
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhazinilam–leuconolam family of natural products: a half century of total synthesis
Fatih
Sirindil
 ,
Jean-Marc
Weibel
,
Patrick
Pale
* and
Aurélien
Blanc
*
,
Jean-Marc
Weibel
,
Patrick
Pale
* and
Aurélien
Blanc
*
Laboratoire de Synthèse, Réactivité Organiques et Catalyse, Institut de Chimie, UMR 7177 – CNRS, Université de Strasbourg, 4 Rue Blaise Pascal, 67070 Strasbourg, France. E-mail: ppale@unistra.fr; ablanc@unistra.fr
First published on 14th June 2022
Abstract
Covering: 1972 to 2021
The rhazinilam family of natural products exhibits a main structure with a stereogenic quaternary carbon and a tetrahydroindolizine core imbedded within a 9-membered macrocycle, imposing axial chirality. This unique architecture combined with their taxol-like antimitotic activities have attracted various attention, especially from synthetic chemists, notably in the past decade. The present review describes the known total and formal syntheses of the members of the rhazinilam family (rhazinilam, rhazinal, leuconolam and kopsiyunnanines), according to the strategy developed.
 Fatih Sirindil | Fatih Sirindil received a Master degree in Drug Design from the University of Strasbourg. During his internship at Harvard University, supervised by Dr Kevin Hodgetts, he worked on lead optimization for treatment of Alzheimer's disease. In 2015, he obtained his PhD in Chemistry under the supervision of Prof. Patrick Pale and Dr Aurélien Blanc. He developed new methods using gold and palladium, and on its application to total synthesis. He is currently a research fellow at the University College of London with Pr. Erik Arstad. His research focuses on methodology development and synthesis of 18F-labelled radiotracer for PET imaging. |
 Jean-Marc Weibel | Jean-Marc Weibel obtained his PhD in chemistry in 1994 from the University Louis Pasteur of Strasbourg under the supervision of Dr Denis Heissler. After a two-years stay at University College of London as a post-doctoral fellow with Prof. William B. Motherwell, he joined Prof. Pale group in the Chemistry Institute of Strasbourg in 1996 as assistant professor and was appointed as full professor in 2012. His research interests focused on the development of new tools for the organic chemist and on their application to the total synthesis of bioactive molecules and analogs. |
 Patrick Pale | Patrick Pale obtained his PhD in 1982 at the University of Champagne with Prof. J. P. Pete and J. Muzart. After a stay in a pharmaceutical company, he joined the group of Prof. L. Ghosez as postdoc. He then got a CNRS position in Prof. J. Chuche group. He obtained a ‘Doctorat d'Etat’ (Habilitation) in 1988, and then a research associate position at Harvard University in the group of Prof. G. Whitesides in 1988–1990. In 1995, he was appointed as full professor at the University of Strasbourg. His scientific interests include total synthesis, carbohydrate chemistry, homo/heterogeneous catalysis and organocatalysis. |
 Aurélien Blanc | Aurélien Blanc obtained his PhD in organic chemistry in 2004 from the University of Geneva (Switzerland) under the supervision of Prof. Christian Bochet. After a postdoctoral stay in the group of Prof. F. Dean Toste at the University of California, Berkeley (USA), he joined Prof. Pale group in 2006 as a CNRS Fellow at the University of Strasbourg (France). His research focuses on homogeneous and heterogeneous (POM) gold catalysis, especially with small heterocycles, and on their application to the synthesis of natural products. |
1 Introduction
The natural product rhazinilam is the first member of a series of alkaloids isolated from Apocynacea plants, which belong to the Aspidosperma alkaloid family and exhibit unprecedented structures and very interesting biological activities (Fig. 1). Rhazinilam was first isolated from the plant Melodinus australis1 and then from Rhazya sp.2 as well as from Kopsia sp.3 and Leuconotis sp.,4 respectively growing in the desert parts of Iran, Iraq and the Arabic peninsula, and in South East Asia. It has also been found in Quebracho, a South American tree.5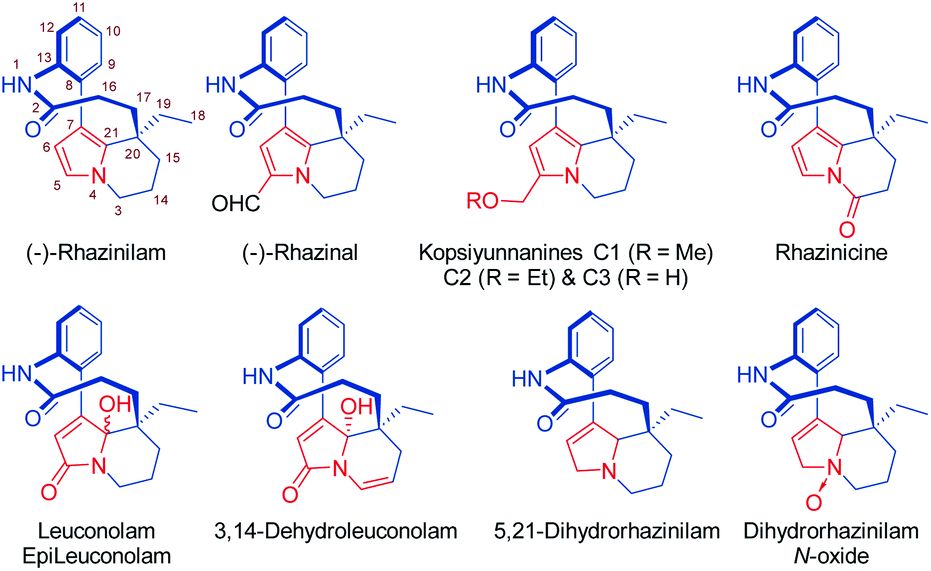 | ||
| Fig. 1 Structures of rhazinilam/leuconolam family of alkaloids including numbering of the rhazinilam structure.14 | ||
Rhazinilam exhibits taxol-like activity at low micromolar range (IC50 0.6–1.2 μM). As various other anticancer agents acting as spindle poisons,6 rhazinilam interacts with microtubules,7 but with a complex mode of action as it both inhibits assembly and disassembly and promotes the formation of abnormal tubulin spirals.7,8 However, only the naturally occurring (−)-enantiomer is active.3a,9
A few other alkaloids related to rhazinilam have also been isolated and characterized. They mostly correspond to oxidized forms of rhazinilam. Rhazinal and rhazinicine are the closest members of this family. They were isolated from Kopsia singapurensis together with other alkaloids.3b,c,10 Both also exhibit tubulin-binding properties, but inferior to that of rhazinilam. Nevertheless, they both showed interesting cytotoxicity toward drug-sensitive and vincristine-resistant KB cells (IC50 0.73 and 4.06 μM for respectively rhazinal and rhazinicine). More recently, the hydroxymethylated derivatives, that correspond to reduced forms of rhazinal, have also been isolated from Kopsia arborea indigenous from Yunnan and thus named kopsiyunnanines C1–3.11 Despite its facile and spontaneous oxidation, the reduced 5,21-dihydrorhazinilam could be isolated and characterized,3a,12 as well as later its N-oxide.4b
More oxidized forms of rhazinilam have also been discovered in other Apocynacea plants growing in Indonesia and peninsular Malaysia. The species Leuconotis griffithii and L. eugenifolia provided the closest members, named leuconolam, and epi-leuconolam.13 A hydrogenated form, the 3,14-dehydroleuconolam, was also isolated.4b In contrast to the rhazinilam members, leuconolam does not exhibit tubulin-related activity.3a
With a stereogenic quaternary carbon and a tetrahydroindolizine core imbedded within a 9-membered macrolactam, which imposes axial chirality, rhazinilam and its close relatives represent a fascinating and challenging target for synthetic chemists. Such a unique structure has thus motivated several total syntheses. Furthermore, the fact that rhazinilam exhibits unique antimitotic activities has led to the development of various analogues as new anticancer drug candidates.9,15
Three short reviews were published relative to rhazinilam total synthesis. Two of them covered rhazinilam syntheses known before 2011 (up to 7 compared to the 20 published now, as summarized in Fig. 2).16 The third only focused on C–H activation with only 3 examples (one for rhazinilam, rhazinal and rhazinicine).17 A book chapter also offered a broader view of the Aspidosperma alkaloid subfamily to which belongs the rhazinilam–rhazinal–leuconolam natural products among others.18 Syntheses were described in chronological order up to mid-2016, together with details on plant origins, spectroscopic characterizations and on the pharmacology of these alkaloids.
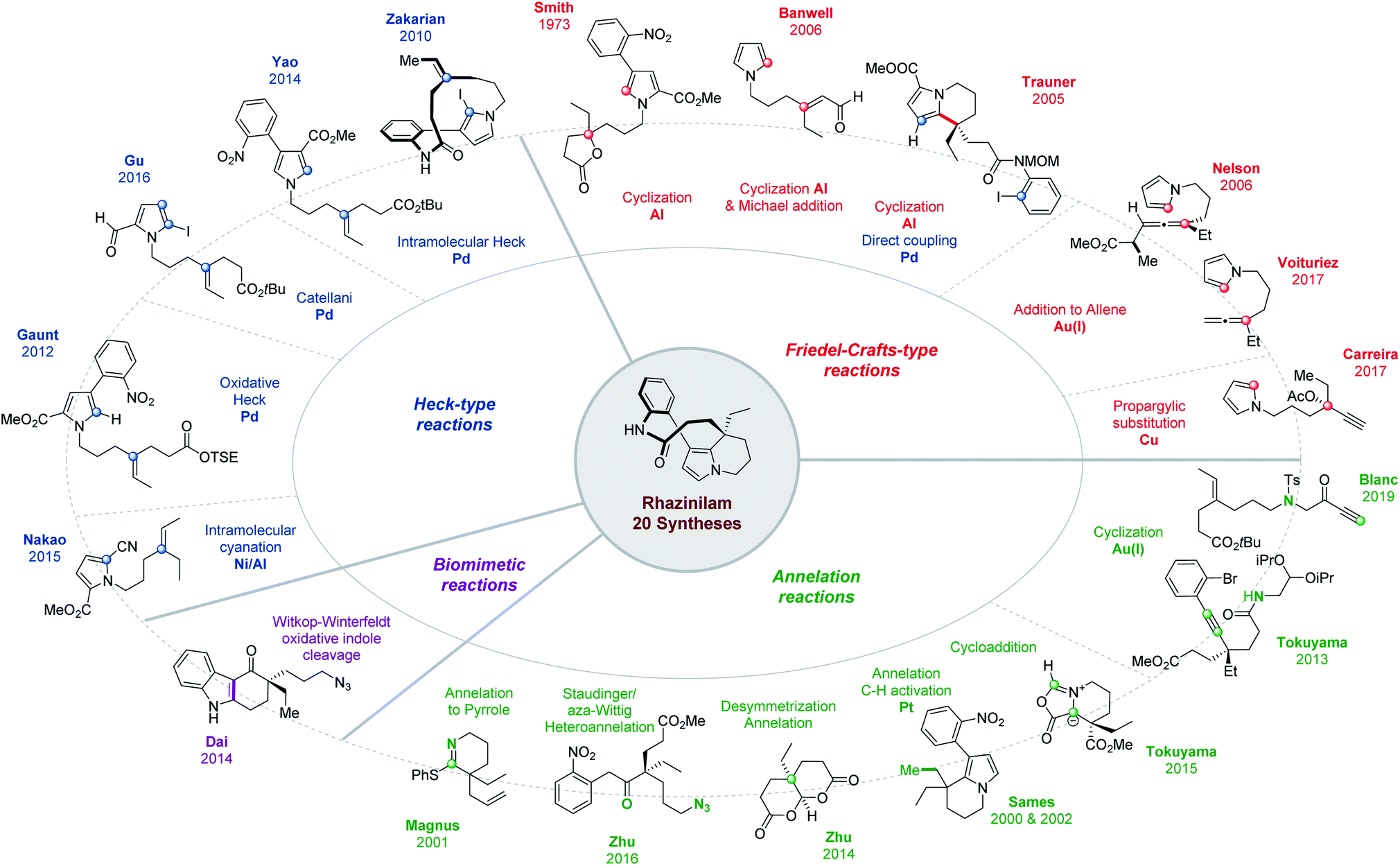 | ||
| Fig. 2 Overview of Rhazinilam syntheses. | ||
As can be seen from Figs. 2 and 3, several total syntheses have now been published for the main members of the rhazinilam family of natural products and few more are emerging, while formal syntheses have also been reported. Therefore, it seems timely to compile and compare them, especially from an organic chemist point of view. Indeed, the various strategies developed to produce such complex structures relied on the most challenging and state-of-the-art synthetic methods (C–H activation, metal-promoted cyclization, oxidative cross-coupling, etc), including various chirality controls. The present review is thus aimed at highlighting these different reactivity and strategic aspects. It is thus organized according to the main strategy used and to the key methods applied.
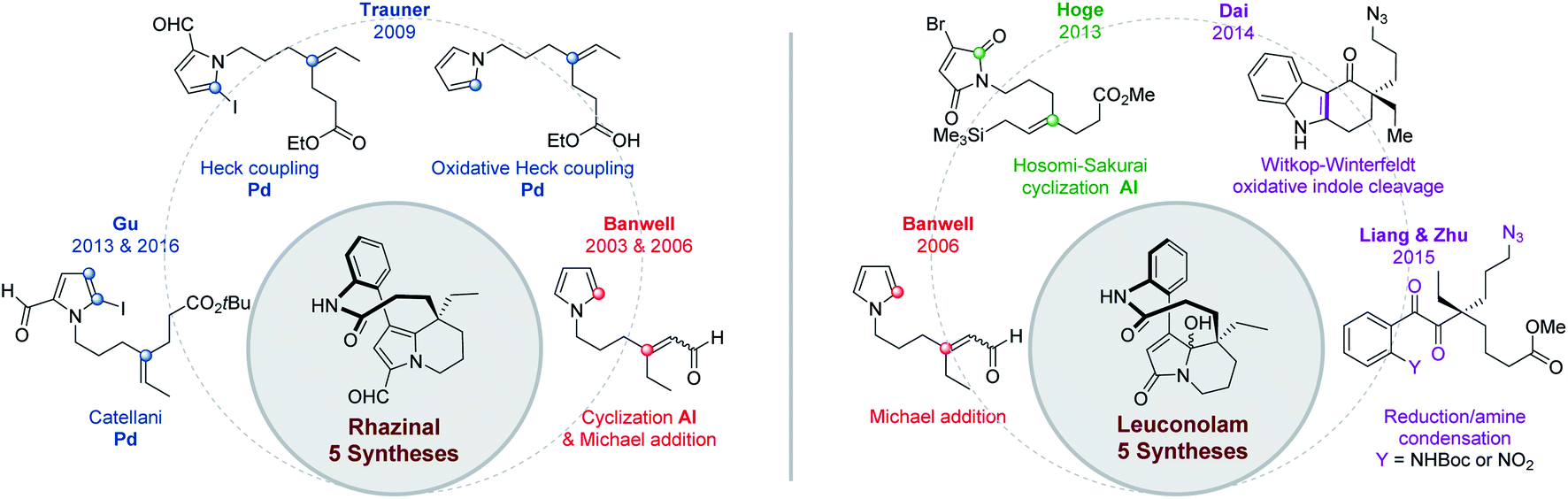 | ||
| Fig. 3 Overview of Rhazinal and Leuconolam total syntheses. | ||
2 Strategies
As mentioned above, the structural complexity of the rhazinilam–rhazinal–leuconolam natural products has induced a surge of creativity both in term of approaches and of transformations which could be applied to their synthesis (see Figs. 2 and 3).Within the various approaches reported in the literature, and besides the few aimed at elucidating their biosynthetic origin, the controlled formation of the tetrahydroindolizine core, the anilinopyrrole motif or the quaternary center have represented the most important challenges. The former sparked off the most variations and innovative methods. We thus focused the present review on the way the tetra-hydroindolizine core was produced. 4 Main methods have been developed to form this central motif carrying the all carbon and stereogenic quaternary centre. The formation of the pyrrole moiety also induced interesting annelation and cycloaddition approaches.
3 Biosynthetically inspired total syntheses
Beside its structural elucidation, rhazinilam arouse a few hemi-syntheses aimed at understanding its biosynthetic pathway and at identifying possible precursors.The first biomimetic synthesis was proposed by Smith19 and coworkers in 1973 based on the fact that rhazinilam gradually accumulated in the basic fractions of plant extracts, suggesting it is formed during extraction and purification from a precursor.2b Suspecting that rhazinilam could be formed from Aspidosperma alkaloids, Smith and coworkers submitted (+)-1,2-dehydroaspidospermidine, also called (+)-eburenine, to oxidative then reductive conditions. They indeed obtained around 30% of (−)-rhazinilam upon m-chloroperoxybenzoic acid (mCPBA) and then ferrous sulfate treatment. They proposed a diaminal intermediate, which could evolve to rhazinilam through elimination (Scheme 1, eqn (1)).
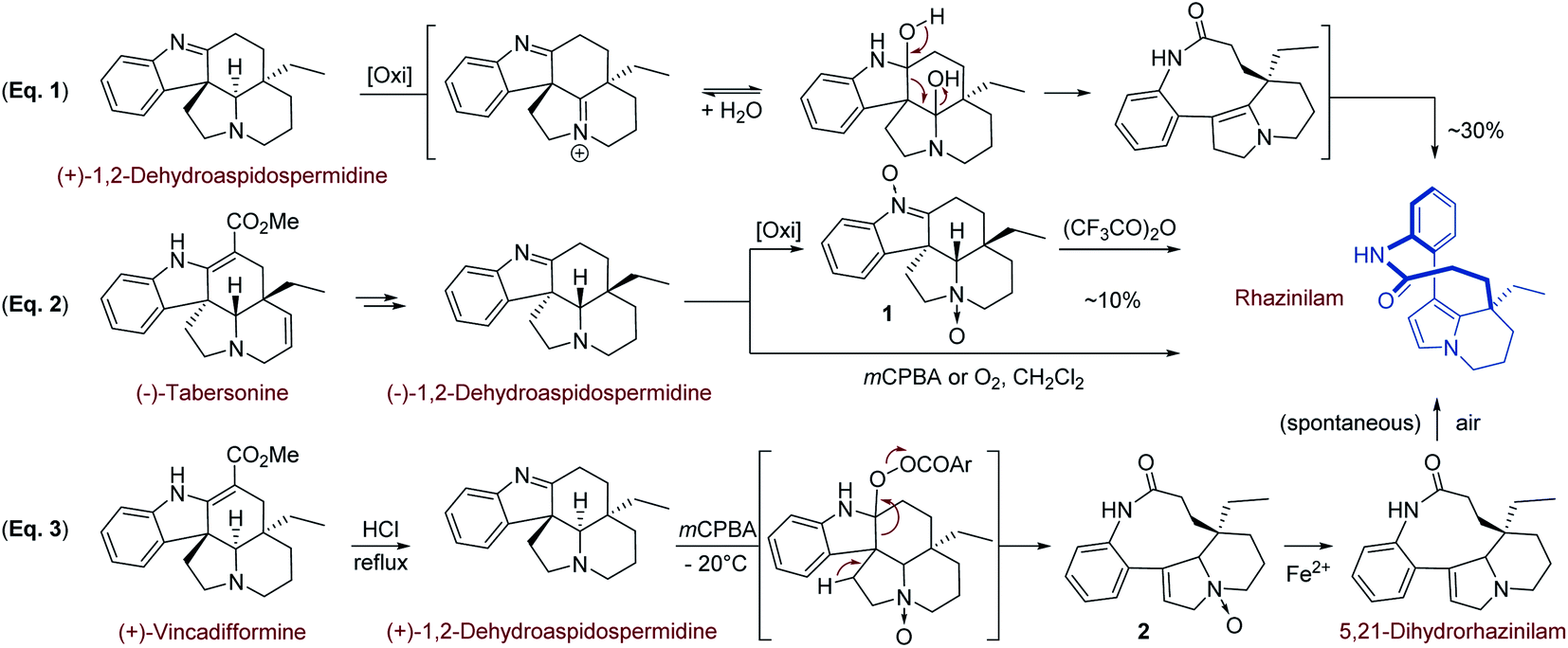 | ||
| Scheme 1 Biomimetic routes to rhazinilam. | ||
As the 5,21-dihydrorhazinilam has been isolated as a minor product together with rhazinilam3a or leuconolam,4a it was envisaged as the actual precursor. Potier and coworkers demonstrated the validity of such biosynthetic route by synthesising rhazinilam from Aspidosperma alkaloid. (−)-Tabersonine could indeed be readily converted to (−)-1,2-dehydroaspidospermidine, which itself under oxidative conditions or its N-oxide upon Polonovski–Potier reaction provided rhazinilam, although in low yields (Scheme 1, eqn (2)).3a
Reinvestigation of these transformations revealed the intermediate formation of (−)-1,2-didehydroaspidospermidine N-oxide 1 and 5,21-didehydrorhazinilam N-oxide 2. Once isolated, the latter led to rhazinilam upon treatment with ferrous sulfate (Scheme 1, eqn (3)).9 Since the starting 1,2-didehydroaspidospermidine can readily be obtained from vincadifformine or tabersonine, the whole biosynthetic pathway was thus elucidated.
More recently, and in a totally different but interesting approach, Daï's group developed a biomimetic but divergent strategy applied to the total synthesis of (±)-rhazinilam and leuconolam, as well as other related natural products (Scheme 2).20 They exploited the biomimetic Witkop–Winterfeldt reaction21 to open the pyrrolyl part, while the resulting diketoamide set the stage for further functionalization. Starting from the commercially available carbazolone 3, the azidoketone 4 was produced in 7 steps and its oxidative cleavage gave the azido hemiaminal 5 after spontaneous transannular cyclization. Azide conversion to acetamide allowed for further cyclization under acidic condition through an iminium intermediate. Subsequent aldolization provided the tetracyclic leuconidine B. Mesylation and elimination afforded melodinine E in 79% yield. The latter was then rearranged to leuconolam in acidic media in 72% yield, through again an iminium intermediate. Finally, DIBAL-H chemoselective reduction of the 5-hydroxypyrrolone moiety of leuconolam furnished (±)-rhazinilam in 74% yield. The latter was thus obtained in 4% yield over 14 steps.
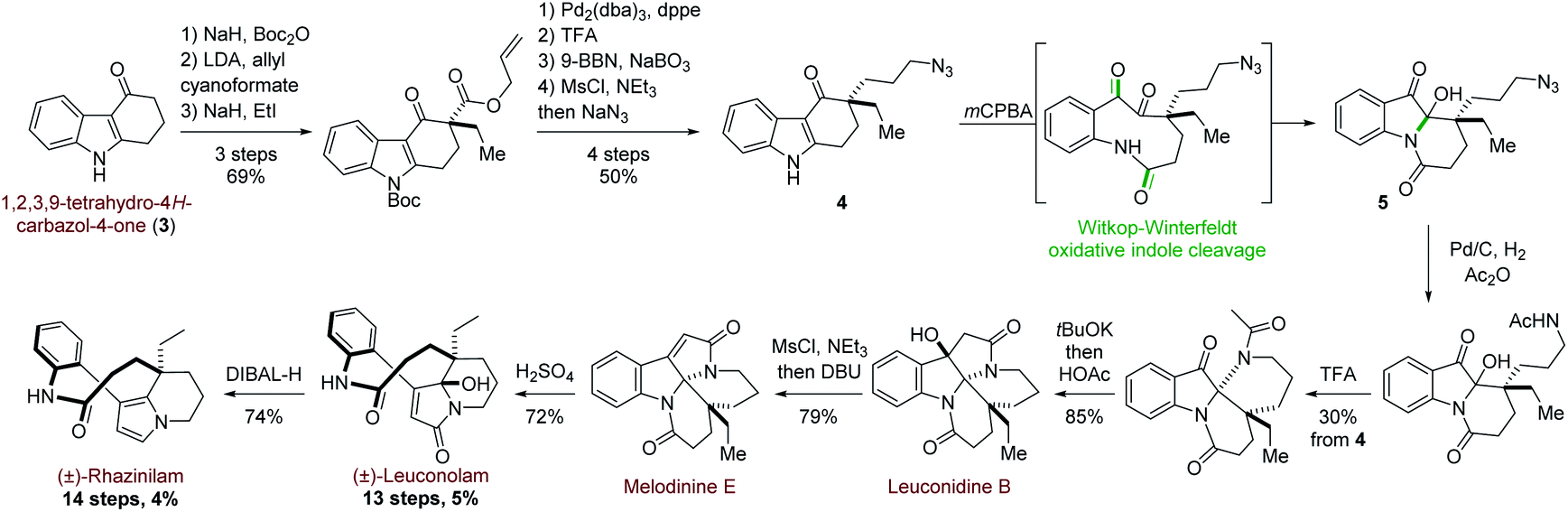 | ||
| Scheme 2 Divergent synthesis of (±)-rhazinilam by Witkop–Winterfeldt oxidative indole cleavage reaction. | ||
In a similary way, (−) and (±)-leuconolam was synthesized by Zhu22 and Liang23 groups from melodinine E or leuconodine B, respectively, after performing their total syntheses. Recently, the Tokuyama's group also achieved the (±)-rhazinilam synthesis in 3 steps from melodinine E via the formation of 6-hydro-21-dehydroxy-leuconolam, which was postulated to be a biosynthetically intermediate of the rhazinilam–leuconolam family.24
4 Total syntheses through Friedel–Crafts-type reactions
With a pyrrole unit in the structure, it is not surprising that several routes have been developed based on the nucleophilic property of such unit. Among them, Friedel–Crafts reactions occupy an historical position, but they also offered rapid accesses to the tetrahydroindolizine core, even in enantioselective way. Furthermore, more recent versions exploited the mildness and efficiency of gold and copper catalysts.4.1 First total synthesis of (±)-rhazinilam
The first total synthesis of (±)-rhazinilam was disclosed in 1973 and relied on N-alkylation and Friedel–Crafts cyclization as key steps. This first achievement required 12 steps to the Smith's group to give the natural product with 7% overall yield (Scheme 3).19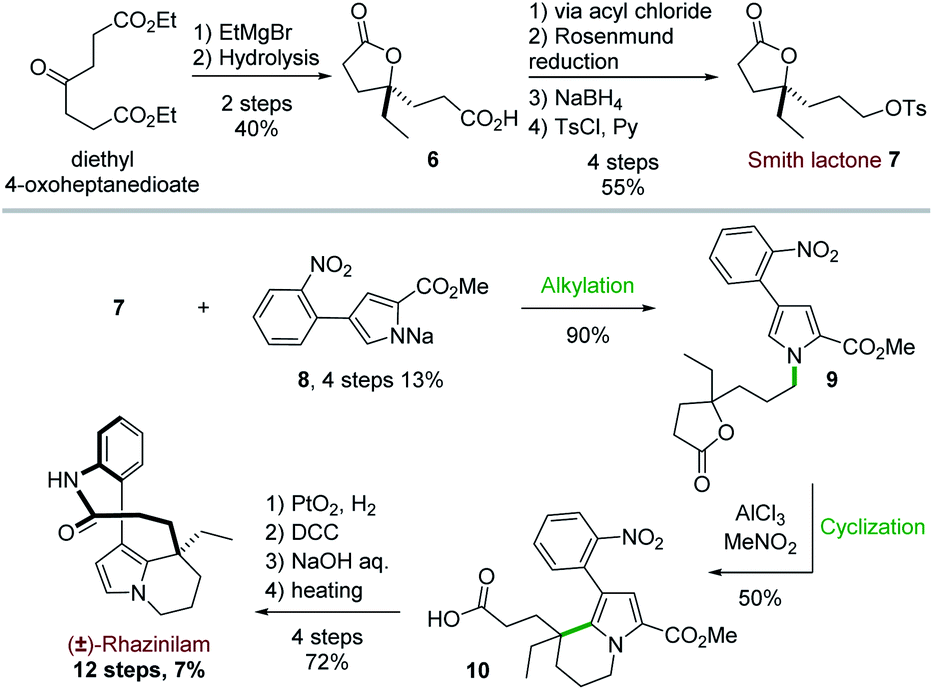 | ||
| Scheme 3 First total synthesis of (±)-rhazinilam. | ||
Starting from diethyl 4-oxoheptanedioate, ethyl magnesium addition and hydrolysis provided the γ-lactone 6 in 40% yield. The lactone acid side chain was then modified through a series of reactions, which converted the acid moiety to the corresponding alcohol and then to its tosylate. The resulting so-called Smith lactone 7 was thus obtained with a 22% overall yield (Scheme 3, top).
Substitution of this tosylate with the pyrrolyl sodium derivative 8 then led to the N-alkylated pyrrole 9 in 90% yield. In the presence of anhydrous aluminium trichloride, the ring opening of the lactone induced an intramolecular cyclization with the pyrrole moiety. This cascade provided the tetrahydroindolizine 10 in 50% yield. The carbomethoxy group at position 2 of the pyrrole 9 directed the cyclization to the desired position by blocking the other nucleophilic pyrrole position. The synthesis was then completed in 4 steps after reduction of the nitro group with Adams catalyst (PtO2) followed by a macrolactamization reaction using dicyclohexylcarbodiimide (DCC). Saponification (aq. NaOH/MeOH) and subsequent decarboxylation under reduced pressure at 240 °C gave the racemic rhazinilam.
4.2 Trauner total synthesis of (±)-rhazinilam
In 2005, the Trauner's group used the Smith strategy to build up the tetrahydroindolizine core of (±)-rhazinilam, but with the simple 2-carboxymethylpyrrole moiety. A nicely designed palladium-catalyzed direct coupling then allowed to stereoselectively form the atropoisomeric macrolactam 13. N-deprotection and decarboxylation provided the natural product, which was thus obtained in 13 steps with 2% overall yield (Scheme 4).25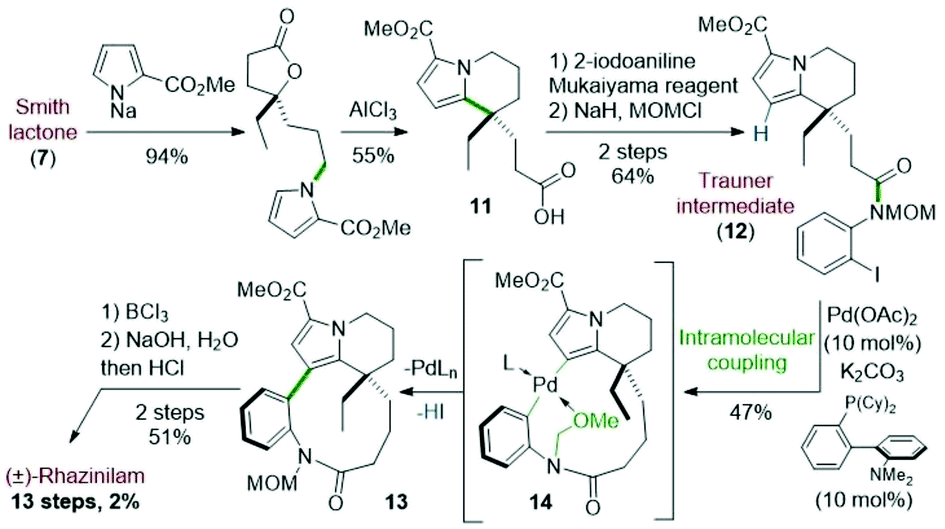 | ||
| Scheme 4 Total synthesis of (±)-rhazinilam proposed by Trauner. | ||
The tetrahydroindolizinyl acid 11 derived from the Smith lactone was converted to the N-protected 2-iodoaniline amide 12, the so-called Trauner intermediate. The latter was engaged in a direct intramolecular coupling catalyzed by Pd(OAc)2/DavePhos, which afforded the macrolactam 13 with 47% yield. The oxidative addition of Pd(0) into the C–I bond initiated the intramolecular cyclization by nucleophilic addition of pyrrole to the Pd(II) centre in a Friedel–Crafts-type reaction. Subsequent deprotonation and final reductive elimination results in the formation of the biaryl bond of the so-formed macrocycle 13. The MOM protective group proved to be essential for this cyclization because the free amide only led to the deiodination product. It may play a stabilizing role after HI elimination (see intermediate 14 in Scheme 4). The total synthesis was then completed in 2 steps from 13 after boron trichloride deprotection of the MOM group, saponification of the pyrrolyl ester and decarboxylation.
4.3 First total synthesis of (±) rhazinal and its extension to an asymmetric version
Thirty years after the first synthesis of (±)-rhazinilam, the Banwell's group reported in 2003 the first total synthesis of (±)-rhazinal, based on a related Friedel–Crafts cyclization. Despite a more linear approach, this synthesis required almost the same number of steps (14) with 4–7% overall yield (Scheme 5).26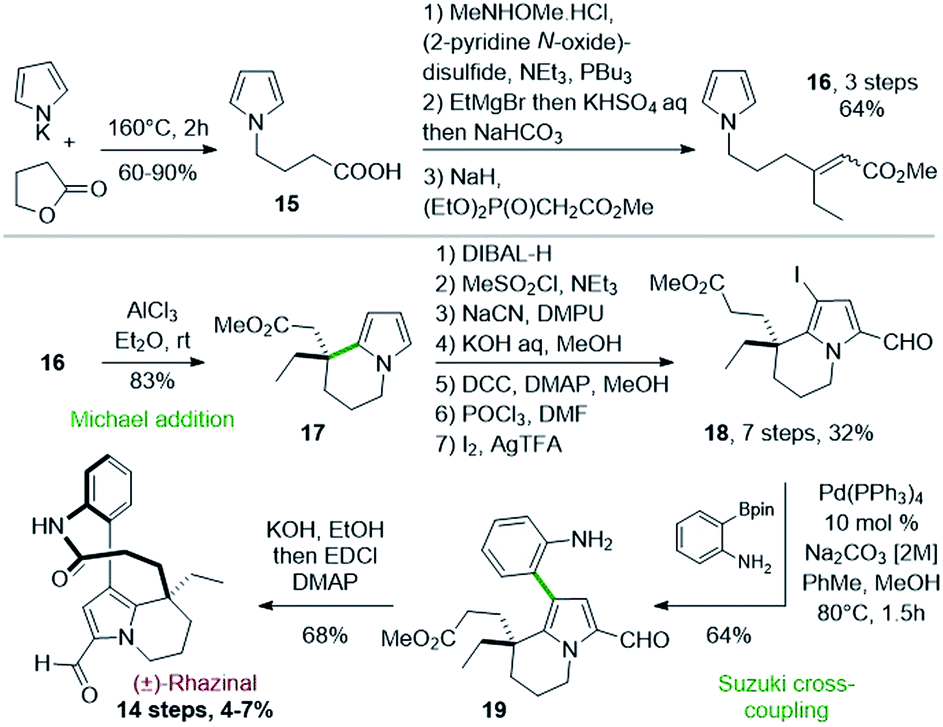 | ||
| Scheme 5 First total synthesis of (±)-rhazinal proposed by Banwell. | ||
The cyclization of a pyrrole N-substituted with a β,β-disubstituted methyl acrylate chain was implemented as the key step. The 4-(pyrrol-1-yl)butanoic acid 15, obtained by heating pyrrolyl potassium salt with γ-butyrolactone, was homologated in a three step sequence. The acid group was converted into a Weinreb amide, its treatment with ethylmagnesium bromide then provided the corresponding ethyl ketone and a Horner–Wadsworth–Emmons (HWE) reaction gave the required acrylate 16 as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z mixture (Scheme 5, top).
:1 E/Z mixture (Scheme 5, top).
This pyrrolo-acrylate 16 was then efficiently cyclized (83% yield) through an intramolecular Friedel–Crafts/Michael addition process upon treatment with 5 equivalents of aluminium chloride. The so-formed tetrahydroindolizine ester 17 was subsequently homologated in 5 steps, formylated and regioselectively iodinated (I2/AgTFA) to the ester 18. Palladium-catalyzed Suzuki–Miyaura cross-coupling between the iodopyrrolyl moiety of 18 and 2-aminophenylboronic pinacol ester afforded the arylated indolizine 19 in 90 minutes with 64% yield. Ester hydrolysis and classical lactamization promoted by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI) gave access to (±)-rhazinal in 68% yield, five years after its extraction and characterization.
Later, the group extended this intramolecular cyclization strategy to the enantioselective synthesis of (−)-rhazinal, (−)-rhazinilam and (−)-leuconolam by using MacMillan's first generation organocatalyst as cyclization promoter (Scheme 6).27 The synthesis started from the previously described pyrrolyl acrylate 16 (1:1 E/Z mixture; Scheme 5, top).
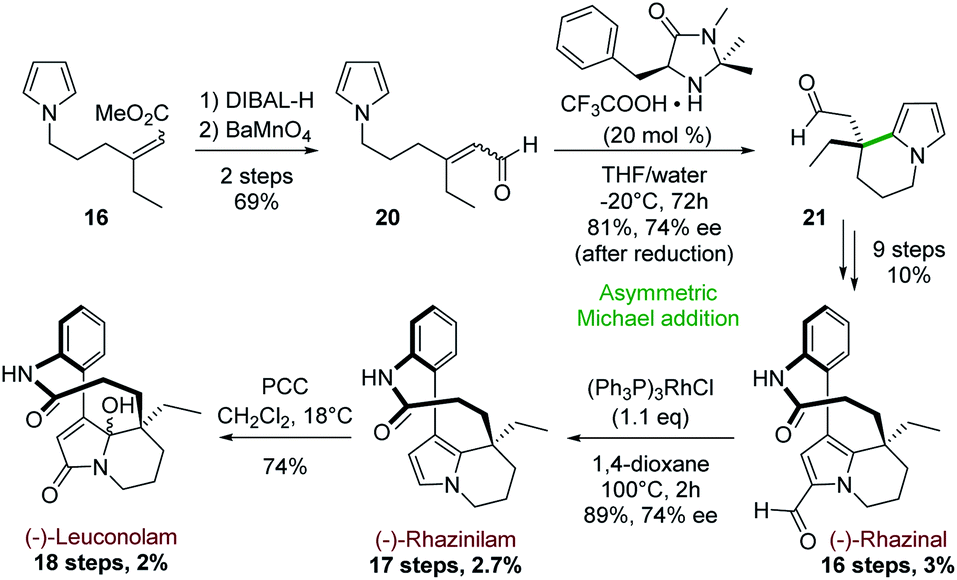 | ||
| Scheme 6 Asymmetric intramolecular Michael addition for the synthesis of (−)-rhazinal and (−)-rhazinilam, and (−)-leuconolam. | ||
The latter was transformed in two steps (69%) to the aldehyde 20 by a sequence of reduction (DIBAL-H) and oxidation of the resulting allylic alcohol using barium manganate (Scheme 6). Prior to cyclization, no effort was made to separate E/Z isomers, because the intermediate iminium ions would undergo rapid interconversion. Indeed, 20 was cyclized in the presence of (5S)-2,2,3-trimethyl-5-phenylmethyl-4-imidazolidinone monotrifluoroacetate (MacMillan's catalyst) and the resulting tetrahydroindolizine 21 was obtained in 81% yield. This unstable aldehyde was directly reduced with sodium borohydride to the stable alcohol, which exhibited an interesting enantiomeric purity (74% ee). The corresponding alcohol was then transformed to (−)-rhazinal according to the previous synthesis (Scheme 5, bottom) without any racemisation. Decarbonylation of (−)-rhazinal with the Wilkinson's catalyst produced (−)-rhazinilam in 89% yield and 74% ee.
Further PCC oxidation of the latter natural product readily produced the (−)-leuconolam and its epimer with 74% yield in a 62:38 ratio.
4.4 Gold(I)-catalyzed cyclization of enantioenriched pyrrolo allene
In 2006, Nelson and coworkers described an asymmetric synthesis of (−)-rhazinilam, in which the key step was catalyzed by gold(I), in 14 steps with the better overall yield to date (22%). This synthesis relied first on an enantiopure β-lactone formation and then on gold-catalyzed intramolecular addition of a pyrrole to an enantioenriched trisubstituted allene derived from the β-lactone (Scheme 7).28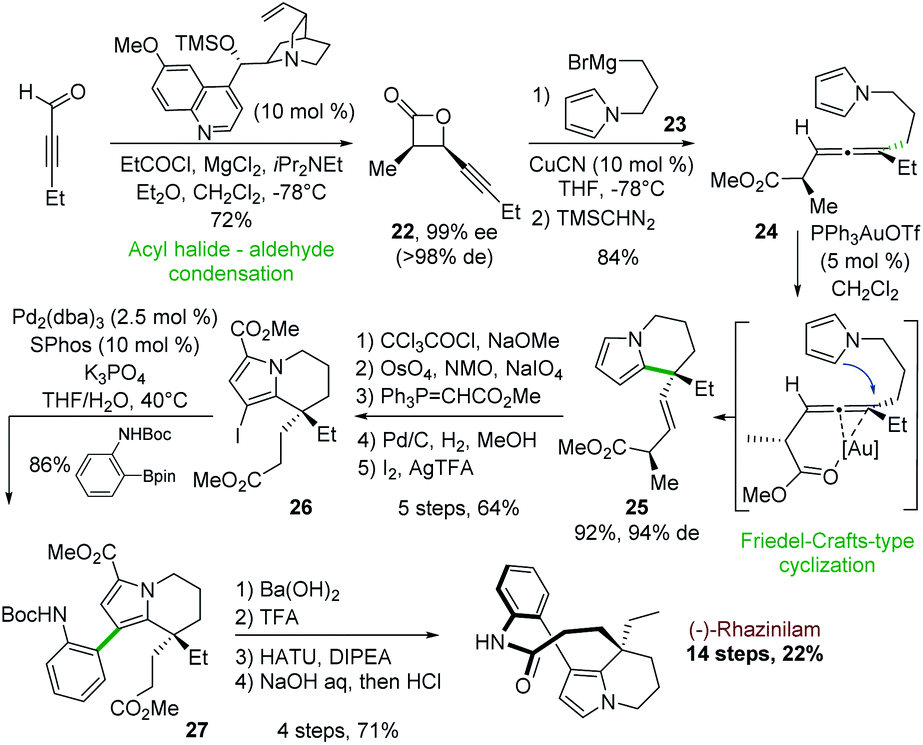 | ||
| Scheme 7 Asymmetric synthesis of (−)-rhazinilam via intramolecular Au-catalyzed cyclization. | ||
The cyclocondensation between 2-pentynal and the chiral ammonium enolate derived from propionyl chloride and O-trimethylsilyl quinine as catalyst led to the enantiopure cis-β-lactone 22 (Scheme 7). A high yielding copper-catalyzed SN2′ nucleophilic substitution with the pyrrole Grignard reagent 23 and subsequent methylation (TMSCHN2) provided the pyrrolyl allene ester 24 as a single diastereoisomer in 84% yield.
The gold(I) complex Ph3PAuOTf was found to be optimal (92%) for the annulation of this pyrrolic allene to tetrahydroindolizine 25 with an efficient chirality transfer (94% d.e). Further functionalization steps (carboxylation, oxidative olefin cleavage, Horner–Wittig homologation, hydrogenation, and iodination) led to compound 26 in 64% yield. Suzuki coupling of the latter with N-Boc aniline boronic ester in the presence of Pd2(dba)3/SPhos gave the arylpyrrole derivative 27 with 86% yield. The latter was then converted in 4 steps (ester saponification, N-Boc deprotection, macrolactamization, decarboxylation) to (−)-rhazinilam with 94% ee.
4.5 Asymmetric gold(I)-catalyzed pyrroloallene cyclization
Following a similar strategy, but with asymmetric gold catalysts, Voituriez and coworkers reported in 2017 another asymmetric synthesis of (−)-rhazinilam (Scheme 8).29 In this approach, the enantioselective cycloisomerization of the achiral pyrroloallene 28 was optimized by employing various chiral ligands with gold(I) catalysts. Phosphoramidite, BINAP and SEGPHOS ligands showed low enantioselectivity (2–41% ee). However, BIPHEP-type ligands induced good enantiomeric excess, with a very important influence of the phosphorus substituents (R in 29) and a slight effect of the silver salt required to activate the catalyst.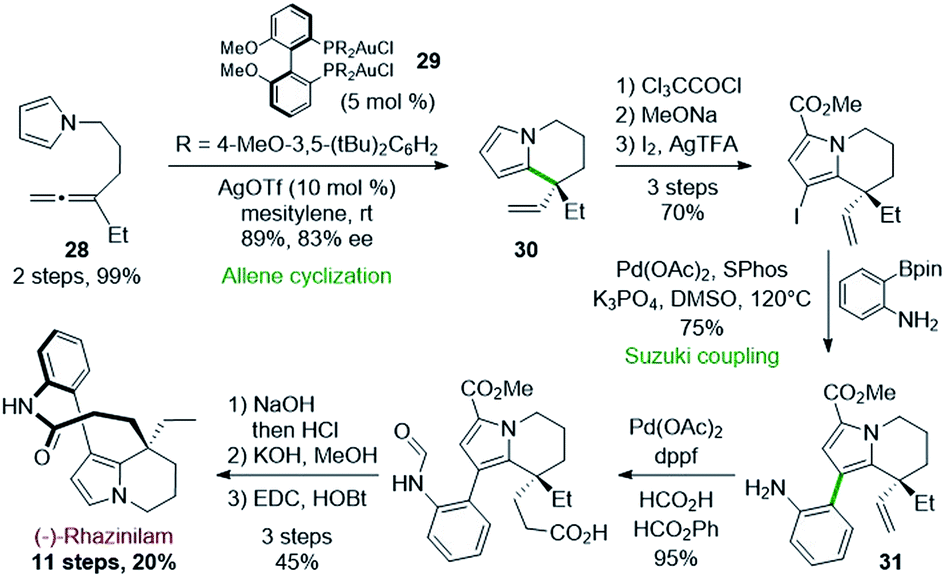 | ||
| Scheme 8 Asymmetric gold(I) complex used for enantioselective allene cyclization. | ||
Thus, the complex (R)-(DTBM-MeO-BiPHEP)(AuCl)229 provided the tetrahydroindolizine 30 with a good 83% ee in high yield. The pyrrole unit of the latter was stabilized towards oxidation by introducing a methoxycarbonyl group.
Further iodination set the stage for a Suzuki coupling with an unprotected 2-aminophenyl boronic ester. This coupling was achieved with 75% efficiency by the Pd(OAc)2/SPhos catalytic system. The so-formed intermediate 31 was then converted to (−)-rhazinilam in 4 steps with 43% yield, through an interesting palladium-catalyzed vinyl–hydrocarboxylation reaction according to Shi's30 conditions.
4.6 Asymmetric copper-catalyzed pyrrolopropargyl acetate cyclization
In a related approach, Carreira and his group reported in 2017 the first synthesis of (−)-rhazinilam based on a copper-catalyzed cyclization of pyrrolopropargylic acetate.31 This synthesis was based on a propargylic substitution using pyrrole as internal nucleophile, probably through an allenic intermediate, catalyzed by a chiral 2,6-bis(oxazolinyl)pyridine ligand (PyBOX) copper complex (Scheme 9). Optimization of such propargylic substitution revealed that enantiopure PyBOX ligand and [(CH3CN)4Cu]PF6 efficiently catalyzed the formation of the ethyl ethynyl tetrahydroindolizine 33 (89% yield, 80% ee) from propargylic acetate 32. The latter was then converted in 4 steps to the Trauner intermediate analogue 34 (Scheme 4) by trapping the corresponding acetylide with ethyl chloroformate, hydrogenation with Adam's catalyst, condensation with 2-iodoaniline in the presence of AlCl3 and finally amide MOM protection. Pd(OAc)2-catalyzed intramolecular coupling of 34 in the presence of 10 equivalents of water (crucial for the reaction success) furnished the MOM protected rhazinilam 35 which led to (−)-rhazinilam after deprotection.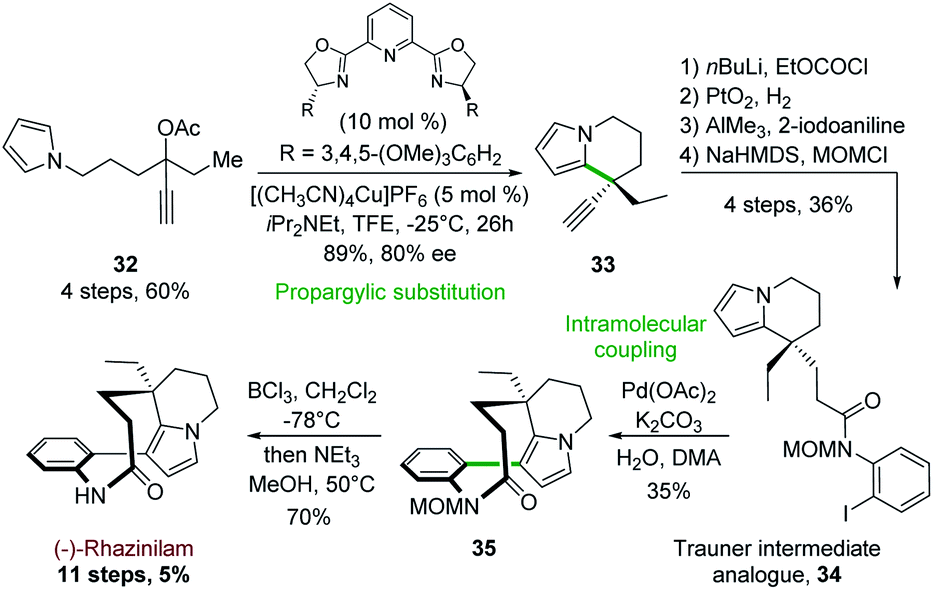 | ||
| Scheme 9 Enantioselective propargylic substitution catalyzed by chiral copper complex applied to the synthesis of (−)-rhazinilam. | ||
5 Total syntheses through Heck-type reactions
The Heck reaction is known to provide a useful and possibly enantioselective way to form (hetero)cyclic systems.32 Not so surprisingly, this method has been applied to the synthesis of the rhazinilam–leuconolam alkaloids, leading to interesting approaches.5.1 Zakarian total synthesis of (−)- and (+)-rhazinilam
One of the most interesting application of such Heck-type cyclization was proposed by Zakarian and Gu in 2010 (Scheme 10).33 Used at the late stage of the synthesis, a palladium-catalyzed transannular cyclization allowed to achieve the asymmetric synthesis of (−)-rhazinilam from optically inactive compounds. The stereochemistry of the quaternary carbon generated during this key step was controlled by the axial chirality introduced during macrolactamization.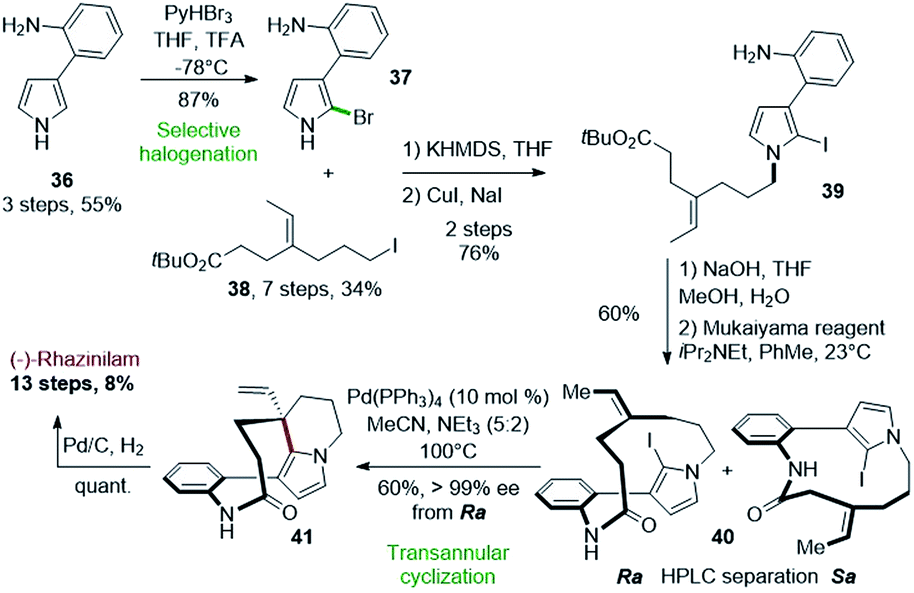 | ||
| Scheme 10 Total synthesis of (−)-rhazinilam proposed by Zakarian via palladium-catalyzed transannular cyclization. | ||
The key intermediate for the latter step, the N-alkylated anilino iodopyrrole 39, was obtained in 3 steps from 2-(1H-pyrrol-3-yl)aniline (36) upon selective bromination (37), N-alkylation with tert-butyl 7-iodo-4-ethylideneheptanoate (38) and halogen exchange (Scheme 10, top). The Mukaiyama reagent, i.e. 2-chloro-1-methylpyridinium iodide, was uniquely effective for the macrolactamization step, providing 40 with 60% yield as a mixture of Ra and Sa atropisomers, separable by chiral HPLC. The iodine in 40 is responsible for axial chirality as the deiodinated analog does not exhibit atropoisomer. Upon HPLC separation, the Ra enantiomer was converted to the ethenyl tetrahydroindolizine 41 by an intramolecular Heck reaction in the presence of palladium tetrakis(triphenylphosphine) in 60% yield with full chirality transfer (>99% ee). Final hydrogenation led quantitatively to (−)-rhazinilam. The latter was thus obtained in 13 steps with 8% overall yield. The same sequence led to (+)-rhazinilam starting from the Sa atropoisomer.
5.2 Yao total synthesis of (±)-rhazinilam
A similar route was proposed by Yao & coworkers in 2014. The Yao's group developed in 2014 a regioselective alkenylation of pyrrole-3-carboxylates based on C–H activation and Heck reaction, and applied it to the total synthesis of (±)-rhazinilam (Scheme 11).34 A specific study showed that the metalation site and thus the regioselectivity of the subsequent Heck alkenylation (C5 or C2) was controlled by the directing alkoxycarbonyl group and the polarity of the solvent (Scheme 11, top). In toluene, alkenylation is oriented to position 2 due to palladium chelation with the alkoxycarbonyl group, but in polar and coordinating medium (DMF/DMSO), this chelation is inhibited, causing electrophilic palladation on the most nucleophilic C5 position.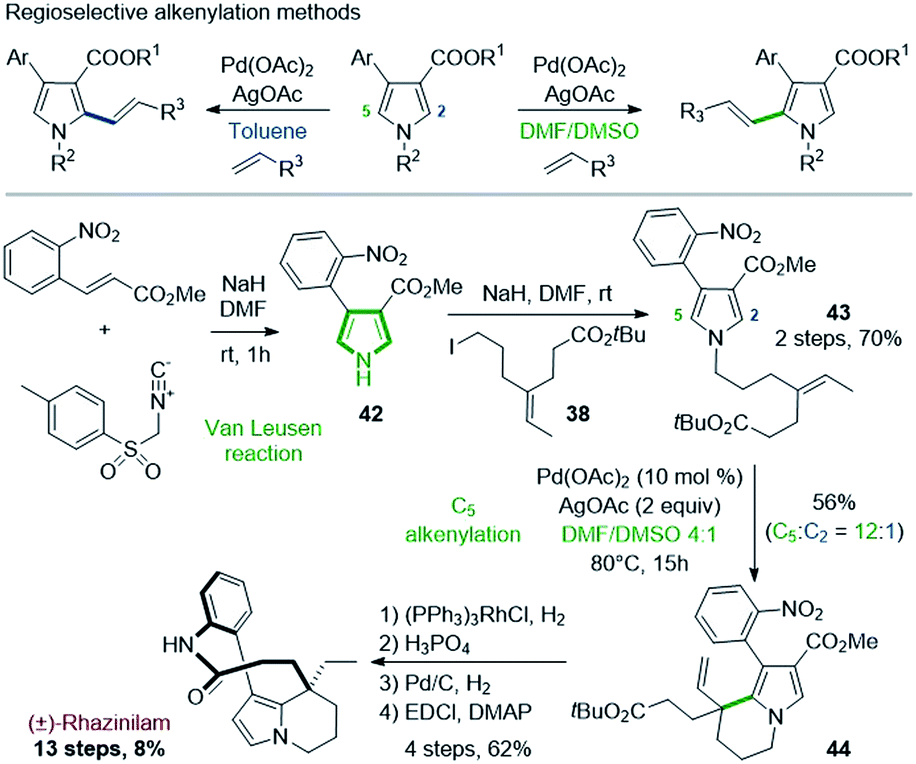 | ||
| Scheme 11 Total synthesis of (±)-rhazinilam via regioselective alkenylation of pyrrole-3-carboxylates. | ||
For the rhazinilam synthesis, an intramolecular version was set up, starting from the alkenyl pyrrole 43. The latter was built in 2 steps. The Van Leusen35 reaction provided the 4-(2-nitrophenyl)pyrrole-3-carboxylate 42, which was then N-alkylated with tert-butyl 7-iodo-4-ethylideneheptanoate (38). The resulting pyrrole 43 was then cyclized in the presence of Pd(OAc)2/AgOAc in DMF:DMSO. Due to the alkene substitution, β-elimination of the carbopalladated species only provided the ethenyl tetrahydroindolizine 44. Although a high regioselectivity was achieved (12:1), 44 was obtained with 56% yield. Decarboxylation, deprotection of the BOC group, reduction, and EDCI-promoted macrolactamization completed the total synthesis.
5.3 Total synthesis of rhazinal and rhazinilam through Catellani reaction
In order to get an even more convergent approach, Gu and coworkers took benefit of the Catellani reaction to develop in 2013 a synthesis of (±)-rhazinal in 12 steps with high overall yield (13%; Scheme 12).36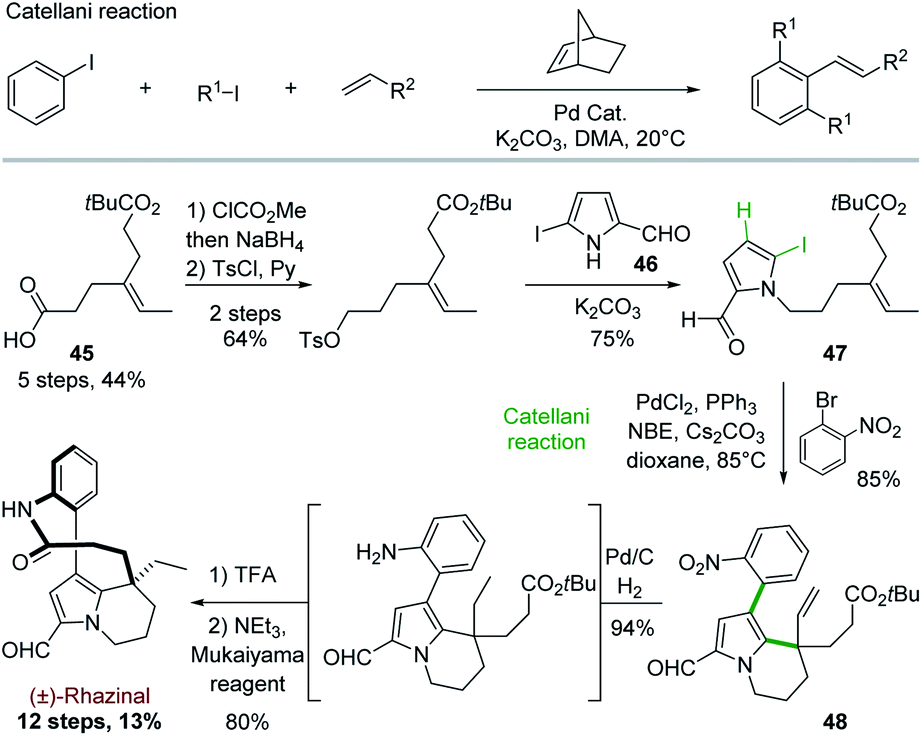 | ||
| Scheme 12 The Catellani reaction and its application to the total synthesis of (±)-rhazinilam proposed by Gu. | ||
The Catellani reaction consists in regioselectively condensing an iodoarene, an aliphatic iodide, and a terminal olefin into 1,2,3-trisubstituted aryl alkene in the presence of palladium and a strained olefin such as norbornene (NBE). This selective arene functionalization at the 1, 2 and 3 positions is based on the different reactivity of palladium(0), (II), and (IV) species formed along a complex catalytic sequence (Scheme 12, top).37
To target (±)-rhazinal, Gu and coworkers chose to implement such multicomponent reaction in an intramolecular way from the N-alkylated 2-formyl-5-iodopyrrole 47 (Scheme 12, bottom). The latter was formed in 3 steps (48%) from half ester3345 by selective reduction to alcohol, tosylation and substitution with the known 2-formyl-5-iodopyrrole 46.38 Upon optimization, the Catellani process allowed the regioselective ortho-arylation/intramolecular Heck coupling of 47, creating in a single step a phenyl–pyrrole bond and a six-membered ring in 85% yield. Further transformation of product 48 led to (±)-rhazinal in 3 steps by hydrogenation (Pd/C, H2) of alkene and nitro groups, tert-butyl deprotection (TFA) and macrolactamization with Mukaiyama's reagent.
Encouraged by this success, Gu and coworkers extended their methodology by proposing, three years later, an asymmetric version of the Catellani process (Scheme 13).39 Shifting from triphenylphosphine to chiral α-aryl tetrahydroquinoline derived phosphoramidite ligand and palladium acetate allowed producing the same key intermediate 48 with high enantiomeric excess (88%), but with lower yield (65%) (Scheme 13, top). The intermediate 48 proved to be a key platform, enabling the total synthesis of (+)-rhazinal as before, but also of (+)-rhazinilam with an 9% overall yield over 13 steps, as well as the total syntheses of the (+)-kopsiyunnanines C1–C3 by reduction of its formyl group prior or after macrolactamization (Scheme 13, bottom).
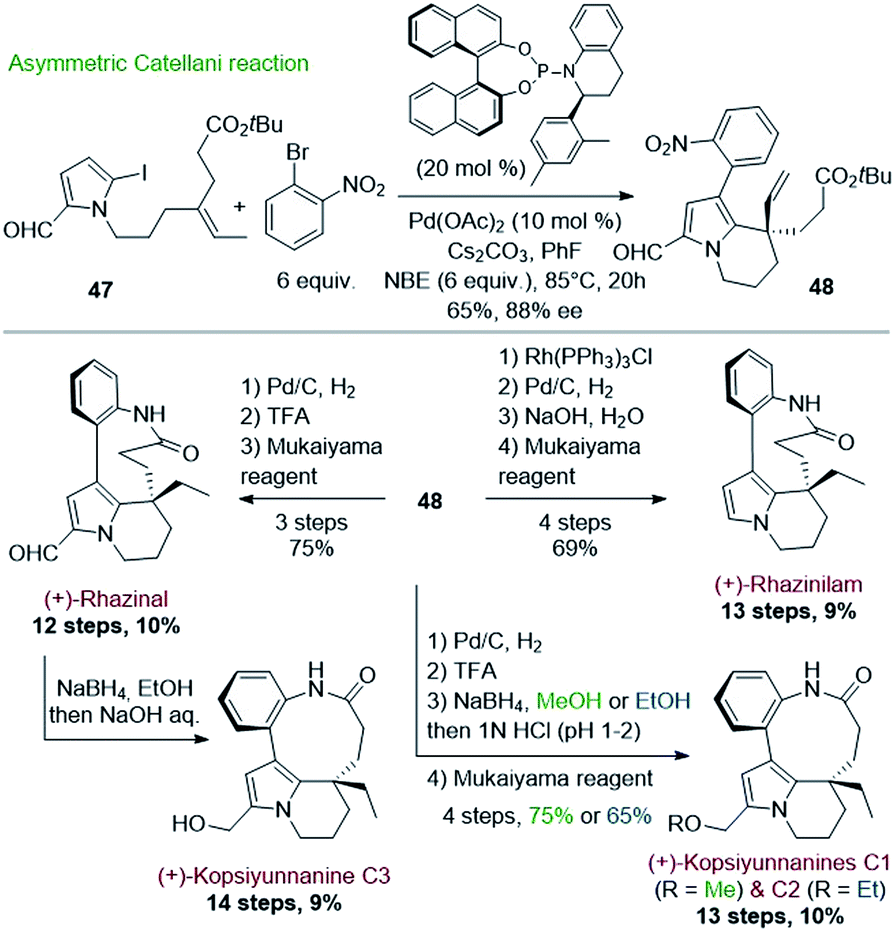 | ||
| Scheme 13 Asymmetric Catellani type reaction for the synthesis of (+)-rhazinilam, (+)-rhazinal, and (+)-kopsiyunnanine C1–3. | ||
5.4 Total syntheses through oxidative Heck reaction
In contrast to classical Heck reactions which required halogenated (hetero)arene or alkene substrates, their oxidative versions allow direct C–H activation, often ortho to a directing group.40The Trauner's group was the first in 2009 to apply such strategy to the synthesis of (±)-rhazinal (Scheme 14).41 The tetrahydroindolizine skeleton was thus directly obtained from the simple N-alkenylated pyrrole 49 upon treatment with Pd(OAc)2/tBuOOH in a peculiar solvent mixture (dioxane/AcOH/DMSO). Vilsmeier–Haack formylation provided the intermediate 50. For comparison purpose, the latter was also produced from a related N-alkenylated iodoformylpyrrole 51 through intramolecular Heck coupling. Although the coupling yield was slightly better (75 vs. 69%), the starting pyrrole required two more steps for its preparation. The total synthesis was then completed through chemoselective vinyl reduction with Crabtree's catalyst, saponification using lithium hydroxide, followed by the same sequence already developed by this group (see Scheme 4).
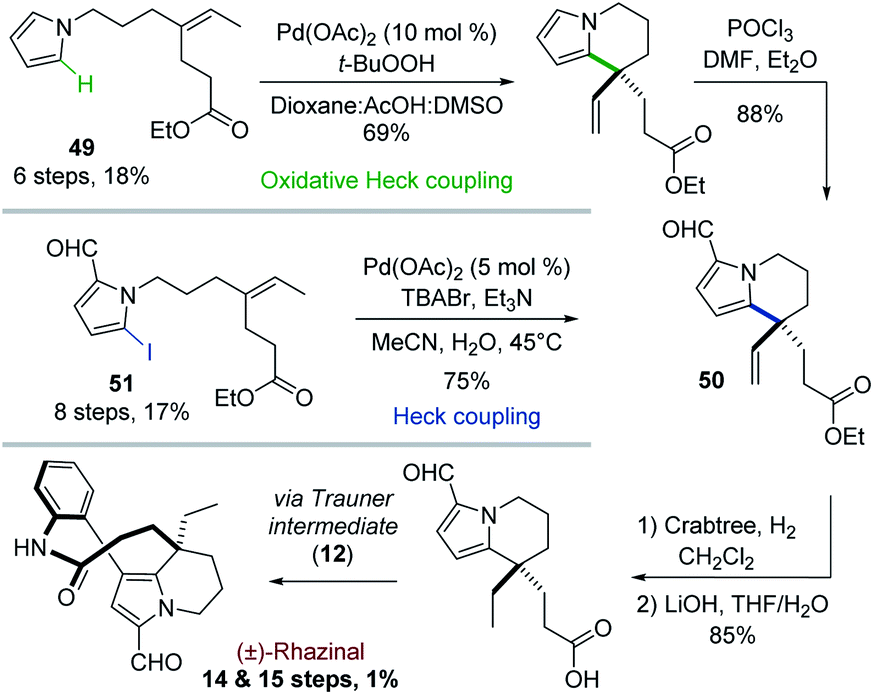 | ||
| Scheme 14 Palladium-catalyzed oxidative Heck coupling for the synthesis of (±)-rhazinal. | ||
In a related approach, Gaunt & coworkers described in 2008 the first total synthesis of rhazinicine (Scheme 15).42 They attempted to build up the tetrahydroindolizine core by using regioselective oxidative Pd(II)-catalyzed C–H alkenylation of pyrroles, controlled by the N-protecting group nature (Scheme 15, top).43 However, the presence of a masking group proved necessary to solve regioselectivity issues.
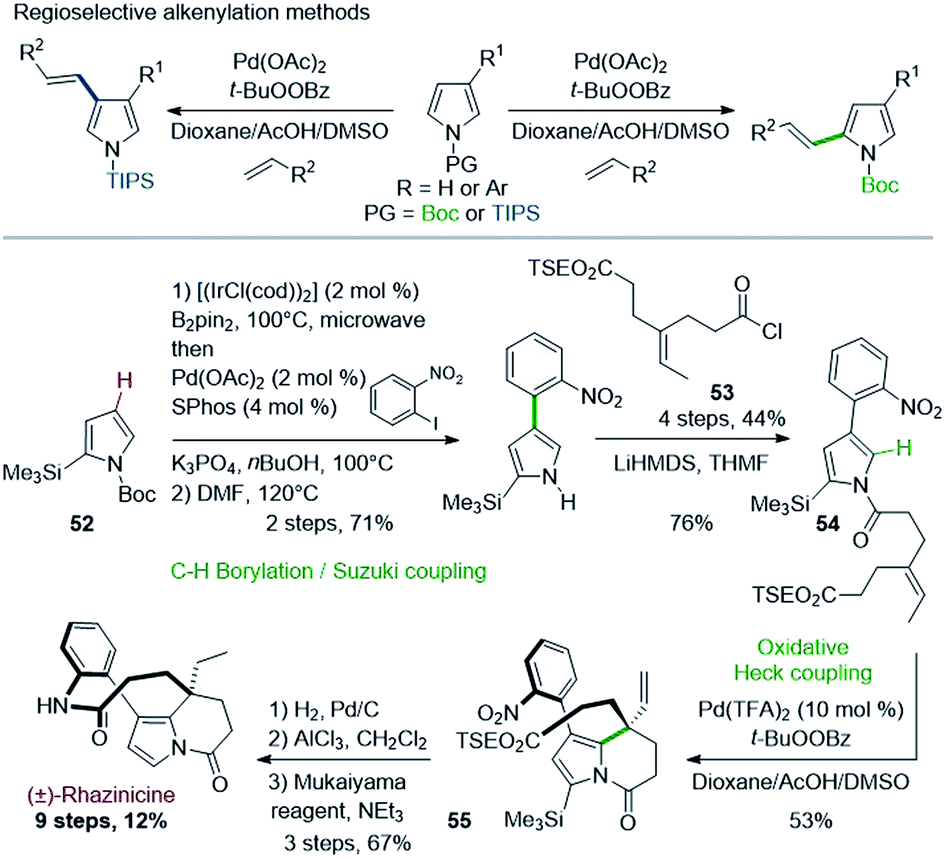 | ||
| Scheme 15 Palladium-catalyzed oxidative Heck coupling for the synthesis of (±)-rhazinicine. | ||
The starting pyrrole 54 was obtained from N-BOC 2-trimethylsilylpyrrole 52 through an interesting one-pot regioselective iridium-catalyzed C–H borylation, Suzuki coupling, followed by BOC deprotection and N-acylation with the acid chloride 53. The oxidative cyclization proceeded as expected, but more efficiently with the more active Pd(TFA)2 catalyst than with Pd(OAc)2. Rhazinicine was then obtained from 55 after hydrogenation (Pd/C), cleavage of both trimethylsilyl and 2-(trimethylsilyl)ethyl (TSE) groups and macrolactamization with the Mukaiyama's reagent.
Later on in 2012, the same group used the oxidative Heck cyclization of N-alkylated 4-(2-nitrophenyl)pyrrole-2-carboxylate 58 as one of the key step of a total synthesis of (±)-rhazinilam (Scheme 16).44 Compound 58 was obtained by N-alkylation of pyrrole 56, prepared through the same one-pot regioselective iridium-catalyzed C–H borylation, Suzuki coupling and BOC deprotection described in Scheme 15 with iodoalkene 57. The oxidative Heck reaction proceeded better under conditions favouring concerted metalation–deprotonation mechanism, i.e. in the presence of base and Pd(OAc)2 under oxygen.45 The synthesis was then fulfilled through the same sequence and a saponification–decarboxylation step.
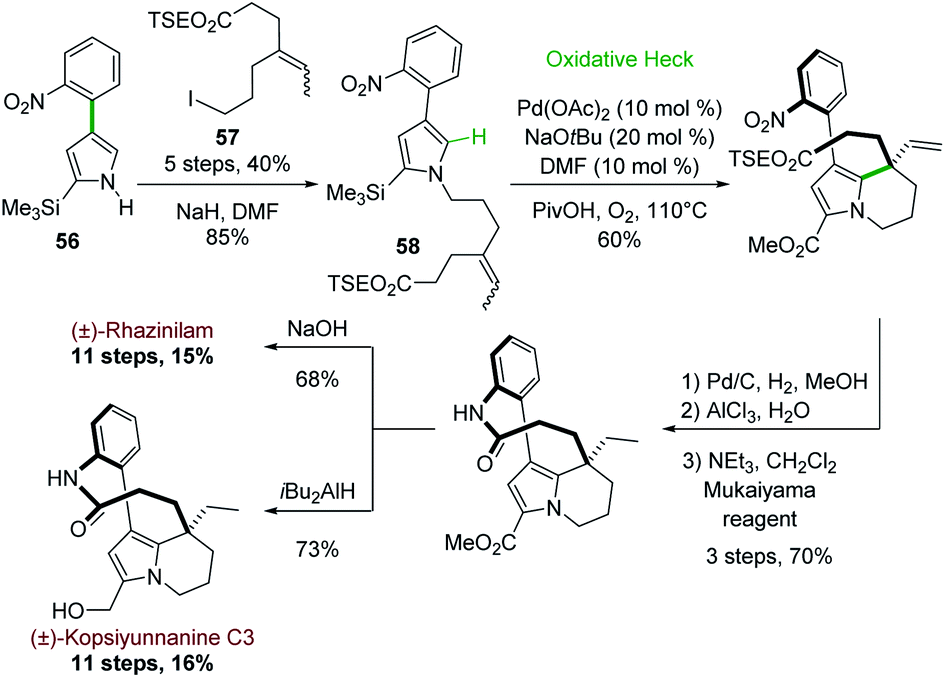 | ||
| Scheme 16 Palladium-catalyzed oxidative Heck coupling for the synthesis of (±)-rhazinilam or (±)-kopsiyuannanine C3. | ||
(±)-Rhazinilam was thus obtained in 11 steps with a rather good overall yield (15%). Switching the last step from saponification to DIBAH reduction readily provided kopsiyunnanine C3 with a similar overall yield (16%).
5.5 Total synthesis through carbocyanation reaction
From a mechanistic point of view, Heck reactions are initiated by Pd(0) insertion into a carbon–halogen bond and propagated via carbopalladation.32c More recent investigations unveiled nickel-catalyzed insertion into carbon–cyanide bond,46 and its extension to carbocyanation processes (Scheme 17, top).47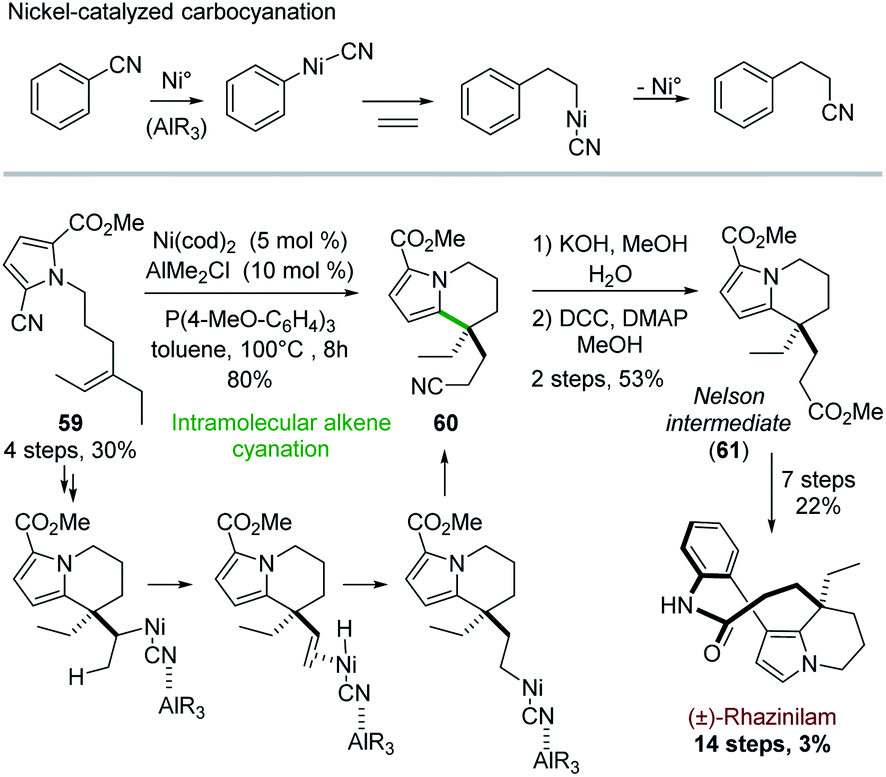 | ||
| Scheme 17 Nickel-catalyzed alkene cyanation for the synthesis of (±)-rhazinilam. | ||
Active in this field, Nakao and coworkers exploited in 2015 this cyanation for building up the rhazinilam tetrahydroindolizine core.48 The intramolecular alkene cyanation of 59 was best catalyzed by nickel and an electron-rich phosphine, cooperatively with alkyl aluminium derivative. The cyano group ended up at the end of the ethylidene chain on compound 60, which implied that β-elimination and re-addition of the so-formed H–[Ni]–CN species occurred (Scheme 17, bottom). Interestingly, the transferred cyano group could then be easily converted to an ester 61, the so-called Nelson intermediate.28 The end of the synthesis followed the Nelson procedure (see Section 4.4 and Scheme 7).
6 Total synthesis through allylative cyclization
The Hosomi–Sakurai allylation has been widely applied in natural product synthesis, especially in the synthesis of cyclic compounds.49 However, a single application of this reaction has so far targeted the rhazinilam–leuconolam family of natural products.In 2013, Hoye and Izgu proposed an interesting intramolecular Hosomi–Sakurai allylation to form the core of leuconolam and then the (±)-leuconolam itself (Scheme 18).50
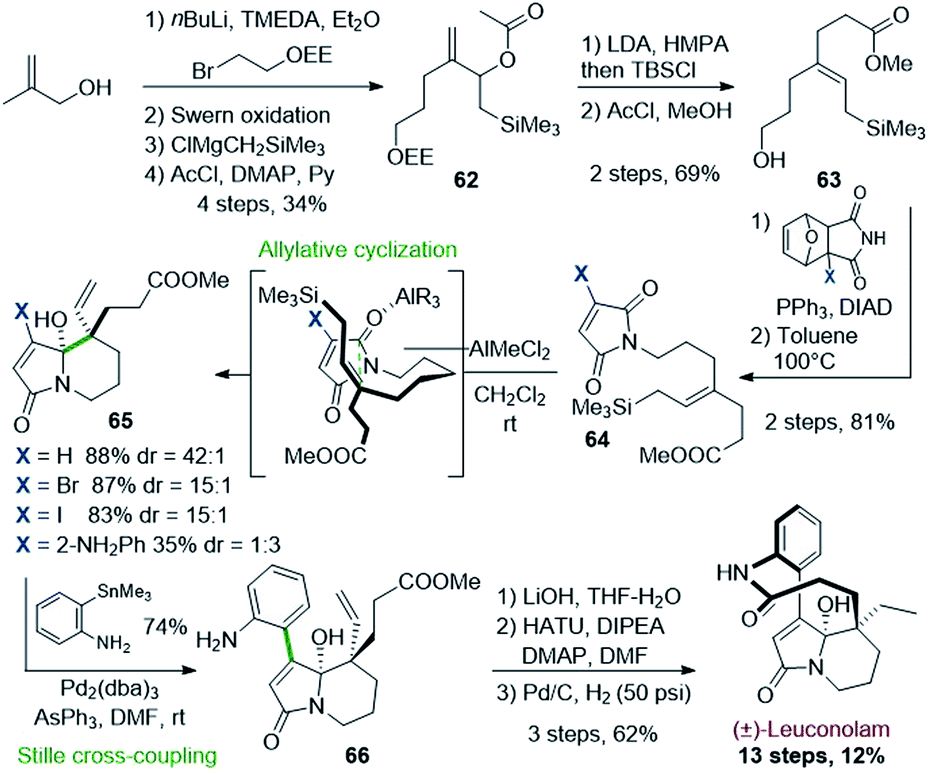 | ||
| Scheme 18 Synthesis of (±)-leuconolam by Hosomi–Sakurai cyclization. | ||
Readily prepared from methallyl alcohol, the silylated allylic acetate 62 was converted to the key E-allyl silane 63 upon Ireland–Claisen rearrangement. A Mitsunobu reaction allowed to graft a furan-protected maleimide. A retro-Diels–Alder reaction unveiled the maleimidyl allylsilane 64 ready for Hosomi–Sakurai cyclization. Upon optimization, dichloromethylaluminium proved to be the most effective Lewis acid to promote the expected cyclization in high selectivity (up to 88%, dr 42:1, X = H). However, the required bicyclic regiosiomer 65 could only be obtained starting from the halogenated maleimide (X = Br, I), as its already arylated version (X = 2-NO2 or NH2Ph) was mostly leading to the opposite regioisomer. The so-formed bicyclic halogenated carbinolamide 65 was then engaged in a Stille cross-coupling affording compound 66 in 74% yield. The completion of the synthesis was achieved in 3 steps after mild saponification, lactamization, and hydrogenation. The natural product was thus obtained over 13 steps with a good overall yield (12%).
7 Total syntheses through pyrrole annelation/formation cyclization reactions
In the syntheses described above, the tetrahydroindolizine core was built up from an already-formed or commercial pyrrole derivative. Alternative routes have been proposed, that relied on cyclization processes, which produced functionalized or substituted pyrroles, already or not embedded into tetrahydroindolizine.7.1 Pyrrole annelation reactions
One of the first examples of such strategy was reported by Magnus and coworkers in 2001 in their synthesis of (±)-rhazinilam (Scheme 19).51 They constructed the tetrahydroindolizine core from a cyclic thioimidate upon N-alkylation and basic treatment, in an electrocyclization process related to Grigg synthesis of pyrroloheterocycles.52 The commercially available 2-piperidinone was converted in 4 steps to the cyclic thioimidate 67 with 44% yield via the formation of the racemic so-called Magnus allyl lactam. N-alkylation of the latter and subsequent cyclization in the presence of substituted allyl bromide 68 and DBU delivered the tetrahydroindolizine 69 in 71% yield. After functionalization of the alkene moiety (oxidative hydroboration, oxidation to aldehyde and then to acid), nitro group reduction and lactamization, (±)-rhazinilam was finally obtained with 8% overall yield over 10 steps.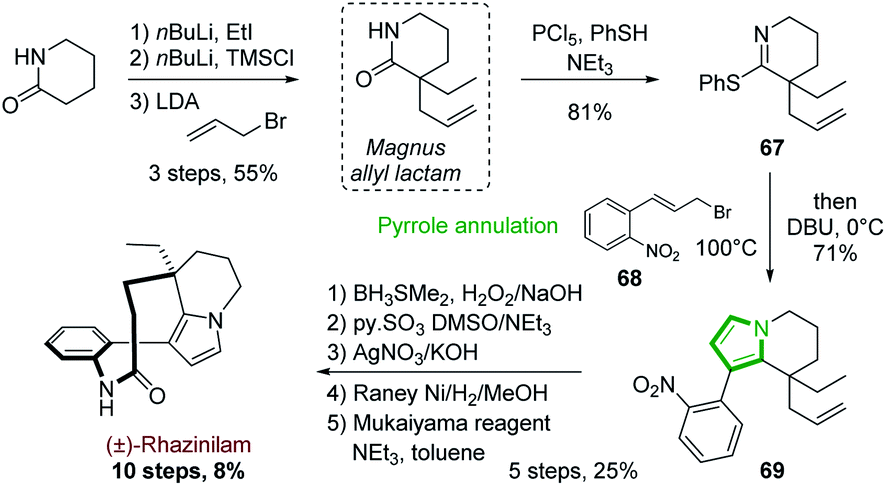 | ||
| Scheme 19 Annulation of pyrrole from cyclic imine proposed for the synthesis of (±)-rhazinilam. | ||
More recently, Stoltz's group described in 2012 an asymmetric route to the Magnus allyl lactam and thus to a formal synthesis of (−)-rhazinilam (Scheme 20).53 They relied on a highly efficient decarboxylative allylic alkylation of lactam, catalyzed by palladium in the presence of a chiral ligand.
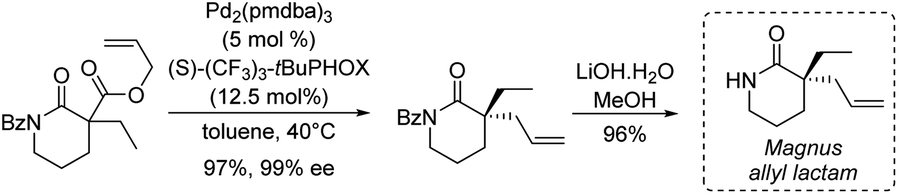 | ||
| Scheme 20 Decarboxylative allylic alkylation of lactam catalyzed with palladium. | ||
Such asymmetric decarboxylative allylic alkylation has also been employed by the group of Zhu in 2016 at the first stage of the (−)-rhazinilam synthesis (Scheme 21).54 This reaction allowed them to obtain an enantioenriched allylic cyclopentanone 70 (86% ee), which was then transformed in 6 steps into the key cyclopentenyl azide 71 by oxidative hydroboration, protection of the resulting alcohol, decarboxylative coupling, deprotection, mesylation and subsequent azidation. Ozonolysis of 71 provided the azido keto ester 72, which was then cyclized to the tetrahydroindolizine 73 through an interesting and efficient one-pot sequence involving Staudinger reduction/aza-Wittig cyclization and N-alkylation/annulation. The first step of this sequence produced a tetrahydropyridine, which upon N-alkylation with bromoacetaldehyde gave the enamine aldehyde A. Intramolecular aldol-type reaction led to indolizinium B, which evolved to 73 after dehydration and tautomerization. Further hydrogenation of the nitro group, saponification and lactamization finally led to (−)-rhazinilam with a high overall yield (16%) over 13 steps.
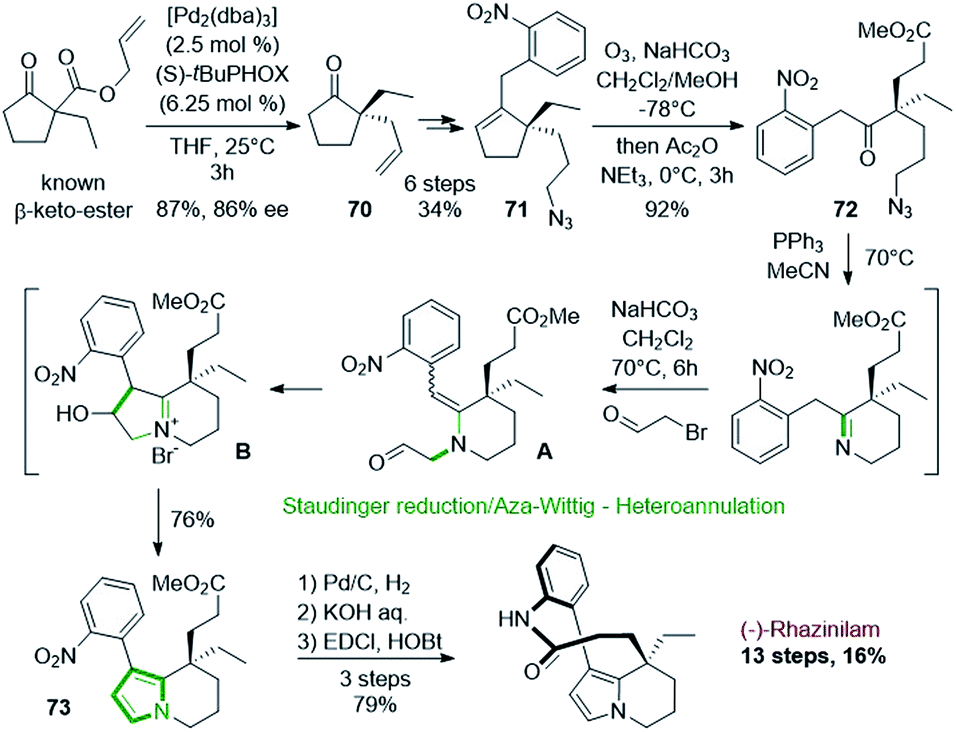 | ||
| Scheme 21 One-pot Staudinger/aza-Wittig-heteroannulation process employed in the synthesis of (−)-rhazinilam. | ||
7.2 Silver-promoted annelation
The first use of a cyclic imine in the synthesis of the rhazinilam alkaloids was actually reported in the second total synthesis of (±)-rhazinilam, reported in 2000, 27 years after the first one (see Section 4.1 and Scheme 3).Based again on Grigg electrocyclization process,52 Sames and coworkers developed a cyclization–aromatization of cyclic iminium salt in the presence of silver carbonate to produce the nitroarylated diethyl tetrahydroindolizine 74 with 70% yield (Scheme 22, top). Sensitive to electrophiles, the pyrrole part was stabilized by a carbomethoxy group (75).55
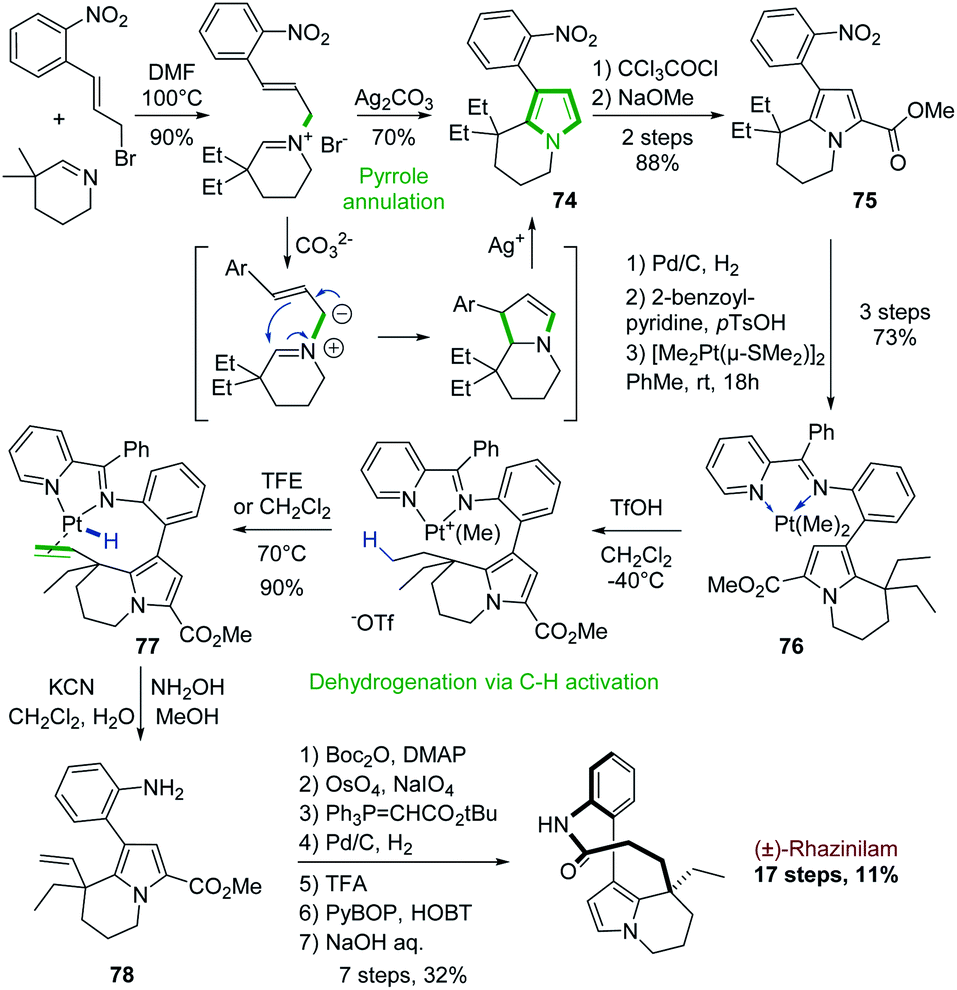 | ||
| Scheme 22 Silver-promoted pyrrole formation and platinum-mediated dehydrogenantion for the synthesis of (±)-rhazinilam. | ||
Beside this annulation reaction, the Sames's group also used another unique approach to further functionalize the so-formed tetrahydroindolizine 75 (Scheme 22, bottom). They carried out a Pt-mediated dehydrogenation of the ethyl chain by C–H activation. For that, the nitro group was reduced (Pd/C, H2) into the corresponding amine, which was converted to a coordinating 2-pyridinylimine. Complexation with [Me2Pt(μ-SMe2)]2 provided the dimethyl platinum complex 76. Upon protonation with trifluoromethanesulfonic acid and methane release, a cationic complex was formed, which evolved on heating in trifluoroethanol to the ethenylplatinum hydride 77 with 90% yield. During this step, C–H activation took place, inducing the selective dehydrogenation of an ethyl fragment followed by a β–H elimination. Platinum decomplexation and subsequent imine hydrolysis provided intermediate 78 which was converted to (±)-rhazinilam after 7 steps via notably a one carbon homologation of the so-formed ethenyl chain. This total synthesis was achieved in 17 steps with a good overall yield of 11%. A stoichiometric amount of platinum complex was nevertheless required.
Two years later, Sames and coworkers proposed an asymmetric version of their initial C–H bond functionalization strategy and they applied it to the synthesis of (−)-rhazinilam (Scheme 23).56 By placing a chiral oxazoline imine 79 instead of the achiral pyridinyl imine, a mixture of diastereoisomeric platinum complexes were produced, but whatever the oxazoline side chain (R = Ph, iPr, tBu, Cy), one major diastereoisomer was formed, with a ratio up to 20:1 for R = tBu (determined by 1H NMR). In these complexes, the two enantiotopic ethyl groups could be differentiated. After separation of the so-formed diastereoisomers by preparative HPLC, the major R enantiomer 78 was obtained with high ee (96%).
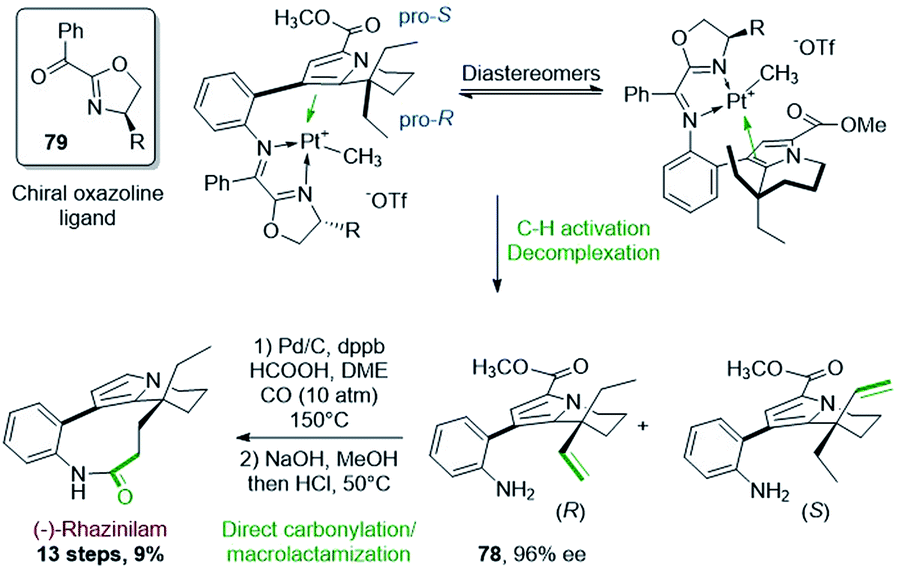 | ||
| Scheme 23 Platinum-mediated dehydrogenation for the enantioselective synthesis of (−)-rhazinilam. | ||
Interestingly, the rather long last sequence (7 steps) of the former synthesis were replaced by a direct hydroformylation–macrolactamization (58%) and methyl ester deprotection–decarboxylation.
A similar cyclization–aromatization of cyclic iminium salt promoted by silver carbonate was also used by the group of Zhu in 2014. They applied it to another enantioselective synthesis of (−)-rhazinilam. The chirality was introduced at the very first stage via an interesting alcohol desymmetrization of achiral bislactones, catalyzed by a chiral imidodiphosphoric acid (Scheme 24).57
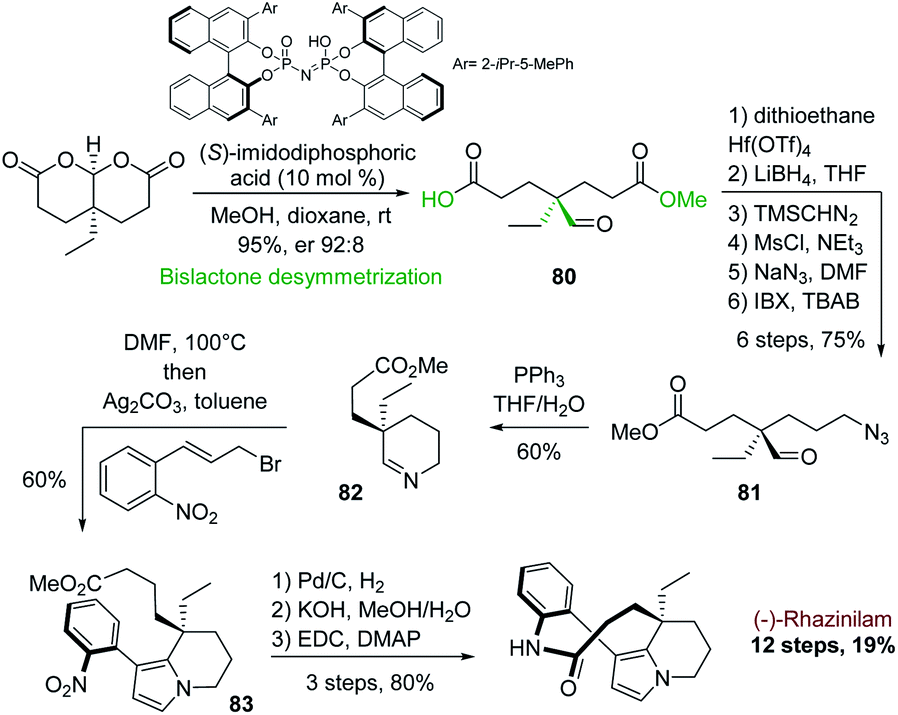 | ||
| Scheme 24 Synthesis of (−)-rhazinilam via enantioselective desymmetrization of bislactone. | ||
This desymmetrization efficiently provided the asymmetric center with excellent enantioselectivity (er. 92:8) and with the right stereochemistry to access to (−)-rhazinilam. The resulting half ester 80 was converted in 6 steps to the azido formyl ester 81, which was converted to cyclic imine 82 by Staudinger/aza-Wittig reaction. Heating this imine in DMF with allyl bromide and then subjecting it to silver carbonate afforded the tetrahydroindolizine 83 with 60% yield. (−)-Rhazinilam was finally obtained in three steps through nitro group reduction, saponification and macrocyclization, achieving the total synthesis in 12 steps with an excellent overall yield of 19%.
7.3 Gold-catalyzed annelation
Since the turn of the century, gold catalysis has raised, providing numerous new and very mild methods in organic synthesis,58 especially in cyclization processes,59 with various applications to total synthesis.60 Unsurprisingly, gold catalysis was also applied to the synthesis of rhazinilam alkaloids, especially for the formation of the pyrrole moiety (see also Sections 4.4 and 4.5).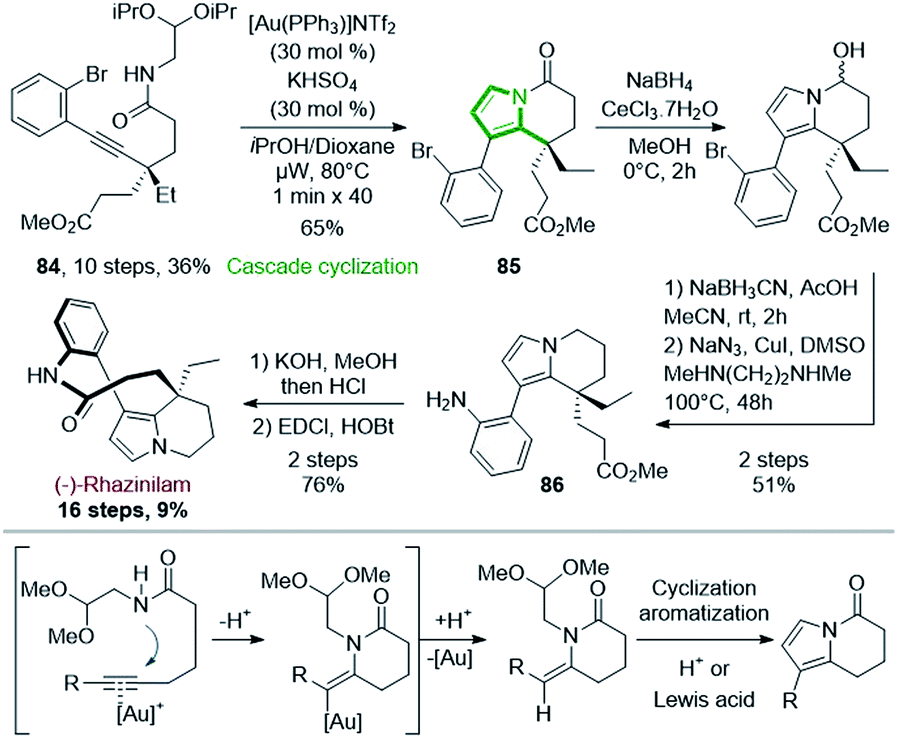 | ||
| Scheme 25 Enantioselective synthesis of (−)-rhazinilam through Au(I)-catalyzed cyclization cascade. | ||
The enantiopure alkynyl amide acetal 84 (>99% ee) was prepared from 2-ethylcyclohexanone in 10 steps with 36% yield.61 After a long optimization process, 84 could be transformed to the tetrahydroindolizinone 85 in good yield (65%) via a double cyclization cascade catalyzed by the Gagosz catalyst (PPh3AuNTf2) under microwave irradiation at 80 °C. In this cascade (Scheme 25, bottom), Au(I) activated the alkyne, so that intramolecular nucleophilic 6-exo-dig addition of nitrogen occurred, giving an enamide while liberating Au(I) (Scheme 25, bottom). The latter then acted as Lewis acid on the acetal moiety, providing an oxonium, which could be attacked by the enamide, leading to a methoxypyrrolidine. Subsequent elimination and aromatization provide the tetrahydroindolizinone scaffold.
The tetrahydroindolizinone 85 was then converted to 86 by stepwise Luche reduction of ketone, NaBH3CN reduction of the resulting hemiaminal and copper mediated aniline formation. Finally, (−)-rhazinilam was reached after saponification of ester and lactamization with 76% yield, but with a reasonable 9% overall yield.
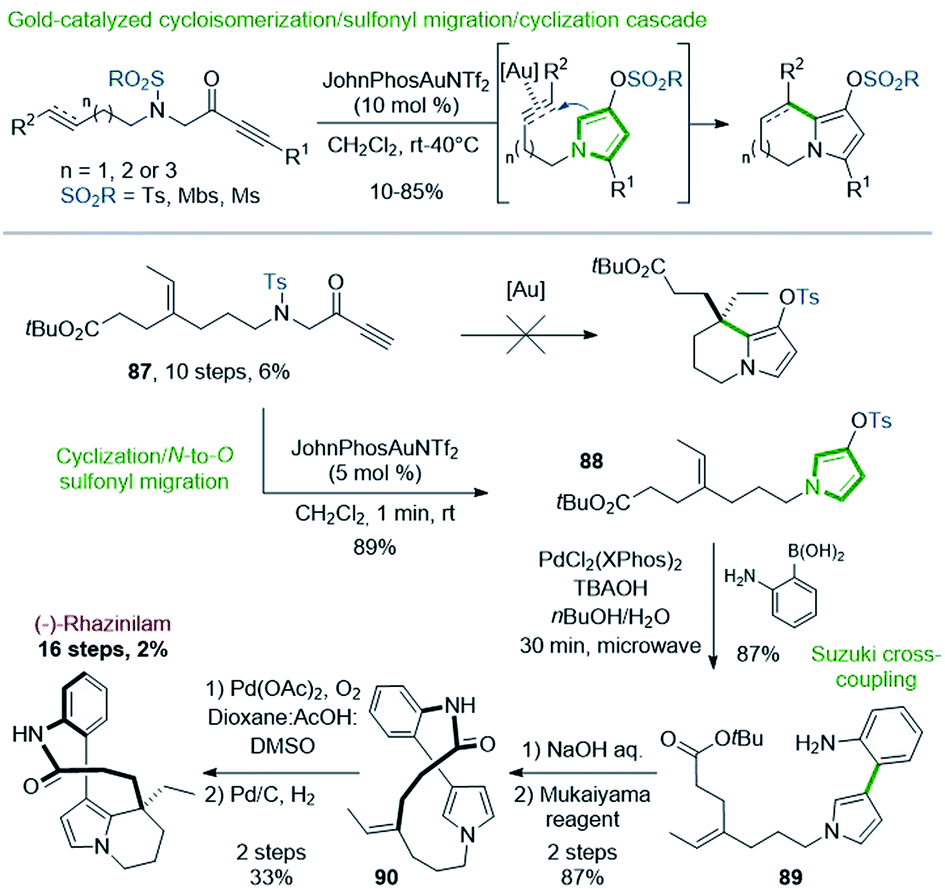 | ||
| Scheme 26 Synthesis of (±)-rhazinilam via gold(I)-catalyzed cyclization/sulfonyl migration. | ||
Preliminary works revealed that pyrrolizine, indolizine or pyrrolo[1,2-a]azepine derivatives were produced by a gold-catalyzed three-step cascade from linear N-alkenyl or alkynyl N-sulfonyl aminopropargyl ketones, via pyrrole intermediates (Scheme 26, top).63 Produced through this cycloisomerization/N-to-O sulfonyl migration/cyclization process, the resulting sulfonylated derivatives could be engaged in further cross-coupling reactions.64
Applied to rhazinilam synthesis, such reaction required the preparation of a key linear precursor 87, readily available in 10 steps from N-tosyl glycine methyl ester. Unfortunately, the gold cascade failed under standard conditions to furnish the expected indolizine derivative from this precursor (Scheme 26, bottom). However, 87, submitted to JohnPhosAuNTf2 under smoother reaction conditions, entered in a cycloisomerization/N-to-O 1,5-sulfonyl migration/cyclization cascade, which gave within only 1 minute the N-alkylated pyrrolyl tosylate 88 with 89% yield. Suzuki coupling of the latter with 2-aminophenylboronic acid under microwave irradiation (30 min) using PdCl2(XPhos)2 precatalyst furnished the aryl-pyrrole 89 in 87% yield.65 Saponification and immediate macrolactamization in the presence of Mukaiyama's reagent provided the 13-membered lactam 90 in 87% yield. The synthesis of (±)-rhazinilam was completed by a regioselective intramolecular oxidative Heck coupling and subsequent hydrogenation of the resulting vinyl chain.
7.4 Pyrrole formation through [3 + 2]-cycloaddition
Another alternative route, in which the pyrrole part of the tetrahydroindolizine core was built up, relies on [3 + 2]-cycloaddition.In 2015, the Tokuyama's group based their (−)-rhazinilam synthesis on a regioselective 1,3-dipolar cycloaddition reaction between a chiral münchnone intermediate and 2-nitrophenylethyne (Scheme 27).66 The chiral precursor of münchnone 91 was prepared from dimethyl D-aspartate in 7 steps with 27% yield, according to described conditions.67 Upon refluxing (140 °C) in acetic anhydride with 2-nitrophenylethyne, the tetrahydroindolizine 92 was obtained in high yield by a [3 + 2] cycloaddition via the münchnone intermediate and a CO2 expulsion-aromatization. Further functionalization (ester reduction, Parikh–Doering oxidation, Wadsworth–Emmonds alkenylation, nitro group reduction, olefin hydrogenation, ester hydrolysis, lactamization) furnished (−)-rhazinilam with 7% overall yield in 14 steps.
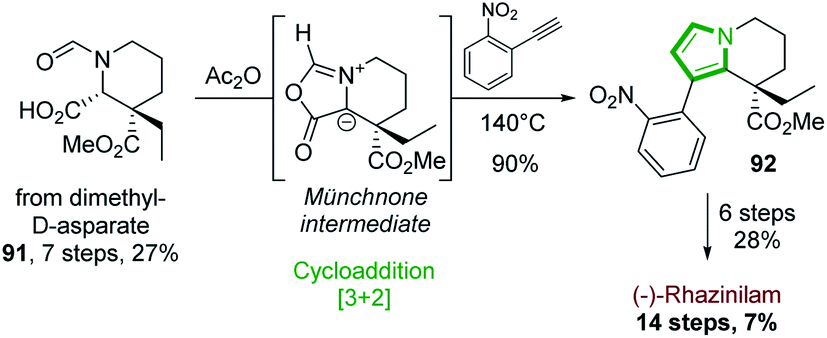 | ||
| Scheme 27 (−)-Rhazinilam obtained by regioselective 1,3-dipolar cycloaddition reaction. | ||
8 Formal syntheses
Besides the total syntheses described above, a few formal syntheses have also been reported. They highlighted interesting approaches to build up the key tetrahydroindolizine core. Two exploited radical cyclization, and one an intramolecular pyrrole amidation.8.1 Radical based cyclization
Although radical reactions, especially cascade reactions/cyclizations,68 have been used in natural product synthesis, only two radical-based routes have been reported for the synthesis of (±)-rhazinal.Proposed by Gansauer and coworkers69 in 2017, a formal (±)-rhazinal synthesis took benefit of the Nugent–Rajanbabu opening of epoxides mediated by in situ generated Ti(III) species (Scheme 28).70
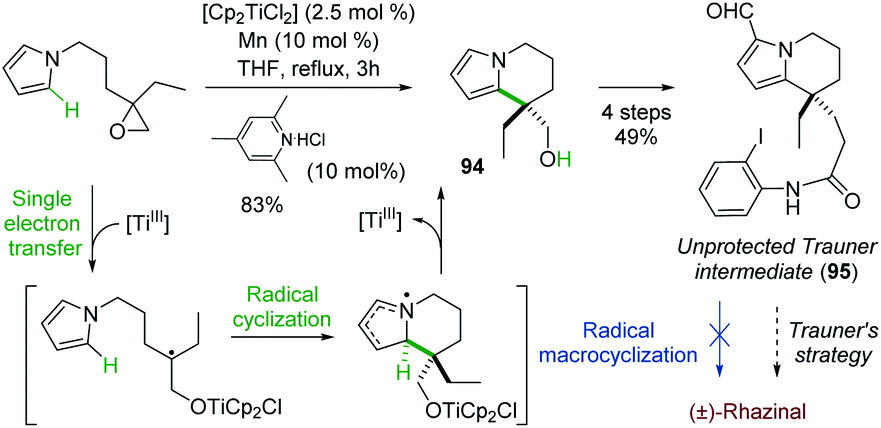 | ||
| Scheme 28 Formal synthesis of (±)-rhazinal through radical-based epoxide opening. | ||
The radical resulting from such Ti(III) epoxide opening added to the neighbouring pyrrole and thus gave in high-yield (83%) the tetrahydroindolizine 94, an intermediate towards (±)-rhazinal described by Banwell (see Scheme 5). Nevertheless, further functionalization of 94 (oxidation, HWE reaction, alkene reduction, Vilsmeier–Haack formylation) enabled to access to compound 95 close to the Trauner intermediate, ready for radical macrocyclization. However, all attempts by iridium photoredox catalysis or by radical chain reaction were unsuccessful. The intermediate 95 thus allowed the formal synthesis of (±)-rhazinal according to Trauner's strategy (Scheme 4).
In 2011, the Miranda group reported the formal synthesis of (±)-rhazinal based on a tandem radical addition/cyclization cascade (Scheme 29).71 The alkenyl 2-formylpyrrole 96 was converted to tetrahydroindolizine 98 under Zard's conditions72 (dilauralyl peroxide in dichloroethane) in the presence of the xanthate 97. Such radical transformation allowed the formation of two carbon–carbon bonds by an intermolecular addition of a radical ethyl acetate onto the double bond followed by cyclization with the pyrrole ring.
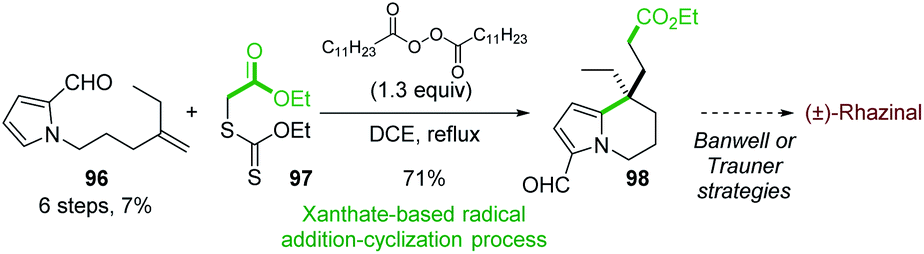 | ||
| Scheme 29 Formal synthesis of (±)-rhazinal by tandem radical addition/cyclization process. | ||
8.2 Intramolecular amidation
In 2015, Chandrasekhar and coworkers73 developed an intramolecular N-acylation of pyrrole in order to prepare the key tetrahydroindolizine intermediate 101 for the synthesis of (±)-rhazinal (Scheme 30) through Trauner or Banwell routes (see Schemes 4 & 5). Starting from the unprotected pyrrole 99 carrying an appropriate ester chain an intramolecular amidation took place in the presence of DBU affording 100 in 88%. Combined reduction of the pyrroloamide and ethyl ester functions with chloroalane provided compound 101 which thus allowed the formal synthesis of (±)-rhazinal.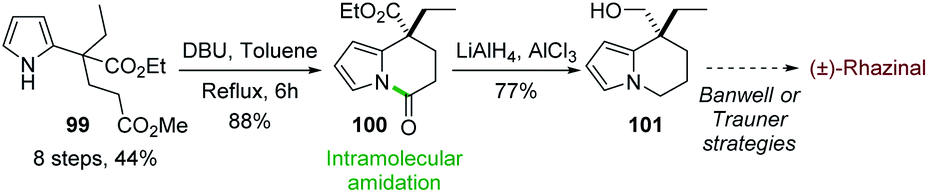 | ||
| Scheme 30 Formal synthesis of (±)-rhazinal via intramolecular amidation. | ||
9 Conclusion
The first total synthesis of rhazinilam was performed by Smith and coworkers in 1973, eight years after its isolation. About thirty years passed before the second total synthesis described in 2000 by the Sames group. But in the last decade (2010–21), several research groups have been attracted by the pronounced biological activities together with the unique structure of rhazinilam and analogues (rhazinal and leuconolam), and not less than 17 total syntheses appeared in the literature.To sum up, there are currently 20 total syntheses of rhazinilam (11 asymmetrically), 5 of rhazinal (2 enantioselectively), 5 of leuconolam (1 enantioselectively) and 2 of kopsiyunnanines, and only one of rhazimicine, including the biomimetic syntheses. Furthermore, a few formal syntheses have been described.
Most of the syntheses required reactions promoted or catalyzed by metal ions or complexes, and only two relied on organocatalysis.
For rhazinilam synthesis, 15 routes involve metal-promoted reactions as key step, while only 5 other routes rely on more classical methods. The maximum overall yield obtained for rhazinilam is 22% in 14 steps.28 For rhazinal, 5 total syntheses, with 4 involving metal-promoted steps and 1 relying on organocatalysis, have been described with overall yields ranging from 1% to 13% over 12 to 16 synthetic steps. Furthermore, three formal syntheses also been reported, two of them are based on radical cyclization. For leuconolam, 2 total syntheses involve metal promoted steps and 1 relies on organocatalysis, a few others are based on more classical conditions. The best overall yield of 12% was achieved over 13 steps.50
As revealed by this review, most newly developed methods have often been applied to the synthesis of one or the other member of the rhazinilam–leuconolam family of natural products. Therefore, more syntheses of these structurally appealing axially chiral natural products will appear with the development of new synthetic methodologies.
10 Author contributions
Fatih Sirindil: writing – original draft Jean-Marc Weibel: validation Patrick Pale: supervision, writing – review & editing Aurélien Blanc: writing – review & editing, visualization.11 Conflicts of interest
There are no conflicts to declare.12 Acknowledgements
We gratefully acknowledge the French Ministry of Higher Education, Research and Innovation and the CNRS for financial support. F. S. thanks the French Ministry of Research for PhD fellowship.13 Notes and references
- H. H. A. Linde, Helv. Chim. Acta, 1965, 48, 1822 CrossRef CAS.
- (a) A. Banerji, P. L Majumder and A. Chatterjee, Phytochemistry, 1970, 9, 1491 CrossRef CAS; (b) K. T. Da Silva, A. H. Ratcliffe, G. F. Smith and G. N. Smith, Tetrahedron Lett., 1972, 10, 913 CrossRef.
- (a) O. Thoison, D. Guenard, T. Sevenet, C. Kan-Fan, J. C. Quirion, H. P. Husson, J. R. Deverre, K. C. Chan and P. Potier, C. R. Acad. Sci., Ser. IIb: Mec., Phys., Chim., Astron., 1987, 304, 157 CAS; (b) T. S. Kam, Y. M. Tee and G. Subramaniam, Nat. Prod. Lett., 1998, 12, 307 CrossRef CAS; (c) G. Subramaniam, O. Hiraku, M. Hayashi, T. Koyano, K. Komiyama and T.-S. Kam, J. Nat. Prod., 2007, 70, 1783 CrossRef CAS PubMed; (d) G. M. T. Robert, A. Ahond, C. Poupat, P. Potier and H. Jacquemin, J. Nat. Prod., 1983, 46, 694 CrossRef CAS.
- (a) S. H. Goh, C. Wei and A. R. M. Ali, Tetrahedron Lett., 1986, 27, 2501 CrossRef CAS; (b) C.-Y. Gan, Y.-Y. Low, N. F. Thomas and T.-S. Kam, J. Nat. Prod., 2013, 76, 957 CrossRef CAS PubMed.
- (a) D. J. Abraham, R. D. Rosenstein, R. L. Lyon and H. H. S. Fong, Tetrahedron Lett., 1972, 10, 909 CrossRef; (b) R. L. Lyon, H. H. S. Fong, N. R. Farnsworth and G. H. Svoboda, J. Pharm. Sci., 1973, 62, 218 CrossRef CAS PubMed.
- (a) M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253 CrossRef CAS PubMed; (b) D. R. Matson and P. T. Stukenber, Mol. Interventions, 2011, 11, 141 CrossRef CAS PubMed.
- B. David, T. Sevenet, M. Morgat, D. Guenard, A. Moisand, Y. Tollon, O. Thoison and M. Wright, Cell Motil. Cytoskeleton, 1994, 28, 317 CrossRef CAS PubMed.
- M. C. Edler, G. Yang, M. K. Jung, R. Bai, W. G. Bornmann and E. Hamel, Arch. Biochem. Biophys., 2009, 487, 98 CrossRef CAS PubMed.
- B. David, T. Sevenet, O. Thoison, K. Awang, M. Pais, M. Wright and D. Guénard, Bioorg. Med. Chem. Lett., 1997, 7, 2155 CrossRef CAS.
- I. Gerasimenko, Y. Sheludko and J. Stöckigt, J. Nat. Prod., 2001, 64, 114 CrossRef CAS PubMed.
- Y. Wu, M. Suehiro, M. Kitajima, T. Matsuzaki, S. Hashimoto, M. Nagaoka, R. Zhang and H. Takayama, J. Nat. Prod., 2009, 72, 204 CrossRef CAS PubMed.
- S. H. Goh and A. R. M. Ali, Tetrahedron Lett., 1986, 27, 2501 CrossRef CAS.
- (a) S. H. Goh, W. Chen and A. R. M. Ali, Tetrahedron Lett., 1984, 25, 3483 CrossRef CAS; (b) C. Wei, A. R. M. Ali, S. H. Goh, E. Sinn and R. J. Butcher, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 349 CrossRef; (c) S. H. Goh, A. R. M. Ali and W. H. Wong, Tetrahedron, 1989, 45, 7899 CrossRef CAS.
- J. Le Men and W. I. Taylor, Experientia, 1965, 21, 508 CrossRef CAS PubMed.
- (a) C. Pascal, J. Dubois, D. Guénard, L. Tchertanov, S. Thoret and F. Guéritte, Tetrahedron, 1998, 54, 14737 CrossRef CAS; (b) E. Pasquinet, P. Rocca, S. Richalot, F. Guéritte, D. Guénard, A. Godard, F. Marsais and G. Quéguiner, J. Org. Chem., 2001, 66, 2654 CrossRef CAS PubMed; (c) C. Dupont, D. Guénard, C. Thal, S. Thoret and F. Guéritte, Tetrahedron Lett., 2000, 41, 5853 CrossRef CAS; (d) O. Baudoin, F. Claveau, S. Thoret, A. Herrbach, D. Guénard and F. Guéritte, Bioorg. Med. Chem., 2002, 10, 3395 CrossRef CAS PubMed; (e) M. Sailu, S. S. Muley, A. Das, P. S. Mainkar and S. Chandrasekhar, Tetrahedron, 2015, 71, 1276 CrossRef CAS.
- (a) O. Baudoin, D. Guenard and F. Gueritte, Mini-Rev. Org. Chem., 2004, 1, 333–341 CrossRef CAS; (b) I. Kholod, O. Vallat, A.-M. Buciumas and R. Neier, Heterocycles, 2010, 82, 917 CrossRef.
- D. Le Floch, N. Goulaut, M. David and P. van de Weghe, ARKIVOC, 2010, 1, 247 Search PubMed.
- M. Pfaffenbach and T. Gaich, Alkaloids–Chem. Biol., 2017, 77, 1 Search PubMed.
- A. H. Ratcliffe, G. F. Smith and G. N. Smith, Tetrahedron Lett., 1973, 14, 5179 CrossRef.
- Y. Yang, Y. Bai, S. Sun and M. Dai, Org. Lett., 2014, 16, 6216 CrossRef CAS PubMed.
- (a) B. Witkop, J. P. Patrick and M. Rosenblum, J. Am. Chem. Soc., 1951, 73, 2641 CrossRef CAS; (b) E. Winterfeldt, Liebigs Ann. Chem., 1971, 23, 745 Search PubMed.
- J. Xu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2015, 137, 6712 CrossRef PubMed.
- Z. Li, Q. Geng, Z. Lv, B. P. Pritchett, K. Baba, Y. Numajiri, B. M. Stoltz and G. Liang, Org. Chem. Front., 2015, 2, 236 RSC.
- A. Umehara, H. Ueda and H. Tokuyama, Tetrahedron, 2021, 79, 131809 CrossRef CAS.
- A. L. Bowie, C. C. Hughes and D. Trauner, Org. Lett., 2005, 23, 5207 CrossRef PubMed.
- M. G. Banwell, A. J. Edwards, K. A. Jolliffe, J. A. Smith, E. Hamel and P. Verdier-Pinard, Org. Biomol. Chem., 2003, 1, 296 RSC.
- M. G. Banwell, D. A. Beck and A. C. Willis, ARKIVOC, 2006, 3, 163 Search PubMed.
- Z. Liu, A. S. Wasmuth and S. G. Nelson, J. Am. Chem. Soc., 2006, 128, 10352 CrossRef CAS PubMed.
- V. Magné, C. Lorton, A. Marinetti, X. Guinchard and A. Voituriez, Org. Lett., 2017, 19, 4794 CrossRef PubMed.
- Y. Wang, W. Ren, J. Li, H. Wang and Y. Shi, Org. Lett., 2014, 16, 5960 CrossRef CAS PubMed.
- A. Shemet and E. M. Carreira, Org. Lett., 2017, 19, 5529 CrossRef CAS PubMed.
- (a) M. M. Meravi, R. Moradi and M. Malnir, Curr. Org. Chem., 2018, 22, 165 CrossRef; (b) P. J. Guiry and D. Kiely, Curr. Org. Chem., 2004, 8, 781 CrossRef CAS; (c) D. Mc Cartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122 RSC; (d) M. Shibasaki, E. M. Vogl and T. Ohshima, Adv. Synth. Catal., 2004, 346, 1533 CrossRef CAS.
- Z. Gu and A. Zakarian, Org. Lett., 2010, 12, 4224 CrossRef CAS PubMed.
- Y. Su, H. Zhou, J. Chen, J. Xu, X. Wu, A. Lin and H. Yao, Org. Lett., 2014, 16, 4884 CrossRef CAS PubMed.
- D. van Leusen, E. van Echten and A. M van Leusen, J. Org. Chem., 1992, 57, 2245 CrossRef CAS.
- X. Sui, R. Zhu, G. Li, X. Ma and Z. Gu, J. Am. Chem. Soc., 2013, 135, 9318 CrossRef CAS PubMed.
- M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed., 1997, 36, 119 CrossRef CAS.
- (a) F. Denat, H. Gaspard-Houghmane and J. J. Dubac, Organomet. Chem., 1992, 423, 173 CrossRef CAS; (b) J. M. Muchowski and P. Hess, Tetrahedron Lett., 1988, 29, 777 CrossRef CAS.
- K. Zhao, S. Xu, C. Pan, X. Sui and Z. Gu, Org. Lett., 2016, 18, 3782 CrossRef CAS PubMed.
- B. Harimi, H. Behzadnia, D. Elhamifar, P. F. Akhavan, F. K. Esfahani and A. Zamani, Synthesis, 2010, 1399 Search PubMed.
- A. L. Bowie and D. Trauner, J. Org. Chem., 2009, 74, 1581 CrossRef CAS PubMed.
- E. M. Beck, R. Hatley and M. J. Gaunt, Angew. Chem., Int. Ed., 2008, 47, 3004 CrossRef CAS PubMed.
- E. M. Beck, N. P. Grimster, R. Hatley and M. J. Gaunt, J. Am. Chem. Soc., 2006, 128, 2528 CrossRef CAS PubMed.
- L. McMurray, E. M Beck and M. J. Gaunt, Angew. Chem., Int. Ed., 2012, 51, 9288 CrossRef CAS PubMed.
- (a) D. García-Cuadrado, A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066 CrossRef PubMed; (b) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS PubMed; (c) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef.
- N. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300 RSC.
- (a) Y. Nakao, Bull. Chem. Soc. Jpn., 2012, 85, 731 CrossRef CAS; (b) Y. Nakao, Top. Curr. Chem., 2014, 346, 33 CrossRef CAS PubMed.
- Y. Yamada, S. Ebata, T. Hiyama and Y. Nakao, Tetrahedron, 2015, 71, 4413 CrossRef CAS.
- (a) D. Schinzer, Synthesis, 1988, 263 CrossRef CAS; (b) J. H. Lee, Tetrahedron, 2020, 76, 131351 CrossRef CAS.
- E. C. Izgu and T. R. Hoye, Chem. Sci., 2013, 4, 2262 RSC.
- P. Magnus and T. Rainey, Tetrahedron, 2001, 57, 8647 CrossRef CAS.
- R. Grigg, P. Myers, A. Somasunderama and V. Sridharana, Tetrahedron, 1992, 48, 9735 CrossRef.
- D. C. Behenna, Y. Liu, T. Yurino, J. Kim, D. E. White, S. C. Virgil and B. M. Stoltz, Nat. Chem., 2012, 4, 130 CrossRef CAS PubMed.
- D. Dagoneau, Z. Xu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 760 CrossRef CAS PubMed.
- J. A. Johnson and D. Sames, J. Am. Chem. Soc., 2000, 122, 6321 CrossRef CAS.
- J. A. Johnson, N. Li and D. Sames, J. Am. Chem. Soc., 2002, 124, 6900 CrossRef CAS PubMed.
- J. B. Gualtierotti, D. Pasche, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2014, 53, 9926 CrossRef CAS PubMed.
- A. M. Echavarren, A. S. K. Hashmi and F. D. Toste, Adv. Synth. Catal., 2016, 358, 1347 CrossRef CAS.
- (a) M. Marin-Luna, O. N. Faza and C. S. Lopez, Front. Chem., 2019, 7, 296 CrossRef CAS PubMed; (b) B. Alcaide and P. Almendros, Acc. Chem. Res., 2014, 47, 939 CrossRef CAS PubMed.
- D. Pflästerer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331 RSC.
- K. Sugimoto, K. Toyoshima, S. Nonaka, K. Kotaki, H. Ueda and H. Tokuyama, Angew. Chem., Int. Ed., 2013, 52, 7168 CrossRef CAS PubMed.
- F. Sirindil, J.-M. Weibel, P. Pale and A. Blanc, Org. Lett., 2019, 21, 5542 CrossRef CAS PubMed.
- F. Sirindil, S. Golling, R. Lamare, J.-M. Weibel, P. Pale and A. Blanc, Org. Lett., 2019, 21, 8997 CrossRef CAS PubMed.
- S. Miaskiewicz, B. Gaillard, N. Kern, J.-M. Weibel, P. Pale and A. Blanc, Angew. Chem., Int. Ed., 2016, 55, 9088 CrossRef CAS PubMed.
- F. Sirindil, R. Pertschi, E. Naulin, D. Hatey, J.-M. Weibel, P. Pale and A. Blanc, ACS Omega, 2022, 7, 1186 CrossRef CAS PubMed.
- K. Sugimoto, Y. Miyakawa and H. Tokuyama, Tetrahedron, 2015, 71, 3619 CrossRef CAS.
- P. L. Feldman and H. J. Rapoport, J. Org. Chem., 1986, 51, 3882 CrossRef CAS.
- (a) K. J. Romero, M. S. Galliher, D. A. Pratt and C. J. Stephenson, Chem. Soc. Rev., 2018, 47, 7851 RSC; (b) C. P. Jasperse, D. P. Curran and T. L. Fevig, Chem. Rev., 1991, 91, 1237 CrossRef CAS.
- (a) S. Hildebrandt, H. Weissbarth and A. Gansäuer, Synthesis, 2017, 49, 2584 CrossRef CAS; (b) S. Hildebrandt and A. Gansäuer, Angew. Chem., Int. Ed., 2016, 55, 9719 CrossRef CAS PubMed.
- (a) W. A. Nugent and T. V. Rajanbabu, J. Am. Chem. Soc., 1988, 110, 8561 CrossRef CAS; (b) For a review, see: A. F. Barrero, J. F. Quilez del Moral, E. M. Sanchez and J. F. Artega, Eur. J. Org. Chem., 2006, 74, 1627 CrossRef.
- E. Paleo, Y. M. Osornio and L. D. Miranda, Org. Biomol. Chem., 2011, 9, 361 RSC.
- A. Biechy and S. Zard, Org. Lett., 2009, 11(13), 2800 CrossRef CAS PubMed.
- M. Sailu, S. S. Muley, A. Das, P. S. Mainkar and S. Chandrasekhar, Tetrahedron, 2015, 71, 1276 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |