
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalming egress of inflammatory monocytes and related septic shock by therapeutic CCR2 silencing using macrophage-derived extracellular vesicles†
Liang
Ding‡
a,
Wenjing
Zhou‡
a,
Jian
Zhang‡
 b,
Qingsong
Tang
a,
Weizhang
Xiao
a,
Ming
Chen
a,
Zhenya
Shen
*a and
Weiqian
Chen
*a
b,
Qingsong
Tang
a,
Weizhang
Xiao
a,
Ming
Chen
a,
Zhenya
Shen
*a and
Weiqian
Chen
*a
aDepartment of Cardiovascular Surgery of the First Affiliated Hospital & Institute for Cardiovascular Science, Suzhou Medical College of Soochow University, Soochow University, Suzhou 215006, China. E-mail: chenweiqian@suda.edu.cn; uuzyshen@aliyun.com
bInstitute of Advanced Magnetic Materials, College of Materials and Environmental Engineering, Hangzhou Dianzi University, Hangzhou 310018, China
First published on 9th February 2022
Abstract
Uncontrolled inflammation, featuring the aggravated mobilization of Ly6Chigh inflammatory monocytes (Mos), may cause high morbidity and mortality in the pathogenesis of sepsis-associated immune disorders. Inspired by the similar membrane protein profile of extracellular vehicles (EVs) and their parent cells, EVs are generated from immortalized bone marrow-derived macrophages (Mps) for Mo/Mp-targeting drug delivery. Compared with MSC-EVs, Mac-EVs are more efficiently internalized by inflammatory Mo/Mps in vitro as well as by septic spleen in vivo. By loading with siRNA targeting the chemokine receptor CCR2, the mediator for chemotaxis of inflammatory Mo/Mps, Mac-EVsiCCR2 not only restrains chemotaxis of inflammatory Mo/Mps but also relieves septic symptoms in mice by limiting the mobilization of splenic inflammatory monocytes and calming the subsequent serum cytokine storm. The current study provides functional evidence for the successful therapeutic targeting of septic inflammatory Mos, mandating the clinical development of CCR2 inhibition in patients with infectious diseases.
Introduction
Inflammation in most cases serves as a beneficial defense mechanism against infection and injury in the body.1,2 However, uncontrolled infections may also cause an overwhelming systemic or local inflammatory response, leading to life-threatening diseases including sepsis3 and the 2019 novel coronavirus disease (SARS-Cov-2; COVID-19).4Inflammatory monocytes (Mos) give rise to classical macrophages (Mps) and promote inflammatory response following a variety of infections.5 We and others have developed a large number of strategies to dampen detrimental inflammation by engineering the inflammatory Mos/Mps (Ly6Chigh in mouse, CD14+CD16− in humans) through specific ablation,6 immuno-metabolic reprogramming7 or secretome modulation.8 Nonetheless, none of these aforementioned interventions are readily available for translational medicine.
Mature Ly6Chigh Mos reside in the subcapsular red pulp of the spleen, from which they rapidly egress and enter the circulation in response to various infections.9 As essential early responders, excessive recruitment or prolonged accumulation of these spleen-derived Ly6Chigh subsets may hinder regression of inflammation and propagate disease progression.
Distinct from the reparative Mo subset (Ly6Clow in mouse, CD14−CD16+ in humans) which depends on fractalkine/(C-X3-C motif) receptor 1 (CX3CR1), the recruitment of inflammatory Mos depends largely on the chemokine/chemokine receptor pair C–C ligand 2 (CCL2)/C–C receptor 2 (CCR2).10 Previous studies have demonstrated reduced severity of inflammation through the genetic deletion of CCR2 in several disease models, leaving reparative Mo/Mps, antigen-presenting cells and other tissue residents unaffected.11–13 Hence, calming Ly6Chigh Mo/Mps via the therapeutic silencing of CCR2 immediately after infection occurs may retard their exaggerated infiltration and benefit multiple inflammatory diseases.
As one kind of cell-based drug-delivery system,14,15 extracellular vehicles (EVs)/exosomes have received increasing attention due to their excellent biocompatible behaviors compared with other synthetic systems. Among them, we and others have linked mesenchymal stem/stromal cell-derived EVs (MSC-EVs) to translational medicine in the context of numerous diseases, including myocardial infarction,16 sepsis,17 stroke,18 and Parkinson's disease,19 due to their availability, quality, and reproducibility. EVs fuse to target cells either with their plasma membranes directly or with the endosomal membranes after endocytic uptake.20 Regarding this, we propose that macrophage-derived EVs (Mac-EVs) may achieve a higher uptake efficiency to Mo/Mps than commonly used MSC-EVs due to the fact that Mac-EVs possess surface membrane properties similar to their parent cells,21 hence suit perfectly for drug delivery to the Mo/Mps system.
In this work, a large number of MSC-EVs and Mac-EVs were readily obtained from bone marrow MSCs and immortalized bone marrow-derived macrophages (iBMDMs), respectively. Intriguingly, Mac-EVs seemed to achieve a higher uptake to inflammatory Mo/Mps than MSC-EVs did. It was further demonstrated that Mac-EVs exhibit excellent capacity of accumulating at the site of the spleen, from which Ly6Chigh inflammatory Mos are dispatched to sites of local infection.9 Mac-EVs were then encapsulated with siCCR2 for therapeutic silencing of CCR2. These siCCR2-loaded Mac-EVs not only blunted in vitro chemotaxis of inflammatory Mo/Mps toward CCL2 but also impaired the in vivo egress of splenic Ly6Chigh Mos and calmed subsequent septic cytokine storm syndrome (Scheme 1). Finally, such a nanobiotechnology platform may be further optimized by loading drugs into Mac-EVs from specified patients for personalized medication.
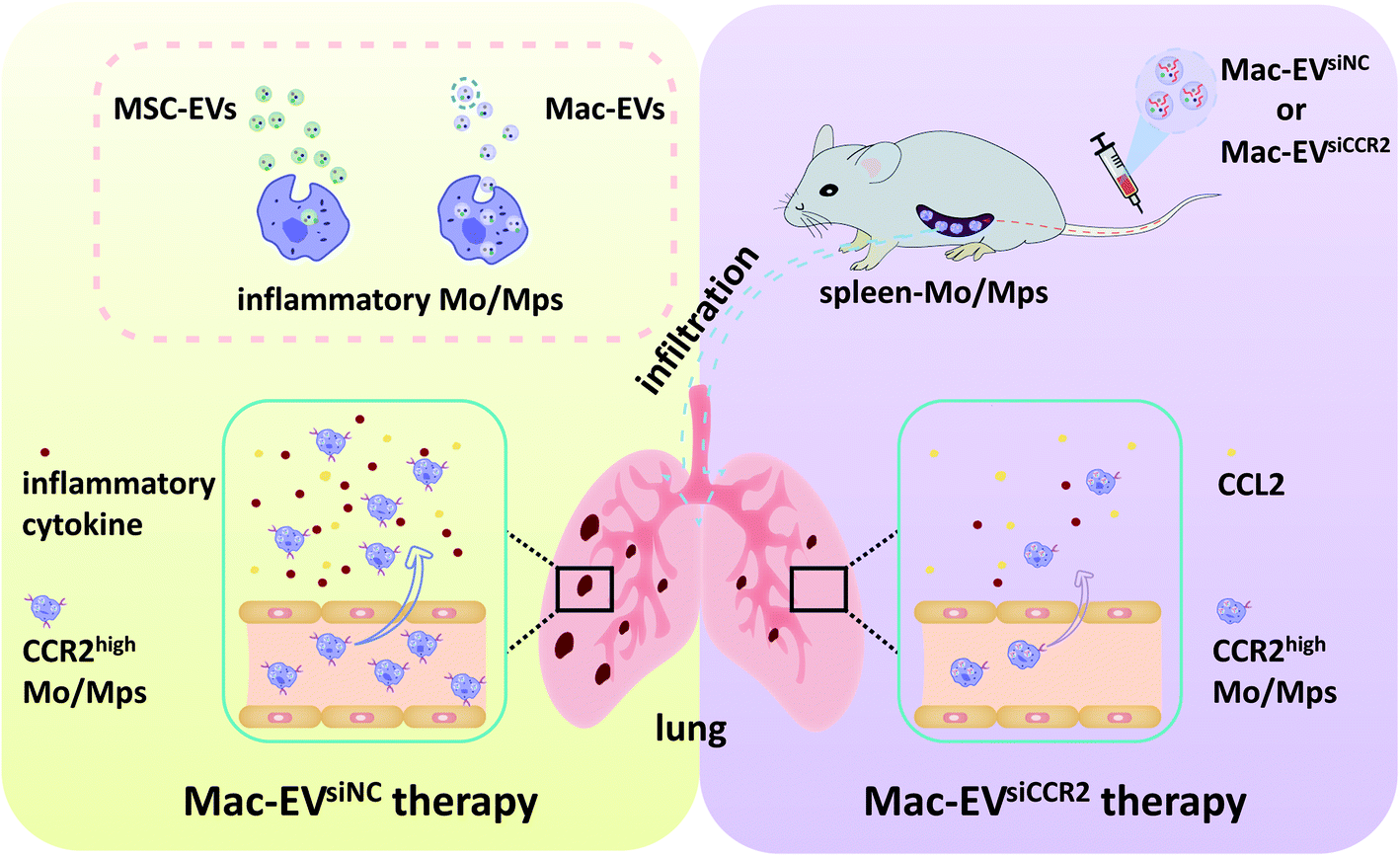 | ||
| Scheme 1 Schematic illustration demonstrating the Mac-EV-based siCCR2 delivery system which may calm the egress of inflammatory monocytes and related septic shock. | ||
Materials and methods
Cell culture and treatments
C57BL/6 mouse-derived immortalized bone marrow-derived macrophage (iBMDM) cells were kindly provided by F. Shao (National Institute of Biological Sciences, China). iBMDM cells were maintained in RPMI 1640 medium (Procell) supplemented with 10% fetal bovine serum FBS, ExCell Bio. Bone marrow-derived MSCs from C57BL/6 mice (Cyagen Biosciences) were maintained with mesenchymal stem cell growth medium (Cyagen Biosciences) supplemented with 10% FBS as we described previously.22 iBMDMs were polarized to inflammatory status with LPS (Sigma, 20 ng mL−1) plus IFN-γ (PeproTech, 20 ng mL−1) for 24 h.Peritoneal macrophages were isolated from male C57BL/6 mice. Briefly, mice were euthanized 72 h after intraperitoneal injection of 1 mL of sterile starch broth (0.3% yeast powder, 1% peptone, 0.5% sodium chloride, and 5% starch) for three consecutive days. Peritoneal cells were then harvested by peritoneal lavage and nonadherent cells were removed 2 h later. Adherent macrophages were maintained in RPMI 1640 supplemented with 15% FBS until use.
EV isolation, qualification and characterization
iBMDM Mo/Mps and MSCs were cultured with EV-free FBS, and EVs were isolated by serial ultracentrifugation as described previously.23,24 Briefly, cell culture supernatants were passed through a 0.22 μm filter and centrifuged at 2000g for 30 min followed by 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min, 4 °C to remove dead cells and cell debris, and finally centrifuged twice at 110000g for 70 min, 4 °C. PBS was used to resuspend purified EVs. EVs were kept either at 80 °C for long-term preservation or at 20 °C for short-term preservation. BCA protein assay was used for EV quantification and EV morphology was analyzed by transmission electron microscopy (TEM, Tecnai). To examine EV size distribution, nanoparticle tracking analysis (NTA) was performed using a ZetaView instrument (Particle Metrix). The expression of EV marker proteins, including CD63 and CD81, was confirmed by flow cytometry.
000g for 30 min, 4 °C to remove dead cells and cell debris, and finally centrifuged twice at 110000g for 70 min, 4 °C. PBS was used to resuspend purified EVs. EVs were kept either at 80 °C for long-term preservation or at 20 °C for short-term preservation. BCA protein assay was used for EV quantification and EV morphology was analyzed by transmission electron microscopy (TEM, Tecnai). To examine EV size distribution, nanoparticle tracking analysis (NTA) was performed using a ZetaView instrument (Particle Metrix). The expression of EV marker proteins, including CD63 and CD81, was confirmed by flow cytometry.
EV labelling and in vitro internalization
MSC-EVs and Mac-EVs were labelled with 1 μM CM-DiI dye (Invitrogen). After sequential incubation at 37 °C and 4 °C, excess dye was removed and labelled EVs were reisolated. Recipient iBMDM Mo/Mps were incubated with CM-DiI-labelled EVs (120 μg mL−1) for 24 h. In vitro internalization was visualized using a confocal microscope (Zeiss LSM880) and quantitated by flow cytometry.EV labelling and in vivo tracking
Indicated EVs were labelled with Vybrant DID (Invitrogen) according to the manufacturer's instructions. Next, Vybrant DID-labeled EVs were intravenously injected via tail vein (100 μg per mouse) 4 h after CLP surgery. Finally, spleens were harvested for ex vivo imaging 24 h after CLP. Fluorescence intensity was monitored using the in vivo imaging system (IVIS) and Living Image software (PerkinElmer).Loading siCCR2 into Mac-EVs
Loading of Mac-EVs with siCCR2 or siNC (GenePharma) was obtained by electroporation with a 4D-Nucleofector™ System (Lonza) as described previously.25 After removing the free-floating siRNAs, EVs were purified and resuspended in PBS and stored at −80 °C. Sequences of siRNA oligonucleotides are as follows: siCCR2 (mus) and 5′-uGcuAAAcGucucuGcAAAdTsdT-3′. Chemically modified siRNA with higher stability and membrane affinity was used in the whole study.Transwell assay
Migration analysis was carried out using Boyden transwell chambers (8.0 μm pore size, Corning). Briefly, EV-infused iBMDM Mo/Mps were seeded at 2 × 105 cells/100 μL in the upper chamber of a 24-well plate and 500 μL of medium containing CCL2 (PeproTech, 100 ng mL−1) was added into the lower chamber. After 4 h of incubation, the remaining cells on the upper side of the insert were wiped off and cells that had migrated to the reverse side of the insert were stained with DAPI (Solarbio) and counted as we described previously.26Animal studies and depletion of Mo/Mps
All experimental procedures were performed according to the Guide for the Care and Use of Laboratory Animals (NIH, 8th edition, 2011) and approved by the Ethics Committee of Soochow University. Mid-grade CLP surgery was performed on 8–12-week-old male C57BL/6 mice as described previously.17 Briefly, the cecum was ligated at half the cecal length and punctured with a 22-gauge needle, and fecal contents were then gently extruded into the peritoneal cavity. After surgery, we resuscitated the mice by subcutaneously injecting prewarmed normal saline (1 mL per mouse). CLP-challenged mice were randomly assigned for tail vein injection of the indicated EVs (100 μg per mouse) 4 h after the operation. Lung, blood, and spleen tissues were collected for subsequent experiments 24 h after CLP surgery. Otherwise, the survival rate was monitored every 24 h for 6 days. Depletion of Mo/Mps was achieved by intraperitoneal injection of Cl2MDP-lipo (ClodronateLiposomes.org) at a dosage of 60 μL per mice one day before CLP operation.Pulmonary vascular leakage and lung wet/dry ratio
Pulmonary vascular leakage was quantified using Evans blue dye. In brief, CLP mice were administered 1% Evans blue solution via tail vein injection. After perfusion 30 min later, lungs were collected and placed in 2 mL of formamide at 60 °C for 24 h to extract Evans blue. Concentration of Evans blue in the supernatant was quantified by measuring absorbance at 620 nm (BIO-TEK) and calculated from a standard curve.Lung samples were weighed immediately after removal (wet weight) and dried for 48 h until a stable dry weight was reached. The ratio of wet weight to dry weight was calculated to quantify the degree of pulmonary edema.
Tissue digestion and flow cytometry
For flow cytometry of EVs, the isolated EVs were prebound to aldehyde/sulfate latex beads (4 μm) before antibody staining. For flow cytometry of pulmonary immune cells, lung tissues were digested in a solution composed of 10 mM HEPES, 10 μg mL−1 DNAse I and 0.4 mg mL−1 collagenase D and 2% FBS for 30 min. For flow cytometry of peripheral blood, red blood cells (RBCs) were lysed using RBC lysis buffer (Biolegend). For antibody staining, anti-CD29-Alexa Fluor 488, anti-CD44-APC, anti-CD45-PE, anti-CD90-FITC, anti-CD117-APC, anti-Sca-1-APC, anti-CD63-PE, anti-CD81-PE, anti-CD11b-APC, anti-Ly6G-Alexa Fluor 488, anti-Ly6C-PE, and anti-CD45-PE (Biolegend or eBioscience) were used. Finally, the stained cells were analyzed by flow cytometry (Millipore Guava easyCyte) and data were analyzed using the FlowJo software.Enzyme-linked immunosorbent assay (ELISA)
Serum cytokine production of IL-1β and IL-6 was measured by ELISA (MultiSciences) according to the manufacturer's protocol as we described previously.27 Optical densities were determined using a multifunctional microplate reader (BIO-TEK) at 450 nm.Reverse transcription (RT)-PCR and quantitative (q) RT-PCR
Total RNA was quantified using an ND2000 spectrophotometer (NanoDrop), and reversed transcribed with HiScript III RT SuperMix Kit (Vazyme). qRT-PCR was carried out using SYBR Premix Ex Taq reaction mix (Takara) on a StepOne Plus real-time PCR system (Applied Biosystems) as we previously reported.28 The expression of target genes was determined by the comparative ΔΔCt method and 18S was used as an internal control gene. Primer sequences are as follows: Ccr2, 5′-TTACACCTGTGGCCCTTATTT-3′ and 5′-CTGAGTAGCAGATGA CCA TGAC-3′; Ccr5, 5′-GCTCCAAGAGATGAGGAAAGAG-3′ and 5′-GAACACAGAGAGCAGTCGTTAT-3′; Cx3cr1, 5′-GAGAGATGGCTCAGTGGTTAG-3′ and 5′-CACAGGAACAGGGAGCTATTT-3′; 18S, 5′-GTAACCCGTTGAACCCCATT-3′ and 5′-CCATCCAATCGGTAGTAGCG-3′.Immunostaining
For fluorescence immunostaining, cells grown on coverslips or frozen tissue sections were routinely fixed, permeabilized and blocked as we described previously.7 Fixed slides were further stained with anti-F4/80 (Abcam, ab6640, 1:100), anti-CD11b (Biolegend, 101201, 1:100), or anti-CCR2 (Abcam, ab203128, 1:100), and detected by fluorescent conjugated secondary antibodies (Yeasen). Nuclei were counterstained with DAPI (Solarbio). Images were captured using a confocal microscope (Zeiss LSM880) and processed using the ZEN software.
Western blot analysis
Protein lysates were processed for western blot analysis following the standard protocol as we described previously.29 The following primary antibodies were used: anti-CCR2 (Abcam, ab203128, 1:1000), anti-CX3CR1 (Proteintech, 13885-1-AP, 1:1000), and anti-β-actin (Sungene). Immunoreactivity was detected by routine enzymatic chemiluminescence (Meilun Biotechnology).
Statistics
Data are shown as mean ± SEM using the GraphPad Prism 8 software. Unless otherwise specified, statistical analysis was performed using unpaired two-tailed Student's t test. Statistical analysis for survival was performed with the log-rank (Mantel–Cox) test. *P < 0.05, **P < 0.01, ***P < 0.001.Results and discussion
Preparation and characterization of MSC-EVs and Mac-EVs
First, MSCs were isolated from the bone marrow of C57BL/6 mice and their surface markers were confirmed according to established criteria. In agreement with our previous report,22 MSCs express CD29, CD44, CD90, and Sca-1 (Fig. 1A) and they are devoid of hematopoietic lineage marker CD45 and progenitor marker CD117 (Fig. 1B). Next, immortalized BMDMs (iBMDM) were also characterized by expressing CD11b and F4/80 (Fig. 1C).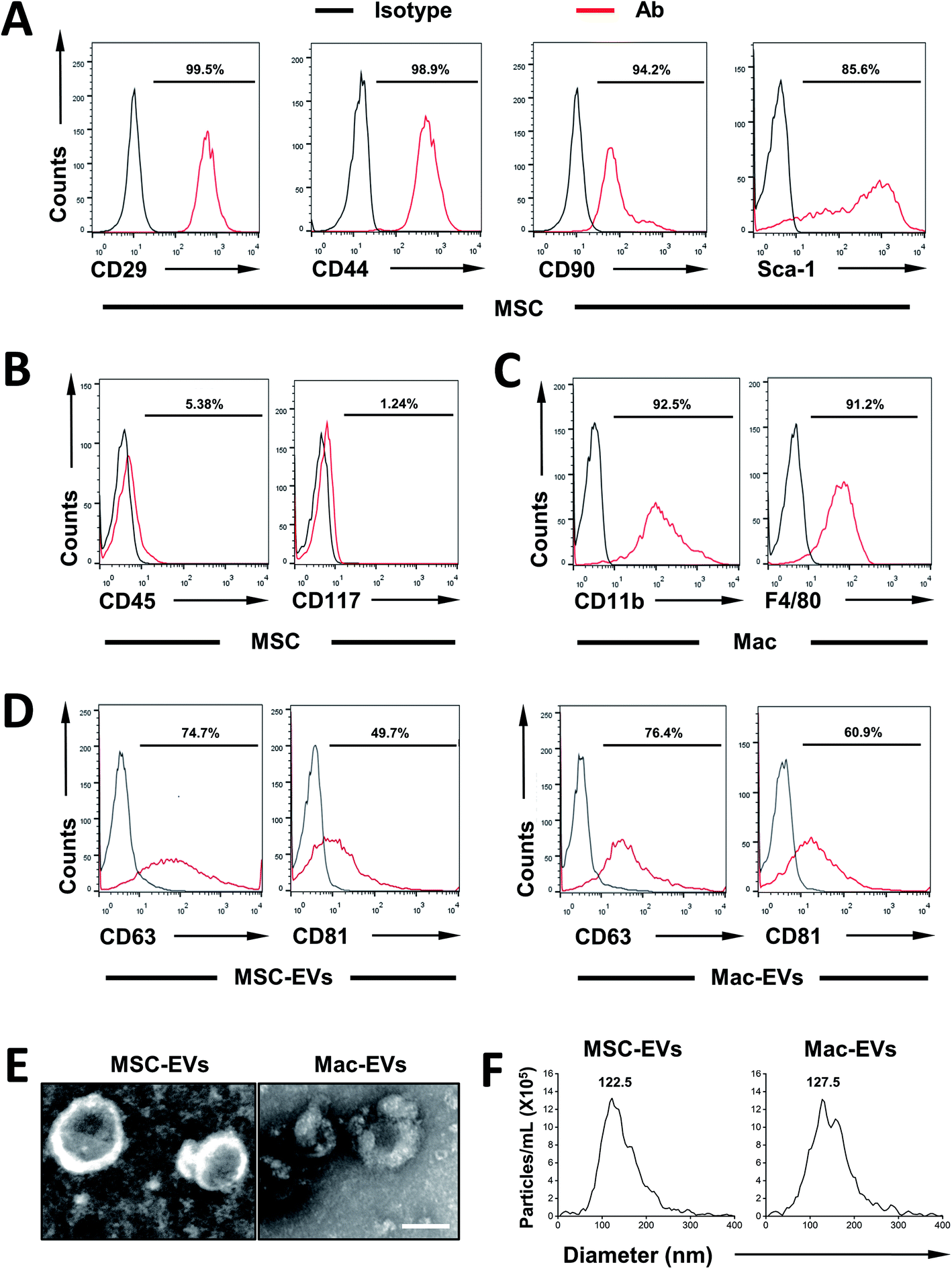 | ||
| Fig. 1 Characterization of MSC-EVs and Mac-EVs. (A–C) Flow cytometry analysis for surface antigens of bone marrow MSCs and Mac. Isotype controls are indicated by black spectra. (D) Expression of EV marker proteins, namely CD63 and CD81, was identified by flow cytometry. (E) Morphology of MSC-EVs and Mac-EVs observed by TEM. Scale bar = 100 nm. (F) Particle size distribution of MSC-EVs and Mac-EVs measured by NTA. | ||
Then, MSC-EVs and Mac-EVs were isolated from bone marrow MSC and iBMDM Mo/Mp culture supernatant respectively. The purified EVs were characterized according to their protein expression, morphological features and particle size. Biological characterization of isolated EVs was done via flow cytometry of common EV marker CD63 and CD81 and both EVs were positive for all the markers tested (Fig. 1D). Besides, the purified EVs showed a spherical shape, with size ranging from 100 to 150 nm (Fig. 1E). Finally, as examined by nanoparticle tracking analysis, both EVs were morphologically homogeneous, with sizes peaking at 122.5 nm and 127.5 nm respectively (Fig. 1F).
In vitro internalization of EVs to inflammatory Mo/Mps
Ly6ChighCCR2+ inflammatory and Ly6ClowCCR2− resident Mos are generally thought to differentiate into inflammatory and reparative Mps, respectively.30 To compare uptake efficiency of MSC-EVs and Mac-EVs to inflammatory Mo/Mps, we labeled both EVs with CM-DiI (red) and cocultured them with LPS/IFN-γ-activated inflammatory iBMDM Mo/Mps for 24 h. It was observed by immunofluorescence analysis that both EVs were able to be internalized by inflammatory Mo/Mps. Importantly, a relatively higher level of fusing to Mo/Mps was observed by Mac-EV application (Fig. 2A). Likewise, flow cytometry further confirmed the superior internalization of Mac-EVs by inflammatory Mo/Mps to that of MSC-EVs (Fig. 2B). Collectively, these results indicated that Mac-EVs have higher affinity toward inflammatory Mo/Mps in vitro. Nevertheless, the receptors/proteins as well as mechanisms by which Mo/Mps internalize Mac-EVs need to be further investigated in detail. | ||
| Fig. 2 In vitro targeting of MSC-EVs and Mac-EVs. (A) Confocal fluorescence image and quantification by the Image J software for CM-DiI-labeled MSC-EVs and Mac-EVs after incubation with inflammatory Mo/Mps (n > 50). Scale bar = 5 μm. (B) Flow cytometry dot plots and quantification for CM-DiI signal in LPS/IFN-γ elicited iBMDMs cocultured with CM-DiI-labeled EVs (n = 4). | ||
In vivo targeting of Mac-EVs to spleen
Previous work described a large monocyte reservoir in the spleen which dispatches these cells to sites of local inflammation.9 We therefore investigated whether Mac-EVs could target spleen in vivo more efficiently. DiD-labeled EVs (100 μg per mice) were injected into septic mice intravenously and spleens were collected and imaged by an ex vivo imaging system 20 h following EV injection. Of note, a much stronger DiD signal was observed in the spleens of DiD-Mac-EV-treated mice than in DiD-MSC-EV-treated mice (7.207 ± 0.335 vs. 9.329 ± 0.366, P < 0.01) (Fig. 3A). Confocal images and flow cytometry of the spleens further confirmed excellent accumulation of Mac-EVs in the splenic reservoir compared with that of MSC-EVs (Fig. 3B and C).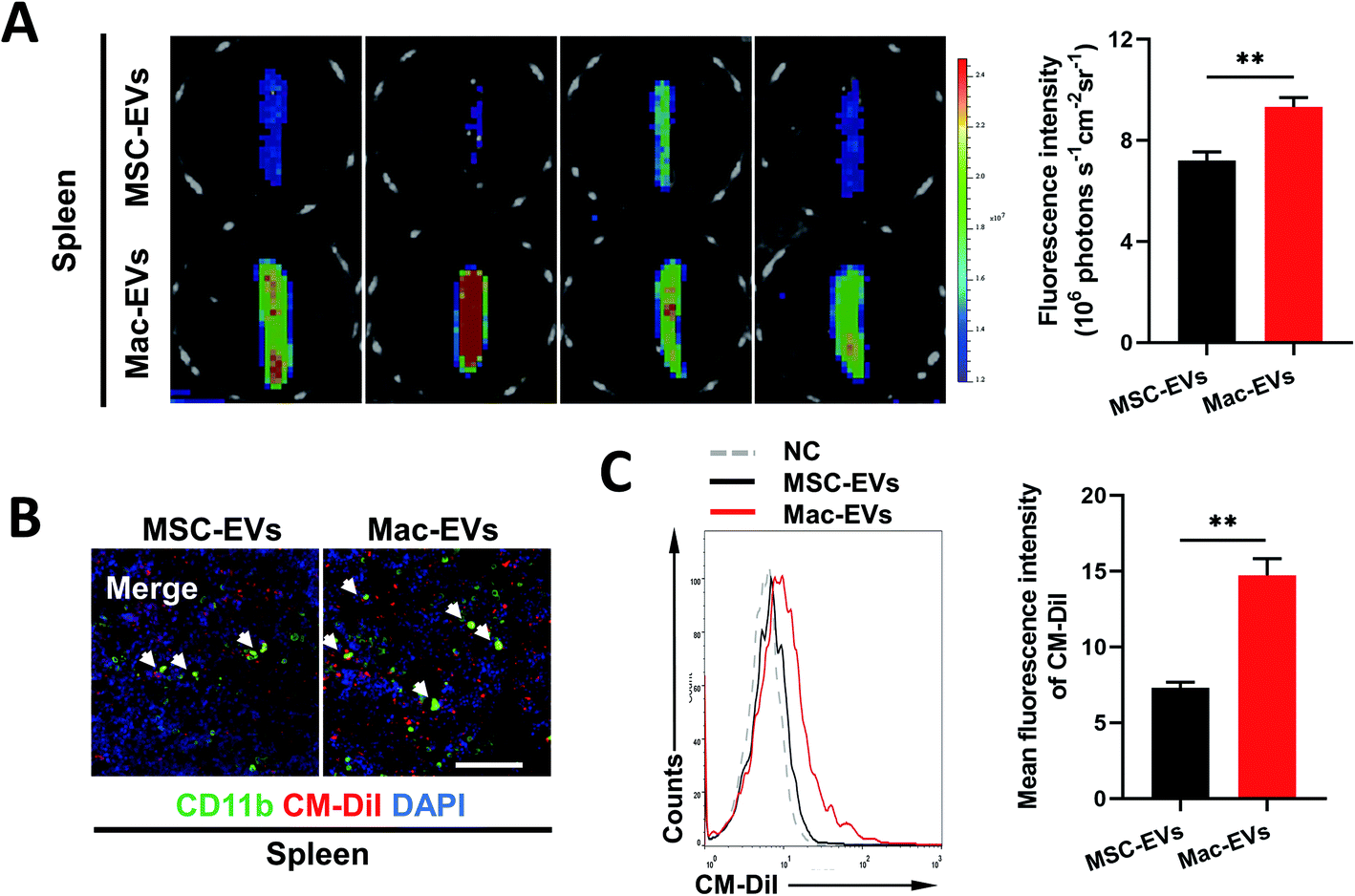 | ||
| Fig. 3 In vivo targeting of MSC-EVs and Mac-EVs. (A) Ex vivo fluorescence imaging of septic spleen after administration of DiD-labeled MSC-EVs and Mac-EVs 20 h following EV injection (n = 4). (B) Immunofluorescence of septic spleen tissue after administration of CM-DiI-labeled MSC-EVs or Mac-EVs 20 h following injection (red, CM-DiI-labeled EVs; green, CD11b; blue, nuclei). Scale bar = 100 μm. (C) Flow cytometry analysis of CM-DiI signal in spleen after intravenous injection of CM-DiI-labeled MSC-EVs or Mac-EVs (n = 5). Data are presented as mean ± SEM. **P < 0.01. | ||
Silencing of CCR2 completely blocks chemotaxis of inflammatory Mo/Mps
Recruitment of inflammatory Ly6Chigh Mo/Mps during inflammation depends largely on the CCR2 receptor.31 Not surprisingly, LPS/IFN-γ stimulation resulted in a detectable elevation in the Ccr2 mRNA level (Fig. 4A). We encapsulated siCCR2 into Mac-EVs for the efficient silencing of CCR2 in Mo/Mps. Although Mac-EVs encapsulated with Cy5-labeled siNC or siCCR2 were comparably internalized by LPS/IFN-γ-elicited iBMDM Mo/Mps (Fig. 4B), exposure of Mo/Mps to Mac-EVsiCCR2 did result in the impaired expression of CCR2 in both mRNA and protein levels (Fig. 4C–E). Consequently, application of Mac-EVsiCCR2 was able to cause a CCL2-induced chemotactic deficit not only in LPS/IFN-γ-activated Mo/Mps (−96.44%, P < 0.001) but also in starch broth-elicited ex vivo peritoneal macrophages (−78.47%, P < 0.001) (Fig. 4F–H). In summary, Mac-EVsiCCR2 not only suppressed CCR2 expression in inflammatory Mo/Mps but also diminished their chemotaxis toward CCL2. | ||
| Fig. 4 Drug loading of siCCR2 to the EVs and in vitro therapeutic effect. (A) Ccr2 expression of iBMDMs after stimulation with LPS/IFN-γ (n = 3–4). (B) Mac-EVs encapsulated with Cy5-labeled siNC or siCCR2 could be comparably internalized by LPS/IFN-γ elicited iBMDM. Scale bar = 20 μm. (C) mRNA expression of Ccr2, Ccr5 and Cx3cr1 in iBMDMs after stimulation with Mac-EVsiNC and Mac-EVsiCCR2 (n = 3–6). (D and E) Protein expression of CCR2 was evaluated by western blot (D) and immunostaining (E). Scale bar = 10 μm. (F–H) Representative images of DAPI staining of transwell migration assay on LPS/IFN-γ-stimulated iBMDMs (F) or starch broth-elicited ex vivo peritoneal macrophages (G). Scale bar = 100 μm; (H) Quantitative analysis of migrated cells in transwell assay (n = 6). Data are presented as mean ± SEM. *P < 0.05, ***P < 0.001, ns = not significant. | ||
Mac-EVs encapsulated with siCCR2 protects against systemic septic shock
To extend our in vitro findings, we next examined whether infusion of Mac-EVsiCCR2 could protect mice from systemic septic shock. Cecal ligation and puncture (CLP) challenged mice were infused with 100 μg of the indicated EVs 4 h after the surgery (Fig. 5A). In agreement with a previous study,17 more than 60% of CLP mice that were treated with Mac-EVsiNC succumbed within 72 h whereas over 60% of mice receiving Mac-EVsiCCR2 therapy survived beyond 72 h (Fig. 5B). Under the protection of Mac-EVsiCCR2, CLP mice produced significantly less serum IL-1β (9.213 ± 1.087 vs. 5.744 ± 0.397, P < 0.05) and IL-6 (1841 ± 238 vs. 1246 ± 76, P < 0.05), lending further support to the anti-inflammatory effects of Mac-EVsiCCR2 (Fig. 5C). | ||
| Fig. 5 Anti-sepsis therapy by Mac-EVsiCCR2 infusion. (A) Schematic diagram of Mac-EVsiCCR2 therapy on CLP mice. Murine sepsis was generated by the mid-grade CLP model. Mice were intravenously infused with Mac-EVsiNC or Mac-EVsiCCR2 4 h post-CLP and serum and lung tissues were collected for experimental analysis 24 h post-CLP. Otherwise, survival rate was monitored every 24 h for 6 days. (B) Survival of CLP mice after the indicated EV therapy (n = 8–9). (C) Serum IL-1β and IL-6 after the indicated treatments were measured 24 h after CLP challenge (n = 5–7). (D) Wet-to-dry ratio of lungs by various treatments as indicated (n = 4–5). (E) H&E-stained lung tissue sections were imaged. Representative H&E-stained lung sections obtained 24 h post-CLP. Scale bar = 50 μm. (F) Pulmonary vascular leakage in lungs was measured with Evans blue at 24 h after CLP (n = 7). Data are presented as mean ± SEM. *P < 0.05. | ||
Lung dysfunction, referred to as acute respiratory distress syndrome, is frequently associated with clinical sepsis.32 Similar to pneumonia induced by COVID-19, septic lungs also exhibit excessive edema, which may be due to exacerbated mucus production and immune cell infiltration. Accordingly, as evidenced by lung wet/dry weight ratio, administration of Mac-EVsiCCR2 greatly alleviated lung edema compared with their Mac-EVsiNC counterparts (Fig. 5D). Moreover, a similar alteration in histopathology was also observed, validating dampened pulmonary edema, alveolar inflammatory cell infiltration, and alveolar injury in septic mice receiving Mac-EVsiCCR2 therapy (Fig. 5E). Finally, as assessed by Evans blue tissue dispersion, Mac-EVsiCCR2 treatment also relieved septic pulmonary vascular leakage more effectively (Fig. 5F). In sum, these results suggested that Mac-EVsiCCR2 might be protective in CLP-induced septic shock; the underlying mechanism might be associated with its inhibitory effect on the release of proinflammatory cytokines.
Mac-EVsiCCR2 infusion prevents egress of inflammatory Mos from the spleen to septic lungs
Ly6Chigh Mo/Mps are potent inflammatory mediators and are believed to be the dominant source of inflammation;31 we therefore assessed accumulation of Ly6Chigh Mo/Mps in septic lungs. Consistent with previous findings, the percentage of pulmonary inflammatory Mo/Mps (Ly6ChighCD11b+Ly6G−) was statistically attenuated by Mac-EVsiCCR2 treatment (1.583% ± 0.272% vs. 0.778% ± 0.167%, P < 0.05, Fig. 6A and B), whereas the percentage of reparative Mo/Mps (Ly6ClowCD11b+Ly6G−) was comparable between the two groups (2.967% ± 0.383% vs. 2.672% ± 0.676%, P > 0.05, Fig. 6A and C), indicating that the silencing of CCR2 prevents accumulation of only inflammatory Mo/Mps in septic lungs. | ||
| Fig. 6 Egress of inflammatory macrophages from the spleen to septic lungs could be calmed by the delivery of siCCR2 using macrophage-derived EVs. (A–C) Representative flow cytometry dot plots (A) and quantification of inflammatory Mo/Mps (Ly6ChighCD11b+Ly6G−, B) and reparative Mo/Mps (Ly6ClowCD11b+Ly6G−, C) in septic lungs obtained 24 h after CLP challenge (n = 6–7). (D) Immunofluorescence of spleen tissue slices of mice after the indicated treatments (red, CM-DiI-labeled EVs; pink, Cy5-labeled siRNAs; blue, nuclei). Scale bar = 20 μm. (E and F) Representative flow cytometry dot plots (E) and quantification (F) of splenic inflammatory Mos (Ly6ChighCD11b+Ly6G−) as a percentage of total cell population (n = 6–7). Data are presented as mean ± SEM. *P < 0.05, ns = not significant. | ||
Mature Ly6Chigh Mos reside in the subcapsular red pulp of the spleen, from which they can be rapidly deployed and mobilized to the site of inflammation.9 To directly visualize in vivo accumulation of Mac-EVs in septic spleen, we encapsulated Cy5-labeled siRNAs into CM-DiI-labeled Mac-EVs and monitored CM-DiI and Cy5 signals 20 h after tail vein injection. Although relatively comparable CM-DiI and Cy5 signals were observed between the two groups (Fig. 6D), an appreciable elevation in the percentage of inflammatory Ly6Chigh Mos in septic spleen, though statistically insignificant, was still obvious following Mac-EVsiCCR2 therapy (0.537% ± 0.041% vs. 0.694% ± 0.110%, P > 0.05, Fig. 6E and F), suggesting that the egress of inflammatory Ly6Chigh Mos from the spleen could be calmed by the delivery of siCCR2 using Mac-EVs.
Mac-EVsiCCR2 mediates sepsis resistance by primarily targeting Mo/Mps
To confirm the requirement of Mo/Mps in our Mac-EVsiCCR2 therapy against sepsis, we selectively depleted Mo/Mps via systemic administration of clodronate liposomes (Cl2MDP-lipo), which causes apoptosis of Mo/Mps (Fig. 7A).31 As expected, treatment with Cl2MDP-lipo effectively eliminated Mo/Mps from both spleen and peripheral blood (Fig. 7B–D). More importantly, in the setting of additional Mo/Mp removal, mice receiving Mac-EVsiCCR2 were similarly sensitive to the CLP challenge compared with those infused with Mac-EVsiNC (Fig. 7E), demonstrating that Mac-EVsiCCR2 mediates sepsis resistance by primarily targeting Mo/Mps.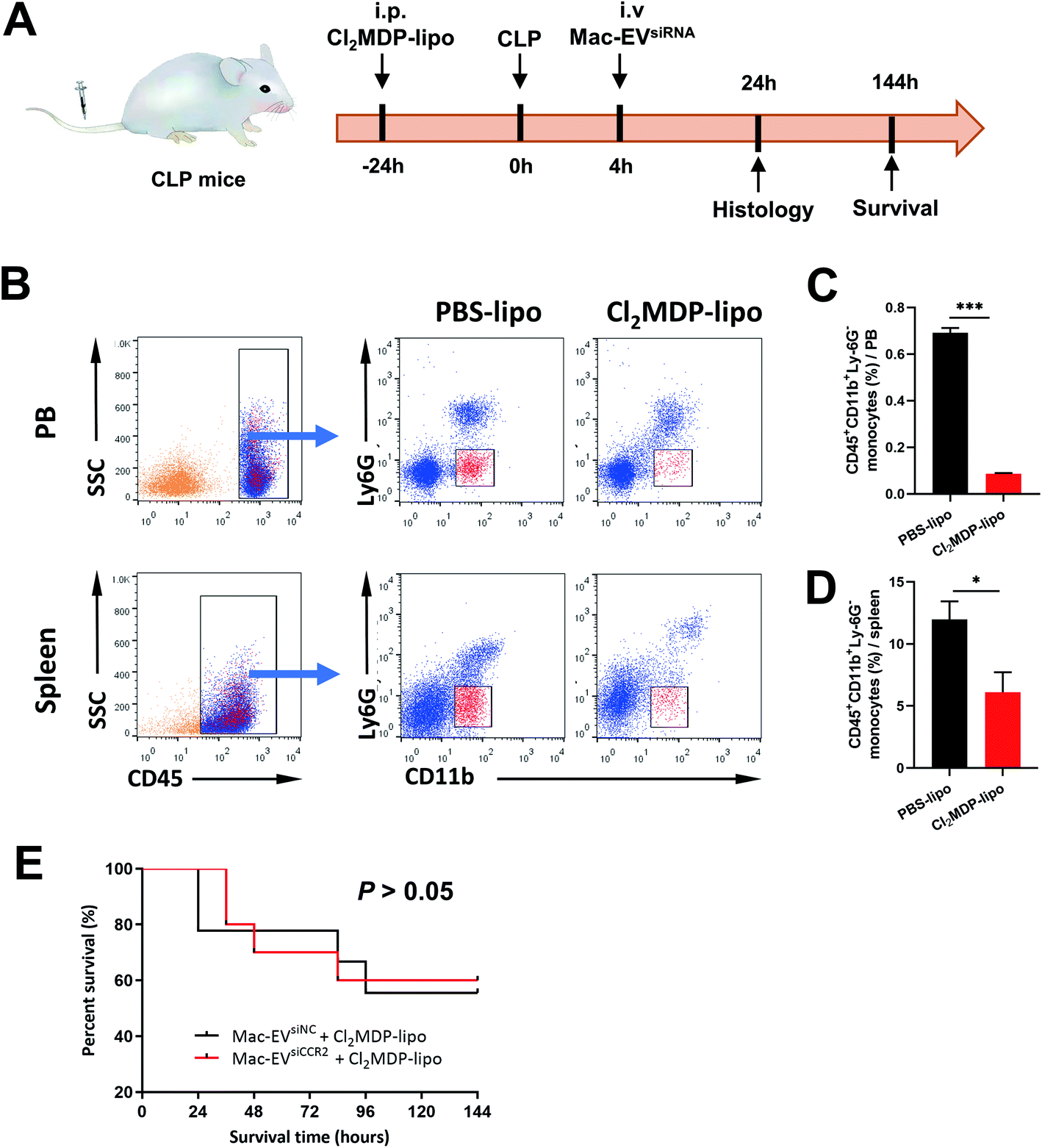 | ||
| Fig. 7 Depletion of Mo/Mps in septic mice removes survival difference by EV therapy. (A) Schematic diagram of Cl2MDP-lipo-induced Mo/Mps depletion model. Cl2MDP-lipo and Mac-EVsiNC or Mac-EVsiCCR2 were infused 24 h pre-CLP and 4 h post-CLP, respectively. Histopathology was determined 24 h post-CLP and survival rate was monitored every 24 h for 6 days. (B–D) Representative flow cytometry dot plots (B) and quantification of Mo/Mps (CD45+CD11b+Ly6G−) in the peripheral blood (PB, C) and spleen (D) following Cl2MDP-lipo or PBS-lipo treatment (n = 3–4). (E) Survival of CLP mice after the indicated EV therapy together with the removal of Mo/Mps (n = 9–10). Data are presented as mean ± SEM. *P < 0.05, ***P < 0.001. | ||
Conclusions
In summary, we have developed a Mac-EV-based drug-delivery platform, mass production of which is easily achieved through immortalized mouse BMDMs. In light of this, we further leveraged Mac-EVs for the delivery of siCCR2 to the splenic monocyte reservoir through intravenous administration, thereby dampening the deployment of this pathological Ly6Chigh subset from the spleen. Our results highlighted that Mac-EVsiCCR2 effectively ablates infiltration of inflammatory Mo/Mps and relieves septic symptoms in mice subsequently. Due to its excellent biocompatibility and ease of preparation, this technology exhibits potential for further clinical translation.Abbreviations
| CCR2 | Chemokine (C–C motif) receptor 2 |
| CX3CR1 | (C-X3-C motif) receptor 1 |
| EVs | Extracellular vehicles |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| MSC | Mesenchymal stem cell |
| PBS | Phosphate-buffered saline |
| PCR | Polymerase chain reaction |
| qRT-PCR | Quantitative RT-PCR |
Author contributions
Conceptualization, WC; methodology, WX, MC, JZ; formal analysis, LD, WZ; investigation, LD, WZ; data curation, LD, WZ; writing – original draft preparation, WC; writing – review and editing, QT, JZ, WC; validation, LD, WZ, JZ; supervision, WC; project administration, ZS, WC; funding acquisition, JZ, ZS, WC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key R&D Program of China (2017YFA0103700), National Natural Science Foundation of China (92168203, 82070363, 81770258, 91839101), National Key Scientific Instrument and Equipment Development Project of China (51927802), Zhejiang Provincial Key Research and Development Program (2019C01121), Start-up Program of Hangzhou Dianzi University (KYS385618067), and the Introduction Project of Clinical Medicine Expert Team for Suzhou (SZYJTD201704).References
- R. Medzhitov, Cell, 2010, 140, 771–776 CrossRef CAS PubMed
.
- R. Medzhitov, Nature, 2008, 454, 428–435 CrossRef CAS PubMed
- M. Singer, C. Deutschman, C. Seymour, M. Shankar-Hari, D. Annane, M. Bauer, R. Bellomo, G. Bernard, J. Chiche, C. Coopersmith, R. Hotchkiss, M. Levy, J. Marshall, G. Martin, S. Opal, G. Rubenfeld, T. van der Poll, J. Vincent and D. Angus, J. Am. Med. Assoc., 2016, 315, 801–810 CrossRef CAS PubMed
- M. Sadarangani, A. Marchant and T. Kollmann, Nat. Rev. Immunol., 2021, 21, 475–484 CrossRef CAS PubMed
- F. Geissmann, M. Manz, S. Jung, M. Sieweke, M. Merad and K. Ley, Science, 2010, 327, 656–661 CrossRef CAS PubMed
- E. Zigmond, C. Varol, J. Farache, E. Elmaliah, A. Satpathy, G. Friedlander, M. Mack, N. Shpigel, I. Boneca, K. Murphy, G. Shakhar, Z. Halpern and S. Jung, Immunity, 2012, 37, 1076–1090 CrossRef CAS PubMed
- P. Zhao, W. Zhou, Y. Zhang, J. Li, Y. Zhao, L. Pan, Z. Shen, W. Chen and J. Hui, J. Cell. Mol. Med., 2020, 24, 2593–2609 CrossRef CAS PubMed
- T. Wissing, E. van Haaften, S. Koch, B. Ippel, N. Kurniawan, C. Bouten and A. Smits, Biomater. Sci., 2019, 8, 132–147 RSC
- F. Swirski, M. Nahrendorf, M. Etzrodt, M. Wildgruber, V. Cortez-Retamozo, P. Panizzi, J. Figueiredo, R. Kohler, A. Chudnovskiy, P. Waterman, E. Aikawa, T. Mempel, P. Libby, R. Weissleder and M. Pittet, Science, 2009, 325, 612–616 CrossRef CAS PubMed
- I. Charo and R. Ransohoff, N. Engl. J. Med., 2006, 354, 610–621 CrossRef CAS PubMed
- J. Osterholzer, M. Olszewski, B. Murdock, G. Chen, J. Erb-Downward, N. Subbotina, K. Browning, Y. Lin, R. Morey, J. Dayrit, J. Horowitz, R. Simon and T. Sisson, J. Immunol., 2013, 190, 3447–3457 CrossRef CAS PubMed
- L. Boring, J. Gosling, M. Cleary and I. Charo, Nature, 1998, 394, 894–897 CrossRef CAS PubMed
- O. Dewald, P. Zymek, K. Winkelmann, A. Koerting, G. Ren, T. Abou-Khamis, L. Michael, B. Rollins, M. Entman and N. Frangogiannis, Circ. Res., 2005, 96, 881–889 CrossRef CAS PubMed
- Y. Guo, D. Wu, X. Zhang, K. Zhang and Y. Luo, Nanoscale, 2021, 13, 16017–16033 RSC
- L. Duan, L. Xu, X. Xu, Z. Qin, X. Zhou, Y. Xiao, Y. Liang and J. Xia, Nanoscale, 2021, 13, 1387–1397 RSC
- Y. Chen, Y. Zhao, W. Chen, L. Xie, Z. Zhao, J. Yang, Y. Chen, W. Lei and Z. Shen, Stem Cell Res. Ther., 2017, 8, 268 CrossRef PubMed
- Y. Song, H. Dou, X. Li, X. Zhao, Y. Li, D. Liu, J. Ji, F. Liu, L. Ding, Y. Ni and Y. Hou, Stem Cells, 2017, 35, 1208–1221 CrossRef CAS PubMed
- H. Xin, Y. Li, Z. Liu, X. Wang, X. Shang, Y. Cui, Z. Zhang and M. Chopp, Stem Cells, 2013, 31, 2737–2746 CrossRef CAS PubMed
- M. Haney, N. Klyachko, Y. Zhao, R. Gupta, E. Plotnikova, Z. He, T. Patel, A. Piroyan, M. Sokolsky, A. Kabanov and E. Batrakova, J. Controlled Release, 2015, 207, 18–30 CrossRef CAS PubMed
- L. Mulcahy, R. Pink and D. Carter, J. Extracell. Vesicles, 2014, 3, 24641 CrossRef PubMed
- Y. Xia, L. Rao, H. Yao, Z. Wang, P. Ning and X. Chen, Adv. Mater., 2020, 32, e2002054 CrossRef PubMed
- H. Shen, G. Cui, Y. Li, W. Ye, Y. Sun, Z. Zhang, J. Li, G. Xu, X. Zeng, Y. Zhang, W. Zhang, Z. Huang, W. Chen and Z. Shen, Stem Cell Res. Ther., 2019, 10, 17 CrossRef CAS PubMed
- Q. Wu, J. Wang, W. Tan, Y. Jiang, S. Wang, Q. Li, X. Yu, J. Tan, S. Liu, P. Zhang, Z. Tiang, Z. Chen, R. Foo and H. Yang, Cell Death Dis., 2020, 11, 354 CrossRef PubMed
- A. Saleh, E. Lázaro-Ibáñez, M. Forsgard, O. Shatnyeva, X. Osteikoetxea, F. Karlsson, N. Heath, M. Ingelsten, J. Rose, J. Harris, M. Mairesse, S. Bates, M. Clausen, D. Etal, E. Leonard, M. Fellows, N. Dekker and N. Edmunds, Nanoscale, 2019, 11, 6990–7001 RSC
- Y. Zhang, Y. Wang, L. Shao, X. Pan, C. Liang, B. Liu, Y. Zhang, W. Xie, B. Yan, F. Liu, X. Yu and Y. Li, J. Cell. Mol. Med., 2020, 24, 695–710 CrossRef CAS PubMed
- W. Chen, S. Wang, J. Xia, Z. Huang, X. Tu and Z. Shen, Mol. Cell. Biochem., 2016, 412, 173–180 CrossRef CAS PubMed
- Y. Xiao, Y. Zhang, Y. Chen, J. Li, Z. Zhang, Y. Sun, H. Shen, Z. Zhao, Z. Huang, W. Zhang, W. Chen and Z. Shen, Hum. Gene Ther., 2019, 30, 286–301 CrossRef CAS PubMed
- J. Wu, J. Wang, X. Zeng, Y. Chen, J. Xia, S. Wang, Z. Huang, W. Chen and Z. Shen, Biochem. Biophys. Res. Commun., 2016, 478, 1179–1184 CrossRef CAS PubMed
- J. Wu, Y. Dong, X. Teng, M. Cheng, Z. Shen and W. Chen, Biochem. Biophys. Res. Commun., 2015, 466, 711–716 CrossRef CAS PubMed
- M. Egawa, K. Mukai, S. Yoshikawa, M. Iki, N. Mukaida, Y. Kawano, Y. Minegishi and H. Karasuyama, Immunity, 2013, 38, 570–580 CrossRef CAS PubMed
- M. Nahrendorf, F. Swirski, E. Aikawa, L. Stangenberg, T. Wurdinger, J. Figueiredo, P. Libby, R. Weissleder and M. Pittet, J. Exp. Med., 2007, 204, 3037–3047 CrossRef CAS PubMed
- S. Fujishima, Inflammation Regener., 2016, 36, 24 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr06922e |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2022 |