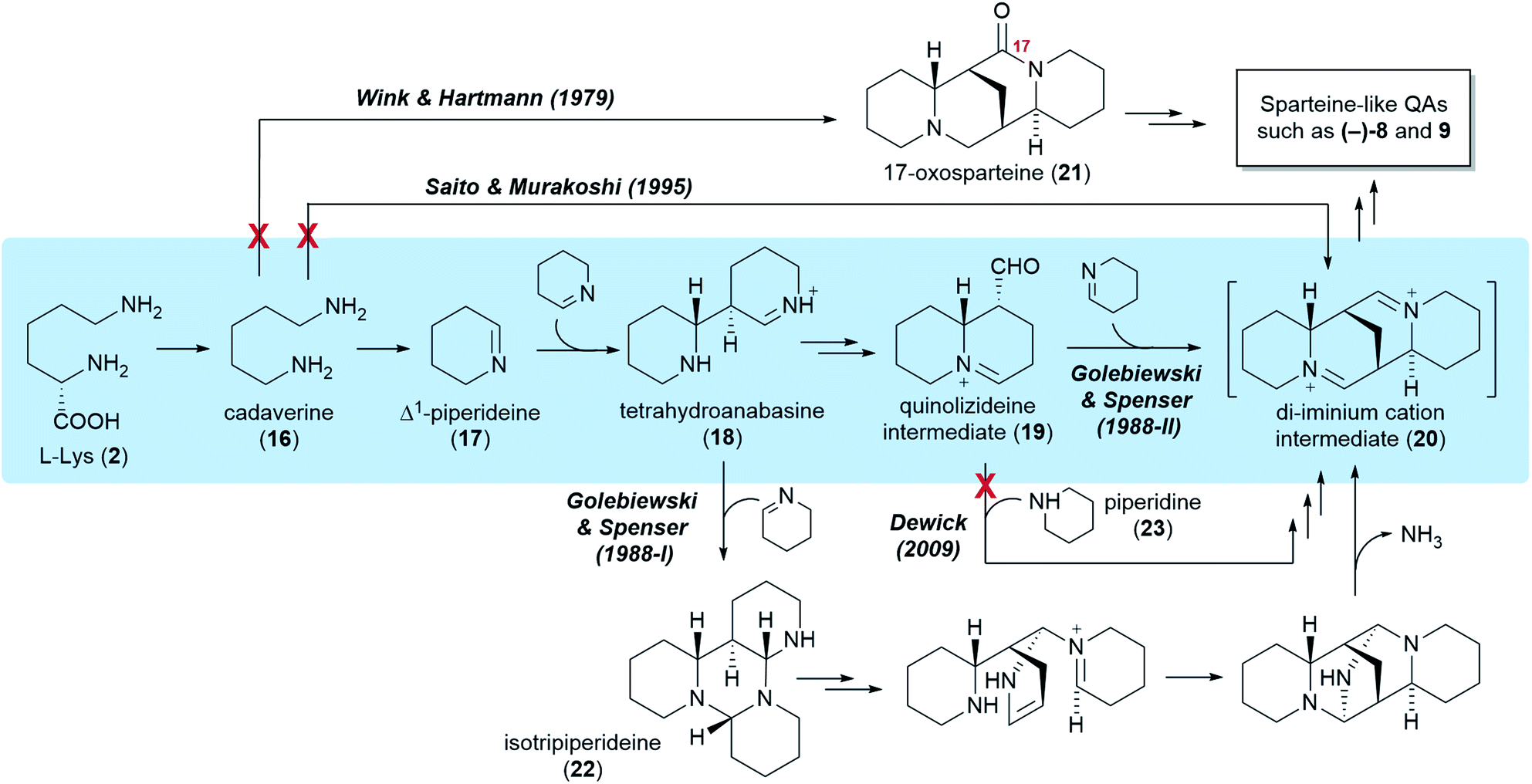 | ||
| Fig. 4 Five different hypotheses for the biosynthesis of the sparteine-like tetracyclic QAs, exemplified by the formation of the most common QA backbone (6R,7S,9S,11S). Double arrows indicate two or more biosynthetic steps. The authors of the different hypotheses are mentioned at the first pathway step unique to their respective hypotheses. Red crosses indicate implausibility based on precursor feeding studies. The preferred hypothesis is highlighted in blue. | ||
The four remaining hypotheses postulate that the first common tetracyclic intermediate is the hypothetical di-iminium cation (20), which can yield sparteine by sequential reduction (Fig. 4). One of these hypothesis was inspired by the 17-oxosparteine model and proposes the single-enzyme conversion of cadaverine (16) into the di-iminium cation (20) via a series of enzyme-bound intermediates (Fig. 4, Saito & Murakoshi, 1995).46 However, the proposed mechanism (not shown) conflicts with the observed labelling patterns of QAs derived from [2-14C]- and [6-14C]Δ1-piperideines, which reveal that the cadaverine carbon that is initially oxidized ends up at positions 6, 11, and 17 of sparteine (Fig. 3E). By contrast, this model predicts that the initially oxidized cadaverine carbon ends up at positions 6, 11, and 10.
The three remaining hypotheses postulate the oxidation of cadaverine (16) to Δ1-piperideine (17) and agree on the stereoselective dimerization of Δ1-piperideine (17) into tetrahydroanabasine (18) as the next step (Fig. 4). One hypothesis from 2009 proposed that the third C5 unit is supplied to the pathway as fully reduced piperidine (23) rather than Δ1-piperideine (17) (Fig. 4, Dewick, 2009).47 However, feeding studies have shown that the third Δ1-piperideine (17) molecule must be incorporated in a regiospecific manner that is not compatible with prior conversion into a symmetrical intermediate such as piperidine (23).42
The two remaining models, both proposed by Golebiewski & Spenser in 1988,26 are the only ones generally consistent with the body of evidence from feeding studies presented in the previous section. The first of these two models suggests that tetrahydroanabasine (18) reacts with Δ1-piperideine (17) to form isotripiperideine (22), which is then modified to yield the di-imunium cation (20) through an unconventional mechanism that eliminates ammonia as the last step (Fig. 4, Golebiewski & Spenser, 1988-I).26,42 The second of these two models postulates the conversion of tetrahydroanabasine (18) to a bicyclic quinolizideine intermediate (19) via hydrolysis, oxidation, and intramolecular Schiff base formation (Fig. 4, Golebiewski & Spenser, 1988-II). This quinolizideine intermediate (19) is then coupled to Δ1-piperideine (17) in a reaction that is analogous to the earlier dimerization of Δ1-piperideine (17). The tetracyclic di-iminium cation (20) is then produced spontaneously by intramolecular Schiff base formation.26
Although both of these models are in general accordance with the precursor feeding studies, its authors favored the one involving the bicyclic quinolizideine intermediate (19) (Fig. 4, Golebiewski & Spenser, 1988-II). As noted by them, this model offers a more satisfactory explanation for the differential incorporation of radioactive precursors into the three C5 units of (+)-lupanine (9) as defined in Fig. 3B. Indeed, a significantly higher incorporation into the third C5 unit (leading to ring D) was observed when feeding L. angustifolius with 14C-labelled DL-lysine or Δ1-piperideine (17) and subjecting the resulting (+)-lupanine (9) to controlled chemical degradation.26 Such distribution of label is better explained by a model in which the third Δ1-piperideine (17) unit is incorporated at a much later biosynthetic step than the dimerization of Δ1-piperideine (17), thus suffering less endogenous dilution en route to the (+)-lupanine (9) product.
6. The preferred biosynthetic hypothesis
We share the predilection of Golebiewski & Spenser for their second biosynthetic model, and we would like to mention two additional justifications. First, this hypothesis seems more biochemically-feasible in so far as it is composed mainly of reaction steps for which there is mechanistic precedence. Second, this hypothesis can explain the biochemical origin of the bicyclic QA (−)-lupinine (4) via sequential reduction of the bicyclic quinolizideine intermediate (19). In this section, we break down the preferred hypothesis in discrete steps and highlight particular mechanistic restrictions imposed by the results of the precursor feeding studies. We also review recent advances on the identity of the first two enzymes in the pathway and speculate on which downstream steps necessitate enzymatic catalysis.6.1 From L-Lys to cadaverine
L-Lysine (2) is converted to cadaverine (16) through the action of lysine decarboxylase (LDC). LDC was first cloned from L. angustifolius using a cDNA library enriched in transcripts from a high-QA cultivar compared to a low-QA cultivar. Heterologously expressed LDCs from L. angustifolius and from two other QA-containing species (Sophora flavescens and Echinosophora koreensis) were able to decarboxylate L-lysine (2) and its 1-carbon shorter analogue L-ornithine with similar catalytic efficiency. Considering that ornithine decarboxylases (ODCs) are more ubiquitous and typically display a marked preference for L-ornithine, it was hypothesized that LDC evolved from ODC through expansion of substrate preference. Remarkably, the same study identified a key amino acid residue likely to be involved in this transition (F344 in LDC from L. angustifolius).48Like ODC, LDC is a pyridoxal phosphate (PLP)-dependent enzyme that can be inhibited by α-difluoromethylornithine.48 Thus, a mechanism similar to that of ODC can be postulated, namely a conventional PLP-dependent decarboxylation with retention of configuration (Fig. 5). The retention of configuration implies that the quinonoid intermediate (24) is protonated at the Re face so that the new proton ends up occupying the same position as the original carboxylate group (Fig. 5).49 Notably, feeding studies using deuterated versions of L-lysine (2) and cadaverine (16) are consistent with the retention of configuration. In particular, the label in (R)- or (S)-(1-2H)cadaverine leads to different patterns of labelling of the tetracyclic QAs (Fig. 3D),39 and the label in L-(2-2H)lysine gives the same labelling pattern as (S)-(1-2H)cadaverine.39 Accordingly, it is very likely that the LDCs cloned from L. angustifolius, S. flavescens, and E. koreensis operate with retention of configuration, although this remains to be shown experimentally.
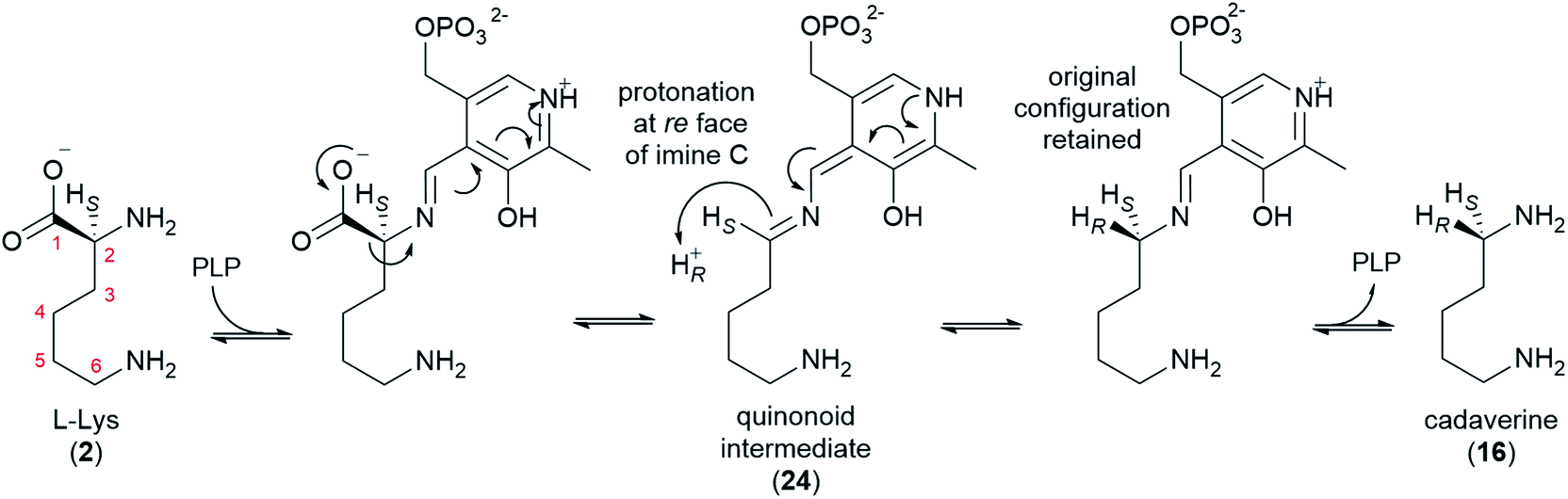 | ||
| Fig. 5 Proposed mechanism for the decarboxylation of L-lysine by LDC in QA biosynthesis. The mechanism accounts for the decarboxylation of L-lysine (2) with retention of the original configuration at C2 to yield cadaverine (16). | ||
6.2 From cadaverine to Δ1-piperideine
The decarboxylation of L-lysine (2) to cadaverine (16) is followed by the oxidative deamination of cadaverine (16) to 5-aminopentanal (26), which likely exists in equilibrium with its cyclized form, Δ1-piperideine (17). The deamination has been proposed to be catalyzed either by a transaminase (using a ketoacid as co-substrate) or a copper amine oxidase (CAO, using O2 as co-substrate). Early claims of a transaminase-like enzyme able to convert cadaverine (16) to 17-oxosparteine (21) in the presence of CAO inhibitors and in the absence of O2 have not been replicated.45,50,51 Moreover, feeding studies have ruled out 17-oxosparteine (21) as a precursor for the other sparteine-like tetracyclic QAs, as previously mentioned.39Recently, a CAO that is tightly co-regulated with LDC was cloned from L. angustifolius. This CAO was shown to possess the three highly conserved L-His residues that chelate the catalytically important Cu2+ ion in canonical CAOs. The enzyme also featured the highly conserved L-Tyr residue that is auto-catalytically converted to a crucial topaquinone residue (25) prior to entering the catalytic cycle. When heterologously expressed in E. coli, this CAO was able to oxidize cadaverine (16) with an unusually high affinity,52 thus supporting a proposed role in QA biosynthesis.
In the oxidation of cadaverine (16) to 5-aminopentanal (26), the carbon atom that is being oxidized loses one of its two protons. According to the feeding experiments described in Section 4, the lost proton must be the pro-S proton, specifically. The evidence can be summarized in two parts as follows: (1) the 14C label at the aldimine carbon of Δ1-piperideine (17) results in the specific labelling of C6, C11, and C17 of (+)-lupanine (9), indicating that these three carbons correspond to the oxidized carbon of 5-aminopentanal (26) (Fig. 3E);42 (2) a single 2H label at Cα of cadaverine (16) gets incorporated into carbons C6, C11, and C17 only if the label is placed at the pro-R position (Fig. 3D).39 The specific abstraction of the pro-S proton from Cα of cadaverine (16) is represented in Fig. 6 in the context of a plausible oxidation mechanism catalyzed by CAO.
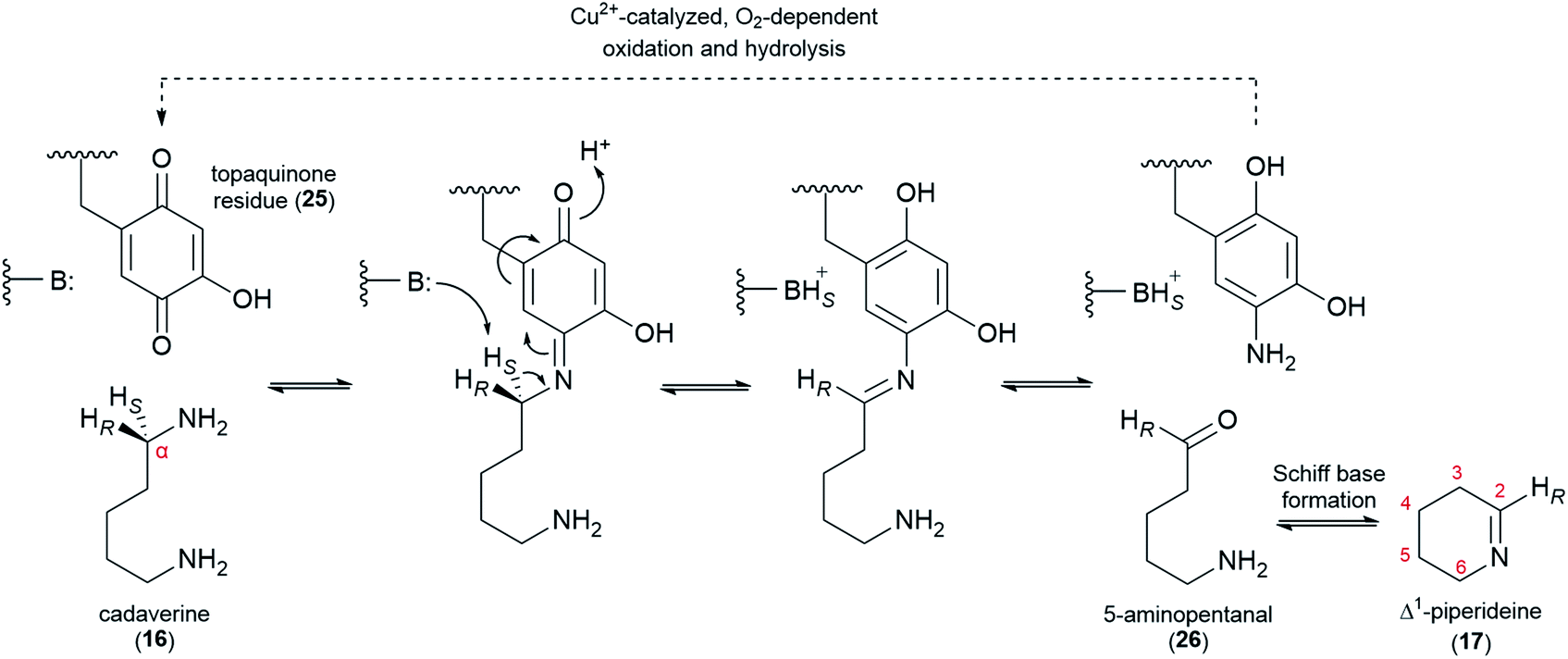 | ||
| Fig. 6 Proposed mechanism for the CAO-dependent oxidation of cadaverine (16) to 5-aminopentanal (26) in QA biosynthesis. Central to the mechanism is the participation of a topaquinone residue (25) that is auto-catalytically generated from an L-Tyr residue prior to catalysis. Only the first half of the catalytic cycle is drawn explicitly; the second half is represented by the dashed arrow. The 5-aminopentanal product (26) likely exists in equilibrium with its cyclized form, Δ1-piperideine (17). B: represents an acid/base residue acting as a base. | ||
6.3 Dimerization of Δ1-piperideine
At physiological pH, Δ1-piperideine (17) dimerizes spontaneously to give tetrahydroanabasine (28),53 which is the next proposed pathway intermediate. The reaction can be thought of as an aldol-like coupling where a molecule of Δ1-piperideine (17) first tautomerizes to Δ2-piperideine (27) and then adds to a second molecule of Δ1-piperideine (17) (Fig. 7A).26 The coupling creates two new stereocenters (numbered in red in Fig. 7A), potentially giving a total of four products: (2R,3′R), (2S,3′S), (2S,3′R), and (2R,3′S). Recent computational chemistry calculations using density-functional theory (DFT) have predicted that the (2R,3′R) and (2S,3′S) isomers should form faster than the (2S,3′R) and (2R,3′S) isomers (Fig. 7B).54 In fact, based on the calculated energy barriers at neutral or acidic conditions (10–20 kcal mol−1), the latter two isomers might form quite slowly if at all. The lack of a method by which to analyze the four tetrahydroanabasine isomers has prevented the experimental validation of these theoretical calculations.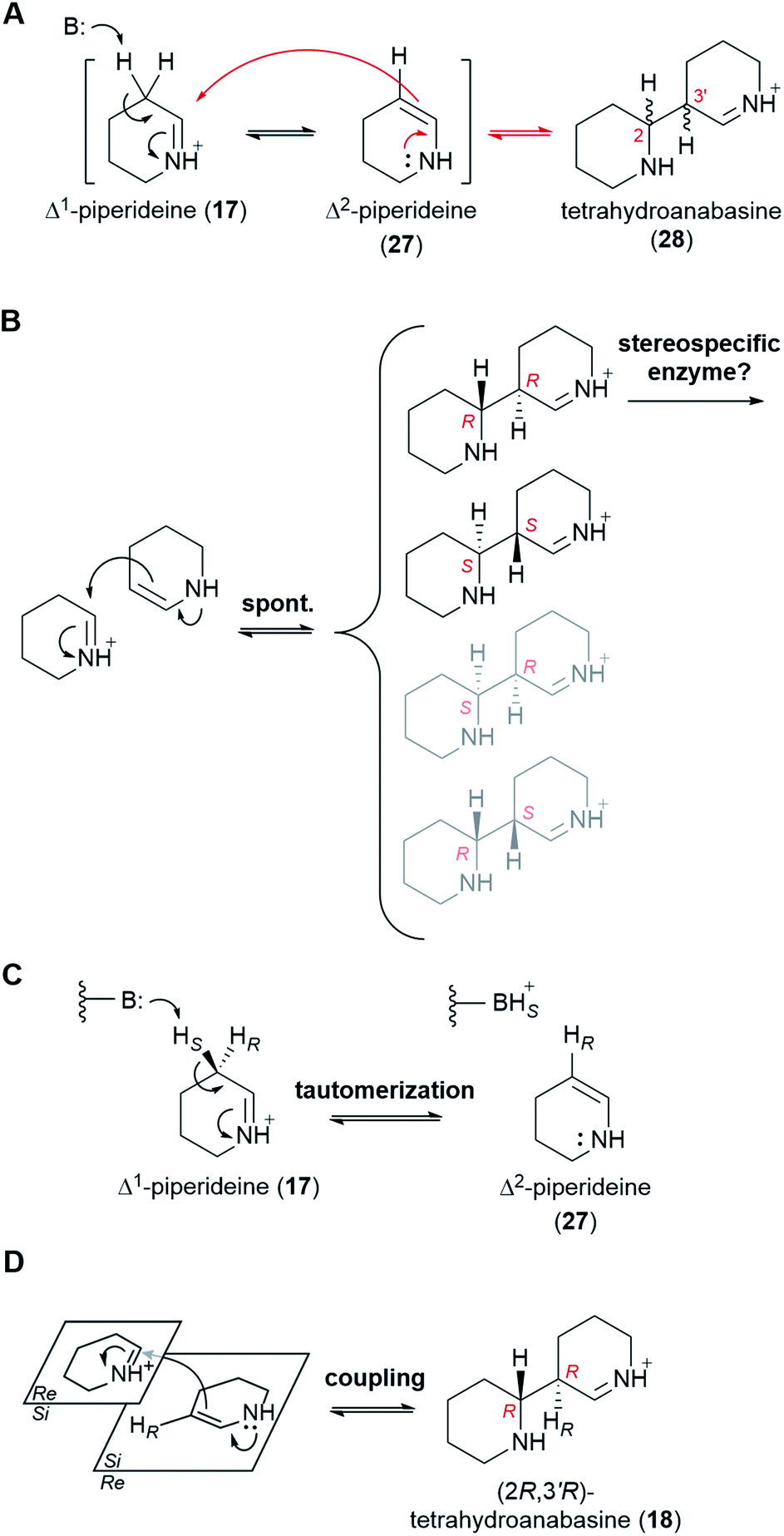 | ||
| Fig. 7 Proposed mechanism for the dimerization of Δ1-piperideine (17) in QA biosynthesis. All reaction steps are shown as equilibria, although it is uncertain to which extent each of the steps is reversible. (A) Possible mechanism for the non-enzymatic dimerization of Δ1-piperideine (17) to give tetrahydroanabasine (28). B: represents a general base. (B) Preferential non-enzymatic formation of the (2R,3′R) and the (2S,3′S) dimerization products as predicted using theoretical DFT calculations. Since only the (2R,3′R) form is used for QA biosynthesis in most species of lupin, a stereospecific enzyme could be responsible for exclusively taking this form further down the QA pathway. (C) Proposed mechanism for the enzymatic tautomerization of Δ1-piperideine (17) to Δ2-piperideine (27). B: represents an acid/base residue acting as a base. (D) Proposed mechanism for the stereoselective coupling of Δ2-piperideine (27) to Δ1-piperideine (17). The enzyme involved in the coupling step could be the same enzyme or a different one than the one catalyzing the preceding tautomerization. | ||
The two newly formed stereocenters in tetrahydroanabasine (18), C2 and C3′, correspond to carbons C6 and C7 of (−)-sparteine [(−)-8], respectively, (Fig. 2A), with C6 tracing back to the imine carbon of Δ1-piperideine (17) that receives the nucleophilic attack during dimerization (Fig. 7A). It is very plausible that the final stereochemistry at C6 of (−)-sparteine [(−)-8] is established at this early step, given that the proton at this position is retained all the way from labelled cadaverine (pro-R proton at Cα) to (−)-sparteine [(−)-8] in feeding experiments (Fig. 3D).39 If so, the configuration of C2 in the tetrahydroanabasine intermediate (28) should be restricted to R. By contrast, the proton at C7 in (−)-sparteine [(−)-8] is not derived from cadaverine (16) (Fig. 3G),44 and this allows for a potential change in configuration of C3′ in tetrahydroanabasine (28) upon proton loss during its conversion to (−)-sparteine [(−)-8]. However, in the case of the bicyclic QA (−)-lupinine (4), which often co-occurs with (−)-sparteine [(−)-8] in lupins, the corresponding proton [pro-R proton at Cβ in cadaverine (16)] is retained at position C1, which is in the R configuration (Fig. 3H).44 Assuming that the pathways toward the bicyclic and tetracyclic QAs diverge after the dimerization step, this provides a crucial insight into the stereochemistry of the dimerization product at this second stereocenter. Altogether, the feeding experiments strongly suggest that only the (2R,3′R) form of tetrahydroanabasine (18) is used for QA biosynthesis in lupins.‡
We envision two possible ways for the exclusive use of (2R,3′R)-tetrahydroanabasine (18) in QA biosynthesis in lupins. First, if the (2R,3′R) and (2S,3′S) forms are in equilibrium with the piperideine monomers (19 and 28), then a stereospecific enzyme metabolizing the (2R,3′R) form exclusively would be enough to ensure a complete conversion into the next pathway intermediate (Fig. 7B). However, we favor a different possibility in which the dimerization is actually an enzymatic process that only produces the (2R,3′R) form. The involvement of an enzyme could explain the often ignored fact that only the pro-R proton at Cβ of cadaverine (16) appears at C1 of (−)-lupinine (4) in feeding experiments (Fig. 3H).44 Indeed, we postulate that an enzyme removes the pro-S proton from Cβ during the first part of the reaction, namely, during the tautomerization to Δ2-piperideine (27) (Fig. 7C). It is not unusual for a spontaneous isomerization to be catalyzed by an enzyme, as enzymes can increase the speed of spontaneous reactions that might otherwise occur at sub-optimal rates in an organism.55 Subsequent coupling of Δ2-piperideine (27) to an unreacted Δ1-piperideine molecule (17) by the same or by a different enzyme would complete the dimerization.§ In order to yield the (2R,3′R) form (18), the coupling enzyme must coordinate an attack from the Si face of Δ2-piperideine (27) to the Si face of Δ1-piperideine (17) (Fig. 7D).¶
6.4 Formation of the quinolizideine intermediate
From (2R,3′R)-tetrahydroanabasine (18), the formation of the quinolizidine core (1) requires the hydrolysis of the imine group, an oxidative deamination, and the formation of a new Schiff base (Fig. 8A).26 The oxidative deamination is central to this process and most certainly requires an enzyme. Evidence of enzymatic involvement was afforded by feeding experiments with labelled cadaverine (16), which revealed that the carbon atom being oxidized {corresponding to C10 of (−)-sparteine [(−)-8]} loses one of its hydrogens selectively during biosynthesis (Fig. 3D). However, in contrast to the oxidative deamination of cadaverine (16) catalyzed by a CAO, the hydrogen lost in this case is the pro-S hydrogen.39 This clear mechanistic difference strongly suggests that this deamination is performed by a different enzyme. Possible candidates include a homolog of the cadaverine-oxidizing CAO as well as an enzyme from a different family, such as an aminotransferase or a FAD-dependent oxidase.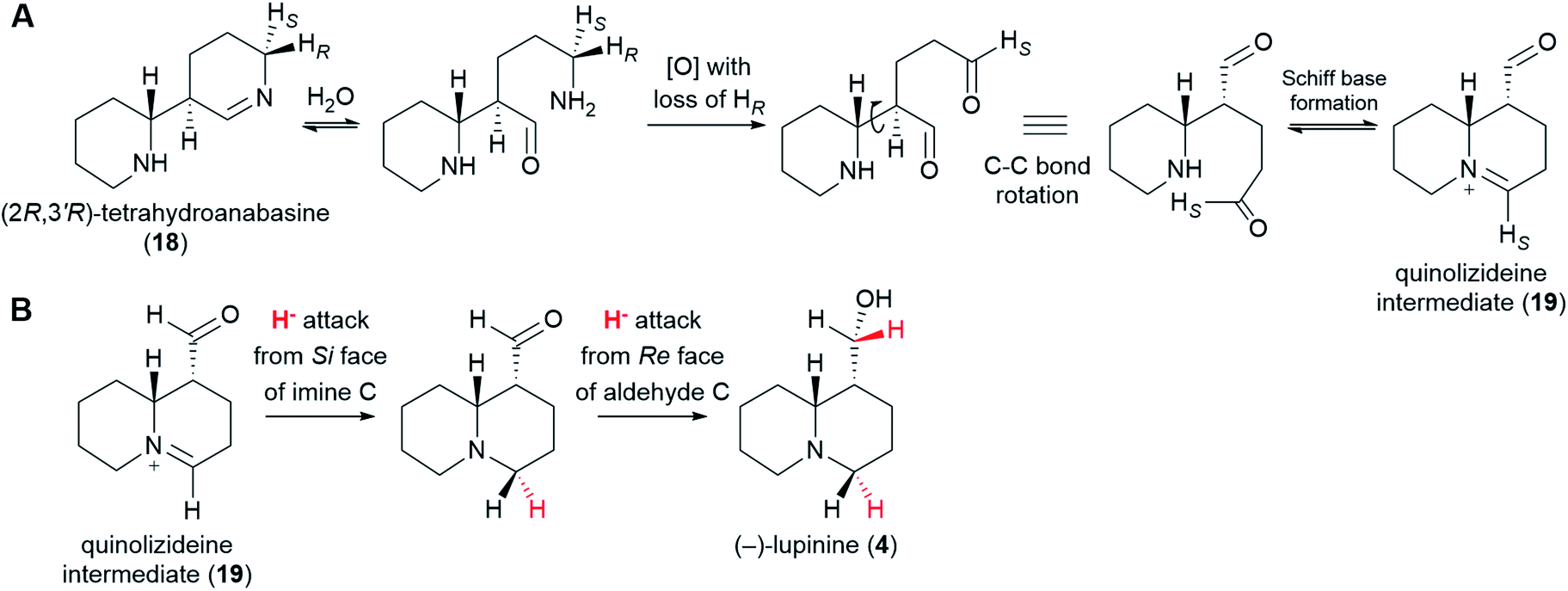 | ||
| Fig. 8 Postulated formation of the quinolizideine intermediate (19) and branching towards the bicyclic QAs. (A) (2R,3′R)-Tetrahydroanabasine (18) undergoes hydrolysis, oxidative deamination, and formation of a new Schiff base to give the quinolizideine intermediate (19). The oxidative deamination occurs with concomitant loss of the pro-R proton and must be enzymatic.39 By contrast, the hydrolysis and the formation of a new Schiff base may be spontaneous under physiological conditions. (B) Proposed conversion of the quinolizideine intermediate (19) to (−)-lupinine (4). The stereochemistry of hydride donation is indicated (inferred from precursor feeding studies).56 The sequence shown indicates a reduction of the imine carbon followed by a reduction of the aldehyde carbon; however, the precise order remains unknown. | ||
With respect to the hydrolysis and the formation of a new Schiff base, both of these processes are likely part of chemical equilibria and may occur spontaneously under physiological conditions. Whether enzymes are used in vivo to increase the rate of these equilibrations remains unknown.
The quinolizideine intermediate (19) is likely to be the last common intermediate between the tetracyclic and the bicyclic QAs. From here, two consecutive, enzyme-catalyzed reductions would afford the bicyclic QA (−)-lupinine (4) (Fig. 8B). The stereochemistry of these reductions has been determined by feeding L. luteus with (R)-(1-2H)cadaverine and (S)-(1-2H)cadaverine and analyzing the resulting (−)-lupinine (4) via2H-NMR (not shown in Fig. 3).56 The results show that the iminium carbon is reduced from the Si face, whereas the aldehyde carbon is reduced from the Re face. The precise order of these reactions remains to be investigated.
6.5 Formation of the tetracyclic di-iminium cation intermediate
In order to form a tetracyclic structure, one last molecule of Δ1-piperideine (17) is thought to be coupled to the quinolizideine intermediate (19). Given the right stereochemistry, the resulting product can undergo intramolecular Schiff base formation leading to the tetracyclic di-iminium cation (20) (Fig. 9).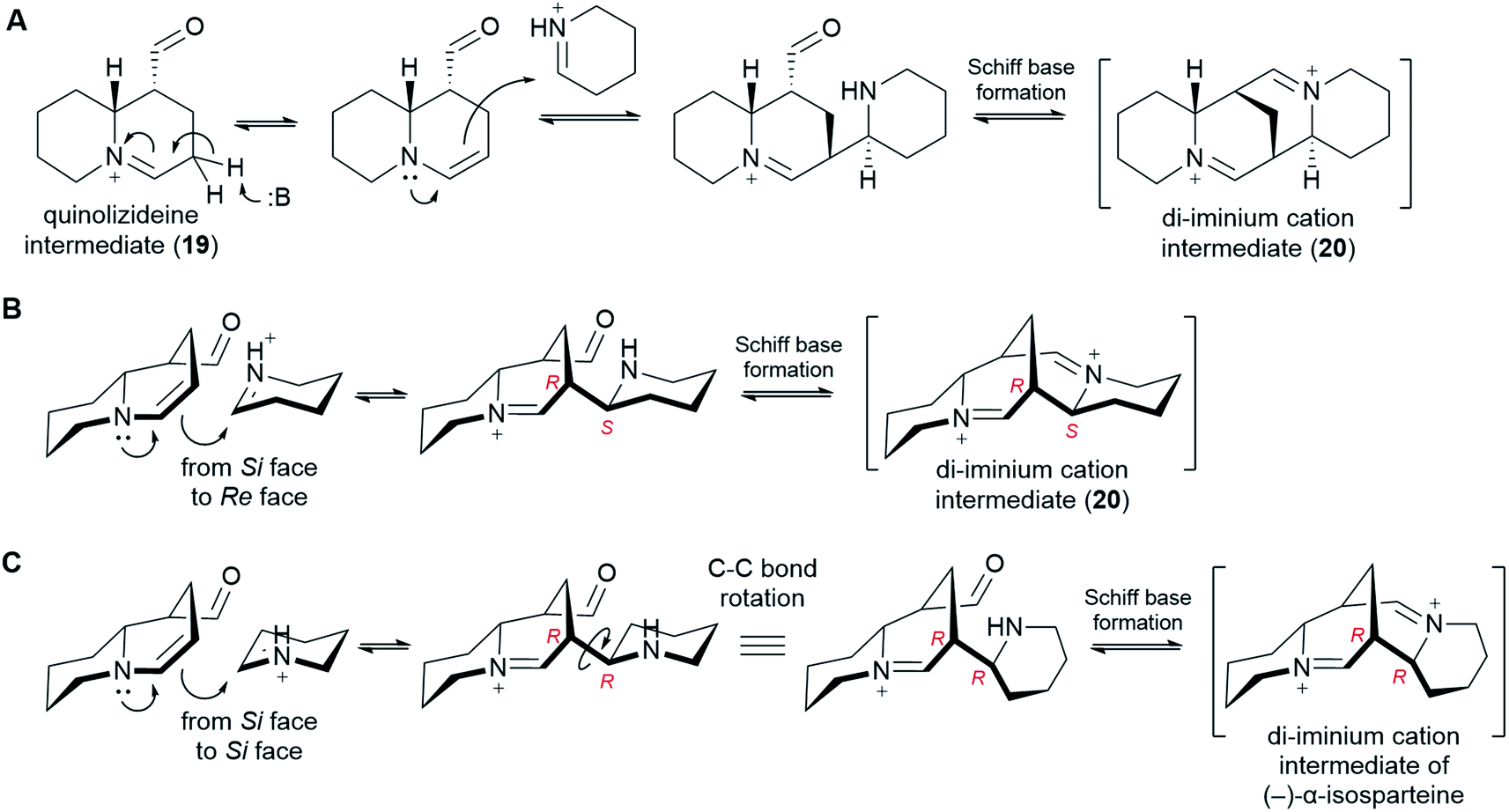 | ||
| Fig. 9 Proposed formation of the tetracyclic di-iminium cation intermediate (20) from the bicyclic quinolizideine intermediate (19). (A) Mechanistic proposal involving tautomerization, stereoselective coupling to Δ1-piperideine and Schiff base formation. (B) 3D representation of the coupling step, specifying the attack from the Si face of the tautomerized quinolizideine intermediate to the Re face of Δ1-piperideine (17). (C) 3D representation of a coupling step that would lead to the tetracyclic backbone of (−)-α-isosparteine (14). | ||
The coupling reaction is similar to the dimerization of Δ1-piperideine (17) described in sub-Section 6.3. By analogy, it has been proposed that the quinolizideine intermediate (19) undergoes base-catalyzed tautomerization to give an enamine that then attacks a protonated Δ1-piperideine (17) molecule (Fig. 9A).26 In the case of the dimerization of Δ1-piperideine (17) (the first coupling event), we noted earlier that the deprotonation at C3 necessary to form Δ2-piperideine (27) occurred stereoselectively, thus strongly arguing for the involvement of an enzyme (Fig. 7C). Selective proton abstraction could be asserted given that the unreacted proton was retained in (−)-lupinine (4) (hydrogen at C1 in Fig. 3H).44 In the case of this second coupling event, however, both of the analogous hydrogens are lost on the way to (−)-sparteine [(−)-8] (hydrogens at C9 in Fig. 3G),44 thus making it impossible to formulate a similar argument. However, given the high likelihood of enzyme involvement in the dimerization of Δ1-piperideine, we postulate that this second coupling event is also enzymatic.
In order to give the right stereochemistry to the product, the coupling enzyme must facilitate an attack from the Si face of the tautomerized quinolizideine intermediate to the Re face of protonated Δ1-piperideine (17) (Fig. 9B). This creates the two stereocenters later to become C9 and C11 in (−)-sparteine [(−)-8] (Fig. 2C).|| To create the (−)-α-isosparteine backbone instead, a backbone that predominates in L. argenteus,3,32 the attack should be performed on the Si face of protonated Δ1-piperideine (Fig. 9C), thus ensuring the opposite configuration in the new stereocenter later to become C11 in (−)-α-isosparteine (14) (Fig. 2C).
6.6 From the di-iminium cation to sparteine
The di-iminium cation (20) is poised to become (−)-sparteine [(−)-8] upon sequential reduction. Biological imine reductions are typically enzyme-catalyzed processes where the universal reducing agent NAD(P)H donates a hydride (H−) at a particular face of an iminium carbon.57 The stereochemistry of hydride donation to the di-iminium cation (20) has been inferred from feeding studies with cadaverine deuterated at Cα. Based on the configuration of the label at positions C10 and C17 in (−)-sparteine [(−)-8] (Fig. 3D),39 it can be inferred that hydride donation must occur at the Re face of both iminium carbons (Fig. 10A). It remains to be shown whether two separate enzymes are required or whether a single enzyme can catalyze both reductions.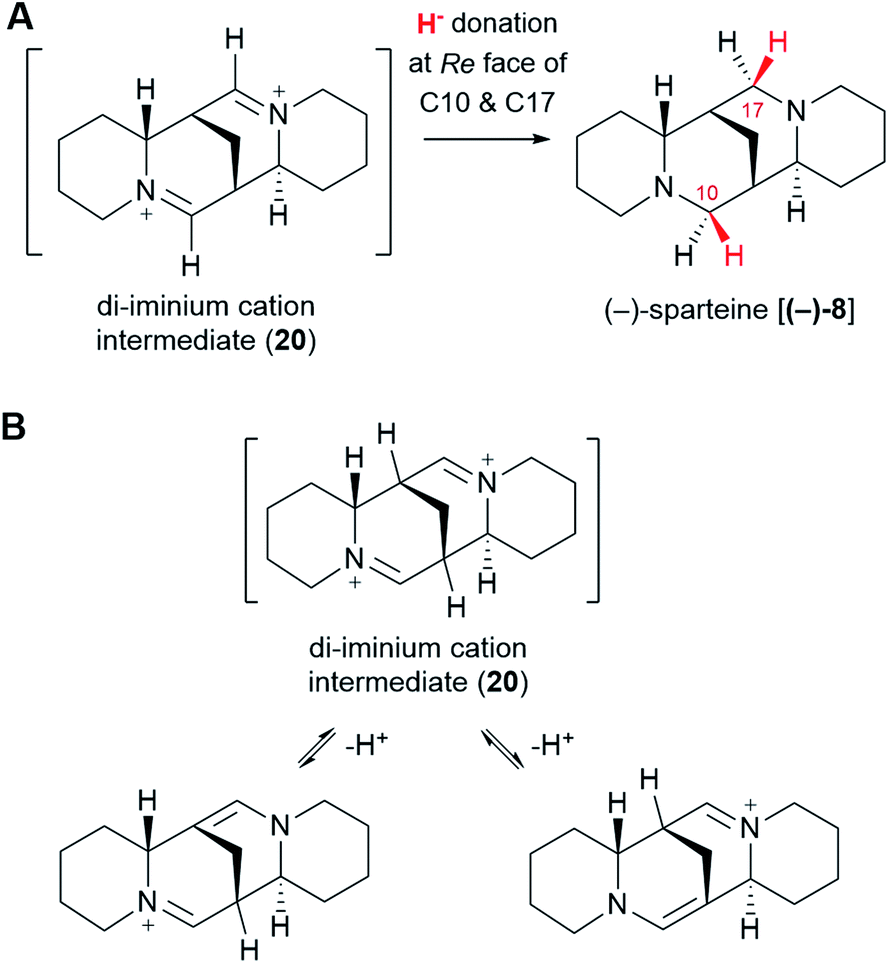 | ||
| Fig. 10 Sequential reduction of the di-iminium cation intermediate (20) to (−)-sparteine [(−)-8]. (A) Stereochemistry of the hydride attack, as inferred from precursor feeding studies. (B) Possible tautomerism of the di-iminium cation (20) that could lead to the observed loss of protons from the bridgehead carbons. | ||
One last interesting result from the feeding studies with cadaverine labelled at Cβ is the loss of the label at the bridgehead carbons C7 and C9 in (−)-sparteine [(−)-8] (Fig. 3G). These two carbons are located next to the iminium carbons of the di-iminium cation (20), and it is tempting to speculate that this position has to do with the loss of label. In particular, it is conceivable that each of the iminium groups is in equilibrium with the corresponding enamine form (Fig. 10B). A rapid establishment of these equilibria prior to reduction could explain the observed loss of the hydrogen atom at these positions. Whether this mechanism or an alternative one is at play remains to be shown experimentally.
7. Potential for pathway elucidation and future directions
The past few decades have witnessed impressive advances in the elucidation of plant-derived alkaloid pathways, for example, those towards the benzylisoquinoline alkaloids (e.g. morphine and codeine),58 the monoterpene indole alkaloids (e.g. vinblastine and vincristine),59 and colchicine.60 Despite these advances, the biosynthesis of several notable alkaloid classes remains obscure. One common theme for these alkaloid classes is the lack of a generally agreed pathway hypothesis that can aid the enzyme discovery efforts. In the present review, we have aimed at closing this particular gap for the sparteine-like tetracyclic QAs by selecting the most likely hypothesis and evaluating the likelihood of enzyme involvement at each pathway step. Our combined proposal is summarized in Fig. 11.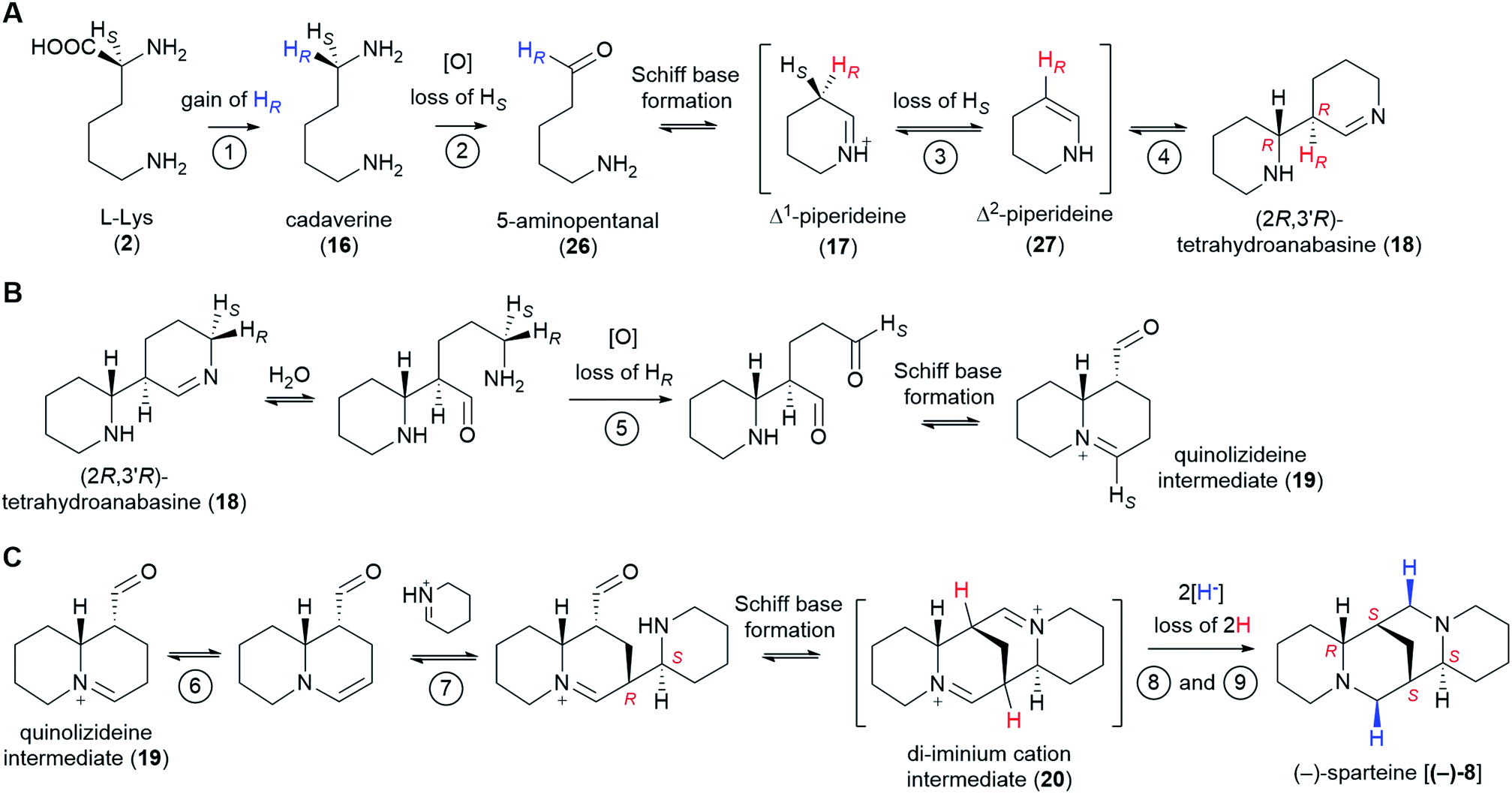 | ||
| Fig. 11 Summary of the proposed QA pathway from L-Lys (2) to (−)-sparteine [(−)-8]. Stereoselective losses/gains of hydrogen are indicated. The configuration of stereocenters is specified only when first produced as well as in the final product. Apparent stereochemical discrepancies with the final (−)-sparteine [(−)-8] product are only due to differences in Cahn–Ingold–Prelog priorities (see footnotes** and ‡). We postulate the involvement of up to 9 enzymes as shown by the encircled numbers. Only the first two enzymes are known: LDC and CAO, respectively. Enzymes 3 and 4 might be the same enzyme or different enzymes acting as part of a tightly bound complex. The same applies to enzymes 6 and 7. Up to two enzymes (enzymes 8 and 9) may be involved in the double reduction of the di-iminium cation intermediate (20) to give (−)-sparteine [(−)-8]. (A) Conversion from L-Lys (2) to (2R,3′R)-tetrahydroanabasine (18). (B) Conversion from (2R,3′R)-tetrahydroanabasine (18) to the quinolizideine intermediate (19). (C) Conversion from the quinolizideine intermediate (19) to (−)-sparteine [(−)-8]. | ||
Recent years have seen the development of a multiplicity of resources for gene discovery in QA-producing species, most notably in lupins. Combined with the curated understanding of the QA pathway presented in this review, the prospects for the full elucidation of the QA pathway are very good. The available resources include high-quality genome drafts for L. angustifolius61–63 and L. albus,64,65 a pan-genome for L. albus,66 and transcriptomic data for a range of lupin species and tissues, as compiled by Kamphuis et al.67 Additionally, transcriptomic data has been generated for Sophora species,68,69 and these are of interest to uncover the pathway towards the matrine-like tetracyclic QAs. However, equivalent resources do not exist for species that primarily accumulate α-pyridone QAs, such as Laburnum anagyroides, which produces the smoking cessation agent (−)-cytisine (12). The fact that the α-pyridone QAs are derived from (+)-sparteine [(+)-8] and not (−)-sparteine [(−)-8] (Fig. 1) increases the interest to target this species for sequencing and QA pathway elucidation.
The sequencing resources mentioned above can be used in different ways for the discovery of QA pathway genes. One powerful way to identify candidates involves the critical inspection of genes that are co-regulated with already known QA genes. Indeed, most plant specialized metabolite pathways are regulated tightly at the transcriptional level, showing distinct expression patterns across organs, tissues, or cell types. This is how the second QA pathway gene CAO was identified in L. angustifolius.52 Another strategy involves mining the published genome drafts for potential QA biosynthetic gene clusters (BGCs) using software such as plantiSMASH.70 While BGCs are less prevalent in plants than in bacteria or fungi, several examples of plant specialized metabolite BGCs have been identified in the past decade.71
Notably, QA pathway genes may also be discovered by uncovering the causative mutations for low-QA phenotypes. In lupin breeding, several different loci are known to control QA levels in L. angustifolius, L. albus, L. luteus and L. mutabilis; however, the identity of the underlying genes remains unknown.72 Candidate genes have been proposed for the iucundus73–75 and pauper76,77 loci, but further work is needed to pinpoint single genes and establish causality. Similarly, mutagenized populations in a high-QA background may yield several low-QA mutant phenotypes, and the causative mutations can be identified in forward genetics approaches. Currently, a mutagenized population in a high-QA background only exists for L. mutabilis.78 It would be of interest to generate analogous mutant populations for the two species with the most sequencing resources, L. angustifolius and L. albus, and subsequently screen for low-QA phenotypes. For both the breeding loci and the eventual new loci obtained via mutagenesis, the underlying genes will likely code for enzymes, regulators, or transporters involved directly or indirectly in the QA pathway.
Once identified, candidate QA pathway genes may be tested and characterized in vitro or in heterologous hosts such as yeast or Nicotiana benthamiana. In addition, there are methods for in planta gene characterization in lupin species. Most notably, gene downregulation in L. angustifolius is now possible using a recently published virus-induced gene silencing (VIGS) method.79 Downregulation of LDC using this method resulted in a marked decrease in QA accumulation in leaves. Stable transformation protocols are also available for L. angustifolius, L. luteus, and L. mutabilis;80–82 however, transformation efficiencies are low. Thus, new and more efficient protocols must be developed before stable transformation can become a viable approach for gene characterization in lupins. An increased focus on such protocols will also enable the use of gene editing technology (e.g. CRISPR/Cas9) in the process of lupin crop improvement.
The full elucidation of the QA pathway will represent an important milestone in the field of alkaloid biosynthesis. It will also contribute to the development of improved lupin crops, as it will allow the creation of new, low-QA varieties with potentially more stable, low-QA phenotypes. Finally, it will allow the cost-effective production of industrially or medicinally important QAs such as (−)-sparteine [(−)-8] or (+)-matrine (6) via synthetic biology.
8. Conclusions
Although several hypotheses have been put forth for the biosynthesis of QAs, only one hypothesis by Golebiewski & Spenser (1988-II)26 is fully consistent with the comprehensive precursor feeding studies carried out in 1964–1994. These feeding studies constitute a solid body of work that further establishes mechanistic constraints on many of the proposed pathway steps. With these constraints in mind, we here propose that between 6 and 9 enzymes catalyze the conversion of L-lysine to (−)-sparteine. The first two pathway enzymes are already known; the rest await discovery. In the face of novel resources for gene discovery and characterization in QA-containing species, the prospects for pathway elucidation are very good. The elucidation of the QA pathway will facilitate the generation of improved lupin varieties and will allow the production of industrially important QAs via synthetic biology.9. Conflicts of interest
There are no conflicts to declare.10. Acknowledgements
We thank the VILLUM Foundation (project 15476), the Novo Nordisk Foundation (projects NNF17OC0027744 and NNF2019OC53580), and the European Union's Horizon 2020 research and innovation programme (Marie Skłodowska-Curie grant agreement 846089) for their generous support.11. Notes and references
- S. Ohmiya, K. Saito and I. Murakoshi, in The Alkaloids: Chemistry and Pharmacology, ed. G. A. Cordell, Academic Press, 1995, vol. 47, pp. 1–114 Search PubMed.
- M. F. Roberts, in Alkaloids: Biochemistry, Ecology, and Medicinal Applications, ed. M. F. Roberts and M. Wink, Springer US, Boston, MA, 1998, pp. 109–146 Search PubMed.
- M. Wink, C. Meißner and L. Witte, Phytochemistry, 1995, 38, 139–153 CrossRef CAS.
- W. Cowling, in Lupins as Crop Plants: Biology, Production, and Utilization, ed. J. S. Gladstones, C. A. Atkins and J. Hamblin, CAB International, 1998, vol. 101, pp. 93–120 Search PubMed.
- M. Wink, L. Witte, T. Hartmann, C. Theuring and V. Volz, Planta Med., 1983, 48, 253–257 CrossRef CAS PubMed.
- P. M. Krishna, K. N. V. Rao, S. Sandhya and D. Banji, Rev. Bras. Farmacogn., 2012, 22, 1145–1154 CrossRef CAS.
- J. Pothier, S.-L. Cheav, N. Galand, C. Dormeau and C. Viel, J. Pharm. Pharmacol., 1998, 50, 949–954 CrossRef CAS.
- A. Di Grande, R. Paradiso, S. Amico, G. Fulco, B. Fantauzza and P. Noto, Eur. J. Emerg. Med., 2004, 11, 119–120 CrossRef PubMed.
- W. A. Cowling and A. Tarr, Aust. J. Agric. Res., 2004, 55, 745–751 CrossRef.
- M. Wink, in Insect-Plant Interactions, ed. E. A. Bernays, CRC Press, Boca Raton, 1992, vol. IV, pp. 131–166 Search PubMed.
- J. Philippi, E. Schliephake, H.-U. Jürgens, G. Jansen and F. Ordon, J. Pestic. Sci., 2016, 89, 359–373 Search PubMed.
- M. Wink, presented in part at the Proceedings of the 6th International Lupin Conference, Temuco, Chile, 1991 Search PubMed.
- J. Senges and L. Ehe, Naunyn-Schmiedeberg's Archives of Pharmacology, 1973, 280, 265–274 CrossRef CAS PubMed.
- F. Villalpando-Vargas and L. Medina-Ceja, Seizure - European Journal of Epilepsy, 2016, 39, 49–55 CrossRef.
- J. Du, J. Li, D. Song, Q. Li, L. Li, B. Li and L. Li, Mol. Med. Rep., 2020, 22, 3659–3666 CAS.
- X. Zhang, G. Hou, A. Liu, H. Xu, Y. Guan, Y. Wu, J. Deng and X. Cao, Cell Death Dis., 2019, 10, 770 CrossRef PubMed.
- J. F. Etter, Arch. Intern. Med., 2006, 166, 1553–1559 CrossRef CAS PubMed.
- N. Walker, C. Howe, M. Glover, H. McRobbie, J. Barnes, V. Nosa, V. Parag, B. Bassett and C. Bullen, N. Engl. J. Med., 2014, 371, 2353–2362 CrossRef.
- D. Hoppe and T. Hense, Angew. Chem., Int. Ed. Engl., 1997, 36, 2282–2316 CrossRef CAS.
- S. K. Ritter, Chem. Eng. News, 2017, 95, 18–20 Search PubMed.
- M. Wink, Phytochemistry, 2003, 64, 3–19 CrossRef CAS PubMed.
- M. F. Wojciechowski, M. Lavin and M. J. Sanderson, Am. J. Bot., 2004, 91, 1846–1862 CrossRef CAS PubMed.
- G. C. Kite, D. Cardoso, N. C. Veitch and G. P. Lewis, S. Afr. J. Bot., 2013, 89, 176–180 CrossRef CAS.
- M. Wink, in Methods in Plant Biochemistry, ed. P. Waterman, Academic Press Limited, 1993, vol. 8, pp. 197–239 Search PubMed.
- H. R. Schütte and H. Hindorf, Justus Liebigs Ann. Chem., 1965, 685, 187–194 CrossRef.
- W. M. Golebiewski and I. D. Spenser, Can. J. Chem., 1988, 66, 1734–1748 CrossRef CAS.
- J. Rana and D. J. Robins, J. Chem. Res., Synop., 1985, 196–197 CAS.
- A. M. Fraser, D. J. Robins and G. N. Sheldrake, J. Chem. Soc., Perkin Trans. 1, 1988, 3275–3279 RSC.
- M. Wink and L. Witte, Planta, 1984, 161, 519–524 CrossRef CAS.
- G. Boschin, P. Annicchiarico, D. Resta, A. D'Agostina and A. Arnoldi, J. Agric. Food Chem., 2008, 56, 3657–3663 CrossRef CAS PubMed.
- M. a. de Cortes Sánchez, P. Altares, M. M. Pedrosa, C. Burbano, C. Cuadrado, C. Goyoaga, M. Muzquiz, C. Jiménez-Martínez and G. Dávila-Ortiz, Food Chem., 2005, 90, 347–355 CrossRef.
- W. J. Keller, B. N. Meyer and J. L. McLaughlin, J. Pharm. Sci., 1983, 72, 563–564 CrossRef CAS.
- R. Greenhalgh and L. Marion, Can. J. Chem., 1956, 34, 456–458 CrossRef CAS.
- M. Carmack, B. Douglas, E. W. Martin and H. Suss, J. Am. Chem. Soc., 1955, 77, 4435 CrossRef CAS.
- L. S. Davis, Arch. Pharm., 1897, 235, 199–217 CrossRef CAS.
- K. Gerhard, Arch. Pharm., 1897, 235, 355–363 CrossRef CAS.
- A. K. Przybył and M. Kubicki, Tetrahedron, 2011, 67, 7787–7793 CrossRef.
- H.-R. Schütte, H. Hindorf, K. Mothes and G. Hübner, Justus Liebigs Ann. Chem., 1964, 680, 93–104 CrossRef.
- W. M. Golebiewski and I. D. Spenser, J. Am. Chem. Soc., 1984, 106, 7925–7927 CrossRef CAS.
- G. Floris and A. Finazzi Agrò, in Encyclopedia of Biological Chemistry, ed. W. J. Lennarz and M. D. Lane, Academic Press, Waltham, 2nd edn, 2013, pp. 87–90 Search PubMed.
- A. M. Fraser and D. J. Robins, J. Chem. Soc., Perkin Trans. 1, 1987, 105–109 RSC.
- W. M. Golebiewski and I. D. Spenser, J. Am. Chem. Soc., 1976, 98, 6726–6728 CrossRef CAS.
- T. Hemscheidt and I. D. Spenser, Can. J. Chem., 1987, 65, 170–174 CrossRef CAS.
- D. J. Robins and G. N. Sheldrake, J. Chem. Soc., Chem. Commun., 1994, 1331–1332 RSC.
- M. Wink and T. Hartmann, Z. Naturforsch., 1979, 34, 704–708 CrossRef CAS PubMed.
- K. Saito and I. Murakoshi, in Studies in Natural Products Chemistry, ed. R. Atta ur, Elsevier, 1995, vol. 15, pp. 519–549 Search PubMed.
- P. M. Dewick, Medicinal Natural Products: a Biosynthetic Approach, John Wiley & Sons, Ltd, 3rd edn, 2009, pp. 328–330 Search PubMed.
- S. Bunsupa, K. Katayama, E. Ikeura, A. Oikawa, K. Toyooka, K. Saito and M. Yamazaki, Plant Cell, 2012, 24, 1202–1216 CrossRef CAS PubMed.
- E. J. Fogle and M. D. Toney, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 1113–1119 CrossRef CAS PubMed.
- M. Wink, T. Hartmann and L. Witte, Z. Naturforsch., C: J. Biosci., 1980, 35, 93–97 CrossRef.
- M. Wink and T. Hartmann, FEBS Lett., 1979, 101, 343–346 CrossRef CAS PubMed.
- T. Yang, I. Nagy, D. Mancinotti, S. L. Otterbach, T. B. Andersen, M. S. Motawia, T. Asp and F. Geu-Flores, J. Exp. Bot., 2017, 68, 5527–5537 CrossRef CAS PubMed.
- C. Schöpf, A. Komzak, F. Braun, E. Jacobi, M.-L. Bormuth, M. Bullnheimer and I. Hagel, Justus Liebigs Ann. Chem., 1948, 559, 1–42 CrossRef.
- H. Sato, M. Uchiyama, K. Saito and M. Yamazaki, Metabolites, 2018, 8, 48 CrossRef PubMed.
- M. Dastmalchi, X. Chen, J. M. Hagel, L. Chang, R. Chen, S. Ramasamy, S. Yeaman and P. J. Facchini, Nat. Chem. Biol., 2019, 15, 384–390 CrossRef CAS PubMed.
- W. Golebiewski and I. Spenser, Can. J. Chem., 1985, 63, 2707–2718 CrossRef CAS.
- J. Mangas-Sanchez, S. P. France, S. L. Montgomery, G. A. Aleku, H. Man, M. Sharma, J. I. Ramsden, G. Grogan and N. J. Turner, Curr. Opin. Chem. Biol., 2017, 37, 19–25 CrossRef CAS PubMed.
- A. Singh, I. M. Menéndez-Perdomo and P. J. Facchini, Phytochem. Rev., 2019, 18, 1457–1482 CrossRef.
- L. Caputi, J. Franke, S. C. Farrow, K. Chung, R. M. E. Payne, T.-D. Nguyen, T.-T. T. Dang, I. S. T. Carqueijeiro, K. Koudounas, T. D. d. Bernonville, B. Ameyaw, D. M. Jones, I. J. C. Vieira, V. Courdavault and S. E. O'Connor, Science, 2018, 360, 1235–1239 CrossRef CAS PubMed.
- R. S. Nett, W. Lau and E. S. Sattely, Nature, 2020, 584, 148–153 CrossRef CAS PubMed.
- J. K. Hane, Y. Ming, L. G. Kamphuis, M. N. Nelson, G. Garg, C. A. Atkins, P. E. Bayer, A. Bravo, S. Bringans and S. Cannon, Plant Biotechnol. J., 2017, 15, 318–330 CrossRef CAS PubMed.
- H. Yang, Y. Tao, Z. Zheng, Q. Zhang, G. Zhou, M. W. Sweetingham, J. G. Howieson and C. Li, PLoS One, 2013, 8, e64799 CrossRef CAS PubMed.
- P. Wang, G. Zhou, J. Jian, H. Yang, D. Renshaw, M. K. Aubert, J. Clements, T. He, M. Sweetingham and C. Li, Plant J., 2021, 105, 1192–1210 CrossRef CAS PubMed.
- B. Hufnagel, A. Marques, A. Soriano, L. Marquès, F. Divol, P. Doumas, E. Sallet, D. Mancinotti, S. Carrere and W. Marande, Nat. Commun., 2020, 11, 1–12 CrossRef PubMed.
- W. Xu, Q. Zhang, W. Yuan, F. Xu, M. M. Aslam, R. Miao, Y. Li, Q. Wang, X. Li and X. Zhang, Nat. Commun., 2020, 11, 1–13 CrossRef PubMed.
- B. Hufnagel, A. Soriano, J. Taylor, F. Divol, M. Kroc, H. Sanders, L. Yeheyis, M. Nelson and B. Péret, Plant Biotechnol. J., 2021, 19, 2532–2543 CrossRef CAS PubMed.
- L. G. Kamphuis, G. Garg, R. Foley and K. B. Singh, Legume Sci., 2021, 3, e77 CAS.
- R. Han, H. Takahashi, M. Nakamura, S. Bunsupa, N. Yoshimoto, H. Yamamoto, H. Suzuki, D. Shibata, M. Yamazaki and K. Saito, Biol. Pharm. Bull., 2015, 38, 876–883 CrossRef CAS PubMed.
- Y. Liang, K. Wei, F. Wei, S. Qin, C. Deng, Y. Lin, M. Li, L. Gu, G. Wei and J. Miao, BMC Plant Biol., 2021, 21, 1–20 CrossRef PubMed.
- S. A. Kautsar, H. G. Suarez Duran, K. Blin, A. Osbourn and M. H. Medema, Nucleic Acids Res., 2017, 45, W55–W63 CrossRef CAS PubMed.
- G. Polturak and A. Osbourn, PLoS Pathog., 2021, 17, e1009698 CrossRef CAS PubMed.
- K. M. Frick, L. G. Kamphuis, K. H. M. Siddique, K. B. Singh and R. C. Foley, Frontiers in Plant Science, 2017, 8, 87 CrossRef PubMed.
- M. Kroc, G. Koczyk, K. A. Kamel, K. Czepiel, O. Fedorowicz-Strońska, P. Krajewski, J. Kosińska, J. Podkowiński, P. Wilczura and W. Święcicki, Sci. Rep., 2019, 9, 2231 CrossRef PubMed.
- P. Plewiński, M. Książkiewicz, S. Rychel-Bielska, E. Rudy and B. Wolko, Int. J. Mol. Sci., 2019, 20, 5670 CrossRef PubMed.
- P. Wang, G. Zhou, J. Jian, H. Yang, D. Renshaw, M. K. Aubert, J. Clements, T. He, M. Sweetingham and C. Li, Plant J., 2021, 105, 1192–1210 CrossRef CAS PubMed.
- S. Rychel and M. Książkiewicz, J. Appl. Genet., 2019, 60, 269–281 CrossRef CAS PubMed.
- C. E. Osorio and B. J. Till, Frontiers in Plant Science, 2022, 12, 795091 CrossRef PubMed.
- R. Galek, E. Sawicka-Sienkiewicz, D. Zalewski, S. Stawiński and K. Spychała, Czech J. Genet. Plant Breed., 2017, 53, 55–62 CrossRef CAS.
- D. Mancinotti, M. C. Rodriguez, K. M. Frick, B. Dueholm, D. G. Jepsen, N. Agerbirk and F. Geu-Flores, Plant Methods, 2021, 17, 1–13 CrossRef PubMed.
- A. Pigeaire, D. Abernethy, P. M. Smith, K. Simpson, N. Fletcher, C.-Y. Lu, C. A. Atkins and E. Cornish, Mol. Breed., 1997, 3, 341–349 CrossRef CAS.
- H. Li, S. Wylie and M. Jones, Plant Cell Rep., 2000, 19, 634–637 CrossRef CAS PubMed.
- P. L. Polowick, N. N. Loukanina and K. M. Doshi, In Vitro Cell. Dev. Biol.: Plant, 2014, 50, 401–411 CrossRef CAS.
Footnotes |
| † To name the isotopically modified compounds used in precursor feeding studies, we here use square brackets surrounding the nuclide symbols to indicate partial labeling and round brackets for isotopically substituted compounds (full or near full labeling). |
| ‡ For readers who have noticed that the equivalence of the (6R,7S,9S,11S) and the (6S,7S,9S,11R) forms of (−)-sparteine [(−)-8] (Fig. 2C and Section 3) virtually enables an alternative biosynthetic route via (2S,3′S)-tetrahydroanabasine, we would like to point out that this alternative route is not supported by the precursor feeding experiments that show preferential incorporation of label into ring D of the (6R,7S,9S,11S) form specifically (Section 4). Such alternative route would lead to preferential incorporation into ring D of the (6S,7S,9S,11R) form, but this ring corresponds to ring A of the (6R,7S,9S,11S) form, as visualized via the 180°-rotation shown in Fig. 2C. |
| § Support for a single-enzyme hypothesis might appear to come from the fact that the two C5 units coupled here are incorporated into the sparteine-like QAs to an equivalent extent.26 This absence of differential extent of labelling, however, may be explained by either the action of a single enzyme catalyzing both proposed steps [without releasing the intermediate Δ2-piperideine (27) into solution] or by two tightly bound enzymes acting as part of a so-called metabolon. |
| ¶ For readers that have noticed that the stereocenter at C3′ in the proposed (2R,3′R)-tetrahydroanabasine intermediate (18) is the R configuration whereas the corresponding stereocenter at C7 in (−)-sparteine [(−)-8] is in the S one, we would like to note that this difference corresponds to a difference in Cahn–Ingold–Prelog priorities rather than a true difference in stereochemistry. |
| || Note that the two newly formed stereocenters depicted in Fig. 9B must be in the (R,S) configuration. The fact that (−)-sparteine [(−)-8] possesses an (S,S) configuration at the corresponding C9 and C11 stereocenters is due to a difference in Cahn–Ingold–Prelog priorities between (−)-sparteine [(−)-8] and the di-iminium cation intermediate (20) at the first stereocenter and does not indicate to a true difference in stereochemistry. |
| ** In this case, (2,2,4,4-2H4)cadaverine was mixed with an unspecified molar amount of radiolabelled [1,5-14C2]cadaverine tracer before being fed to L. luteus. |
| This journal is © The Royal Society of Chemistry 2022 |