
A study on the role of plasmonic Ti3C2Tx MXene in enhancing photoredox catalysis†
Guanshun
Xie
a,
Chuang
Han
b,
Fei
Song
a,
Yisong
Zhu
a,
Xuanyu
Wang
a,
Jialin
Wang
a,
Zhenjun
Wu
c,
Xiuqiang
Xie
 *a and
Nan
Zhang
a
*a and
Nan
Zhang
a
aCollege of Materials Science and Engineering, Hunan University, Changsha 410082, P. R. China. E-mail: xiuqiang_xie@hnu.edu.cn
bDepartment of Chemistry, University of Cincinnati, USA
cCollege of Chemistry and Chemical Engineering, Hunan University, P. R. China
First published on 10th November 2022
Abstract
Engineering the spatial separation and transfer of photogenerated charge carriers has been one of the most enduring research topics in the field of photocatalysis due to its crucial role in determining the performances of photocatalysts. Herein, as a proof-of-concept, Ti3C2Tx MXene is coupled with a typical heterojunction of TiO2@CdS through a co-assembly strategy to boost electron pumping towards improving the photocatalytic efficiency. In addition to the band alignment-mediated electron transfer in TiO2@CdS–Ti3C2Tx heterojunctions, the plasmon-induced electric field enhancement of Ti3C2Tx is found to cooperate with the electron-reservoir role of Ti3C2Tx to extract photoinduced electrons. The synergistic dual functions of Ti3C2Tx promote multichannel electron transfer in TiO2@CdS–Ti3C2Tx hybrids to improve the photocatalytic efficiency. These results intuitively show that there is a wide scope to manipulate the spatial separation and transfer of photoinduced electrons by cultivating the fertile ground of Ti3C2Tx toward boosting the efficiency of solar-to-chemical conversion.
 Xiuqiang Xie | Dr Xiuqiang Xie is now an associate professor in the College of Materials Science and Engineering, Hunan University. He received his PhD degree from the University of Technology Sydney in 2017 under the supervision of Prof. Guoxiu Wang. He has been a visiting PhD student supervised by Prof. Yury Gogotsi at Drexel University, where he started his research on MXenes. His current research interests include the optical properties and the assembly methods of MXenes for sustainable technologies, such as photocatalysis and hybrid alkali-ion capacitors. |
1. Introduction
Photoredox catalysis holds great promise as an alternative to traditional synthetic pathways and is an ideal strategy to solve the growing global energy crisis.1–3 The bulk recombination of photoinduced charge carriers has been a long-standing challenge fundamentally restricting the solar-to-chemical conversion efficiency over photocatalysts.4–8 Under this background, engineering appropriate cocatalysts to steer photo-induced electron transfer has been an enduring research theme.9–15MXene is a large family of two-dimensional (2D) transition metal carbides, nitrides, and carbonitrides with the general formula of Mn+1XnTx (n = 1, 2, or 3), where M is an early transition metal, X represents carbon and/or nitrogen, and Tx represents surface functionalities (such as –OH, –O and/or –F).16,17 Since Ti3C2Tx MXene has been prepared successfully for the first time in 2011,18 its chemical and structural versatility, such as a 2D lamellar structure with exposed metastable transition metals, hydrophilicity, and high electrical conductivity,19 endows Ti3C2Tx with great promise in the field of photocatalysis.20–23 Thus far, enormous endeavors have been predominantly focused on using Ti3C2Tx to construct single electron transfer channels based on the Schottky effect to improve the charge carrier separation efficiency toward boosting the photocatalytic performance.24–26 Very recently, metallic Ti3C2Tx MXene has been found to exhibit the attracting localized surface plasmon resonance (LSPR) effect,27–30 which results from its high carrier density (∼8 × 1021 cm−3)31 and strong absorption of electromagnetic waves.32 The strong electron–phonon collision in Ti3C2Tx with a large absorption cross-section and an ultrathin-atomic-layer structure enables efficient light absorption across a wide spectrum in the visible-near-infrared (NIR) region,33–36 which is not valid for other two-dimensional materials, such as reduced graphene oxide (RGO). Compared to traditional plasmonic metals,37–39 Ti3C2Tx generates hot carriers with a rather slow relaxation due to a longer electronic free mean path.36 Furthermore, in addition to modulating the geometrical features, it is also possible to tune the LSPR effect of Ti3C2Tx by controlling the population of Tx moieties compatible with most polar semiconductor photocatalysts,40–43 which is not possible for classical plasmonic metals. These merits underline the possibilities of using Ti3C2Tx as a cocatalyst to enhance the photocatalytic performance of semiconductors by taking advantage of the potential LSPR effect, which, however, has been rarely explored.
Herein, as a proof-of-concept, we report on the application of Ti3C2Tx to enhance the charge carrier separation of TiO2@CdS as a typical heterojunction photocatalyst. Specifically, CdS is photodeposited on the surface of TiO2 nanosheets to construct the typical heterojunction of TiO2@CdS. Ti3C2Tx MXene is then coupled with TiO2@CdS via an electrostatic self-assembly approach. It is found that the introduction of Ti3C2Tx MXene not only serves as an “electron reservoir” resulting from the Schottky junction effect, but also enhances the near electric field due to the LSPR effect. The dual functions of Ti3C2Tx MXene synergized with the band-alignment mediated electron transfer in TiO2@CdS afford multichannel electron transfer in the ternary TiO2@CdS–Ti3C2Tx hybrids. This results in the enhancement of the solar-to-chemical conversion efficiency compared to the traditional TiO2@CdS binary composite.
2. Results and discussion
The schematic illustration for the preparation of TiO2@CdS–Ti3C2Tx composite photocatalysts is shown in Fig. 1a, in which CdS is photodeposited on the surface of TiO2 nanosheets to construct the typical heterojunction of TiO2@CdS. Then Ti3C2Tx MXene is coupled with TiO2@CdS via an electrostatic self-assembly approach. Typically, CdS is photodeposited on ultra-thin 2D TiO2 nanosheets to generate TiO2@CdS (TC) binary heterostructures. By increasing the feedstock of TiO2 while keeping the photodeposition of CdS unchanged, TiO2@CdS composites with different TiO2/CdS ratios (denoted as TC-1, TC-2, and TC-3) have been prepared. The zeta potentials of TC-1, TC-2, and TC-3 are calculated to be 25, 20, and 15 mV, respectively (Fig. S1a†), while Ti3C2Tx is negatively charged with a zeta potential of −41 mV (Fig. S1b†). Therefore, the electrostatic interactions between TC and Ti3C2Tx enable the construction of TiO2@CdS–Ti3C2Tx composites. The zeta potential of TC-MX increases positively with the increase of the Ti3C2Tx amount due to the neutralization of the surface charge (Fig. S1c†). The resulting composites are labelled as TC-0.1MX, TC-0.2MX, and TC-1.0MX, corresponding to TC-MX samples with Ti3C2Tx weight ratios of 0.1%, 0.2%, and 1.0%, respectively. | ||
| Fig. 1 Fabrication of TC-MX ternary heterostructures. (a) Schematic illustration for the preparation of TC-MX ternary heterostructures. The morphologies and textural structures of the samples. TEM images of (b) TiO2, (c) TC-2 and (d) TC-0.1MX; the insets are the corresponding HRTEM images. (e) HAADF-STEM image of TC-0.1MX with the corresponding Ti, C, O, Cd, and S EDX mapping results. | ||
The crystal phase structures of the samples have been investigated by X-ray diffraction (XRD). As shown in Fig. S2,† it is clear that pure TiO2, TC, and TC-MX composites all feature the characteristic diffraction peaks of anatase TiO2 (JCPDS no. 73-1764),44 and the characteristic diffraction peaks of hexagonal CdS (JCPDS no. 80-0006)45 are observed for TC and TC-MX composites, indicating that CdS has been successfully photodeposited on the surface of TiO2. As summarized in Table S1,† the intensity ratio between the TiO2 and CdS diffraction peaks increases with the increase of the TiO2 feedstock, which indicates that the weight ratio of TiO2 increases in the order TC-1 < TC-2 < TC-3. According to the ICP results, the TiO2/CdS mass ratios are calculated to be 2.71, 3.97, and 6.17 for TC-1, TC-2, and TC-3, respectively, following the order TC-1 < TC-2 < TC-3. This is consistent with the XRD results. In the TC-MX composites, the absence of the characteristic diffraction peaks of Ti3C2Tx can be ascribed to the very weak diffraction peak of Ti3C2Tx itself and the relatively low contents of Ti3C2Tx.
The morphologies and textural structures of the samples have been investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 1b and Fig. S3,† bare TiO2 exists as 2D nanosheets with an average width of 50 nm. The lattice spacing in the inset of Fig. 1b is 0.19 nm, corresponding to the (200) crystal plane of anatase TiO2. The SEM images of TC-1, TC-2, and TC-3 are shown in Fig. S4.† It is seen that TC composites still maintain the 2D morphology of TiO2, and the surface of the samples becomes rough due to the coating of CdS. Fig. 1c shows the TEM image of TC-2. It shows that nanoparticles are deposited onto the 2D structured TiO2 nanosheets. These nanoparticles have a lattice spacing of 0.338 nm (the inset of Fig. 1c), corresponding to the (002) facet of hexagonal CdS. In addition, a lattice spacing of 0.19 nm is also observed, which corresponds to the (200) crystal plane of anatase TiO2. The high-angle annular dark-field (HAADF) scanning TEM (STEM) image of TC-2 and the corresponding energy dispersive X-ray (EDX) maps of Ti, O, Cd, and S are presented in Fig. S5.† It can be seen that the mapping of Ti and O shares a similar margin to that of Cd and S, confirming that CdS is generally distributed on the surface of TiO2. These results suggest the successful deposition of CdS onto TiO2. Then Ti3C2Tx is introduced to the binary TiO2@CdS composites. Ti3C2Tx is characteristic of 2D nanosheets with a lateral size of hundreds of nanometers, as shown in Fig. S6.† The 2D structure can be clearly distinguished in the SEM image of TC-0.1MX (Fig. S7†). The TEM image provides further information of the microscopic structure of the sample. As shown in Fig. 1d, a lattice spacing of 1.2 nm is observed, which corresponds to the (002) crystal plane of Ti3C2Tx, indicating that TiO2@CdS composites are successfully loaded onto the surface of the Ti3C2Tx nanosheet. The HAADF STEM image of TC-0.1MX and the corresponding EDX maps of Ti, C, O, Cd, and S are presented in Fig. 1e. It can be seen that Ti shares a similar margin to C, confirming that the substrate is the Ti3C2Tx nanosheet. In addition, Ti exhibits a slightly enhanced concentration in the regions identical to the O element, which further indicates that TiO2 is dispersed on the substrate of Ti3C2Tx. It is also discernable that the signals of Cd and S accumulate in the TiO2 region. These results demonstrate that TiO2@CdS structures interact with the Ti3C2Tx substrate driven by electrostatic forces.
The chemical composition and surface states of the samples have been investigated by X-ray photoelectron spectroscopy (XPS). As shown in the XPS survey spectra of TC-0.1MX (Fig. S8a†), Cd, S, Ti, C, and O are presented, which is in coincidence with the EDX mapping results. As shown in Fig. S8b,† C 1s is calibrated with 284.6 eV. The peak at 281.9 eV is ascribed to the C–Ti bond of Ti3C2Tx, and the peaks at 286.4 and 288.9 eV are ascribed to the C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of the existing defects in the carbon layers of Ti3C2Tx.46 The binding energies at 529.8 and 531.1 eV are assigned to the O 1s peak of TiO247 and the hydroxyl terminations (–OH) in Ti3C2Tx (Fig. S8c†), respectively.48 As shown in Fig. S8d,† two peaks at 464.2 (Ti 2p1/2) and 458.6 eV (Ti 2p3/2) are assigned to Ti(IV) in TiO2,47 while the other two at 462.2 (Ti 2p1/2) and 457.2 eV (Ti 2p3/2) are from the Ti element in Ti3C2Tx,46,48 indicating the successful introduction of Ti3C2Tx. As for TC-2, the binding energies at 404.5 eV and 411.2 eV are assigned to Cd 3d5/2 and Cd 3d3/2, respectively (Fig. 2a). And the binding energies at 160.8 eV and 162.1 eV are assigned to S 2p3/2 and S 2p1/2, respectively47 (Fig. 2b). Noteworthily, the binding energies of Cd and S XPS peaks in TC-0.1MX exhibit a positive shift of 0.2 eV compared to TC-2 (Fig. 2a and b), indicating a decrease of the electron density in CdS. These changes should be ascribed to the electron transfer from CdS to Ti3C2Tx due to the Schottky effect between the semiconductor CdS and metallic Ti3C2Tx.
O bonds of the existing defects in the carbon layers of Ti3C2Tx.46 The binding energies at 529.8 and 531.1 eV are assigned to the O 1s peak of TiO247 and the hydroxyl terminations (–OH) in Ti3C2Tx (Fig. S8c†), respectively.48 As shown in Fig. S8d,† two peaks at 464.2 (Ti 2p1/2) and 458.6 eV (Ti 2p3/2) are assigned to Ti(IV) in TiO2,47 while the other two at 462.2 (Ti 2p1/2) and 457.2 eV (Ti 2p3/2) are from the Ti element in Ti3C2Tx,46,48 indicating the successful introduction of Ti3C2Tx. As for TC-2, the binding energies at 404.5 eV and 411.2 eV are assigned to Cd 3d5/2 and Cd 3d3/2, respectively (Fig. 2a). And the binding energies at 160.8 eV and 162.1 eV are assigned to S 2p3/2 and S 2p1/2, respectively47 (Fig. 2b). Noteworthily, the binding energies of Cd and S XPS peaks in TC-0.1MX exhibit a positive shift of 0.2 eV compared to TC-2 (Fig. 2a and b), indicating a decrease of the electron density in CdS. These changes should be ascribed to the electron transfer from CdS to Ti3C2Tx due to the Schottky effect between the semiconductor CdS and metallic Ti3C2Tx.
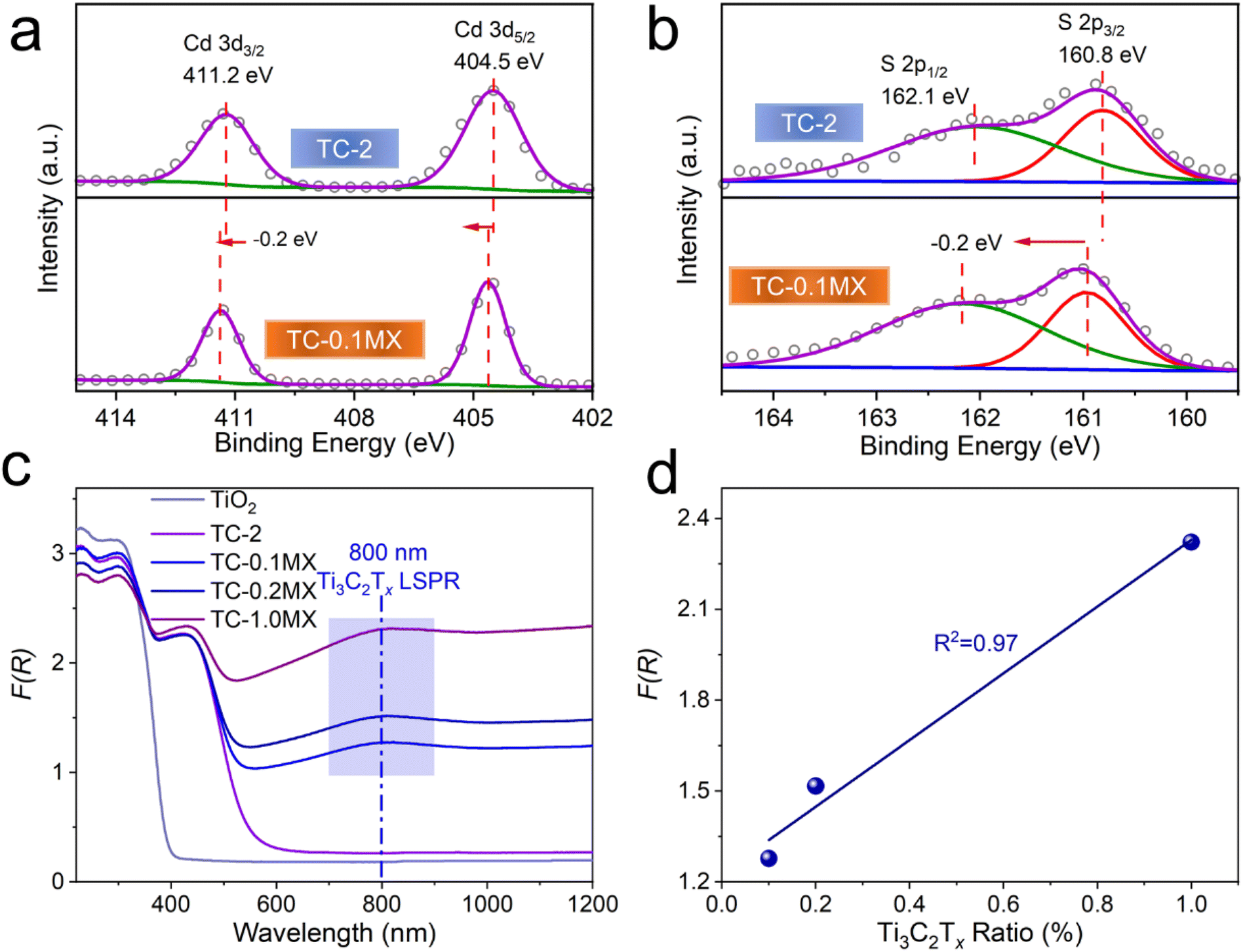 | ||
| Fig. 2 XPS spectra of TC-2 (top) and TC-0.1MX (bottom): (a) Cd 3d, (b) S 2p. (c) UV-vis diffuse reflectance spectra (DRS) of the samples and (d) plots of absorption at a wavelength of 800 nm versus the weight ratio of Ti3C2Tx in the TC-MX composites. | ||
The optical properties of the samples are shown in Fig. 2c. TC-2 exhibits an extended absorption edge to 541 nm compared to TiO2 due to the presence of CdS. Notably, with the addition of Ti3C2Tx, the absorption of the ternary TC-MX composites in the region of 541–1200 nm intensifies compared to TC-2 and increases with the Ti3C2Tx content (Fig. 2c). In particular, it is observed that a distinct absorption peak located at about 800 nm appears in the spectra of TC-MX composites. This peak can be ascribed to the LSPR absorption of Ti3C2Tx.28,30 Compared to the pure Ti3C2Tx aqueous colloids presenting at 780 nm (Fig. S9†), the LSPR peak of Ti3C2Tx in TC-MX composites exhibits a red-shift, which should be ascribed to the high refractive index of the TiO2 matrix since the LSPR absorption band closely relates to the dielectric constant of the environment.49,50 Furthermore, the intensity of the absorption peak located at about 800 nm increases almost linearly as a function of the loading of Ti3C2Tx in the TC-MX samples, as shown in Fig. 2d. Such an optical absorption feature of Ti3C2Tx is distinctively different from its 2D analogue of RGO where only a broad absorption band ranging from the ultraviolet (UV) to the NIR region was observed, as shown in Fig. S10.† In contrast, it is similar to that of traditional metal nanostructures featuring the LSPR effect, such as Au, and Ag.37–39 The XPS and light absorption results suggest that the introduction of Ti3C2Tx affords not only electron interactions between the semiconductors, but also an intriguing LSPR feature. This interesting result has motivated us to evaluate the photocatalytic performances of the samples and investigate the corresponding role of Ti3C2Tx.
The photocatalytic performances of the samples have been evaluated through the anaerobic selective reduction of aromatic nitro compounds to their corresponding amino compounds, based on the formula expressed in Fig. 3a.51,52 For the aqueous selective reduction of 4-nitroaniline (4-NA) to 4-phenylenediamine (4-PDA) with the addition of ammonium formate as a hole scavenger and N2 purging under visible-NIR light irradiation (Fig. S11†), TC-2 exhibits enhanced photoactivity compared to TC-1 and TC-3, as shown in Fig. 3b. According to the Kubelka–Munk function based on the DRS results, the bandgap energy of CdS is calculated to be 2.00 eV (Fig. S12†). The valence band energy (EVB) of CdS determined by the XPS valence band spectrum is 1.03 eV (Fig. S13†). Consequently, the conduction band energy (ECB) is calculated to be −0.97 eV. Considering that the ECB of TiO2 is calculated to be −0.27 eV (Fig. S14†), the photogenerated electron transfer from CdS to TiO2 is thermodynamically permissible. This widely explored band alignment effect improves the separation efficiency of photogenerated electron–hole pairs and thus the photoactivity.53 However, the photoactivity of TC-3 with the further increased TiO2 ratio is lower than that of TC-2. Photocurrent measurements have been conducted to investigate the charge carrier separation of TC composites. As shown in Fig. S15,† the photocurrent densities follow the order TC-2 > TC-1 > TC-3, which agrees well with the photocatalytic activities. These results show that TC-2 with a decent TiO2 ratio has the best effect on the spatial separation and transfer of electron–hole pairs in the binary TC system. The introduction of Ti3C2Tx with appropriate ratios into TC-2 leads to the further improved photoactivity. It can be seen from Fig. 3c that the ternary TC-0.1MX exhibits better photoactivity than TC-2 for the reduction of 4-NA to 4-PDA. Compared to TC-0.1MX, TC-0.2MX and TC-1.0MX with further increased Ti3C2Tx weight ratios exhibit decreased photoactivity. A similar phenomenon has been widely reported previously, which could be ascribed to the “light-shielding effect” of Ti3C2Tx.25 For comparison, TC-2 has also been hybridized with RGO of 0.1 at% and 0.2 at%, which are denoted as TC-0.1RGO and TC-0.2RGO, respectively. The photocatalytic performances of these samples have also been evaluated under the same conduction conditions as those for TC-MX. As shown in Fig. S16,† both TC-0.1MX and TC-0.2MX exhibit enhanced photocatalytic activity compared to the counterparts of TC-0.1RGO and TC-0.2RGO. In addition, control experiments have been performed for the photoreduction of nitroaromatics as shown in Fig. S17.†
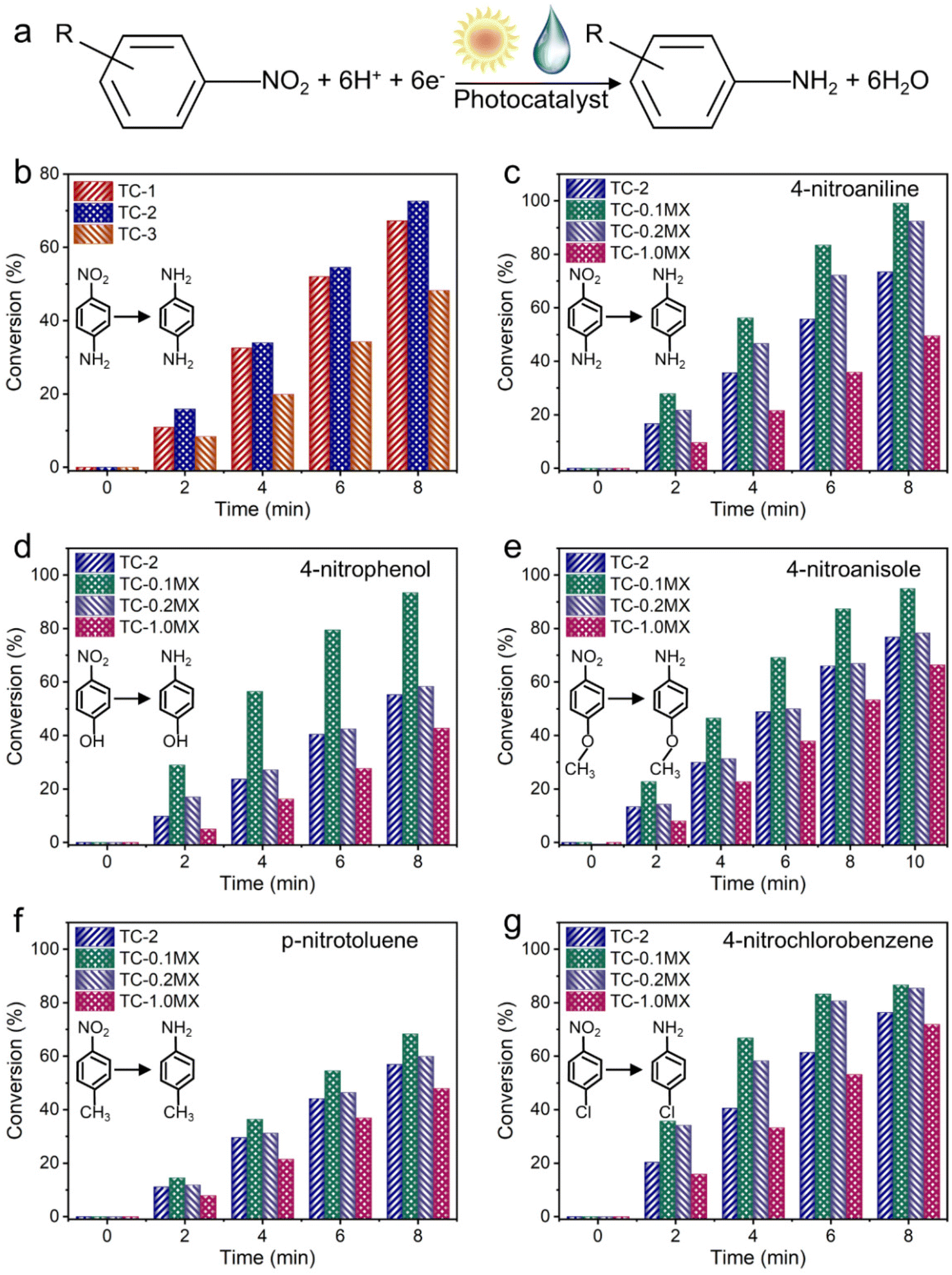 | ||
| Fig. 3 Photocatalytic performances of different samples. (a) The formula for the photocatalytic reduction of nitroaromatics to the corresponding amino compounds. (b) Photocatalytic activities of TC composites for the selective reduction of 4-nitroaniline (4-NA) to 4-phenylenediamine (4-PDA) in water with the addition of ammonium formate as a hole scavenger and N2 purging in water under visible-NIR light irradiation (λ > 420 nm). Photocatalytic activities of TC-2, TC-0.1MX, TC-0.2MX and TC-1.0MX for the selective reduction of (c) 4-nitroaniline, (d) 4-nitrophenol, (e) 4-nitroanisole, (f) p-nitrotoluene, and (g) 4-nitrochlorobenzene. | ||
Specifically, experiments without photocatalysts or light irradiation show no conversion, indicating that the reaction is a photocatalytic process. The photocatalytic reduction of 4-NA over TC-0.1MX decreased dramatically without N2, indicating that the inert atmosphere is indispensable for the photocatalytic reduction of 4-NA.54,55 Furthermore, when employing K2S2O8 as a scavenger for photogenerated electrons, only weak activity has been observed, confirming that the photoreduction reaction of nitroaromatics is decisively driven by electrons.50 In addition, the experiment in the dark excludes the influence of 4-NA adsorption on the photocatalytic performance (Fig. S18†). Fig. S19† displays the resilience of the photocatalytic activity of the TC-0.1MX composite toward the selective reduction of 4-NA under visible-NIR irradiation, which remains relatively stable after three cycles. It can be seen from Fig. S20 and S21† that the morphology and crystal phase structure of TC-0.1MX remained unchanged after three cycles of photocatalytic reaction, indicating that the material has good stability. An identical progressive photoactivity variation has also been found for the selective reduction of a wide range of aromatic nitro compounds, including 4-nitroaniline, 4-nitrophenol, 4-nitroanisole, p-nitrotoluene, and 4-nitrochlorobenzene, as shown in Fig. 3d–g. These confirm the essential role of Ti3C2Tx in enhancing the photocatalytic performance of the composites.
To gain deep insight into the underlying mechanism of Ti3C2Tx enhanced photoactivity, a series of joint electrochemical measurements, optical analysis, and simulations have been carried out. The previous XPS results (Fig. 2a and b) show that there is an obvious Schottky effect between CdS and Ti3C2Tx, which can be understood by the band structure analysis. As discussed above, the conduction band (ECB) of CdS determined by the DRS and the XPS valence band spectrum results is at −0.97 eV. Ultraviolet photoelectron spectra (UPS) have been employed to measure the work function of Ti3C2Tx. As shown in Fig. S22,† the work function of metallic Ti3C2Tx is calculated to be 4.31 eV, indicating that it is feasible for highly electrically conductive Ti3C2Tx (2000 S cm−1) to accept the electrons photogenerated from CdS and construct Schottky heterojunctions, thus inhibiting the recombination of photogenerated charge carriers. In addition, the EF of Ti3C2Tx is calculated to be −0.13 V (versus NHE), which is more positive than the EF of CdS. This suggests that it is feasible for Ti3C2Tx to accept the photogenerated electrons on CdS. Mott–Schottky plots have been measured to ascertain the detail of the energy band of TC-2 and TC-0.1MX composites. As shown in Fig. S23,† the Mott–Schottky plots with positive slopes are observed, which is consistent with the typical feature of n-type semiconductors.56 The flat band potential (EFB) of TC-2 is −0.54 V versus Ag/AgCl (equivalent to −0.34 V versus standard hydrogen electrode, NHE), which shifts positively to −0.37 V versus Ag/AgCl (equivalent to −0.17 V versus NHE) for TC-0.1MX. Considering that the EFB measured by the Mott–Schottky relationship is equal to the Fermi level (EF) for n-type semiconductors,56 the more positive EFB of TC-0.1MX than that of TC-2 should result from the Fermi-level equilibration of the different components, which further confirms the “electron reservoir” effect of Ti3C2Tx due to the Schottky contact.
The LSPR absorption peak of Ti3C2Tx has been identified from the aforementioned DRS results. The interesting light conversion capability by the LSPR effect of Ti3C2Tx has intrigued the exploration of photothermal conversion and photodetectors,32,36 while the application of photocatalysis has not been known clearly. This motivates us to investigate the contribution of the LSPR effect to the enhanced photoactivity of the ternary TC-MX composites compared to the binary TC composites. It has been recognized that the traditional LSPR metals are capable of enhancing photocatalysis by hot electron injection.57 In this regard, while keeping other reaction parameters unchanged, the photocatalytic performance for the reduction of 4-NA has been tested under the irradiation of λ > 780 nm, by which only Ti3C2Tx LSPR is excited. Under such conditions, only 4% of 4-NA is converted over TC-0.1MX (Fig. S24†), which is far less than that over TC-0.1MX (100%) under the visible-NIR light irradiation of λ > 420 nm (Fig. 4a), suggesting that the hot electron injection effect by Ti3C2Tx has a negligible effect on promoting the photocatalytic reduction of 4-NA. Otherwise, when the same weight of pure Ti3C2Tx in TC-0.1MX acts as a direct catalyst, almost no photoactivity is observed for the probe reaction (Fig. 4a), which suggests that the plasmonic Ti3C2Tx does not serve as the direct photocatalyst in our current reaction system. Previous investigation reports that the Ti3C2Tx plasmon-induced thermalization increases the local temperature to 66 °C.28 Although the reaction temperature has been controlled at room temperature by circulating water in our current research, the possible local temperature elevation due to the efficient light-to-heat conversion of plasmonic Ti3C2Tx remains suspicious for enhancing the photocatalysis. In this circumstance, control experiments at elevated temperatures have been performed. Fig. 4b shows that the photocatalytic activity of TC-2 decreases with the increase of reaction temperature, which may be ascribed to the increased probability of photogenerated charge carrier recombination as the temperature increases.58 This result excludes the influence of Ti3C2Tx assisted photothermal catalysis on promoting the photoactivity of TC-0.1MX composites. Considering that the LSPR effect can result in enhanced electric fields, the finite-difference time-domain (FDTD) simulations were further carried out to simulate the electric field (|E|/|E0|) distributions based on the model shown in Fig. S25.† As displayed in Fig. 4c, a significant enhancement of the electric field for TC-0.1MX is observed compared to that of TC-2, which matches well with the FDTD results for bare Ti3C2Tx (Fig. S26†). These results confirm that the electric field enhancement originates from Ti3C2Tx. It is noted that the electric field enhancement is mainly observed on the edge of Ti3C2Tx. The reason could be that the free carriers are confined to the edge of the nanosheet by photon excitation in the direction parallel to Ti3C2Tx.34,37 Furthermore, Fig. 4d shows the Raman spectroscopy of TC-2 and TC-0.1MX. The peaks at 143, 218, 390, 472, 514, and 636 cm−1 correspond to the anatase TiO2 Raman modes of Eg, Eg, B1g, B1g, A1g, and Eg,59 respectively, and the peak at 305 cm−1 belongs to the CdS Raman mode of LO.60 It is noteworthy that the Raman intensity of TC-0.1MX is stronger than that of TC-2. According to the principle of surface-enhanced Raman spectroscopy (SERS), the plasmon-induced local electromagnetic field can significantly enhance the intensity of Raman signals.61 Consequently, compared with TC-2, the enhancement of Raman signals in TC-0.1MX further corroborates the Ti3C2Tx plasmon-induced local electromagnetic field, which can promote the separation of electron–hole pairs in nearby semiconductors, thereby leading to a higher photocatalytic activity. Moreover, the Eg modes of TC-0.1MX have a slight blue shift compared to TC-2, which is ascribed to the carrier density modulation induced by charge transfer between TC-2 and Ti3C2Tx.62–64 Furthermore, the photo-induced charge carrier density over the composites has been estimated by the Mott–Schottky analysis under 1 sun irradiation. The photo-induced charge carrier density (ND) is calculated to be 3.7 × 1019 cm−3 and 7.5 × 1019 cm−3 from the slope of 1/C2vs. V for TC-2 and TC-0.1MX, respectively (Fig. S27†). Thus, the joint experiments indicate that the LSPR enhancement mechanisms should be interpreted in terms of the electric field enhancement effect, which has a significant positive effect on the separation of photogenerated carriers. To the best of our knowledge, this is the first report on the electric field enhancement induced by the LSPR effect of Ti3C2Tx in the field of photocatalysis. Studies have shown that the shape, size, layer thickness and the surface terminal groups of Ti3C2Tx have a great impact on the LSPR absorption band of Ti3C2Tx, and the LSPR light absorption band of Ti3C2Tx can reach about 2400 nm.40 This further unleashes the possibilities of using the LSPR effect to develop efficient Ti3C2Tx-based plasmonic materials to enhance photoredox catalysis.
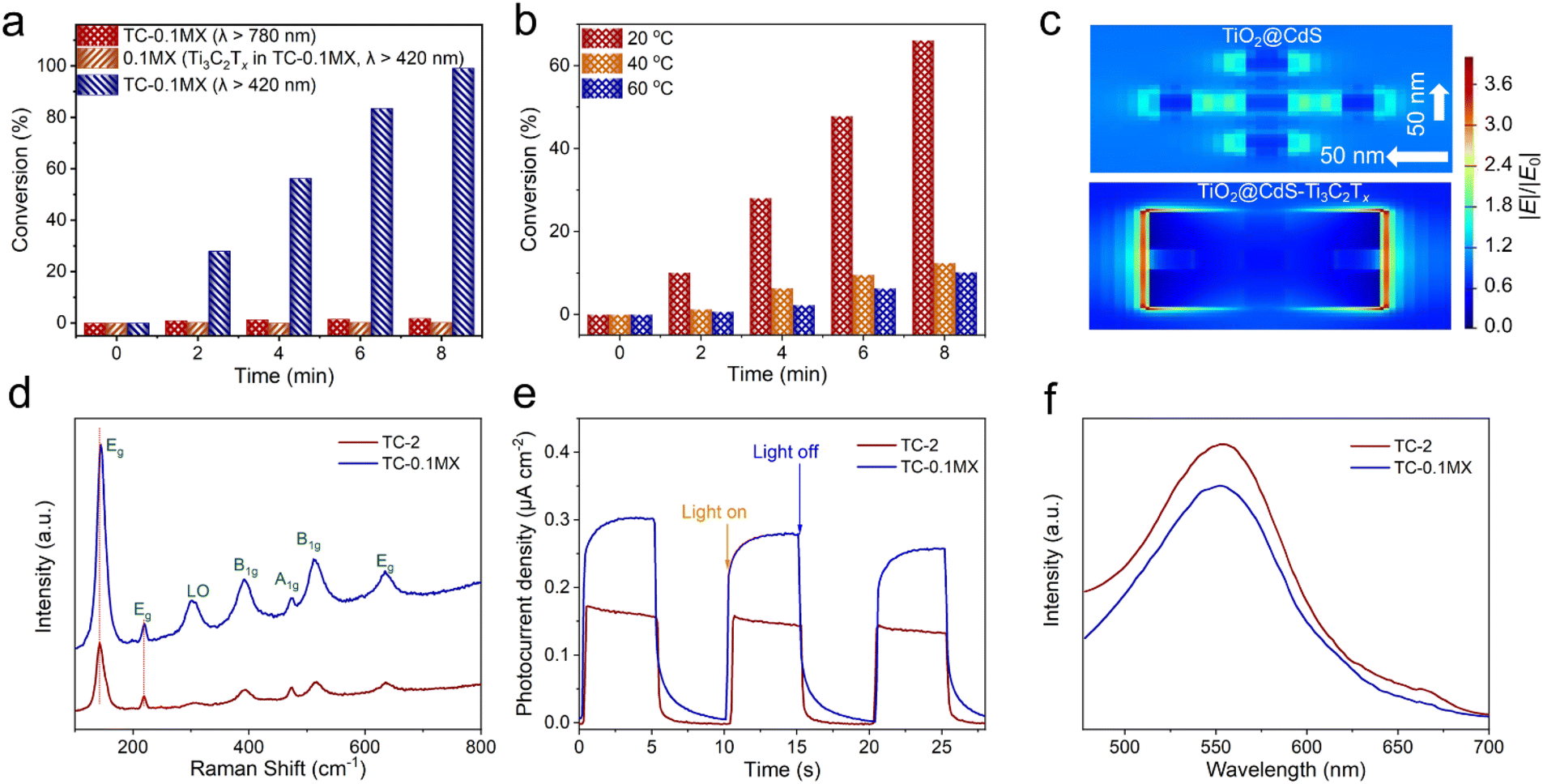 | ||
| Fig. 4 Mechanism analysis of Ti3C2Tx-enhanced photocatalysis. (a) Photocatalytic activities for the selective reduction of 4-NA. (b) Photoactivities of TC-0.1MX for the selective reduction of 4-NA under visible-NIR light irradiation (λ > 420 nm) at different temperatures. (c) Local electric field distributions of TiO2@CdS and TiO2@CdS–Ti3C2Tx composites calculated by the finite-difference time-domain (FDTD) method. (d) Raman spectra of TC-2 and TC-MX composites. (e) Photocurrent densities of TC-2, TC-0.1MX under visible-NIR light irradiation (λ > 420 nm). (f) Photoluminescence (PL) emission spectra of TC-2 and TC-0.1MX. | ||
As shown in Fig. 4e, the photocurrent intensity of TC-0.1MX is higher than that of TC-2, suggesting that the separation of photogenerated charge carriers over TC-0.1MX is more efficient than that over TC-2. This is further verified by the weaker photoluminescence (PL) intensity of TC-0.1MX than that of TC-2 (Fig. 4f), confirming the inhibited recombination of photogenerated charge carriers by Ti3C2Tx. As shown in Fig. S28,† the average fluorescence lifetime increases from 0.53 ns (TC-2) to 0.62 ns (TC-0.1MX), confirming that the introduction of Ti3C2Tx to TC can efficiently accelerate the spatial charge separation and inhibit the charge recombination, and thus prolong the lifetime of photogenerated charge carriers. The open circuit photovoltage (OCP) decay technique has been used to measure the lifetime of photoelectrons under visible-NIR light irradiation (Fig. S29†), from which it is clearly seen that the photoelectron lifetime of TC-0.1MX is longer than that of binary TC-2. In addition, the Nyquist diagrams show that the TC-0.1MX electrode has a smaller semicircle at high frequencies compared to TC-2 (Fig. S30†), suggesting that the introduction of metallic Ti3C2Tx contributes to the enhanced charge transfer process. This is further verified by the cyclic voltammetry (CV) results, where TC-0.1MX exhibits a higher current density than TC-2 (Fig. S31†). Based on these results, the introduction of Ti3C2Tx affords not only the “electron reservoir” effect, but also local electric field enhancement due to the LSPR effect, which cooperatively enhance the separation and transfer of photogenerated charge carriers.
As such, compared to the unidirectional electron transfer controlled by the band-alignment effect in the binary TiO2@CdS, additional transfer pathways of photogenerated electrons in CdS have been established for the ternary TiO2@CdS–Ti3C2Tx. The multichannel electron transfer pathway in TiO2@CdS–Ti3C2Tx is consolidated by in situ irradiated X-ray photoelectron spectroscopy (ISI-XPS). As shown in Fig. 5a, for the experiment without light irradiation, two XPS peaks located at 404.8 and 411.5 eV are observed, which are assigned to Cd 3d5/2 and Cd 3d3/2, respectively.47 Under light irradiation, the Cd 3d XPS peaks exhibit a positive shift of 0.4 eV. Analogously, a positive shift of the S 2p peaks has also been observed after the visible-NIR light irradiation (Fig. 5b), suggesting the electron depletion on CdS. For the Ti 2p XPS spectrum without light irradiation, two peaks at 464.5 (Ti 2p1/2) and 458.8 eV (Ti 2p3/2) are assigned to Ti(IV) in TiO2, while the other two at 462.9 (Ti 2p1/2) and 457.5 eV (Ti 2p3/2) are from the Ti element in Ti3C2Tx![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48 (Fig. 5c). Meanwhile, two peaks at 531.5 and 530.0 eV are attributed to the hydroxyl terminations (–OH) in Ti3C2Tx and the lattice O in TiO2,48 as shown in Fig. 5d. Under visible-NIR light irradiation, there is an apparent negative shift of the binding energies of Ti 2p XPS peaks, i.e., −0.1 eV for TiO2 and −0.3 eV for Ti3C2Tx. Similar decreased binding energies are observed for the O 1s XPS peaks upon visible-NIR light irradiation, indicating the accumulation of electrons on both TiO2 and Ti3C2Tx under visible-NIR light irradiation. These shifts of binding energies provide direct evidence of the photogenerated electron transfer pathways across the TC-0.1MX interface under the same visible-NIR light irradiation conditions as the photocatalytic reaction. Specifically, the photogenerated electrons from the irradiated CdS can migrate to both TiO2 and Ti3C2Tx, thus affording multichannel electron transfer pathways for the separation of photogenerated electron–hole pairs toward enhancing the photoactivity. The electron density difference is further calculated to study the charge transfer process in the TC-0.1MX interface, as shown in Fig. 5e and Fig. S32.† The green and yellow regions depict electron accumulation and depletion regions, respectively. The charge accumulation primarily occurs at the interfaces between CdS and Ti3C2Tx, and between CdS and TiO2. The obvious electron transfer process can also be observed (isovalue = 0.03) in Fig. 5e. According to the Hirshfeld charge analysis,65 1.67 e− can transfer from CdS to Ti3C2Tx, and 0.13 e− can transfer from CdS to TiO2, respectively. It is anticipated that under light irradiation, the photoinduced electrons will accumulate in Ti3C2Tx and TiO2. This further confirms the multichannel electron transfer pathway in the hole–electron charge separation process and is consistent with the ISI-XPS characterization.
48 (Fig. 5c). Meanwhile, two peaks at 531.5 and 530.0 eV are attributed to the hydroxyl terminations (–OH) in Ti3C2Tx and the lattice O in TiO2,48 as shown in Fig. 5d. Under visible-NIR light irradiation, there is an apparent negative shift of the binding energies of Ti 2p XPS peaks, i.e., −0.1 eV for TiO2 and −0.3 eV for Ti3C2Tx. Similar decreased binding energies are observed for the O 1s XPS peaks upon visible-NIR light irradiation, indicating the accumulation of electrons on both TiO2 and Ti3C2Tx under visible-NIR light irradiation. These shifts of binding energies provide direct evidence of the photogenerated electron transfer pathways across the TC-0.1MX interface under the same visible-NIR light irradiation conditions as the photocatalytic reaction. Specifically, the photogenerated electrons from the irradiated CdS can migrate to both TiO2 and Ti3C2Tx, thus affording multichannel electron transfer pathways for the separation of photogenerated electron–hole pairs toward enhancing the photoactivity. The electron density difference is further calculated to study the charge transfer process in the TC-0.1MX interface, as shown in Fig. 5e and Fig. S32.† The green and yellow regions depict electron accumulation and depletion regions, respectively. The charge accumulation primarily occurs at the interfaces between CdS and Ti3C2Tx, and between CdS and TiO2. The obvious electron transfer process can also be observed (isovalue = 0.03) in Fig. 5e. According to the Hirshfeld charge analysis,65 1.67 e− can transfer from CdS to Ti3C2Tx, and 0.13 e− can transfer from CdS to TiO2, respectively. It is anticipated that under light irradiation, the photoinduced electrons will accumulate in Ti3C2Tx and TiO2. This further confirms the multichannel electron transfer pathway in the hole–electron charge separation process and is consistent with the ISI-XPS characterization.
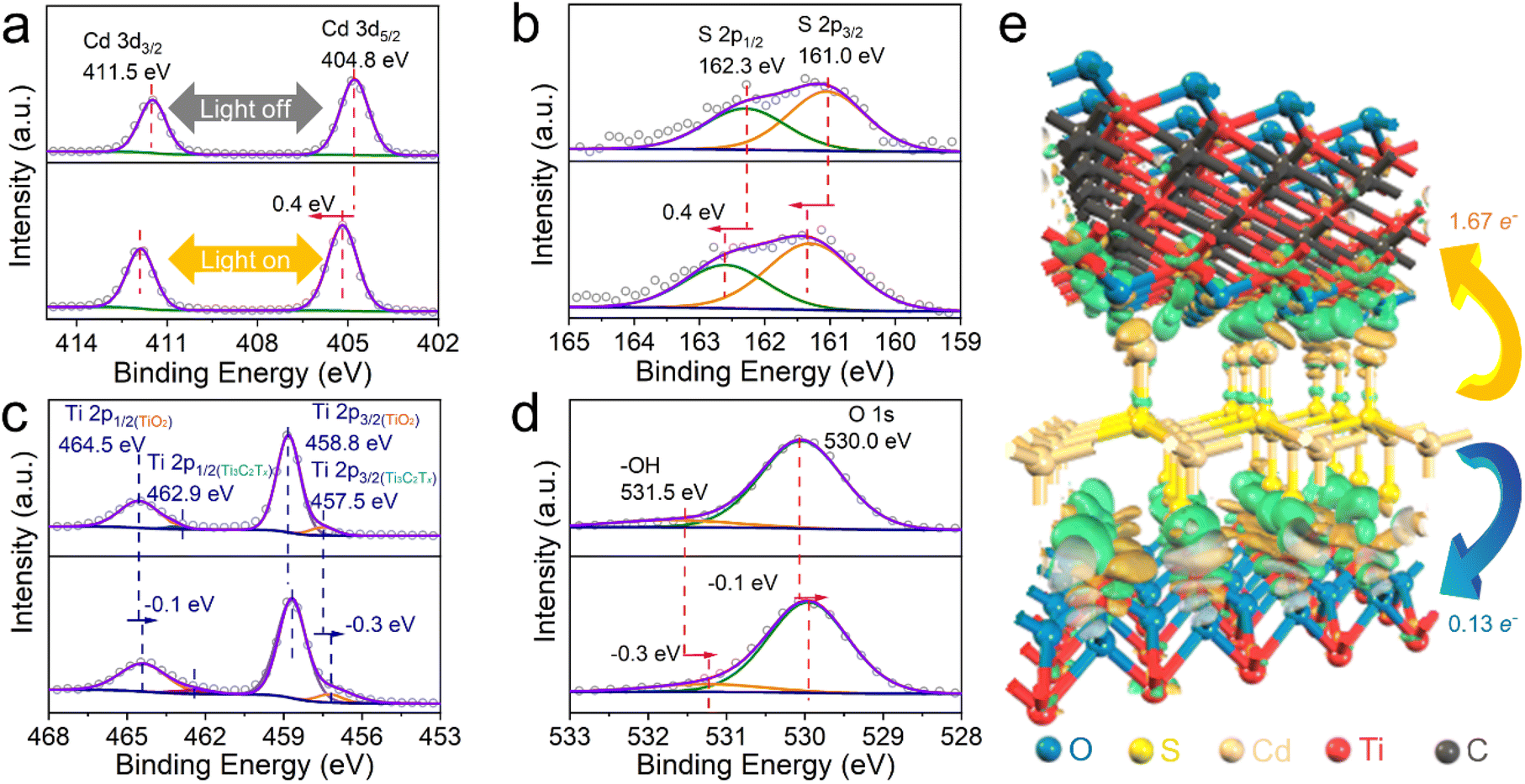 | ||
| Fig. 5 ISI-XPS characterization and DFT calculations of TC-0.1MX. (a) Cd 3d, (b) S 2p, (c) Ti 2p, and (d) O 1s XPS spectra in the dark (top) and under visible-NIR light irradiation of λ > 420 nm (bottom). (e) The electron density difference of TC-MX. Forest green and yellow regions indicate accumulation and depletion of electrons, respectively. | ||
Based on the above analysis, a mechanism of multichannel charge transfer enhanced photoactivity for the reduction of nitroaromatics over the ternary TiO2@CdS–Ti3C2Tx is proposed, as illustrated in Fig. 6. Under visible-NIR light irradiation (λ > 420 nm), photoinduced electron–hole pairs are generated in CdS, which follow band alignment-mediated charge transfer as the electrons in the CB of CdS are transferred to the CB of TiO2. Meanwhile, Ti3C2Tx acts as the “electron reservoir” due to the Schottky junction between CdS and Ti3C2Tx, which further captures and transfers the excited electrons in the CB of CdS, resulting in an efficient spatial separation of photoinduced charge carriers. Besides, the LSPR effect of Ti3C2Tx enables local electric field enhancement to further boost the separation of charge carriers in CdS. As a result, the photogenerated electrons produced by CdS are efficiently extracted and the photocatalytic activity toward the reduction of nitroaromatics is enhanced.
 | ||
| Fig. 6 Illustration of the proposed reaction mechanism for the photocatalytic reduction of nitroaromatics over TC-0.1MX under visible-NIR light irradiation (λ > 420 nm) with the addition of ammonium formate (AF) as a hole scavenger and N2 purging in water. | ||
3. Conclusions
In summary, taking TiO2@CdS as a typical heterostructured photocatalyst, we have demonstrated Ti3C2Tx as a dual-functional cocatalyst for steering photogenerated electron flow for enhanced photoredox catalysis. Our investigations clearly reveal that Ti3C2Tx with a high work function behaves as an “electron reservoir” due to the Schottky-junction effect, which is beneficial for suppressing the recombination of photogenerated electron–hole pairs. Of particular interest is that Ti3C2Tx exhibits the typical LSPR effect, which induces local electric field enhancement to boost the separation efficiency of electron–hole pairs. With these advantages integrated, the introduction of Ti3C2Tx affords multiple electron transfer pathways in the TiO2@CdS–Ti3C2Tx composite photocatalysts. Our findings shed light on the knowledge of the versatility of Ti3C2Tx in enhancing photoredox catalysis, making it much more viable for constructing multicomponent photocatalysts with multichannel charge transfer pathways for the activation and reduction of appealing small molecules, such as CO2 and N2.4. Experimental methods
4.1. Materials
All used chemicals were obtained from commercial suppliers without further purification. Sublimed sulfur (S8), cadmium chloride (CdCl2·2.5H2O), methanol (CH4O), ethanol (C2H6O), hydrochloric acid (HCl), ammonium formate (AF), and N,N-dimethylformamide (DMF) were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Butyl titanate (TBT), hydrogen fluoride (HF) and lithium fluoride (LiF) were obtained from Aladdin Industrial Corporation. Titanium aluminium carbide 312 (Ti3AlC2) powder was obtained from FoShan XinXi Technology Co., Ltd. Deionized (DI) water used throughout the whole experiments was obtained from ultrapure water production apparatus. Fluorine-doped tin oxide (FTO)-coated glass substrates were purchased from Wuhan Jinge-Solar Energy Technology Co., Ltd.4.2. Catalyst preparation
4.3. Material characterization
For zeta potential measurements, 1 mg samples were added to 1 mL DI water, and sonicated to disperse evenly. The above solution was dropped into the Malvern zeta potential sample cell and zeta potential measurements of the samples were conducted on a Zetasizer Nano ZSP. The X-ray diffraction patterns of the samples were measured on a MiniFlex 600 X-ray diffractometer using Ni-filtered Cu Kα radiation at a scan rate of 5° min−1. The mass proportions of Ti and S in TC samples were measured by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) (Agilent, ICP-OES 730). Raman spectra were recorded on a laser confocal inverted microscope Raman spectrometer (inVia-reflex) with a 633 nm laser as an excitation source. Scanning electron microscopy (SEM, S4800, Hitachi S-4800) was performed to determine the morphology of the samples. TEM images, high-resolution TEM (HRTEM) images, and elemental mapping results were obtained on a field emission transmission electron microscope with spherical aberration correction of the objective lens (Titan G2 60-300) at an acceleration voltage of 300 kV. X-ray photoelectron spectroscopy (XPS) measurements were performed using an AXIS SUPRA, and ultraviolet photoelectron spectroscopy (UPS) measurements were performed with He I (21.22 eV) as a monochromatic light source. In situ irradiated X-ray photoelectron spectroscopy (ISI-XPS) was conducted on a Thermo Fisher Escalab 250xi with an Al Kα radiation hemispherical electron energy analyzer and a 300 W Xe arc lamp (Microsolar300A, Beijing Perfect Light Co., Ltd) with a 420 nm cut-off filter being placed ca. 20 cm away from the prepared samples during the ISI-XPS characterization to investigate the electron density changes of the samples under visible-NIR light irradiation (λ > 420 nm). All binding energies were calibrated with the C 1s peak at 284.6 eV. The electrical conductivity of the MXene films was measured using a four-point probe setup (4 PROBES TECH) with a uniform 1.0 mm probe spacing. A Shimadzu UV-vis near-infrared spectrometer was used to measure the optical properties of the samples. The photoluminescence (PL) spectra for samples were analyzed on an Edinburgh FLS9800 with an excitation wavelength of 375 nm.4.4. Electrochemical measurements
The electrochemical analysis was carried out in a conventional three electrode cell, which uses an Ag/AgCl electrode as the reference electrode and a Pt plate as the counter electrode on an electrochemical workstation (Shanghai Chenhua, CHI600E). Fluorine-doped tin oxide (FTO) glasses were used as the substrate for the working electrode, which were cleaned by sonication in absolute ethanol for 30 min and dried at 60 °C. The conductive surface of the FTO glass was covered with cellophane with a 0.5 mm hole near the end. 5 mg of sample was ultrasonically dispersed in 0.5 mL of DMF, and then 20 μl of the above dispersions was dropped on the pretreated FTO glass. After drying at room temperature, the cellophane was removed and the uncoated area of the FTO glass was protected with nail polish glue. The photocurrent measurement was performed under visible-NIR light irradiation (>420 nm), and the open circuit photovoltage decay (OCP) measurements were conducted under visible-NIR light irradiation for 60 s and then the subsequent decay of photovoltage was monitored for 50 s with the light turned off, and the Mott–Schottky (M–S) experiments were obtained in 0.2 M Na2SO4 aqueous solution at 300 Hz. The electrochemical impedance spectroscopy (EIS) experiments were performed in 0.5 M KCl containing 0.01 M K3[Fe(CN)6]/K4[Fe(CN)6] in a frequency range from 1 Hz to 100 kHz under open-circuit potential conditions. Cyclic voltammograms (CV) were collected in the same electrolyte at a scan rate of 0.09 mV s−1.4.5. FDTD simulations and theoretical calculations
The electric field simulation was performed using the Finite Differential Time Domain (FDTD) software package (Lumpolishing Solutions, Inc.). The refractive indices of Ti3C2Tx, TiO2, and CdS were obtained from previously reported data.66 The total field/scattered field (TFSF) light source was used as the incident field in the simulation area. The wavelength of the incident field was consistent with that of the experimental radiation source. The perfect matching layer (PML) boundary conditions were used in the simulation. The length of Ti3C2Tx in the long direction is 340 nm and the width is 205 nm. TiO2@CdS is a square with a length of 55 nm and a thickness of 3.81 nm. For the model of TiO2@CdS–Ti3C2Tx, the middle position of each of the four edges and the center of the Ti3C2Tx sheet is loaded with TiO2@CdS.The density functional theory (DFT) calculations are carried out using the CASTEP code2 for this work.65 The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional is used to describe the electronic exchange and correlation effects,67 and the plane-wave cutoff is tested and set to 571.4 eV. The self-consistent field (SCF) tolerance is 1 × 10–6 eV. The core electrons are treated with ultrasoft pseudo-potentials.
4.6. Photoactivity measurements
40 mg of catalyst and 40 mg of ammonium formate (AF) were mixed with 20 mL of aromatic nitro compounds (50 ppm) in a glass reactor. Before irradiation, the mixture was stirred for 30 min and bubbled with N2 for another 30 min in the dark to ensure the adsorption–desorption equilibrium between the sample and the reactant. In a typical test, the glass reactor was irradiated with a 300 W Xe arc lamp (PLS-XSE300d) with a 420 nm cut-off filter and the distance between the lamp holder and the photocatalytic reactor was 0.5 cm. 2 mL of the sample solution was collected at regular time intervals, centrifuged (8000 rpm) and the supernatant was analyzed using a UV-vis spectrophotometer (Shimadzu, UV-1780).The photocatalytic performance of the aromatic nitro compounds was calculated using the conversion equation as follows:
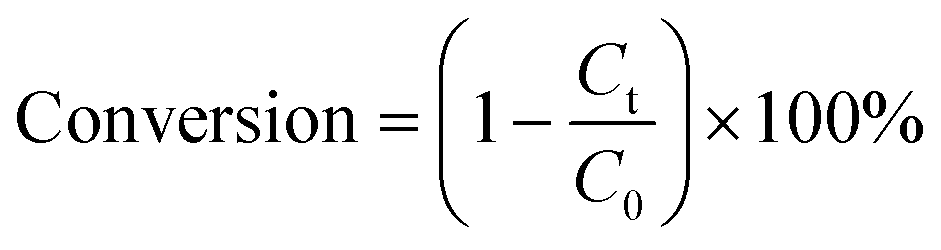
Author contributions
N. Zhang and X. Xie conceived the research. G. Xie, X. Wang and J. Wang prepared photocatalysts and conducted all the experiments. C. Han performed the FDTD calculations. F. Song, Y. Zhu and Z. Wu offered help to analyse the characterization results. G. Xie, X. Xie and N. Zhang wrote and revised the manuscript. All authors provided critical feedback and assisted during manuscript preparation.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52272295, 52071137, 51977071, 51802040, and 21802020), the Science and Technology Innovation Program of Hunan Province (2021RC3066 and 2021RC3067) and the Natural Science Foundation of Hunan Province (2020JJ3004 and 2020JJ4192). N. Zhang and X. Xie also acknowledge the financial support of the Fundamental Research Funds for the Central Universities.References
- S. Li, J. Xiong, X. Zhu, W. Li, R. Chen and G. Cheng, J. Mater. Sci. Technol., 2022, 101, 242–263 CrossRef.
- Z. Q. Wei, S. Hou, X. Lin, S. Xu, X. C. Dai, Y. H. Li, J. Y. Li, F. X. Xiao and Y. J. Xu, J. Am. Chem. Soc., 2020, 142, 21899–21912 CrossRef CAS PubMed.
- K. L. J. Prather, Nat. Catal., 2020, 3, 181–183 CrossRef.
- W. Lei, T. Zhou, X. Pang, S. Xue and Q. Xu, J. Mater. Sci. Technol., 2022, 114, 143–164 CrossRef.
- C. Wan, J. Jin, X. Wei, S. Chen, Y. Zhang, T. Zhu and H. Qu, J. Mater. Sci. Technol., 2022, 124, 102–108 CrossRef.
- Z. Jiang, B. Cheng, Y. Zhang, S. Wageh, A. A. Al-Ghamdi, J. Yu and L. Wang, J. Mater. Sci. Technol., 2022, 124, 193–201 CrossRef.
- Q. Xu, S. Wageh, A. A. Al-Ghamdi and X. Li, J. Mater. Sci. Technol., 2022, 124, 171–173 CrossRef.
- Z. Jin, J. Li, Y. Zhang, D. Liu, H. Ding, B. B. Mamba, A. T. Kuvarega and J. Gui, J. Mater. Sci. Technol., 2022, 125, 38–50 CrossRef.
- X. Ren, C. Wei, Y. Sun, X. Liu, F. Meng, X. Meng, S. Sun, S. Xi, Y. Du, Z. Bi, G. Shang, A. C. Fisher, L. Gu and Z. J. Xu, Adv. Mater., 2020, 32, 1–13 Search PubMed.
- Y. Cui, X. Tan, K. Xiao, S. Zhao, N. M. Bedford, Y. Liu, Z. Wang, K. H. Wu, J. Pan, W. H. Saputera, S. Cheong, R. D. Tilley, S. C. Smith, J. Yun, L. Dai, R. Amal and D. W. Wang, ACS Energy Lett., 2020, 5, 3560–3568 CrossRef CAS.
- W. Xi, K. Wang, Y. Shen, M. Ge, Z. Deng, Y. Zhao, Q. Cao, Y. Ding, G. Hu and J. Luo, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- X. Sun, S. R. Dawson, T. E. Parmentier, G. Malta, T. E. Davies, Q. He, L. Lu, D. J. Morgan, N. Carthey, P. Johnston, S. A. Kondrat, S. J. Freakley, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2020, 12, 560–567 CrossRef CAS PubMed.
- C. Chen, Y. Zhu, M. Tian, Y. Chen, Y. Yang, K. Jiang and S. Gao, Nano Energy, 2021, 81, 105623 CrossRef CAS.
- J. Song, S. Liu, C. Yang, G. Wang, H. Tian, Z. Zhao, R. Mu and J. Gong, Appl. Catal., B, 2020, 263, 118367 CrossRef CAS.
- J. Lu, J. Wang, Q. Zou, D. He, L. Zhang, Z. Xu, S. He and Y. Luo, ACS Catal., 2019, 9, 2177–2195 CrossRef CAS.
- J. Zhang, N. Kong, S. Uzun, A. Levitt, S. Seyedin, P. A. Lynch, S. Qin, M. Han, W. Yang, J. Liu, X. Wang, Y. Gogotsi and J. M. Razal, Adv. Mater., 2020, 32, 2001093 CrossRef CAS PubMed.
- C. E. Shuck, A. Sarycheva, M. Anayee, A. Levitt, Y. Zhu, S. Uzun, V. Balitskiy, V. Zahorodna, O. Gogotsi and Y. Gogotsi, Adv. Energy Mater., 2020, 22, 1901241 CAS.
- Y. Li, X. Chen, Y. Sun, X. Meng, Y. Dall’Agnese, G. Chen, C. Dall’Agnese, H. Ren, S. Sasaki, H. Tamiaki and X. F. Wang, Adv. Mater. Interfaces, 2020, 7, 1902080 CrossRef CAS.
- T. S. Mathis, K. Maleski, A. Goad, A. Sarycheva, M. Anayee, A. C. Foucher, K. Hantanasirisakul, C. E. Shuck, E. A. Stach and Y. Gogotsi, ACS Nano, 2021, 15, 6420–6429 CrossRef CAS PubMed.
- X. Han, L. An, Y. Hu, Y. Li, C. Hou, H. Wang and Q. Zhang, Appl. Catal., B, 2020, 265, 118539 CrossRef CAS.
- R. Xiao, C. Zhao, Z. Zou, Z. Chen, L. Tian, H. Xu, H. Tang, Q. Liu, Z. Lin and X. Yang, Appl. Catal., B, 2020, 268, 118382 CrossRef CAS.
- F. He, B. Zhu, B. Cheng, J. Yu, W. Ho and W. Macyk, Appl. Catal., B, 2020, 272, 119006 CrossRef CAS.
- X. Xie and N. Zhang, Adv. Funct. Mater., 2020, 30, 2002528 CrossRef CAS.
- J. Ran, G. Gao, F. T. Li, T. Y. Ma, A. Du and S. Z. Qiao, Nat. Commun., 2017, 8, 13907 CrossRef CAS PubMed.
- Y. Yang, Z. Zeng, G. Zeng, D. Huang, R. Xiao, C. Zhang, C. Zhou, W. Xiong, W. Wang, M. Cheng, W. Xue, H. Guo, X. Tang and D. He, Appl. Catal., B, 2019, 258, 117956 CrossRef CAS.
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- J. Xuan, Z. Wang, Y. Chen, D. Liang, L. Cheng, X. Yang, Z. Liu, R. Ma, T. Sasaki and F. Geng, Angew. Chem., 2016, 128, 14789–14794 CrossRef.
- X. Wu, J. Wang, Z. Wang, F. Sun, Y. Liu, K. Wu, X. Meng and J. Qiu, Angew. Chem., Int. Ed., 2021, 60, 9416–9420 CrossRef CAS PubMed.
- H. Xu, A. Ren, J. Wu and Z. Wang, Adv. Funct. Mater., 2020, 30, 2000907 CrossRef CAS.
- J. Xuan, Z. Wang, Y. Chen, D. Liang, L. Cheng, X. Yang, Z. Liu, R. Ma, T. Sasaki and F. Geng, Angew. Chem., 2016, 128, 14789–14794 CrossRef.
- A. Miranda, J. Halim, M. W. Barsoum and A. Lorke, Appl. Phys. Lett., 2016, 108, 33102 CrossRef.
- R. Li, L. Zhang, L. Shi and P. Wang, ACS Nano, 2017, 11, 3752–3759 CrossRef CAS PubMed.
- X. Fan, Y. Ding, Y. Liu, J. Liang and Y. Chen, ACS Nano, 2019, 13, 8124–8134 CrossRef CAS PubMed.
- K. Chaudhuri, M. Alhabeb, Z. Wang, V. M. Shalaev, Y. Gogotsi and A. Boltasseva, ACS Photonics, 2018, 5, 1115–1122 CrossRef CAS.
- H. Lin, X. Wang, L. Yu, Y. Chen and J. Shi, Nano Lett., 2017, 17, 384–391 CrossRef CAS PubMed.
- D. B. Velusamy, J. K. El Demellawi, A. M. El Zohry, A. Giugni, S. Lopatin, M. N. Hedhili, A. E. Mansour, E. Di Fabrizio, O. F. Mohammed and H. N. Alshareef, Adv. Mater., 2019, 31, 1807658 CrossRef PubMed.
- N. Zhang, C. Han, X. Fu and Y. J. Xu, Chem, 2018, 4, 1832–1861 CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- S. Peng, J. M. McMahon, G. C. Schatz, S. K. Gray and Y. Sun, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14530–14534 CrossRef CAS PubMed.
- J. K. El Demellawi, S. Lopatin, J. Yin, O. F. Mohammed and H. N. Alshareef, ACS Nano, 2018, 12, 8485–8493 CrossRef CAS PubMed.
- D. Magne, V. Mauchamp, S. Célérier, P. Chartier and T. Cabioc'h, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 201409 CrossRef.
- V. Mauchamp, M. Bugnet, E. P. Bellido, G. A. Botton, P. Moreau, D. Magne, M. Naguib, T. Cabioc'h and M. W. Barsoum, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 235428 CrossRef.
- X. Wang, C. Garnero, G. Rochard, D. Magne, S. Morisset, S. Hurand, P. Chartier, J. Rousseau, T. Cabioc'h, C. Coutanceau, V. Mauchamp and S. Célérier, J. Mater. Chem. A, 2017, 5, 22012–22023 RSC.
- X. Yu, X. Jin, X. Chen, A. Wang, J. Zhang, J. Zhang, Z. Zhao, M. Gao, L. Razzari and H. Liu, ACS Nano, 2020, 14, 13876–13885 CrossRef CAS PubMed.
- Q. Ruan, X. Ma, Y. Li, J. Wu, Z. Wang, Y. Geng, W. Wang, H. Lin and L. Wang, Nanoscale, 2020, 12, 20522–20535 RSC.
- W. Yuan, L. Cheng, Y. An, S. Lv, H. Wu, X. Fan, Y. Zhang, X. Guo and J. Tang, Adv. Sci., 2018, 5, 1700870 CrossRef PubMed.
- J. Low, B. Dai, T. Tong, C. Jiang and J. Yu, Adv. Mater., 2019, 31, 1807920 CrossRef PubMed.
- Y. Li, X. Deng, J. Tian, Z. Liang and H. Cui, Appl. Mater. Today, 2018, 13, 217–227 CrossRef.
- Z. Zhang, A. Li, S. W. Cao, M. Bosman, S. Li and C. Xue, Nanoscale, 2014, 6, 5217–5222 RSC.
- L. Yuan, B. Weng, J. C. Colmenares, Y. Sun and Y.-J. Xu, Small, 2017, 13, 1702253 CrossRef PubMed.
- N. Zhang, S. Xie, B. Weng and Y. J. Xu, J. Mater. Chem. A, 2016, 4, 18804–18814 RSC.
- C. Yu, X. Xie and N. Zhang, Catal. Sci. Technol., 2021, 11, 425–443 RSC.
- Z. Jiang, K. Qian, C. Zhu, H. Sun, W. Wan, J. Xie, H. Li, P. K. Wong and S. Yuan, Appl. Catal., B, 2017, 210, 194–204 CrossRef CAS.
- V. N. Rao, N. L. Reddy, M. M. Kumari, P. Ravi, M. Sathish, K. M. Kuruvilla, V. Preethi, K. R. Reddy, N. P. Shetti, T. M. Aminabhavi and M. V. Shankar, Appl. Catal., B, 2019, 254, 174–185 CrossRef.
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194 CrossRef PubMed.
- J. Y. Li, Y. H. Li, F. Zhang, Z. R. Tang and Y. J. Xu, Appl. Catal., B, 2020, 269, 118783 CrossRef CAS.
- Y. C. Chen, Y. K. Hsu, R. Popescu, D. Gerthsen, Y. G. Lin and C. Feldmann, Nat. Commun., 2018, 9, 232 CrossRef PubMed.
- R. R. Ishiki, H. M. Ishiki and K. Takashima, Chemosphere, 2005, 58, 1461–1469 CrossRef PubMed.
- M. C. Ceballos-Chuc, C. M. Ramos-Castillo, J. J. Alvarado Gil, G. Oskam and G. Rodríguez-Gattorno, J. Phys. Chem. C, 2018, 122, 19921–19930 CrossRef CAS.
- M. L. Lin, M. Miscuglio, A. Polovitsyn, Y. C. Leng, B. Martín-García, I. Moreels, P. H. Tan and R. Krahne, J. Phys. Chem. Lett., 2019, 10, 399–405 CrossRef CAS PubMed.
- G. Yu, J. Qian, P. Zhang, B. Zhang, W. Zhang, W. Yan and G. Liu, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- E. Cortés, P. G. Etchegoin, E. C. Le-Ru, A. Fainstein, M. E. Vela and R. C. Salvarezza, J. Am. Chem. Soc., 2013, 135, 2809–2815 CrossRef PubMed.
- W. X. Wang, S. H. Liang, T. Yu, D. H. Li, Y. B. Li and X. F. Han, J. Appl. Phys., 2011, 109, 07C501 CrossRef.
- Z. Mao, W. Song, X. Xue, W. Ji, L. Chen, J. R. Lombardi and B. Zhao, J. Phys. Chem. C, 2012, 116, 26908–26918 CrossRef CAS.
- Z. Li, Y. Yan, S. Xu, H. Zhou, M. Xu, L. Ma, M. Shao, X. Kong, B. Wang, L. Zheng and H. Duan, Nat. Commun., 2022, 13, 147 CrossRef CAS PubMed.
- A. Miranda, J. Halim, A. Lorke and M. W. Barsoum, Mater. Res. Lett., 2017, 5, 322–328 CrossRef CAS.
- C. Zhan, Y. Xu, L. Bu, H. Zhu, Y. Feng, T. Yang, Y. Zhang, Z. Yang, B. Huang, Q. Shao and X. Huang, Nat. Commun., 2021, 12, 6261 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05983e |
| This journal is © The Royal Society of Chemistry 2022 |