
Persistent luminescence nanoparticles functionalized by polymers bearing phosphonic acid anchors: synthesis, characterization, and in vivo behaviour†
Thomas
Lécuyer
 *a,
Nicolas
Bia
b,
Pierre
Burckel
c,
Cédric
Loubat
b,
Alain
Graillot
b,
Johanne
Seguin
a,
Yohann
Corvis
a,
Jianhua
Liu
a,
Lucie
Valéro
a,
Daniel
Scherman
a,
Nathalie
Mignet
a and
Cyrille
Richard
*a
*a,
Nicolas
Bia
b,
Pierre
Burckel
c,
Cédric
Loubat
b,
Alain
Graillot
b,
Johanne
Seguin
a,
Yohann
Corvis
a,
Jianhua
Liu
a,
Lucie
Valéro
a,
Daniel
Scherman
a,
Nathalie
Mignet
a and
Cyrille
Richard
*a
aUniversité de Paris, CNRS, INSERM, UTCBS, Unité de Technologies Chimiques et Biologiques pour la Santé, Faculté de Pharmacie, 75006 Paris, France. E-mail: thomas.lecuyer@alumni.chimie-paristech.fr; cyrille.richard@u-paris.fr
bSpecific Polymers, ZAC Via Domitia 150 Avenue des Cocardières, 34160 Castries, France
cInstitut de Physique du Globe de Paris (IPGP), Université de Paris, France
First published on 15th December 2021
Abstract
Optical in vivo imaging has become a widely used technique and is still under development for clinical diagnostics and treatment applications. For further development of the field, researchers have put much effort into the development of inorganic nanoparticles (NPs) as imaging probes. In this trend, our laboratory developed ZnGa1.995O4Cr0.005 (ZGO) nanoparticles, which can emit a bright persistent luminescence signal through the tissue transparency window for dozens of minutes and can be activated in vivo with visible irradiation. These properties endow them with unique features, allowing us to recover information over a long-time study with in vivo imaging without any background. To target tissues of interest, ZGO must circulate long enough in the blood stream, a phenomenon which is limited by the mononuclear phagocyte system (MPS). Depending on their size, charge and coating, the NPs are sooner or later opsonized and stored into the main organs of the MPS (liver, spleen, and lungs). The NPs therefore have to be coated with a hydrophilic polymer to avoid this limitation. To this end, a new functionalization method using two different polyethylene glycol phosphonic acid polymers (a linear one, later named lpPEG and a branched one, later named pPEG) has been studied in this article. The coating has been optimized and characterized in various aqueous media. The behaviour of the newly functionalized NPs has been investigated in the presence of plasmatic proteins, and an in vivo biodistribution study has been performed. Among them ZGOpPEG exhibits a long circulation time, corresponding to low protein adsorption, while presenting an effective one-step process in aqueous medium with a low hydrodynamic diameter increase. This new method is much more advantageous than another strategy we reported previously that used a two-step PEG silane coating performed in an organic solvent (dimethylformamide) for which the final hydrodynamic diameter was twice the initial diameter.
Introduction
Optical in vivo imaging has become a widely used technique to identify biological functions and pathology mechanisms, for both in vitro and in vivo models, and is under development for clinical diagnostics and treatments.1 While fluorescent molecules have already reached this achievement, they still have a few disadvantages, such as chemical stability, targeting, and hydrophobicity.2 To overcome these issues and take advantage of the tremendous benefits of optical imaging, researchers have made efforts towards the development of inorganic nanoparticles (NPs) as imaging probes.3,4 While they are bigger than molecules and proteins, yet smaller than cells, NPs induce new interaction pathways.5 Their tunability in terms of size, shape, and optical properties combined with their stability have endowed them with great advantages upon molecular probes.6Following this trend, our laboratory recently developed ZnGa1.995O4Cr0.005 nanoparticles or ZGO.7,8 This material was found to be attractive, due to both its bright near-infrared (λem = 700 nm) persistent luminescence and the possibility of its activation in vivo with visible irradiation. It is noteworthy that ZGO nanoparticles can emit a persistent luminescence signal through the tissue transparency window for dozens of minutes, allowing for performing of in vivo imaging without any background. This allows imaging with a high signal to noise ratio, constituting a promising alternative to the actual commercially available near infrared optical probes.7 More than these unique optical properties, PLNPs have also shown large functionalization capabilities for active targeting.9 Finally, the absence of in vitro and in vivo toxicity of ZGO or its constituents has been observed,10–14 and the degradation of ZGO in artificial lysosomal fluids has been proved.15 Lately, nanocomposites using ZGO have also been developed for imaging16 or detection.17
Despite these advantages, some fundamental challenges hamper NP deployment in clinics, and so is the case with ZGO.18 One of these is the uptake by the mononuclear phagocyte system (MPS), in which NPs are rapidly cleared out of the bloodstream into the liver, spleen, lungs or bone marrow, and they show nonspecific binding to nontargeted or healthy areas.5 This capture is mainly due to the adsorption of plasma proteins facilitating the recognition of the NPs by the macrophages, also known as opsonization.19 Concerns about NP toxicity often arise because of this MPS accumulation.20 Aggregation can lead to NP entrapment in the liver, lungs, or other organs due to capillary occlusion. To avoid this uptake, functionalization processes have been developed to passivate the surface of the NPs.21
In our laboratory, ZGOs were successfully functionalized with polyethylene glycol (PEG), thanks to a two-step synthesis in an organic solvent using silane as an anchor group.22 PEG is inexpensive, versatile and FDA approved for many applications.23 PEG chains modify the layer in interfaces with the biological fluids and nanoparticles, by creating a steric effect and annihilating the electrostatic interaction, and therefore reducing the opsonization process, while the unfunctionalized ZGO is opsonized in a few minutes.7 PEG coating has also been shown to improve the luminescence properties24 and hemocompability.14 Nevertheless, this functionalization process doubles the hydrodynamic diameter of the ZGO, which might increase the protein recognition and hence affect their biodistribution. Furthermore, the silane anchor has been shown to be easily degraded in the presence of reactive oxygen species (ROS).25
For these reasons, novel PEG functionalization is needed. In this case, polyphosphonic acid PEG is a new PEG derivative which has been shown to delay the opsonization of iron oxide NPs.26 These statistical poly(methacrylate) based copolymers bearing (i) PEG lateral chains and (ii) phosphonic acid anchoring allow a new coating strategy and geometry. The phosphonic acid group has already been employed to immobilize organic or organometallic molecules on the surface of metal oxides and has already been used for numerous applications,27 such as functionalization of quantum dots,28 IONPs,29 and UCNPs,30 and as an anchoring group for dye-sensitized solar cells.31 In this work we have evaluated two different PEGylated phosphonic acid polymers with the same chain length and we have developed a one-step synthesis in aqueous solution leading to NPs with smaller hydrodynamic diameters than that in the two-step process described above and used in many studies. A complete characterization has been done and the stability of ZGO coated with both polymers in the medium used for in vivo injection has been investigated. Finally, after determining the number of adsorbed proteins, biodistribution studies in mice have shown a good circulation time (a few hours) in the bloodstream, and the results have been compared to our previously reported two-step PEG silane coating strategy. This new functionalization strategy, advantageously performed in aqueous medium, could be a much safer alternative for in vivo studies and industrial development.
Results and discussion
Synthesis and characterization
The ZGO nanoparticles have been prepared as previously described, by a two-step method with the first step being a hydrothermal treatment from the constituting nitrated ions followed by a 5 h calcination at 750 °C.7 This last step confers to the material its persistent luminescence properties, after both UV and near infrared visible light excitation. The nanoparticles were then extracted from the bulk suspension by selective centrifugation after hydroxylation in hydrochloric acid. We finally obtained stable NPs in aqueous solution with a mean hydrodynamic diameter of 80 nm (called ZGOOH). After injection, the ZGOOH NPs are rapidly trapped by the MPS and so further functionalization is needed to have a longer circulation time in the bloodstream. To this end, PEGylation of the NPs has been investigated,22 and realized as an effective and efficient surface modification process for in vivo use. A two-step reaction has previously been optimized, with the first step being the grafting of aminopropyltriethoxysilane (APTES) followed by activation of polyethylene glycol with N-hydroxysuccinimide (referred to as PEG), and both steps were performed in dimethylformamide (DMF) (Fig. S1†). Such PEGylated ZGO, or ZGOPEG, was obtained with a mean hydrodynamic diameter of 145 ± 15 nm leading to a ∼5 h circulation time in the bloodstream. Nevertheless, the use of organic solvents and the large increase of size have left room for improvement.To do so, we developed herein a new functionalization strategy of ZGO with a new copolymer. The poly(poly(ethylene glycol methacrylate)-stat-dimethyl(methacryoyloxy) phosphonic acid) (poly(PEGMA-stat-MAPC1 acid, referred to as pPEG), was prepared as reported in the literature.32 It consists of two ester moieties: one carrying a PEG lateral chain of 5 kDa (red rectangle in Fig. 1A) and the second a phosphonic acid anchor (blue rectangle in Fig. 1A). Phosphonic acid is a functional group of interest for many current fields of research that include medicine (solubilization of bioactive compounds, agonist agents), and materials sciences (surface chemistry of electrodes and microfluidic devices).33 To compare its effectiveness, a linear PEG polymer with a mono-phosphonic acid anchor (referred to as lpPEG, Fig. 1B) and PEG silane (Fig. 1C), with the same PEG chain length, have also been used as controls.
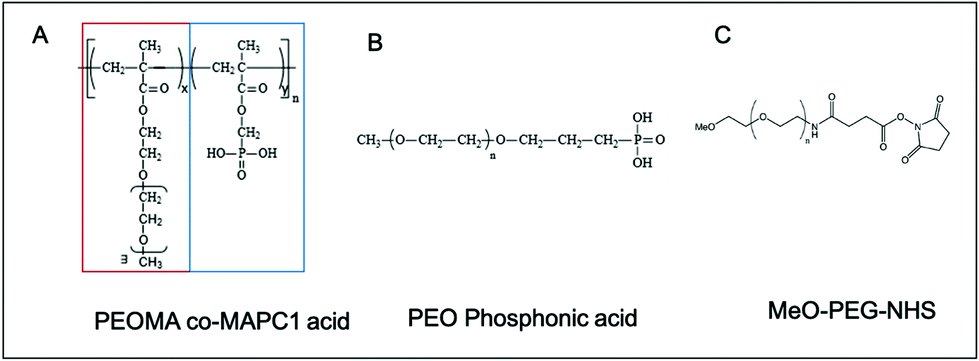 | ||
| Fig. 1 Semi-developed formula of (A) branched pPEG, (B) linear lpPEG and (C) PEG. pPEG has the characteristic of being composed of a repeat of a PEG chain (red rectangle) and a phosphonic acid anchor group (blue rectangle) on each monomer. | ||
Phosphonic acid possesses two main characteristics to be a valuable anchor group. Firstly, this group possesses two acidic protons, with pKa values ranging between 1–3 and 5–8 depending on the attached group,34 and therefore has a good attack site for nucleophilic substitution (SN1 and SN2).33 Secondly, a strong complexation of divalent cations by phosphonic acid has been seen.35,36 Two mechanisms of linkage between ZGO and the polymers are possible. In both cases a large coating of the surface is expected. Because of the numerous anchors of pPEG (n = 13), the polymer can interact with the surface several times, and therefore can “reticulate or attach” all around the NP. This could lead to a denser coating and therefore to a lower protein adsorption.
Functionalization of ZGO by pPEG and lpPEG has been performed in aqueous solution. The influence of several parameters such as the reaction time, the ZGO to polymer mass ratio, and the temperature has been investigated. The products of the reactions have been characterized by dynamic light scattering (DLS) (hydrodynamic diameter and zeta potential) and thermogravimetric analyses (TGA). Indeed, the main goal was to obtain the smallest possible hydrodynamic diameter with the densest coating, which implied a maximum passivation of the surface (zeta-potential close to zero) and a high weight loss percentage by TGA. This weight loss percentage was determined between 150 and 550 °C, a range in which only the organic part of the nanoparticles, i.e., the coating could degrade. An example of these analyses is shown in Fig. S2†.
Finally, the best conditions for functionalization with both polymers have been obtained using a 2/1 mass ratio in polymer/NPs in 5 mM hydrochloric acid (pH 2.5) at room temperature for 16 hours, as shown below (Fig. 2):
After several centrifugations and washing with water, the obtained ZGOlpPEG and ZGOpPEG have been physico-chemically characterized (Fig. 3). First, the luminescence measured using a Biospace photon imager showed similar results for the three pegylated samples (Fig. 3A). In terms of size, ZGOpPEG exhibited a small increase in hydrodynamic diameter (+30 nm), which was even smaller for ZGOlpPEG (+20 nm), with a narrow size distribution (Fig. 3B). Both the zeta potential and TGA confirmed the success of the functionalization (Fig. 3C). The zeta potential decreased for ZGOpPEG and ZGOlpPEG (−2.5 and −0.5 mV, respectively) compared to that for ZGOOH (+15 mV) showing a good passivation of the surface charge. Similarly, TGA showed a high amount of coating for both ZGOpPEG (8.6%) and ZGOlpPEG (5.7%) compared to the ZGOOH control (1.4%). Furthermore, the presence of the grafted coating has been demonstrated using 31P NMR on ZGOpPEG and ZGOlpPEG (Fig. S3,† cf comments). The behavior of the newly coated NPs in biological fluids was then investigated to evaluate the possibility of their use in in vivo imaging.
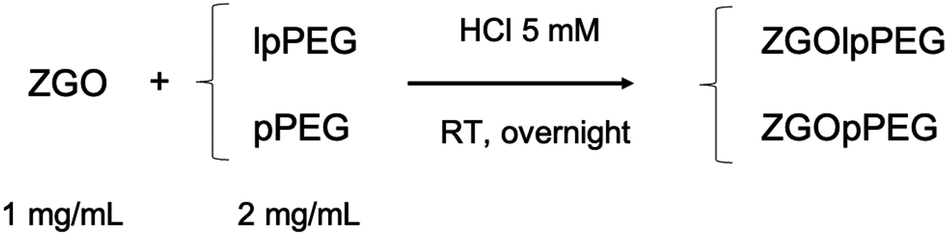 | ||
| Fig. 2 Optimized functionalization reaction of ZGO with lpPEG and pPEG. | ||
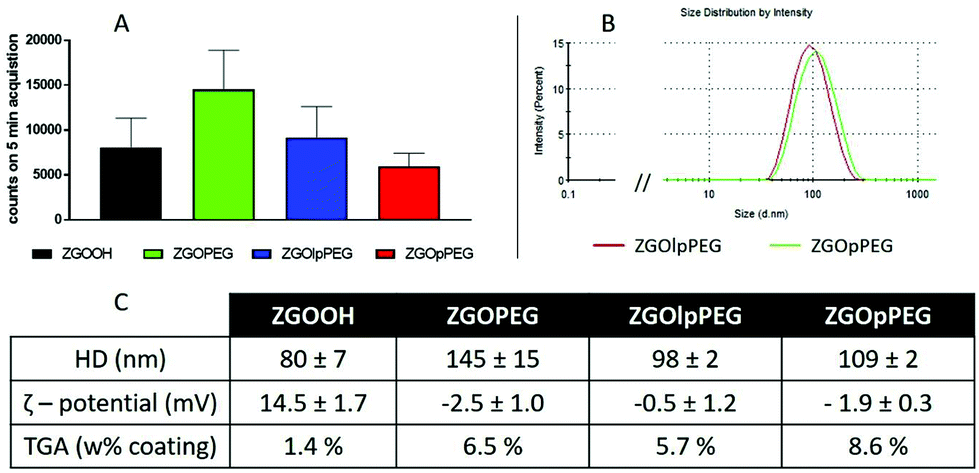 | ||
| Fig. 3 Characterization of the different ZGOs after functionalization. Luminescence properties after UV excitation (A) and size distribution of ZGOpPEG and ZGOlpPEG (B) were investigated. A summary of the different characterization processes is presented in the table (C). | ||
Characterization in in vivo like media
Before injecting the NPs, their colloidal stability in the injection medium was investigated. Indeed, an aggregation on the injection site could lead to severe injuries by local blocking of the bloodstream, and therefore enabling ZGO to circulate and accumulate in the tissue of interest. To assess their stability in 5% glucose, their hydrodynamic diameter and their polydispersity index have been recorded over time (Fig. 4).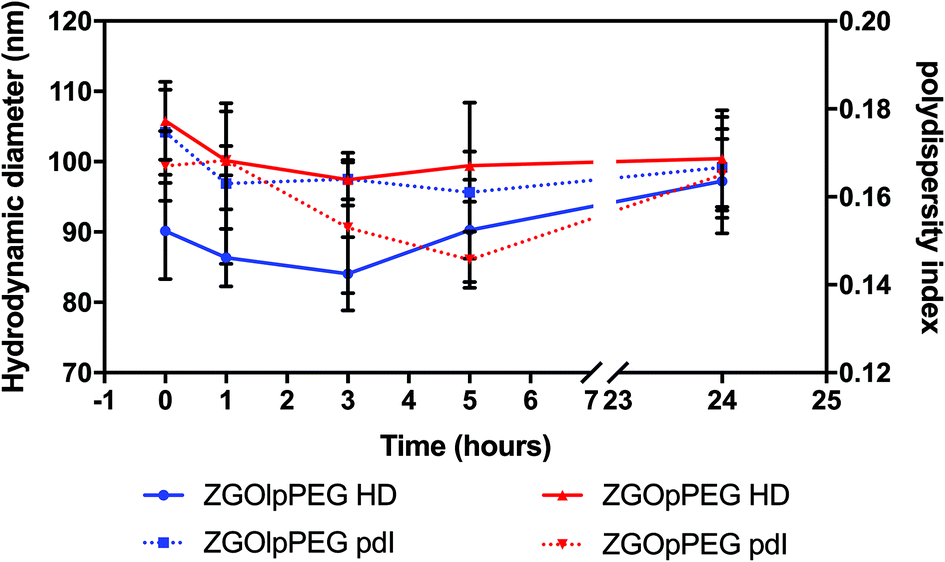 | ||
| Fig. 4 Evolution of the hydrodynamic diameter (continuous line) and polydispersity index (dashed line) of ZGOlpPEG (blue) and ZGOpPEG (red) in 5% glucose injection medium (n = 3). | ||
The same behavior was observed for both ZGOlpPEG and ZGOpPEG. First it can be observed that NPs are stable in 5% glucose with a mean diameter around 100 nm. It is interesting to notice that ZGOlpPEG shows larger variability than ZGOpPEG. This stability can be seen by looking at the evolution of the pdI. It remained stable in around 15% glucose up to 24 h, which indicates no sign of aggregation. In any case, a viable stability of the NPs in the injection medium has been proven, and their in vivo use can be considered.
After injection of the NPs into the bloodstream, ZGO will be in contact with the plasma proteins. These proteins are likely to adsorb on ZGO and therefore change their colloidal characteristics. To study these surface changes, the hydrodynamic diameter of the functionalized NPs, dispersed into mouse serum in 5% glucose (1/1 volume ratio), has been analysed by DLS (Fig. 5).
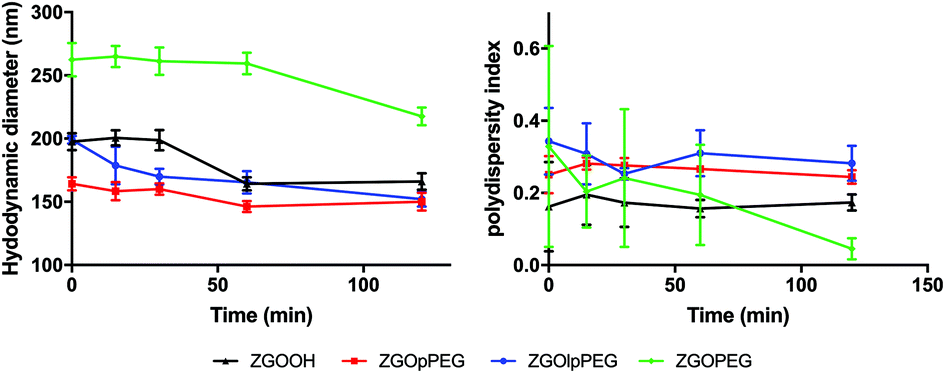 | ||
| Fig. 5 Evolution of the hydrodynamic diameter (left) and polydispersity index (right) of ZGOlpPEG (blue line), ZGOPEG (green line), ZGOOH (black line) and ZGOpPEG (red line) in diluted mouse serum in 5% glucose (n = 3). | ||
The difference between ZGOPEG and the two phosphonic acid-based PEG polymers is clear. While ZGOPEG presents a mean HD increase of 150 nm (100% increase) compared to the stock solution (Fig. 3C), a 50 nm increase is observed for ZGOpPEG (around 50%) and 80 nm for ZGOlpPEG (around 80%), while the unfunctionalized ZGOOH presents a 100 nm increase (125%). These observations indicate a clear limitation of the adsorption of plasma proteins on the surface of ZGOlpPEG and more especially on ZGOpPEG.32 The colloidal stability of the NPs in mouse serum can also be assessed by looking at the evolution of the pdI over time. Except for ZGOPEG, the pdI is found to be constant up to 2 h, showing low variability. This observation is coherent with the low evolution of the HD observed before.
The quantification of adsorbed proteins can be achieved using a colorimetric Bradford test.37 This test consists in incubating the NPs in serum for 2 h and finally adding the Bradford reacting agent, which changes the absorbance properties when in contact with proteins. The concentration of adsorbed proteins on the NPs can then be quantified using a calibration curve (Fig. S4†). The results obtained for each surface chemistry is shown below (Fig. 6). The results exhibit significant differences between ZGOOH and both ZGOPEG, as already described,22 and ZGOpPEG. The quantity of bound proteins per mg of ZGO is two-times lower (around 25 ng mg−1) than for the unfunctionalized ZGO (around 50 ng mg−1). ZGOlpPEG contains a larger number of adsorbed proteins than ZGOOH. This result could be explained by the lower density of the coating determined by TGA.
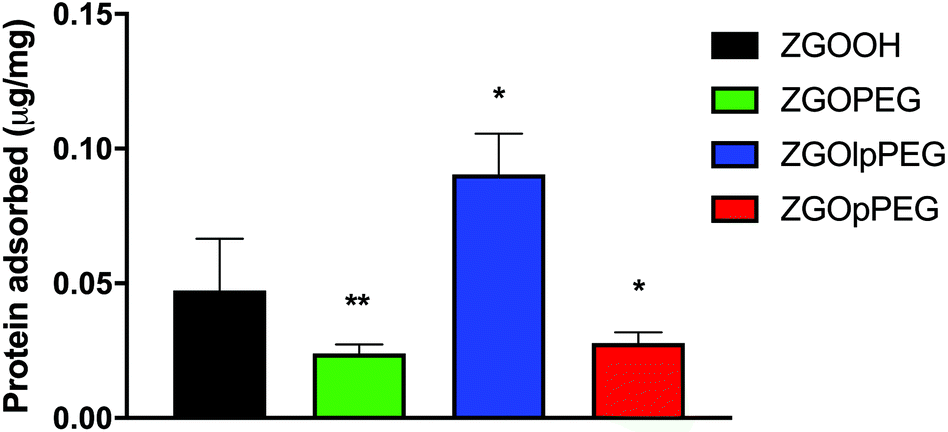 | ||
| Fig. 6 Bradford test showing the concentration of plasma proteins adsorbed on the surface of the different ZGOs (n = 5). Statistical differences with ZGOOH have been determined with the Mann–Whitney test (*: p < 0.1; **: p < 0.01). | ||
Biodistribution study in mice
Each functionalized ZGO had been administered to three healthy mice. A suspension of NPs at 10 mg mL−1 in 5% glucose was prepared and placed in a syringe. To excite the NPs, the syringe was placed under a UV lamp for 1 min. Finally, 0.2 mL of the suspension was parenterally injected into the retro-orbital site. The luminescence was monitored continuously during the first hour by repeated 10 min acquisitions. Once the persistent luminescence signal became low, the mice were re-excited for 2 min under visible light before the different point measurements at longer times.An overview of the biodistribution study is shown below (Fig. 7). The acquisition just after injection (T0) shows that both ZGOPEG and ZGOpPEG are well distributed inside the body. This can be due to the small HD variability in 5% glucose (Fig. 4). Nevertheless, the amount of ZGOlpPEG going in the bloodstream seems to circulate. On the other hand, ZGOpPEG exhibits an intense signal coming from the upper part of the mice. The longer time point study shows a similar behavior: the maximum intensity for ZGOlpPEG continues to come from the injection site, while both ZGOPEG and ZGOpPEG circulate. An accumulation in the liver seems to occur 4 h after injection and is confirmed after 6 h. To sum up, we can affirm that ZGO have been successfully functionalized by pPEG and such NPs can circulate in the bloodstream freely for more than 4 h and seem to present the same characteristic as ZGOPEG.
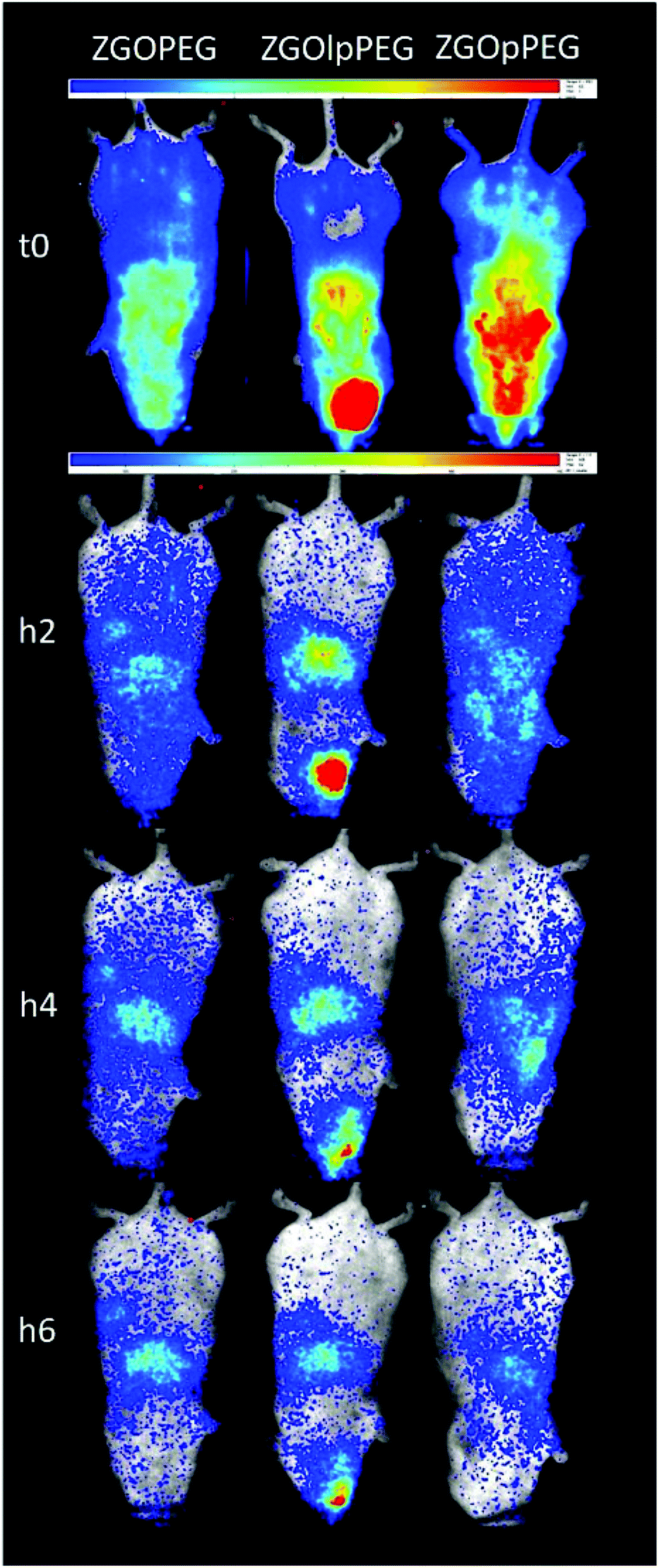 | ||
| Fig. 7 Biodistribution of the differently coated nanoparticles in healthy mice. At t0 the luminescence was recorded after ex vivo UV excitation (scale: 0.5–5) and then the signal was followed after in vivo visible light excitation (scale: 0.05–0.5). | ||
To have a better understanding of the biodistribution, the mice were sacrificed 6 h after injection and the main organs (liver, lungs, kidneys, spleen, heart, and blood) were collected. Ex vivo excitation of the tissues was performed to detect more precisely the provenance of the luminescence signal (Fig. 7). As already described,6 ZGOPEG is essentially maintained in the liver and the spleen 6 h after injection like ZGOlpPEG, as seen at the early time points. In any case, no luminescence signal was observed in the blood, suggesting a total clearance of ZGO from the bloodstream. In contrast, ZGOpPEG does not show the same behavior. The recorded luminescence signal originates from the liver and the spleen, but it is also distributed in the lungs and the kidneys.
To quantify these observations, the luminescence signal coming from the tissues has been counted with a 5 min acquisition, and the resulting photon counts have been normalized by the mass of each organ (Fig. 8).
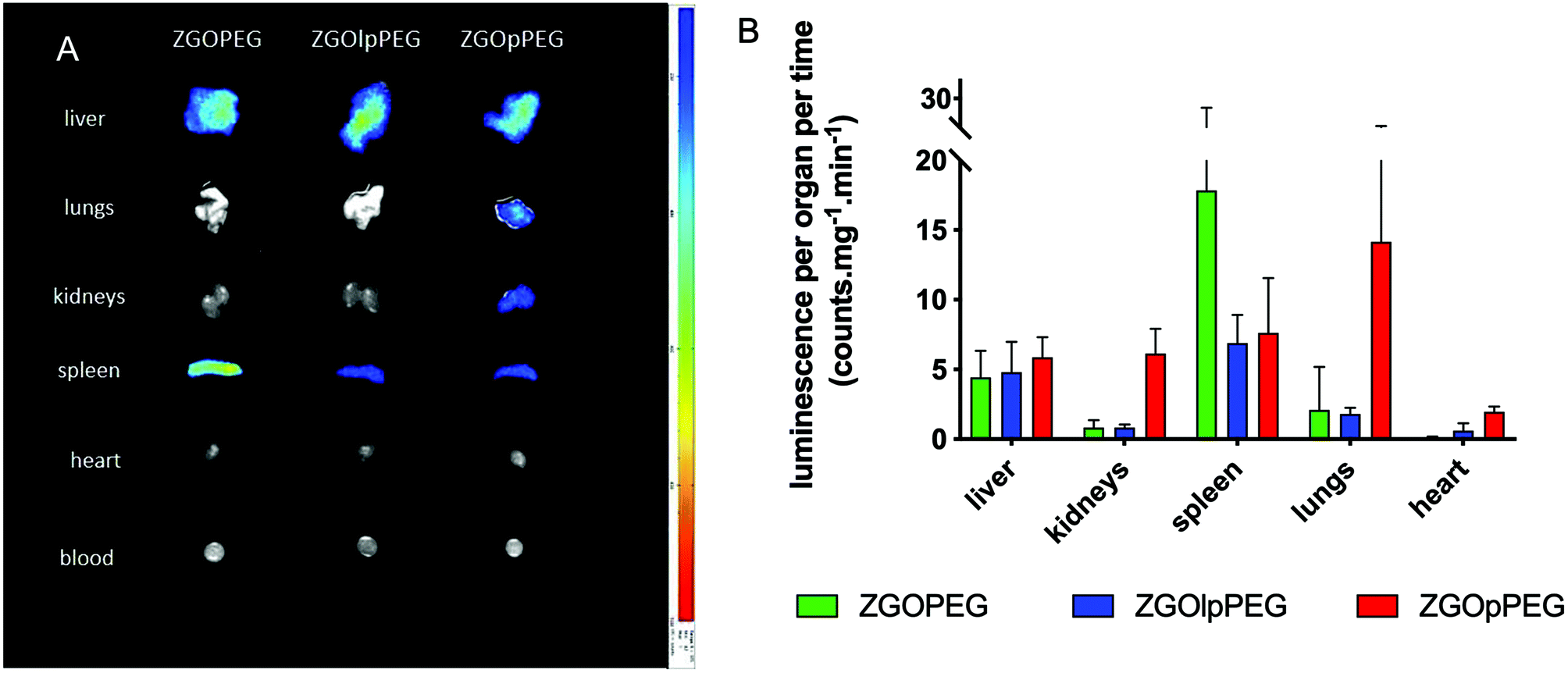 | ||
| Fig. 8 Ex vivo luminescence of the different organs 6 h after injection (left) and quantification of the luminescence signal after ex vivo visible light excitation (right). | ||
As observed before, the signal of luminescence in the liver is the same for the three NPs. The spleen also presents a large uptake of the ZGOs, because of its active role in the MPS. The observation made before is confirmed by quantification. ZGOpPEG is well present in the kidneys, the lungs and more surprisingly in the heart. These results suggest either a continuous circulation in the blood or new cellular interactions. Following these observations, ICP-MS quantifications of the zinc and gallium content in the organs have been performed. The obtained concentrations have been normalized in comparison with the initial injected dose (Fig. 9). As seen before, NPs are mainly present in the liver and spleen, but a larger percentage of ZGOpPEG is observed in the lungs and blood. This result is consistent with a longer circulation time in the bloodstream. On the other hand, a larger quantity of ZGOpPEG in the kidneys is also observed, which was already noted with ex vivo luminescence.
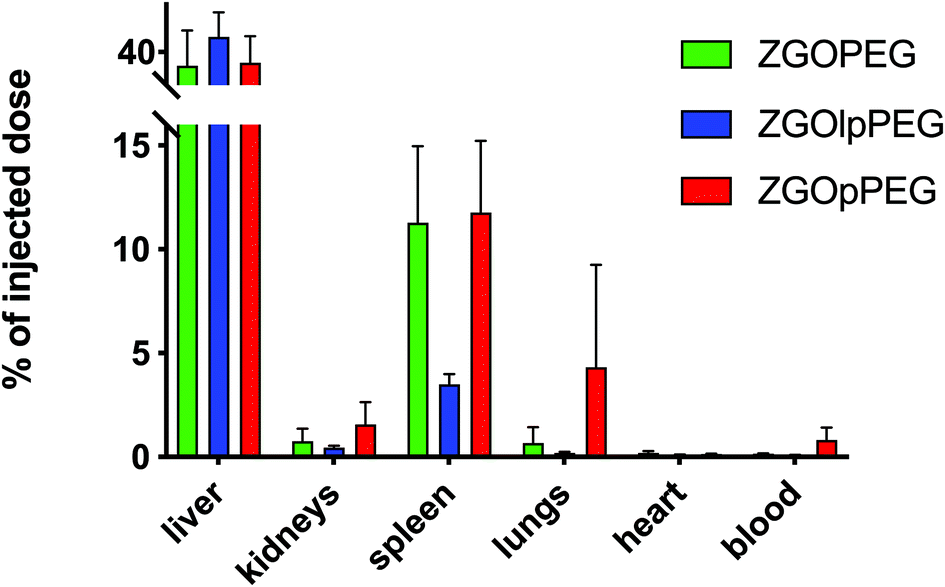 | ||
| Fig. 9 ICP-MS quantification of ZGO content in each organ depending on the coating. Results have been normalized in comparison with the initial injected dose. | ||
From these results, we can see that the structure of the pPEG offers new possibilities. Because of the number of phosphonic acid anchors per polymer, it is unlikely that each one of them binds to the ZGO. Therefore, some of the anchor sites could be in direct interaction with the biological environment. The phosphonic acid has been investigated for its role in various biological processes and has been shown to be able to fix active binding sites, such as tyrosine phosphatases.38 ZGOpPEG could link to different proteins or induce different interactions with cells in contrast to ZGOPEG. Therefore, new investigations could be envisioned such as the passive targeting of kidneys using ZGOpPEG.
Experimental
Chemicals were obtained from Sigma-Aldrich, Fluka or Alfa-Aesar. Alpha-methoxy-omega N-hydroxysuccinimide poly(ethylene glycol) PEG MW 5.000 Dalton was bought from Iris Biotech GmbH. Water refers to Millipore water.Polymer synthesis
Preparation of 1. MAPC1 ester (1.25 g, 6.0 mmol), MPEG5K (30 g, 6.0 mmol), and AIBN (0.312 g, 1.9 mmol) were added along with 60 mL of methylethyl ketone (MEK) into a two-necked round-bottom flask. The mixture was heated at 70 °C under an argon atmosphere in a thermostatic oil bath for 24 hours leading to 100% conversion. The conversion was monitored by 1H NMR spectroscopy. MEK was evaporated and the terpolymer was dissolved in a small volume of THF before precipitation in cold ether. A white powder (PEGMA5k–MAPC1ester) was obtained in 85% yield.
1H NMR (300 MHz, CDCl3) δ (ppm): 4.49–3.91 (–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–O–C
2–O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 3.90–3.27 (–O–C2–C2–O), 3.8–3.91 (OP–(OC3)2), 3.22 (–CH2–O–C3), 2.8–0.8 (C(C3)–C2).
O), 3.90–3.27 (–O–C2–C2–O), 3.8–3.91 (OP–(OC3)2), 3.22 (–CH2–O–C3), 2.8–0.8 (C(C3)–C2).
31P NMR (CDCl3, 300 MHz) δ (ppm): 21.7 (O![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) –(OCH3)2).
–(OCH3)2).
Preparation of 2. The previously obtained copolymer (PEGMA5k–MAPC1ester, 22.2 g) was dissolved in a minimum of dichloromethane (100 mL) and degassed with argon. Bromotrimethylsilane (3.7 g, 24 mmol) was added dropwise into the reactive media under inert and dry conditions. The solution was stirred for 8 hours, and then the solvent was removed with a rotatory evaporator. Ethanol was then added in excess (30 mL) to complete the ethanolysis. Finally, the targeted terpolymer was precipitated in cold ether leading to the PEGMA5k–MPh final product in 81% yield. 1H NMR (300 MHz, CDCl3) δ (ppm): 4.33–3.27 (C
2–O–CO, C2–C2–O), 3.23 (CH2–O–C3), 2.5–0.8 (C(C3)–C2).
31P NMR (CDCl3, 300 MHz) δ (ppm): 18.6 (O–(OH)2).
Preparation of 3. In a two-necked round-bottom flask vinyl–PEG (10 g, 1.98 mmol), and diethyl phosphite (5.5 g, 39.7 mmol, 20 eq.) were added, and the solution was heated at 110 °C. tert butyl peroxypivalate (23 mg, 9.92 10−2 mmol) was added dropwise into the reactive media. The reaction mixture was left at 110 °C for half an hour, and then diethyl phosphite was evaporated. The mixture was solubilized in a minimum of dichloromethane for precipitation in cold ether. The powder was filtered to obtain a white powder, PEG5K–Ph ester (8.9 g, 83% yield).
1H NMR (300 MHz, D2O) δ (ppm): 4.17–4.02 ((OP–C2–CH3)2), 3.76–3.51 (–O–C2–C2–O), 3.37 (–O–CH2–CH2–O–C3), 1.75–1.95 (–O–C2–C2–PO), 1.31 ((OP–CH2–C3)2.
31P NMR (D2O, 300 MHz) δ (ppm): 32.3 (O–(O–CH2–CH3)2).
Preparation of 4. The previously obtained polymer (PEG5K–Ph ester, 5 g, 0.97 mmol) was dissolved in a minimum of dichloromethane (30 mL) and degassed with argon. Bromotrimethylsilane (0.31 g, 2 mmol, 2.1 eq.) was added dropwise into the reactive media under inert and dry conditions. The solution was stirred for 8 hours, and then the solvent was removed with a rotatory evaporator. Ethanol (30 mL) was then added in excess to complete the ethanolysis. Finally, the targeted terpolymer was precipitated in cold ether leading to 4.4 g of PEG5K–Ph final product in 88% yield.
1H NMR (300 MHz, D2O) δ (ppm): 3.48–3.80 (–O–C2–C2–O), 3.33 (–O–CH2–CH2–O–C3), 1.64–1.88 (–O–C2–C2–PO).
31P NMR (D2O, 300 MHz) δ (ppm): 29.45 (O–(OH)2).
ZGO synthesis
ZnGa2O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cr3+ nanoparticles were synthesized by the hydrothermal method developed in our lab. First, gallium nitrate was prepared by reacting 8.94 mmol of gallium oxide with 20 mL concentrated nitric acid (35 wt%) under hydrothermal conditions at 150 °C for 24 hours. Then, a mixture of 0.04 mmol of chromium nitrate and 8.97 mmol of zinc nitrate in 10 mL of water was added to the previous solution of gallium nitrate under vigorous stirring. The pH of the resulting solution was adjusted to 7.5 with an ammonia solution (30 wt%), stirred for 3 hours at room temperature, and transferred into a 45 mL Teflon-lined stainless-steel autoclave for 24 h at 120 °C. The resulting compound was washed several times with water and ethanol before drying at 60 °C for 2 hours. The dry white powder was finally sintered in air at 750 °C for 5 hours. Hydroxylation was performed by basic wet grinding of the powder (500 mg) for 15 minutes, with a mortar and pestle in 2 mL of 5 mM HCl solution, and vigorously stirred overnight at room temperature at 10 mg mL−1 in 5 mM HCl. Nanoparticles with a diameter of 80 nm were first selected from the whole polydisperse colloidal suspension by centrifugation (SANYO MSE Mistral 1000 at 4500 rpm for 10 minutes) and collected in the supernatant (assessed by dynamic light scattering). The supernatants were gathered and concentrated to obtain the final ZGOOH stock suspension.
:Cr3+ nanoparticles were synthesized by the hydrothermal method developed in our lab. First, gallium nitrate was prepared by reacting 8.94 mmol of gallium oxide with 20 mL concentrated nitric acid (35 wt%) under hydrothermal conditions at 150 °C for 24 hours. Then, a mixture of 0.04 mmol of chromium nitrate and 8.97 mmol of zinc nitrate in 10 mL of water was added to the previous solution of gallium nitrate under vigorous stirring. The pH of the resulting solution was adjusted to 7.5 with an ammonia solution (30 wt%), stirred for 3 hours at room temperature, and transferred into a 45 mL Teflon-lined stainless-steel autoclave for 24 h at 120 °C. The resulting compound was washed several times with water and ethanol before drying at 60 °C for 2 hours. The dry white powder was finally sintered in air at 750 °C for 5 hours. Hydroxylation was performed by basic wet grinding of the powder (500 mg) for 15 minutes, with a mortar and pestle in 2 mL of 5 mM HCl solution, and vigorously stirred overnight at room temperature at 10 mg mL−1 in 5 mM HCl. Nanoparticles with a diameter of 80 nm were first selected from the whole polydisperse colloidal suspension by centrifugation (SANYO MSE Mistral 1000 at 4500 rpm for 10 minutes) and collected in the supernatant (assessed by dynamic light scattering). The supernatants were gathered and concentrated to obtain the final ZGOOH stock suspension.
Preparation of PEG-coated nanoparticles (silane two-step strategy)
The PEGylation of ZGOOH nanoparticles was performed by a two-step synthesis already described in the literature. 5 5 mg of ZGOOH was washed with water and twice with DMF before being suspended in 2 mL of DMF. After the addition of 20 μL of (3-aminopropyl)triethoxysilane (APTES), the solution was briefly sonicated and stirred for 6 hours at ambient temperature. The resulting particles were washed twice with DMF and resuspended in 2 mL of DMF. 50 mg of NHS-activated PEG (MeO–PEG5kDa–NHS) were added and the solution was stirred overnight at 90 °C and then washed twice with DMF before drying.Preparation of lpPEG and pPEG-coated nanoparticles
Typically, 5 mg of ZGOOH were washed with 5 mM HCl and placed in a round bottom flask at 1 mg mL−1. Then, 10 mg of the corresponding polymer was added, and the mixture was briefly sonicated. The reaction took place overnight with vigorous stirring. The obtained functionalized NPs were then washed with water.Characterization of nanoparticles
400 rpm for 15 minutes at room temperature). The absence of unbound proteins in the last supernatant was verified with a Bradford assay. 10 μL of a 1 mg mL−1 suspension of washed nanoparticles in 5% glucose were transferred into a 96-well plate (6 wells per sample). Next, 200 μL of the Coomassie blue dye reagent (Bio-Rad) was added to each well, and the plate was incubated at 37 °C for 10 minutes. The absorbance at 595 nm was measured using a plate reader (Tecan Infinite F200 Pro).
In vivo biodistribution studies
All experiments involving mice were approved by the French Comité d’éthique en expérimentation animale N°034 and by the French Ministry of Research APAFIS #8519-20 16090514387844.In vivo and ex vivo luminescence signal acquisition
10 mg of dried ZGOlpPEG, ZGOpPEG and ZGO–PEG were redispersed in 1 mL of 5% glucose. 200 μL (equivalent to 2 mg of nanoparticles) were collected with a syringe and excited for 2 minutes under a 365 nm UV lamp. Three mice were chemically anesthetized with isoflurane gas. The nanoparticles were then injected intravenously in the retro orbital area. During reexcitation and acquisition with an Optima (Biospace) camera, the mice were placed on their back and kept asleep with isoflurane. Luminescence was acquired with the Optima camera (Biospace) at different post-injection times for 10 minutes. For acquisition times longer than 1 hour after injection, nanoparticles were re-excited for 2 minutes under a LED (>515 nm lamp) before acquisition. After 6 h, the mice were sacrificed and the organs were collected, rinsed with mQ water, and placed on a black plate. The tissues were excited for 2 min with visible light. The count of luminescence was performed exactly 1 min after the end of the excitation, with a 5 min acquisition time. The results were been normalized by the corresponding mass of tissue.ICP quantification
Organ samples kept at −80 °C were thawed and then weighed directly from the tube. The different samples were put in the presence of 5 mL of distilled concentrated nitric acid (70%) for 24 h at 90 °C using an Analab mineralization device. The solution was then allowed to cool, and the acid was diluted by adding 45 mL of mQ water. 50 μL of the solution were then taken and placed in 4.95 mL of mQ water for 66Zn and 69Ga analysis with the HR-ICP-MS Element II from ThermoScientific (PARI platform).Conclusions
A new functionalization strategy has been developed using statistical copolymers bearing PEG lateral chains and multi-phosphonic acid anchoring groups. The coating was optimized, and good colloidal stability was obtained in the injection medium. The incubation of ZGOpPEG in mouse serum exhibited a low adsorption of plasma proteins confirmed by both DLS and Bradford test. Finally, the biodistribution study in mice showed a circulation time in blood of more than 4 h, comparable to ZGOPEG. Furthermore, differences in organ biodistributions have been reported.Finally, in this work we have developed a one-step reaction in aqueous medium with a lower amount of polymer used, a higher coating density, and controlled size distribution, while observing long circulation time after injection in the bloodstream. This allows us to consider new potentials for these ZGO NPs and new targeting strategies. Furthermore, this new functionalization strategy, only realized in aqueous medium, is a much safer alternative for in vivo studies.
Author contributions
T. Lécuyer was the principal contributor on the experimental work. N. Bia oversaw the polymer syntheses, under the supervision of A. Graillot and C. Loubat. J. Seguin brought expertise with in vivo imaging and Y. Corvis with thermogravimetric analysis. J. Liu helped with the synthesis of ZGO and L. Valero with DLS analysis. P. Burckel oversaw the ICP-MS measurements. Finally, D. Scherman, N. Mignet and C. Richard oversaw this work and brought funding to support this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Agence Nationale de la Recherche (ANR-14-CE08-0016-01). The authors would like to thank the PARI platform for ICP-MS measurements, and the LIOPA platform for the in vivo imaging. The authors would like to thank M. Ilyess Zouaoui for the graphical work.Notes and references
- S. H. Yun and S. J. J. Kwok, Nat. Biomed. Eng., 2017, 1, 0008 CrossRef CAS PubMed.
- J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–634 CrossRef CAS PubMed.
- S. Bouccara, G. Sitbon, A. Fragola, V. Loriette, N. Lequeux and T. Pons, Curr. Opin. Biotechnol., 2015, 34, 65–72 CrossRef CAS PubMed.
- T. Lécuyer, E. Teston, G. Ramirez-Garcia, T. Maldiney, B. Viana, J. Seguin, N. Mignet, D. Scherman and C. Richard, Theranostics, 2016, 6, 2488–2524 CrossRef PubMed.
- J. P. M. Almeida, A. L. Chen, A. Foster and R. Drezek, Nanomedicine, 2011, 6, 815–835 CrossRef CAS PubMed.
- J. Key and J. F. Leary, Int. J. Nanomed., 2014, 9, 711–726 Search PubMed.
- T. Maldiney, A. Bessière, J. Seguin, E. Teston, S. K. Sharma, B. Viana, A. J. J. Bos, P. Dorenbos, M. Bessodes, D. Gourier, D. Scherman and C. Richard, Nat. Mater., 2014, 13, 418–426 CrossRef CAS PubMed.
- J. Liu, T. Lécuyer, J. Seguin, N. Mignet, D. Scherman, B. Viana and C. Richard, Adv. Drug Delivery Rev., 2019, 138, 193–210 CrossRef CAS PubMed.
- T. Maldiney, M. U. Kaikkonen, J. Seguin, Q. le Masne de Chermont, M. Bessodes, K. J. Airenne, S. Ylä-Herttuala, D. Scherman and C. Richard, Bioconjugate Chem., 2012, 23, 472–478 CrossRef CAS PubMed.
- G. Ramírez-García, S. Gutiérrez-Granados, M. A. Gallegos-Corona, L. Palma-Tirado, F. d'Orlyé, A. Varenne, N. Mignet, C. Richard and M. Martínez-Alfaro, Int. J. Pharm., 2017, 532, 686–695 CrossRef PubMed.
- I. Sekler, S. L. Sensi, M. Hershfinkel and W. F. Silverman, Mol. Med., 2007, 13, 337–343 CAS.
- S. Choi, B. E. Britigan and P. Narayanasamy, Antimicrob. Agents Chemother., 2017, 61, e02505–e02516 CAS.
- S. Terpilowska and A. K. Siwicki, Chemosphere, 2018, 201, 780–789 CrossRef CAS PubMed.
- Y. Jiang, Y. Li, C. Richard, D. Scherman and Y. Liu, J. Mater. Chem. B, 2019, 7, 3796–3803 RSC.
- T. Lécuyer, M.-A. Durand, J. Volatron, M. Desmau, R. Lai-Kuen, Y. Corvis, J. Seguin, G. Wang, D. Alloyeau, D. Scherman, N. Mignet, F. Gazeau and C. Richard, Nanoscale, 2020, 12, 1967–1974 RSC.
- E. Teston, T. Maldiney, I. Marangon, J. Volatron, Y. Lalatonne, L. Motte, C. Boisson-Vidal, G. Autret, O. Clément, D. Scherman, F. Gazeau and C. Richard, Small, 2018, 14, 1800020 CrossRef PubMed.
- H. Liu, H. Yin, T. Yang, H. Ding and Y. Dong, Analyst, 2020, 145, 7412–7420 RSC.
- M. Hofmann-Amtenbrink, D. W. Grainger and H. Hofmann, Nanomedicine, 2015, 11, 1689–1694 CrossRef CAS PubMed.
- V. Pareek, A. Bhargava, V. Bhanot, R. Gupta, N. Jain and J. Panwar, J. Nanosci. Nanotechnol., 2018, 18, 6653–6670 CrossRef CAS PubMed.
- J. Li, X. Chang, X. Chen, Z. Gu, F. Zhao, Z. Chai and Y. Zhao, Biotechnol. Adv., 2014, 32, 727–743 CrossRef CAS PubMed.
- M. Zhu, G. Nie, H. Meng, T. Xia, A. Nel and Y. Zhao, Acc. Chem. Res., 2013, 46, 622–631 CrossRef CAS PubMed.
- T. Maldiney, M. Rémond, M. Bessodes, D. Scherman and C. Richard, J. Mater. Chem. B, 2015, 3, 4009–4016 RSC.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- L. Fu, J. Wang, N. Chen, Q. Ma, D. Lu and Q. Yuan, Chem. Commun., 2020, 56, 6660–6663 RSC.
- A. Li, H. P. Luehmann, G. Sun, S. Samarajeewa, J. Zou, S. Zhang, F. Zhang, M. J. Welch, Y. Liu and K. L. Wooley, ACS Nano, 2012, 6, 8970–8982 CrossRef CAS PubMed.
- V. Baldim, N. Bia, A. Graillot, C. Loubat and J. Berret, Adv. Mater. Interfaces, 2019, 6, 1801814 CrossRef CAS.
- J.-M. Rueff, M. Poienar, A. Guesdon, C. Martin, A. Maignan and P.-A. Jaffres, J. Solid State Chem., 2016, 236, 236 CrossRef CAS.
- R. Gomes, A. Hassinen, A. Szczygiel, Q. Zhao, A. Vantomme, J. C. Martins and Z. Hens, J. Phys. Chem. Lett., 2011, 2, 145–152 CrossRef CAS.
- G. Ramniceanu, B.-T. Doan, C. Vezignol, A. Graillot, C. Loubat, N. Mignet and J.-F. Berret, RSC Adv., 2016, 6, 63788–63800 RSC.
- R. Li, Z. Ji, J. Dong, C. H. Chang, X. Wang, B. Sun, M. Wang, Y.-P. Liao, J. I. Zink, A. E. Nel and T. Xia, ACS Nano, 2015, 9, 3293–3306 CrossRef CAS PubMed.
- J. L. Braid, U. Koldemir, A. Sellinger, R. T. Collins, T. E. Furtak and D. C. Olson, ACS Appl. Mater. Interfaces, 2014, 6, 19229–19234 CrossRef CAS PubMed.
- V. Torrisi, A. Graillot, L. Vitorazi, Q. Crouzet, G. Marletta, C. Loubat and J.-F. Berret, Biomacromolecules, 2014, 15, 3171–3179 CrossRef CAS PubMed.
- C. M. Sevrain, M. Berchel, H. Couthon and P.-A. Jaffrès, Beilstein J. Org. Chem., 2017, 13, 2186–2213 CrossRef CAS PubMed.
- L. D. Freedman and G. O. Doak, Chem. Rev., 1957, 57, 479–523 CrossRef CAS.
- M. Montalti, S. Wadhwa, W. Y. Kim, R. A. Kipp and R. H. Schmehl, Inorg. Chem., 2000, 39, 76–84 CrossRef CAS PubMed.
- A. Graillot, D. Bouyer, S. Monge, J.-J. Robin, P. Loison and C. Faur, J. Hazard. Mater., 2013, 260, 425–433 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- E. M. B. Fonseca, D. B. B. Trivella, V. Scorsato, M. P. Dias, N. L. Bazzo, K. R. Mandapati, F. L. de Oliveira, C. V. Ferreira-Halder, R. A. Pilli, P. C. M. L. Miranda and R. Aparicio, Bioorg. Med. Chem., 2015, 23, 4462–4471 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr07114a |
| This journal is © The Royal Society of Chemistry 2022 |