
RNA cancer nanomedicine: nanotechnology-mediated RNA therapy
Bijan Emiliano
Ferdows†
,
Dylan Neal
Patel†
,
Wei
Chen
*,
Xiangang
Huang
 ,
Na
Kong
and
Wei
Tao
*
,
Na
Kong
and
Wei
Tao
*
Center for Nanomedicine and Department of Anesthesiology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA. E-mail: wchen45@bwh.harvard.edu; wtao@bwh.harvard.edu
First published on 12th January 2022
Abstract
It has been demonstrated that RNA molecules—mRNA, siRNA, microRNA, and sgRNA—regulate cancer-specific genes, and therefore, RNA-based therapeutics can suppress tumor progression and metastasis by selectively upregulating and silencing these genes. However, the innate defense mechanisms (e.g., exonucleases and RNases) involving the human immune system catalyze the degradation of exogenous RNAs. Thus, nonviral nanoparticles have been employed to deliver therapeutic RNAs for effective cancer gene therapy. In this minireview, we highlight efforts in the past decade to deliver therapeutic RNAs for cancer therapy using novel nanoparticles. Specifically, we review nanoparticles, including lipid, polymer, inorganic, and biomimetic materials, which have been employed to deliver therapeutic RNAs and evoke tumor suppressing responses. Finally, we discuss the challenges and considerations that may accelerate the clinical translation of nanotechnology-mediated RNA therapy.
 Wei Tao | Prof. Wei Tao is a Farokhzad Family Distinguished Chair for Innovation and a Principal Investigator in the Center for Nanomedicine, and a Faculty Member in the Department of Anesthesiology, Perioperative, and Pain Medicine at Brigham and Women's Hospital, Harvard Medical School. He is also the first Chaired Professor as an assistant professor in the history of this institution. His primary research interest is the design and synthesis of functional biomaterials, revealing nano-bio interactions (mechanism studies), and exploring their different biomedical applications. His research thrust includes fundamental materials innovation as well as their various applications in translational medicine. |
1. Introduction
Therapeutic ribonucleic acids (RNAs), such as messenger RNA (mRNA), small interfering RNA (siRNA), small noncoding RNA (microRNA), and single guide RNA (sgRNA), have tremendous tumor suppressing capabilities because they can upregulate the expression of target tumor suppressor genes or suppress the expression of target oncogenes.1,2 These therapeutics are favorable to protein-based ones because RNA-based therapeutics are less difficult to design and less expensive to produce en masse.3,4 Furthermore, RNA-based cancer therapy is minimally toxic and does not confer drug-resistance to tumors, these being the adverse effects of drug chemotherapy. However, the human immune system has innate defense mechanisms (e.g., those involving exonucleases and RNases) that catalyze the degradation of exogenous RNAs and therefore diminish the efficacy of RNA-based therapy.3 It follows that the development of nonviral nanoparticles for the delivery of RNA-based cancer therapeutics can substantially improve the efficacy of cutting-edge cancer gene therapy, because nanoparticles can markedly improve the chemical stability and pharmacokinetic profiles of the encapsulated RNAs.5This minireview sheds light on the therapeutic potential of four distinct types of RNA—mRNA, siRNA, microRNA, and sgRNA—to upregulate and restore the expression of tumor suppressor genes as well as to suppress the expression of oncogenes using novel nanoparticle delivery platforms. We highlight the state-of-the-art studies on the delivery of RNA-based cancer therapeutics using lipid, polymer, inorganic, and biomimetic nanoparticles. Recently, RNA lipoplexes composed of the well-known lipid carriers DOTMA and DOPE were clinically tested for the delivery of several antigen (NY-ESO-1, MAGE-A3, tyrosinase, and TPTE)-encoding mRNA transcripts into tumor-associated antigen presenting cells (APCs).6 The RNA lipoplexes inflamed the tumor microenvironment and induced strong effector and memory T-cell responses in melanoma patients. Impressively, the authors reported that the RNA lipoplexes mediated the potent IFNα-dependent rejection of metastatic tumors. All things considered, nanoparticle-mediated RNA therapies have the potential to improve the accuracy of cancer treatment and target specific signaling pathways and the efficacy of pre-existing drug chemotherapies.
2. Therapeutic RNAs for cancer gene therapy
mRNA is a single-stranded molecule of RNA, which is translated into proteins with numerous diverse functions. Similar to therapeutic DNA, therapeutic mRNA can upregulate the expression of tumor suppressing proteins in malignant cells. What distinguishes mRNA-based cancer therapies from comparable DNA-based ones is their convenient in vitro synthesis, consistent and predictable regulation of protein expression, and minimal risk of insertional mutagenesis. Other therapeutic mRNAs encoding viral antigens take effect in antigen APCs—dendritic cells (DCs) and macrophages—where they are translated into protein antigens and subsequently processed into peptide epitopes. The epitopes bind to major histocompatibility complex (MHC) molecules, and together, they are transported to the cell surface. At the cell surface, the epitopes induce strong effector and memory T cell immune responses, suppressing tumor progression.6 However, the unstable and immunogenetic nature of in vitro transcription (IVT) mRNA may diminish the biological viability of mRNA-based cancer therapies. It is necessary, therefore, to modify the structural elements of IVT mRNA, including the 5′ cap, 5′ untranslated region (5′ UTR), protein-encoding open reading frame (ORF), 3′ UTR, and 3′ poly(A) tail, for optimal stability and immunogenicity.2,7In contrast to therapeutic mRNAs, therapeutic siRNAs and microRNAs downregulate the expression of tumor promoting proteins in malignant cells and APCs. Endogenous precursor siRNA (pre-siRNA) and precursor microRNA (pre-microRNA) bind to Dicer RNase, which cleaves pre-siRNA and pre-microRNA into double-stranded siRNA and microRNA, respectively. The resultant siRNA and microRNA bind to endonuclease Argonaute-2 (AGO2), composed of RNA-induced silencing complexes (RISCs). While therapeutic siRNAs confer specific mRNA binding ability to RISCs, therapeutic microRNAs confer general mRNA binding ability to RISCs because they target sequences located in the 3′ UTR or the intronic (i.e., non-coding) regions of mRNA. It follows that an siRNA-based RISC can only bind to and cleave one target mRNA, downregulating the expression of one protein; a microRNA-based RISC, on the other hand, can potentially bind to and cleave hundreds of target mRNAs, downregulating the expression of entire signaling pathways.8 Another application of therapeutic microRNAs is the inhibition of endogenous oncogenic microRNAs (oncomiRs). Evidence suggests that 52.5% of microRNA genes are located in tumor-associated and fragile genomic regions.9 Therefore, the synthesis of single-stranded microRNA antagonists (antagomiRs), which target oncomiRs by Watson–Crick base pairing before the oncomiRs can bind to and cleave mRNAs, represents a promising development in cancer gene therapy.10
Recently, clustered regularly interspaced short palindromic repeat (CRISPR) technologies have aroused considerable scientific interest because they have the potential to cleave and perform multiplex targeting of tumor-associated genomic regions.11 The CRISPR-Cas9 gene-editing system requires therapeutic sgRNA, which binds to the target genomic region and the Cas9 endonuclease by base pairing at the 5′ and 3′ ends, respectively. After sgRNA binds to the genomic target, each of the nuclease domains of Cas9 makes a nick, resulting in a specific double-stranded break in the DNA. The non-homologous end joining (NHEJ) DNA repair pathway introduces insertion-deletions (INDELs) in the exonic (i.e., coding) regions of DNA that mutate the coding frame, and on occasion, install a premature termination codon (PTC); the INDEL-induced mutagenesis of the genomic target downregulates the expression of tumor-promoting proteins in malignant cells and APCs.
3. Nonviral RNA-based nanotherapeutics for cancer treatment
The human immune system has innate defense mechanisms (e.g., exonucleases and RNases) that catalyze the degradation of exogenous RNAs and, therefore, hinder the adoption of therapeutic RNAs for in vivo cancer treatment; the short half-lives of naked RNAs only exacerbate this hindrance. Nanoparticle-mediated delivery platforms can uniquely address these challenges.12–17 Specifically, nonviral nanoparticles not only defend therapeutic RNAs from enzymatic degradation but also accumulate preferentially in the tumor tissues via the enhanced permeability and retention (EPR) effect and attenuate off-target toxicological complications.18 Additionally, nanoparticles can be engineered to release therapeutic RNAs in response to environmental triggers, including the acidic tumor microenvironments and target cell endosomes.19 To improve the cell and, in some instances, nucleus-penetrating ability of nanoparticles, they can be modified with cell-specific targeting ligands and moieties (Fig. 1).20 Finally, nonviral nanodelivery vehicles have diminished immunogenicity and enhanced genetic payloads compared to viral vectors and therefore hold great promise for assisting RNA-based cancer therapies.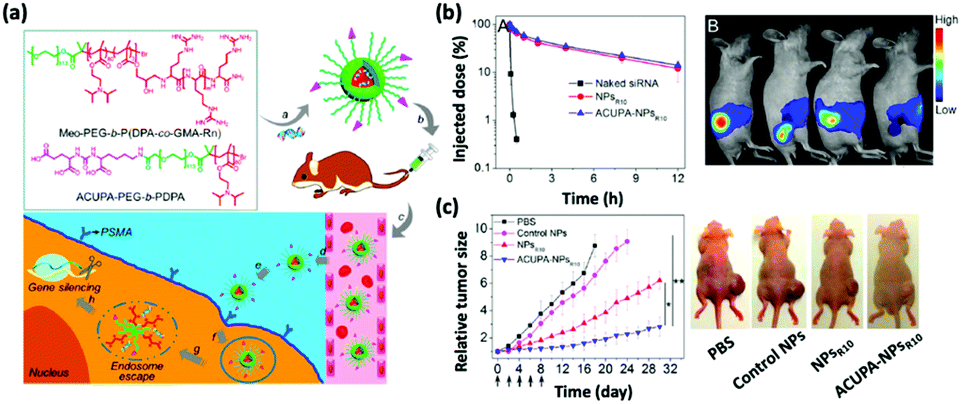 | ||
| Fig. 1 (a) Chemical structures of the oligoarginine-functionalized pH-responsive polymer and prostate cancer cell-specific polymer ACUPA-PEG-b-PDPA and a schematic illustration of the multifunctional envelope-type nanoparticle platform for in vivo prostate cancer cell specific siRNA delivery and therapy. (b) The nanoparticle platform can improve the pharmacokinetic profile of the encapsulated siRNA as revealed by the serum concentration of the various formulations and the fluorescence images. (c) Time-dependent relative tumor size of the tumor-bearing nude mice after various treatments and the photograph of the treated mice at day 18. This figure has been adapted/reproduced from ref. 20 with permission from the American Chemical Society, copyright 2017. | ||
Lipid and polymer-based nanoparticles are among the most rigorously studied nonviral vehicles for the delivery of therapeutic RNAs.19,21 Lipids confer favorable biocompatibility, biodegradability, and natural cell-penetrating ability to nanoparticles because they represent the basic building blocks of the cell membrane. The adoption of cationic lipids for RNA-based cancer therapies is promising because they can bind to therapeutic RNAs by electrostatically attracting the negatively charged RNA phosphate groups. Likewise, the adoption of ionizable lipids is promising because they can increase the osmotic pressure of the endosome and consequently rupture the vesicle by attenuating the declining endosomal pH.22 However, the recurrent payload leaks as well as the facile kidney and liver clearance of lipid-based NPs limit their in vivo delivery of therapeutic RNAs.23 Nanoparticles composed of synthetic polymers, such as polyethyleneimine (PEI), can also release therapeutic RNAs in response to environmental triggers (e.g., the decreased pH of the endosome).24 In addition, the surfaces of polymeric nanoparticles can be functionalized with cell-specific targeting ligands and moieties, such as S,S-2-[3-[5-amino-1-carboxypentyl]ureido]pentanedioic acid (ACUPA) and tumor cell-targeting and penetrating-peptide-amphiphile (TCPA), further supporting the promise of polymeric nanoparticle-assisted RNA delivery for cancer treatment.20,25
The development of cutting-edge inorganic and biomimetic nanoparticles for RNA-based cancer therapy is noteworthy because these nanoparticles possess distinct physicochemical properties. The physicochemical properties of inorganic nanoparticles, in some instances, provide diagnostic and therapeutic functions, which augment the efficacy of cancer treatments. Gold-iron oxide nanoparticles (GIONs) enhance the detection of tumor progression by the combination of computed tomography (CT), magnetic resonance imaging (MRI), photoacoustic imaging (PAI), and surface enhanced Raman spectroscopy (SERS) techniques. Furthermore, GIONs represent a species of photothermal agents that efficiently damage malignant cells by photothermal ablation.26 The GION surface is conveniently covalently functionalized with thiols and oligonucleotides—as in self-assembled monolayers (SAMs)—which bind therapeutic RNAs for transport to the damaged malignant cells and therefore invigorate a synergistic effect for potent cancer therapy.27 Despite the promise of the aforementioned physicochemical properties, the biocompatibility, biodegradability, and toxicity of inorganic nanotherapeutics must be further investigated prior to their clinical translation. On the other hand, biomimetic nanomaterials including extracellular vesicles (EVs) provide remarkable biocompatibility and biodegradability because they share physicochemical properties with endogenous cells.28 Exosomes and microvesicles are bilayer-coated EVs released from the membranes of cells for intercellular communication. Therefore, exosome and microvesicle-based nanoparticles can both carry therapeutic RNAs and penetrate target cells naturally for enhanced RNA-based cancer treatment.
3.1. Nanoparticles for mRNA delivery
The restoration of tumor suppressing proteins is a compelling therapeutic strategy for cancer treatment.29 The phosphatase and tensin homolog (PTEN) is a lost or mutated tumor suppressor gene in half of the metastatic castrate-resistant prostate cancer (mCRPC) patients; PTEN inhibits the P13K-AKT signal transduction pathway, which is overexpressed in mCRPC patients and promotes the survival, proliferation, and migration of malignant cells.30 Recent evidence suggests that the loss of PTEN protein expression correlates with a high Gleason score and accelerated metastasis.31 Islam et al. engineered a self-assembling hybrid nanoparticle, comprising a cationic lipid-mimic (G0-C14) and a synthetic polymer (PLGA) core, with a lipid–polyethylene glycol (PEG) shell for the delivery of therapeutic PTEN mRNA into PTEN-null prostate cancer cells (Fig. 2).32 The investigators systematically injected the 120 nm PTEN-mRNA nanoparticles into prostate cancer-bearing mice every three days for six total treatments and reported significant inhibition of tumor progression in 43 days after tumor induction.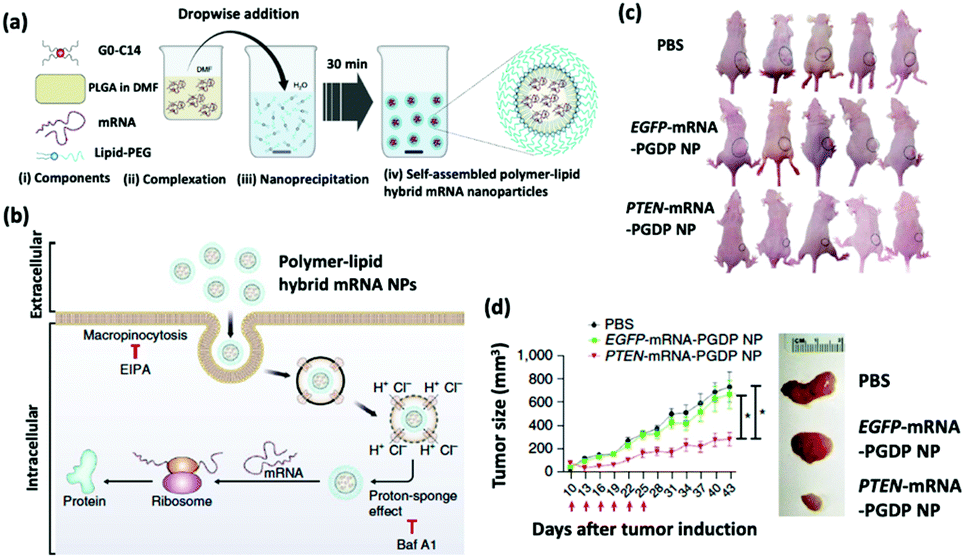 | ||
| Fig. 2 (a) Self-assembly process of hybrid lipid–polymer nanoparticles comprising cationic G0-C14, anionic mRNA, and PLGA; nanoparticles were coated with lipid-PEG. (b) Schematic illustration of the mechanism of cellular uptake and intracellular transport of the hybrid mRNA nanoparticles. (c) Whole-body images of mice bearing PC3 xenograft tumors after various treatments. (d) Time-dependent tumor size measurements show the in vivo therapeutic efficacy of PTEN-mRNA-PGDP nanoparticles. This figure has been adapted/reproduced from ref. 32 with permission from Springer Nature, copyright 2018. | ||
The same group subsequently engineered another self-assembling lipid–polymer hybrid nanoparticle for the delivery of therapeutic p53 mRNA into p53-null hepatocellular carcinoma (HCC) and non-small cell lung cancer (NSCLC) cells. The p53 tumor suppressor gene is defective in 36% of HCC and 68% of NSCLC patients.33 In the nucleus, p53 promotes the transcription of pro-apoptotic proteins, such as BCL-2-associated X (BAX) and p53 upregulated modulator of apoptosis (PUMA). Additionally, in the cytoplasm, p53 inhibits the activation of pro-survival autophagy, which is responsible for multidrug resistance (MDR) in cancer. Kong et al. demonstrated that the nanoparticle-assisted restoration of p53 protein expression can suppress tumor progression by simultaneously inducing apoptosis and sensitizing p53-null cancer cells to everolimus, a potent chemotherapy drug.34 The investigators administered p53-mRNA nanoparticle—composed of G0-C14, hydrophobic redox-responsive poly(disulfide amide) (PDSA), and two lipid-PEG compounds—injections and everolimus concurrently into p53-null HCC and NSCLC tumor-bearing mice every three days for six total treatments. The concurrent administration of the p53-mRNA nanoparticle (125 nm) and everolimus significantly enhanced therapeutic efficacy against p53-null HCC tumors compared to the treatment with p53-mRNA nanoparticle injections or everolimus alone; the combination treatment even successfully regressed metastatic NSCLC tumors.
As stated above, some therapeutic mRNAs encode viral antigens that induce strong effector and memory T cell immune responses at the surfaces of APCs. The chemical modification of such mRNA transcripts is necessary to circumvent the degradation machinery in vivo. However, the modification of mRNA transcripts with alternative naturally occurring nucleotides, such as pseudouridine (Ψ), 5-methylcytidine (5meC) and N1-methylpseudouridine (m1Ψ), silences their immunogenicity and diminishes their self-adjuvant effect because they fail to trigger type I interferon (IFN) induction.35 Therefore, Islam et al. designed an adjuvant-pulsed mRNA nanovaccine, comprising ovalbumin (OVA) mRNA and the toll-like receptor (TLR) 7/8 agonist C16-R848 (R848), with a lipid-PEG shell to restore the innate immune activation of DCs and subsequent T cell priming.36 Compared to an adjuvantless OVA-mRNA vaccine, the adjuvant-pulsed OVA-mRNA vaccine significantly enhanced the proliferation of cytotoxic T lymphocytes (CTLs) as well as the infiltration of CTLs in the tumor bed. Remarkably, the adjuvant-pulsed OVA-mRNA vaccine reduced lymphoma and prostate tumor growth by 84% and 60%, respectively.
3.2. Nanoparticles for siRNA delivery
Recent advances in nanoparticle development have allowed for the targeted delivery of siRNA to specific genes and pathways in macrophages, T-cells, and even tumor cells themselves. The anti-colony stimulating factor-1 receptor (anti-CSF-1R) is a receptor tyrosine kinase that has been employed to induce apoptosis in CSF-1R+ tumor-associated macrophages (TAMs) and improve the ratio of CD8+ T-cells to CD4+ cells in cancer patients. Qian et al. developed a nanoparticle platform comprising of alpha-peptides and a M2-like macrophage binding peptide to deliver anti-CSF-1R siRNA to M2-like TAMs; the authors reported that the nanoparticles significantly limited the survival of M2-like pro-tumor TAMs in melanoma tumor microenvironments.37In addition, programmed death-ligand 1 (PD-L1) is a protein overexpressed in tumor cells to “trick” the immune system to not attack the tumor. Dai et al. designed a nanoparticle platform for silencing immune resistance triggered by the overexpression of PD-L1 via the delivery of siRNA and a mitochondrion-targeting photosensitizer directly to tumor cells.38 To achieve this goal, the researchers synthesized micelleplexes using two types of polymers and siRNA: PEG-CDM-PDEA and PEI-PDEA loaded with siPD-L1. The in vitro and in vivo results reveal that the 43 nm micelleplexes not only induced an efficient immune response by photodynamic therapy but also induced a subsequent siRNA-mediated anti-tumor immune response. All things considered, the micelleplexes induced a synergistic effect for potent cancer treatment.
TWIST-related protein 1 is another example of an overexpressed protein involved in metastatic carcinomas. Shahin et al. employed hyaluronic acid (HA)-modified mesoporous silica nanoparticles (MSNs) which can target CD44 on cancer cells and deliver siRNA-419, to silence TWIST protein expression in ovarian cancer cells.39 In recurrent ovarian tumor models, the silencing of TWIST protein expression depressed the levels of chemotherapy resistance. Consequently, siTWIST-HA and cisplatin-loaded MSN (120 nm) treatment resulted in significantly diminished tumor size and fewer metastases compared to the cisplatin-loaded MSN treatment alone.
Transforming growth factor β (TGF-β) has long been a target in cancer therapy, specifically in glioblastoma treatment; the overexpression of TGF-β in tumor cells can result in an immunosuppressive environment, which nullifies chemotherapy. To overcome this challenge, Qiao et al. developed a zwitterionic lipid (distearoyl phosphoethanol-amine-polycarboxybetaine lipid)-based nanoparticle (42 nm) platform for the delivery of the chemotherapeutic temozolomide and anti-TGF-β siRNA.40 In addition, this nanoformulation efficiently crossed a synthetic blood–brain barrier to target glioblastoma cells via receptor-mediated transcytosis. The delivery strategy enhanced both the cytotoxicity of temozolomide and gene silencing efficiency of siTGF-β. It follows that the anti-TGF-β siRNA and temozolomide-loaded nanoparticles significantly reduced the tumor's ability to induce an immunosuppressive microenvironment, and therefore, prolonged the life expectancy of glioma-bearing mice. Xu et al. employed a different nanoparticle platform—mannose-modified lipid-protamine-hyaluronic acid (∼40 nm)—for the delivery of anti-TGF-β siRNA to B16F10 melanoma tumor cells.41 The authors reported a 50% decrease in the expression of TGF-β and an increase in the count of CD8+ T-cells in the late stage tumor microenvironment. The lipid–protamine nanoparticles significantly inhibited tumor growth by 52% compared to the vaccine treatment alone.
Finally, Bromodomain 4 (BRD4) has been investigated as a therapeutic target for cancer treatment. For example, Xu et al. created a multistage pH-responsive nanoplatform to deliver siRNA against BRD4 in a tumor microenvironment.25 The nanoplatform consists of hydrophobic poly(2-(hexamethylenediamine)ethyl methacrylate) (PHMEMA), which has a sharp pH responsiveness for the delivery of siRNA in the acidic tumor microenvironment, and a PEG outer shell to prolong its circulation in the blood; together, PHMEMA and the PEG outer shell improve nanoparticle accumulation in the tumor tissues. A cell-targeting RGD peptide was derivatized on the nanoplatform surface for enhanced tumor targeting and cellular uptake. The nanoplatform (72 nm) decreased the tumor size and weight in a tumor-bearing mouse model by 5-fold and 4-fold, respectively.
3.3. Nanoparticles for microRNA delivery
The robust nanoparticle-mediated delivery strategy is promising to prevent the degradation of microRNA by nucleases in the circulation and peripheral tissues. The mouse double minute 2 (MDM2) oncogene downregulates the aforementioned p53 tumor suppressor gene, which is defective in 68% of NSCLC patients.32 It follows that the amplification of MDM2 inhibits the transcription of pro-apoptotic proteins, such as BAX and PUMA, and promotes the activation of pro-survival autophagy. Therefore, MDM2 is considered a compelling target for cancer treatment. Moro et al. further demonstrated that the MIR660 gene, which can downregulate MDM2, is downregulated in NSCLC patients.42 The investigators, therefore, developed coated cationic lipid nanoparticles entrapping microRNA-660 (CCL660), which they administered intraperitoneally to mice with lung cancer Patient Derived Xenografts (PDXs). Following eight semiweekly injections, the CCL660 nanoparticles (123 nm) effectively restored the expression of microRNA-660 in PDX-bearing mice and reduced tumor growth by 50%—compared to the controls—in mice bearing two independent NSCLC PDXs.Fifteen percent of breast cancer patients are diagnosed with triple-negative breast cancer (TNBC), which is immunohistochemically characterized by poor human epidermal growth factor receptor 2 (HER2), estrogen receptor, and progesterone receptor expression.43 To establish an effective treatment strategy through the activation of protein kinase C Gong et al. derived exosomes expressing disintegrin and metalloproteinase 15 (A15-EXOs) for the co-delivery of doxorubicin (DOX) chemotherapy and cholesterol-modified microRNA-159 (CHO-microRNA-159) to TNBC cells in vitro and in vivo.44 The A15-EXOs (179 nm) carrying DOX and CHO-microRNA-159 together induced synergistic tumor suppressing effects in TNBC tumor-bearing mice compared to those carrying DOX or CHO-microRNA-159 alone.
The inhibition of oncomiRs represents another promising application of therapeutic microRNAs. The upregulation of oncomiR-21 has been reported in patients diagnosed with brain cancer, breast cancer, and HCC (Fig. 3a).45–47 This oncomiR is known to suppress the expression of apoptotic proteins, such as B cell lymphoma 2 (Bcl-2), and it is thus responsible for chemotherapy resistance. JC Bose et al. manipulated tumor cell-derived extracellular vesicles (TEVs) functionalized with GIONs for the delivery of anti-microRNA-21 (Fig. 3b).26 (Recall that GIONs provide CT and thermal ablation capabilities for simultaneous cancer imaging and therapy.) Ultimately, the delivery of anti-microRNA-21 by TEV-GIONs (34 nm) in combination with low doses of DOX significantly attenuated DOX resistance in breast cancer cells and eliminated these cells 3 times more effectively than DOX alone.
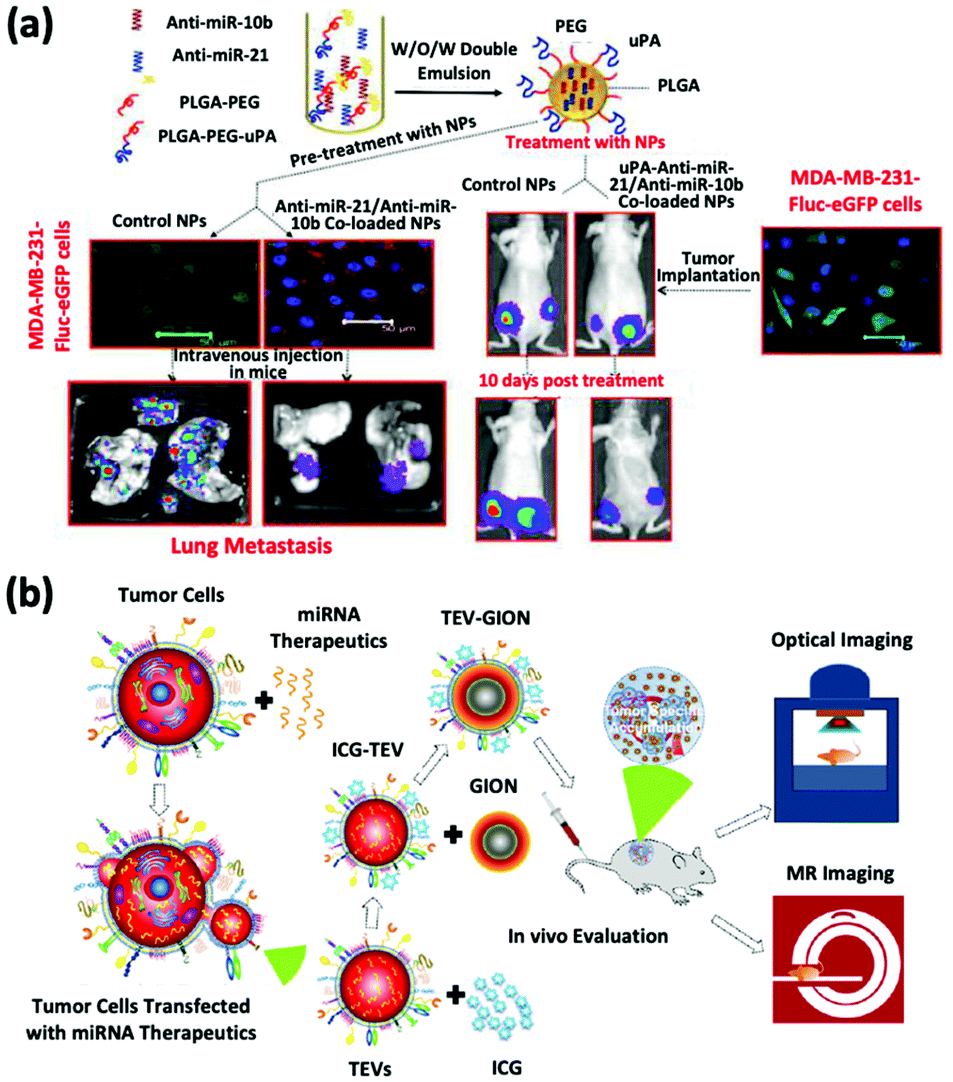 | ||
| Fig. 3 (a) Synthetic strategies of dual-siRNA-loaded polymeric nanoparticles for achieving triple negative breast cancer therapy. This figure has been adapted/reproduced from ref. 47 with permission from the American Chemical Society, copyright 2015. (b) Schematic illustration of the synthetic strategy of tumor cell-derived extracellular vesicle-coated nanocarriers for the delivery of microRNA and bioimaging. This figure has been adapted/reproduced from ref. 26 with permission from the American Chemical Society, copyright 2018. | ||
3.4. Nanoparticles for sgRNA delivery
sgRNA delivery is a prevailing therapy which inhibits the expression of proteins that create immunosuppressive environments by knocking out the respective genes. The inhibition of PLK1, polo-like kinase 1, has been shown to result in tumor cell apoptosis. To achieve this goal, Rosenblum et al. constructed a nanoparticle platform selected from a library that they created using a novel class of ionizable amino lipids based on hydrazine, hydroxyl-amine, and ethanolamine linkers with a linoleic fatty acid chain and an amine head group to deliver Cas9 mRNA and sgRNAs to an aggressive orthotopic glioblastoma (Fig. 4).48 A single intracerebral injection of the CRISPR-lipid nanoparticles against PLK1 (sgPLK1-cLNPs) (81 nm) enabled up to ∼70% gene editing in vivo, which caused tumor cell apoptosis, inhibited tumor growth by 50%, and improved survival by 30%. To reach disseminated tumors, cLNPs were also engineered for antibody-targeted delivery. Remarkably, the intraperitoneal injections of EGFR-targeted sgPLK1-cLNPs caused their selective uptake into disseminated ovarian tumors, enabled up to ∼80% gene editing in vivo, inhibited tumor growth, and increased survival by 80%.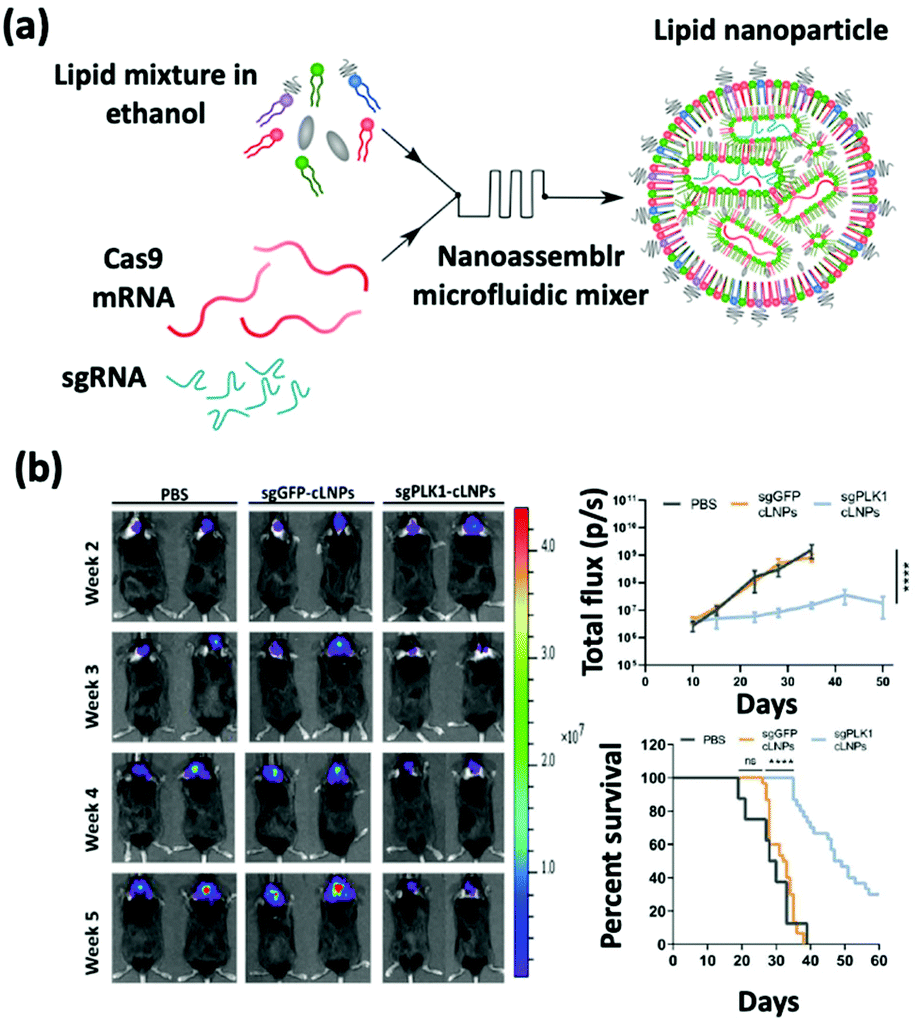 | ||
| Fig. 4 (a) Schematic illustration of the synthesis of Cas9 mRNA and sgRNA-loaded lipid nanoparticles by a nanoassembly microfluidic mixer. (b) Bioluminescence images show that the nanoformulation treatment can inhibit tumor growth and enhance the mouse survival rate. This figure has been adapted/reproduced from ref. 48 with permission from the American Association for the Advancement of Science, copyright 2020. | ||
Similar to PLK1, programmed cell death ligand-1 (PD-L1) is a cytokine which can create an immunosuppressive environment, and thus has been considered a promising therapeutic target. Cheng et al. developed a 200 nm nanoparticle platform using a double emulsion method for complexing plasmids with stearyl polyethylenimine (stPEI) to form human serum albumin (HSA) for the delivery of CRISPR into CT26 cells.49 The treatment by this formulation silenced the expression of PD-L1, thus inhibiting both the proliferation of antigen-specific T cells and the apoptosis of regulatory T cells (Tregs) in the lymph nodes, thereby activating the immune system to attack tumors. To silence PD-L1, Liu et al. created a virus-like nanoparticle (VLN) consisting of a surface-thiolated mesoporous silica nanoparticle (MSN-SH) core; these pores were locked by conjugating a ribonucleoprotein (RNP) to MSN-SH via disulfide bonds (RMSN), and a lipid shell containing PEG2000-DSPE (20 nm). These VLNs were used for the co-delivery of sgRNA targeting the PD-L1 encoding gene (sgPD-L1) and axitinib (Axi), a small molecule inhibitor of tyrosine kinase, into B16F10 murine melanoma cells in mouse models.50 The systemic administration of this nanoformulation achieved an effective CRISPR/Cas9-based PD-L1 knockout in cancer cells, which disrupted the PD-1/PD-L1 pathway and reinvigorated the exhausted T cells to suppress the tumor. Furthermore, VLNs also achieved the delivery of Axi into the tumor, leading to the reduction in the Tregs population in the tumor microenvironment. Reducing the immunosuppressive Tregs further unleashed T cell mediated antitumor immunity, and eventually enhanced the tumor growth inhibition.
PCSK9 is a gene which has been shown to induce progression of a tumor. To knock out this gene, Wei et al. developed C12-200 and MC3 lipid nanoparticles for mediated delivery of the Cas9/sgRNA ribonucleoprotein complex into tumor cells for effective PCSK9 inhibition.51 The study first showed the ability to edit genes in HeLa cells in vitro. The C12-200-DOT-10 CRISPR-Cas9 nanoparticles showed 66.9% indices, which are higher than that of the RNAiMAX positive control of 41.9%. In addition, the 5A2-DOT-50 lipid nanoparticles showed great efficiency in targeting and editing genes in mouse lungs. The administration of lipid nanoparticles into mice significantly decreased PCSK9 protein levels in mouse liver tissue and the serum. Importantly, the lipid nanoparticles show the ability to target multiple genes and hold the ability to deliver a broad range of sgRNA to tumor sites.
4. Conclusions & prospects
Due to their ability to inhibit tumor progression, RNA-based therapies have attracted great interest in the field of cancer nanotherapy. In this minireview, mRNA-based therapeutics were demonstrated to successfully upregulate tumor suppressing proteins and arrest the development of metastases. Conversely, siRNA and microRNA-based therapeutics were demonstrated to silence genes that cause an immunosuppressive environment in tumor cells and tumor associated macrophages. Finally, when coupled with Cas9 exonuclease, sgRNA was shown to target and edit oncogenes.The delivery of RNAs for cancer therapy by nanoparticles in pre-clinical studies has been shown to be efficient and practical. The potential for RNA cancer therapy is observed in some of the few clinical trials that are being conducted. According to a study by Tabernero et al., two siRNAs (VEGF siRNA and KSP siRNA) carried by lipid nanoparticles targeting kinesin spindle proteins were shown to have stimulated regression in cancer patients.52 All treated patients had detectable amounts of each siRNA present in tumor biopsies. The study showed that siRNA cancer therapy is a safer and more direct treatment compared to conventional chemotherapy. The therapy also showed correlation in tumor regression and reduced the likelihood of metastases. A substantial takeaway from this study is the demonstrated efficacy of nanotechnology-mediated RNAi's ability to regulate gene expression. This novel clinical study shows a promising future for nanoparticle-assisted RNA therapies in the battle against cancer.52 Subsequently, comprehensive evaluation of the biodistribution, potential toxicity, and clearance of nanoparticles of interest is necessary before their clinical application in cancer therapies. In addition, the industrial-scale production of these nanoparticles under Good Manufacturing Practice (GMP) with batch-to-batch reproducibility is important for clinical translation. These considerations are of great importance for the development of patient-specific personalized nanotherapy. Nevertheless, the success of numerous nanoparticle-based delivery strategies in preclinical and clinical studies of cancer offers significant promise for the use of nanoparticles in the treatment of cancer patients.
Author contributions
B. E. F., W. C., and W. T. conceived the manuscript. B. E. F. and D. N. P. co-wrote the draft. W. T., and W. C. refined the draft. All authors discussed and edited the manuscript before its submission.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work is supported by Harvard Medical School/Brigham and Women's Hospital Department of Anesthesiology-Basic Scientist Grant (No. 2420 BPA075, W. T.), and the US METAvivor Early Career Investigator Award (No. 2018A020560, W. T.). W. T. is a recipient of the Khoury Innovation Award (No. 2020A003219), The Gillian Reny Stepping Strong Center for Trauma Innovation Breakthrough Innovator Award (113548), and the American Heart Association (AHA) Collaborative Sciences Award (No. 2018A004190). W. T. also received a start-up package (for three years) from the Department of Anesthesiology, Perioperative and Pain Medicine to establish his independent research laboratory at Harvard Medical School and Brigham and Women's Hospital.References
- G. J. Goodall and V. O. Wickramasinghe, Nat. Rev. Cancer, 2021, 21, 22–36 CrossRef CAS PubMed.
- M. A. Oberli, A. M. Reichmuth, J. R. Dorkin, M. J. Mitchell, O. S. Fenton, A. Jaklenec, D. G. Anderson, R. Langer and D. Blankschtein, Nano Lett., 2017, 17, 1326–1335 CrossRef CAS PubMed.
- I. Barbieri and T. Kouzarides, Nat. Rev. Cancer, 2020, 20, 303–322 CrossRef CAS PubMed.
- J. Conde, N. Oliva, M. Atilano, H. S. Song and N. Artzi, Nat. Mater., 2016, 15, 353–363 CrossRef CAS PubMed.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed.
- L. M. Kranz, M. Diken, H. Haas, S. Kreiter, C. Loquai, K. C. Reuter, M. Meng, D. Fritz, F. Vascotto, H. Hefesha, C. Grunwitz, M. Vormehr, Y. Hüsemann, A. Selmi, A. N. Kuhn, J. Buck, E. Derhovanessian, R. Rae, S. Attig, J. Diekmann, R. A. Jabulowsky, S. Heesch, J. Hassel, P. Langguth, S. Grabbe, C. Huber, Ö. Türeci and U. Sahin, Nature, 2016, 534, 396–401 CrossRef PubMed.
- Q. Xiong, G. Y. Lee, J. Ding, W. Li and J. Shi, Nano Res., 2018, 11, 5281–5309 CrossRef CAS PubMed.
- J. K. W. Lam, M. Y. T. Chow, Y. Zhang and S. W. S. Leung, Mol. Ther.–Nucleic Acids, 2015, 4, e252 CrossRef CAS PubMed.
- G. A. Calin, C. Sevignani, C. D. Dumitru, T. Hyslop, E. Noch, S. Yendamuri, M. Shimizu, S. Rattan, F. Bullrich, M. Negrini and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2999–3004 CrossRef CAS PubMed.
- U. K. Sukumar, R. J. C. Bose, M. Malhotra, H. A. Babikir, R. Afjei, E. Robinson, Y. Zeng, E. Chang, F. Habte, R. Sinclair, S. S. Gambhir, T. F. Massoud and R. Paulmurugan, Biomaterials, 2019, 218, 119342 CrossRef CAS PubMed.
- Q. Liu, J. Cai, Y. Zheng, Y. Tan, Y. Wang, Z. Zhang, C. Zheng, Y. Zhao, C. Liu, Y. An, C. Jiang, L. Shi, C. Kang and Y. Liu, Nano Lett., 2019, 19, 7662–7672 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- W. Tao, A. Yurdagul, N. Kong, W. Li, X. Wang, A. C. Doran, C. Feng, J. Wang, M. A. Islam, O. C. Farokhzad, I. Tabas and J. Shi, Sci. Transl. Med., 2020, 12, eaay1063 CrossRef CAS PubMed.
- W. Chen, M. Schilperoort, Y. Cao, J. Shi, I. Tabas and W. Tao, Nat. Rev. Cardiol., 2021 DOI:10.1038/s41569-021-00629-x.
- X. Huang, G. Wu, C. Liu, X. Hua, Z. Tang, Y. Xiao, W. Chen, J. Zhou, N. Kong, P. Huang, J. Shi and W. Tao, Nano Lett., 2021, 21, 9706–9714 CrossRef CAS PubMed.
- S. Guo, K. Li, B. Hu, C. Li, M. Zhang, A. Hussain, X. Wang, Q. Cheng, F. Yang, K. Ge, J. Zhang, J. Chang, X. Liang, Y. Weng and Y. Huang, Exploration, 2021, 1, 35–49 CrossRef.
- X. Huang, C. Liu, N. Kong, Y. Xiao, A. Yurdagul, I. Tabas and W. Tao, Nat. Protoc., 2022 DOI:10.1038/s41596-021-00665-4.
- W. Chen, C. A. Glackin, M. A. Horwitz and J. I. Zink, Acc. Chem. Res., 2019, 52, 1531–1542 CrossRef CAS PubMed.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed.
- X. Xu, J. Wu, Y. Liu, P. E. Saw, W. Tao, M. Yu, H. Zope, M. Si, A. Victorious, J. Rasmussen, D. Ayyash, O. C. Farokhzad and J. Shi, ACS Nano, 2017, 11, 2618–2627 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- M. M. Billingsley, N. Singh, P. Ravikumar, R. Zhang, C. H. June and M. J. Mitchell, Nano Lett., 2020, 20, 1578–1589 CrossRef CAS PubMed.
- S. Chen, Y. Y. C. Tam, P. J. C. Lin, M. M. H. Sung, Y. K. Tam and P. R. Cullis, J. Controlled Release, 2016, 235, 236–244 CrossRef CAS PubMed.
- X. Ke, L. Shelton, Y. Hu, Y. Zhu, E. Chow, H. Tang, J. L. Santos and H. Q. Mao, ACS Appl. Mater. Interfaces, 2020, 12, 35835–35844 CrossRef CAS PubMed.
- X. Xu, P. E. Saw, W. Tao, Y. Li, X. Ji, M. Yu, M. Mahmoudi, J. Rasmussen, D. Ayyash, Y. Zhou, O. C. Farokhzad and J. Shi, Nano Lett., 2017, 17, 4427–4435 CrossRef CAS PubMed.
- R. JC Bose, S. Uday Kumar, Y. Zeng, R. Afjei, E. Robinson, K. Lau, A. Bermudez, F. Habte, S. J. Pitteri, R. Sinclair, J. K. Willmann, T. F. Massoud, S. S. Gambhir and R. Paulmurugan, ACS Nano, 2018, 12, 10817–10832 CrossRef CAS PubMed.
- M. Oishi, J. Nakaogami, T. Ishii and Y. Nagasaki, Chem. Lett., 2006, 35, 1046–1047 CrossRef CAS.
- K. O'Brien, K. Breyne, S. Ughetto, L. C. Laurent and X. O. Breakefield, Nat. Rev. Mol. Cell Biol., 2020, 21, 585–606 CrossRef PubMed.
- N. Pardi, M. J. Hogan, F. W. Porter and D. Weissman, Nat. Rev. Drug Discovery, 2018, 17, 261–279 CrossRef CAS PubMed.
- P. McCall, C. J. Witton, S. Grimsley, K. V. Nielsen and J. Edwards, Br. J. Cancer, 2008, 99, 1296–1301 CrossRef CAS PubMed.
- T. L. Lotan, B. Gurel, S. Sutcliffe, D. Esopi, W. Liu, J. Xu, J. L. Hicks, B. H. Park, E. Humphreys, A. W. Partin, M. Han, G. J. Netto, W. B. Isaacs and A. M. De Marzo, Clin. Cancer Res., 2011, 17, 6563–6573 CrossRef CAS PubMed.
- M. A. Islam, Y. Xu, W. Tao, J. M. Ubellacker, M. Lim, D. Aum, G. Y. Lee, K. Zhou, H. Zope, M. Yu, W. Cao, J. T. Oswald, M. Dinarvand, M. Mahmoudi, R. Langer, P. W. Kantoff, O. C. Farokhzad, B. R. Zetter and J. Shi, Nat. Biomed. Eng., 2018, 2, 850–864 CrossRef CAS PubMed.
- E. Cerami, J. Gao, U. Dogrusoz, B. E. Gross, S. O. Sumer, B. A. Aksoy, A. Jacobsen, C. J. Byrne, M. L. Heuer, E. Larsson, Y. Antipin, B. Reva, A. P. Goldberg, C. Sander and N. Schultz, Cancer Discovery, 2012, 2, 401–404 CrossRef PubMed.
- N. Kong, W. Tao, X. Ling, J. Wang, Y. Xiao, S. Shi, X. Ji, A. Shajii, S. T. Gan, N. Y. Kim, D. G. Duda, T. Xie, O. C. Farokhzad and J. Shi, Sci. Transl. Med., 2019, 11, 1–17 Search PubMed.
- R. Verbeke, I. Lentacker, L. Wayteck, K. Breckpot, M. Van Bockstal, B. Descamps, C. Vanhove, S. C. De Smedt and H. Dewitte, J. Controlled Release, 2017, 266, 287–300 CrossRef CAS PubMed.
- M. A. Islam, J. Rice, E. Reesor, H. Zope, W. Tao, M. Lim, J. Ding, Y. Chen, D. Aduluso, B. R. Zetter, O. C. Farokhzad and J. Shi, Biomaterials, 2021, 266, 120431 CrossRef CAS PubMed.
- Y. Qian, S. Qiao, Y. Dai, G. Xu, B. Dai, L. Lu, X. Yu, Q. Luo and Z. Zhang, ACS Nano, 2017, 11, 9536–9549 CrossRef CAS PubMed.
- L. Dai, K. Li, M. Li, X. Zhao, Z. Luo, L. Lu, Y. Luo and K. Cai, Adv. Funct. Mater., 2018, 28, 1707249 CrossRef.
- S. A. Shahin, R. Wang, S. I. Simargi, A. Contreras, L. Parra Echavarria, L. Qu, W. Wen, T. Dellinger, J. Unternaehrer, F. Tamanoi, J. I. Zink and C. A. Glackin, Nanomedicine, 2018, 14, 1381–1394 CrossRef CAS PubMed.
- C. Qiao, J. Yang, Q. Shen, R. Liu, Y. Li, Y. Shi, J. Chen, Y. Shen, Z. Xiao, J. Weng and X. Zhang, Adv. Mater., 2018, 30, 1705054 CrossRef PubMed.
- Z. Xu, Y. Wang, L. Zhang and L. Huang, ACS Nano, 2014, 8, 3636–3645 CrossRef CAS PubMed.
- M. Moro, D. Di Paolo, M. Milione, G. Centonze, V. Bornaghi, C. Borzi, P. Gandellini, P. Perri, U. Pastorino, M. Ponzoni, G. Sozzi and O. Fortunato, J. Controlled Release, 2019, 308, 44–56 CrossRef CAS PubMed.
- W. D. Foulkes, I. E. Smith and J. S. Reis-Filho, N. Engl. J. Med., 2010, 20, 1938–1948 CrossRef PubMed.
- C. Gong, J. Tian, Z. Wang, Y. Gao, X. Wu, X. Ding, L. Qiang, G. Li, Z. Han, Y. Yuan and S. Gao, J. Nanobiotechnol., 2019, 17, 93 CrossRef PubMed.
- J. S. Ananta, R. Paulmurugan and T. F. Massoud, Mol. Pharm., 2015, 12, 4509–4517 CrossRef CAS PubMed.
- S. Mullick Chowdhury, T. Y. Wang, S. Bachawal, R. Devulapally, J. W. Choe, L. Abou Elkacem, B. K. Yakub, D. S. Wang, L. Tian, R. Paulmurugan and J. K. Willmann, J. Controlled Release, 2016, 238, 272–280 CrossRef CAS PubMed.
- R. Devulapally, N. M. Sekar, T. V. Sekar, K. Foygel, T. F. Massoud, J. K. Willmann and R. Paulmurugan, ACS Nano, 2015, 9, 2290–2302 CrossRef CAS PubMed.
- D. Rosenblum, A. Gutkin, R. Kedmi, S. Ramishetti, N. Veiga, A. M. Jacobi, M. S. Schubert, D. Friedmann-Morvinski, Z. R. Cohen, M. A. Behlke, J. Lieberman and D. Peer, Sci. Adv., 2020, 6, abc9450 CrossRef PubMed.
- W. J. Cheng, L. C. Chen, H. O. Ho, H. L. Lin and M. T. Sheu, Int. J. Nanomed., 2018, 13, 7079–7094 CrossRef CAS PubMed.
- Q. Liu, C. Wang, Y. Zheng, Y. Zhao, Y. Wang, J. Hao, X. Zhao, K. Yi, L. Shi, C. Kang and Y. Liu, Biomaterials, 2020, 258, 120275 CrossRef CAS PubMed.
- T. Wei, Q. Cheng, Y. L. Min, E. N. Olson and D. J. Siegwart, Nat. Commun., 2020, 11, 3232 CrossRef CAS PubMed.
- J. Tabernero, G. I. Shapiro, P. M. LoRusso, A. Cervantes, G. K. Schwartz, G. J. Weiss, L. Paz-Ares, D. C. Cho, J. R. Infante, M. Alsina, M. M. Gounder, R. Falzone, J. Harrop, A. C. S. White, I. Toudjarska, D. Bumcrot, R. E. Meyers, G. Hinkle, N. Svrzikapa, R. M. Hutabarat, V. A. Clausen, J. Cehelsky, S. V. Nochur, C. Gamba-Vitalo, A. K. Vaishnaw, D. W. Y. Sah, J. A. Gollob and H. A. Burris, Cancer Discovery, 2013, 3, 406–417 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |