
Understanding Cu(II)-based systems for C(sp3)–H bond functionalization: insights into the synthesis of aza-heterocycles†‡
Marc
Camats
a,
Isabelle
Favier
a,
Sonia
Mallet-Ladeira
b,
Daniel
Pla
 *a and
Montserrat
Gómez
*a
*a and
Montserrat
Gómez
*a
aLaboratoire Hétérochimie Fondamentale et Appliquée, CNRS UMR 5069, Université Toulouse 3 – Paul Sabatier, 118 route de Narbonne, 31062 Toulouse Cedex 09, France. E-mail: pla@lhfa.fr; gomez@chimie.ups-tlse.fr
bInstitut de Chimie de Toulouse, CNRS UAR 2599, 118 Route de Narbonne, Toulouse 31062 Cedex 09, France
First published on 26th November 2021
Abstract
Herein we report the synthesis of imidazo[1,5-a]pyridine heterocycles via a Cu(II)-mediated functionalization of α′-C(sp3)–H bonds of pyridinylaldimines and subsequent cyclization. This strategy exploits the inherent directing ability of heteroleptic aldimine and pyridine groups in the substrate yielding the C–H functionalization of α′-methylene groups in a regioselective fashion over distant methyl or methylene groups in β or γ positions. The observed correlation between the nature of the anionic ligands (halide vs. carboxylate) bonded to copper and the chemoselectivity of the C(sp3)–H activation process points to a concerted metalation–deprotonation pathway prior to cyclization to furnish the corresponding imidazo[1,5-a]pyridine derivative. This copper-mediated C(sp3)–H bond functionalization reaction works for a variety of substrates incorporating linear alkyl chains (from 3 to 12 carbon atoms), and good functional group tolerance (aryl, ether and ester groups). Cu-Catalyzed C(sp2)–H cyanation on the imidazole ring can then take place selectively under oxidative conditions.
Introduction
In contrast to 4d and 5d transition metal catalysts (e.g. Pd, Pt, Ru, Rh),1 first row transition metals have received less attention in direct functionalization methodologies of unactivated C–H bonds.2 Among them, copper occupies a preferential place due to its abundance, low toxicity and versatility in terms of reactivity as evidenced by the many reports on C(sp2)–H activation reactions,2g,3 in particular those using bidentate ligands.4 Since Fujiwara's early reports on the C(sp3)–H aminomethylation of alkanes catalyzed by Cu(II) salts,5 the advent of direct functionalization methodologies for the selective transformation of unactivated C(sp3)–H bonds has enabled novel synthetic routes by means of C–C cross-dehydrogenative reactions,6 carbonylative7 and decarboxylative couplings,6e,8 aminations,3b,9 amidations,10 alkoxylations,11 acetoxylations12 and phosphorothiolations,13 among the most relevant transformations.3a,6f,14The use of bidentate ligands to direct functionalization of C(sp3)–H bonds in a regioselective fashion has afforded promising results for the functionalization of such recalcitrant bonds.3a,4a,c,15 From a mechanistic point of view, organometallic C–H bond activation, ligand-assisted functionalization and single electron-transfer pathways have been postulated in the process (Fig. 1).16 In particular, the C(sp3)–H activation in α position to a nitrogen atom has been described to undergo via the formation of an iminium group arising from a radical cation. This reactivity has thus far been applied in Cu-catalyzed annulation of N-heteroaryl aldehydes17 or ketones9c,18 with alkylamines and aminoacids via C(sp3)–H amination. The affinity of heterocyclic aldimines for electrophilic Cu(II)-based catalysts represents a straightforward and atom economical manner to accomplish remote C(sp3)–H bond activation. Thus, we envisioned that the imidazo[1,5-a]pyridine scaffold could be formed via C(sp3)–H bond functionalization of aldimines bearing pendant saturated hydrocarbon chains. In this work, we report the direct synthesis of 3-substituted imidazo[1,5-a]pyridines via a Cu-mediated C(sp3)–H activation/cyclization in excellent yield and regioselectivity, thus providing novel methods which are complementary to established strategies.
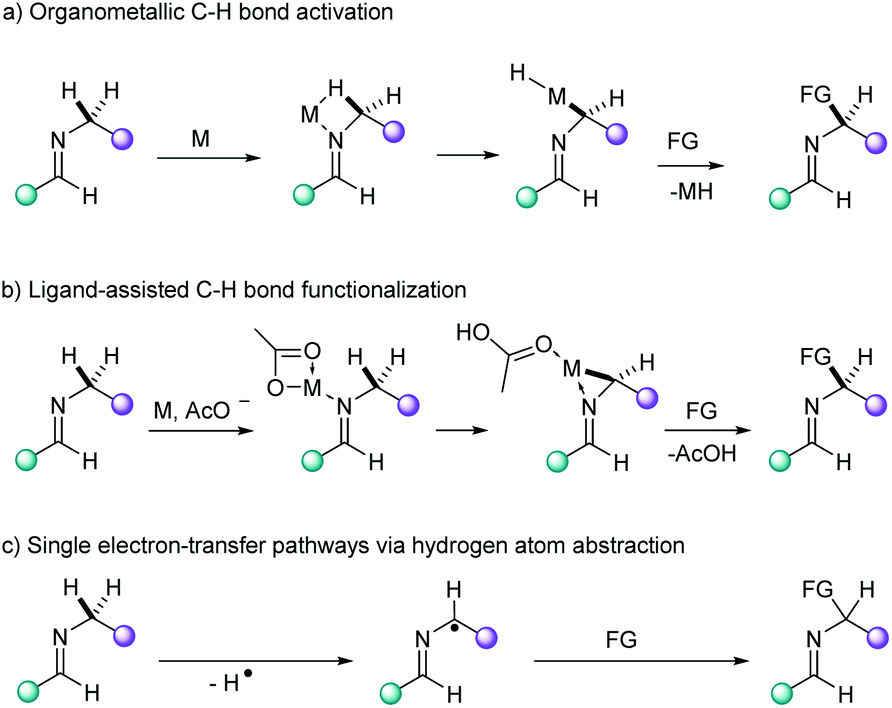 | ||
| Fig. 1 C(sp3)–H bond activation pathways of aldimines. | ||
Diversity-oriented synthesis19 strategies to explore the chemical space around privileged leads such as purine,20 imidazo[1,2-a]pyridine,3f pyrazolo[1,5-a]pyrimidine,21 pyrazolo[1,5-a]pyridine22 and pyrrolo[2,1-f][1,2,4]triazine,23 found in numerous natural products and drug scaffolds (Fig. 2) prompted us to explore novel synthetic methodologies for the synthesis of isosteric scaffolds.24 In particular, imidazopyridine derivatives are garnering increased attention in recent years as tryptophan-2,3-dioxygenase and indoleamine 2,3-dioxygenase inhibitors.25
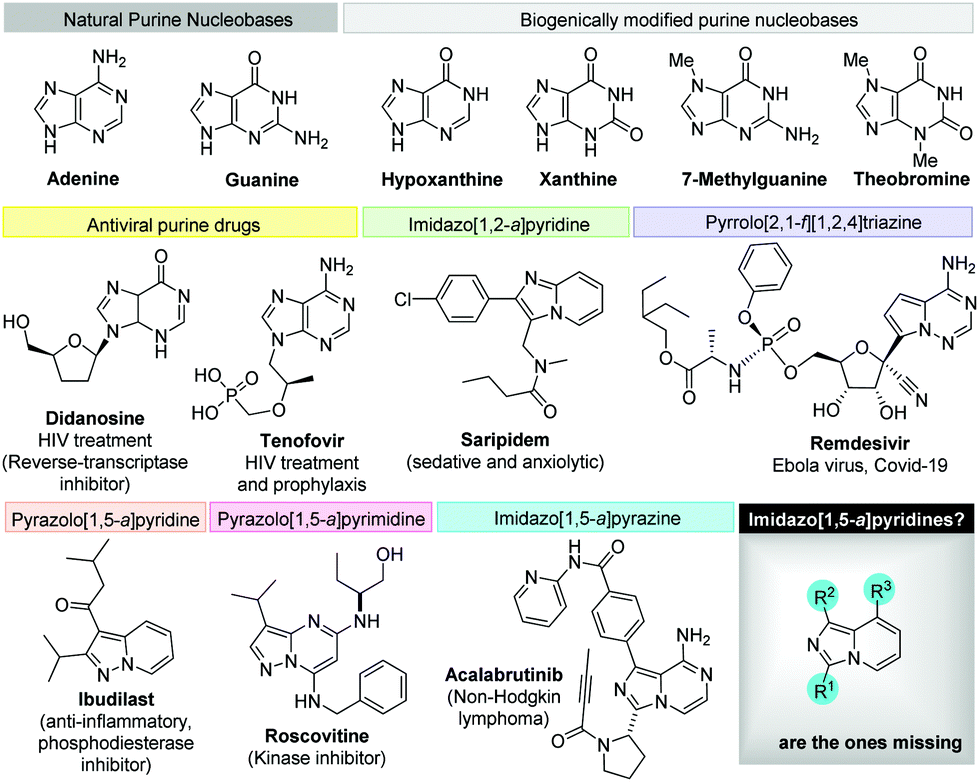 | ||
| Fig. 2 Purine nucleobases and drugs of interest featuring structurally related heterocyclic scaffolds. | ||
Thus, medicinal chemistry approaches to access variations on such fundamental building blocks in a rapid and efficient manner could provide important breakthroughs in translational science. In this regard, the functionalization of recalcitrant C–H bonds represents a powerful and sustainable transformation in organic synthesis, affording new entries to such valuable motifs of potential biological interest.26 In this work, we focus on the use of Cu-promoted C–H bond functionalization strategies towards the preparation of new compounds with untapped potential.
Results and discussion
We prepared original aldimines by condensation of 3-fluoro-2-pyridinecarboxaldehyde and the corresponding primary amine in 2-methyltetrahydrofuran and in the presence of sodium sulfate, affording the expected products 1a–1k in high yields (>89%, Scheme 1), together with an ease of analysis by 19F-NMR.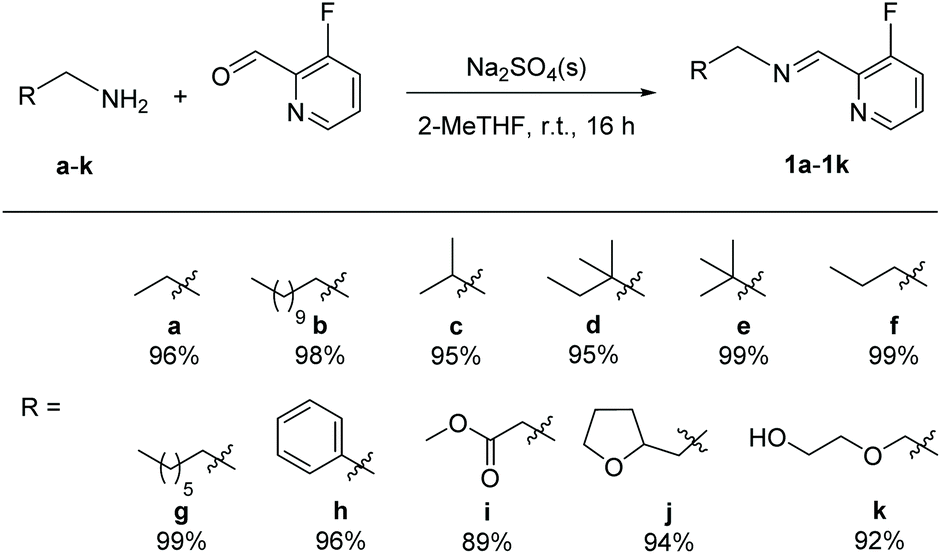 | ||
| Scheme 1 Synthesis of aldimines 1a–1k (isolated yields in brackets). | ||
Our attempts towards the functionalization of pyridinylaldimine substrate 1a began with the use of superstoichiometric amounts of CuBr2 (1.5 equiv.) in CHCl3 or CH3CN at 100 °C under microwave irradiation, resulting in the formation of Cu(II) coordination complexes (no C–H bond activation was observed). Thus, the bimetallic CuA was isolated and further crystallized by reaction of CuBr2 with 1a in CH2Cl2 at room temperature. Similar results were obtained with 1b and 1c (Scheme 2).
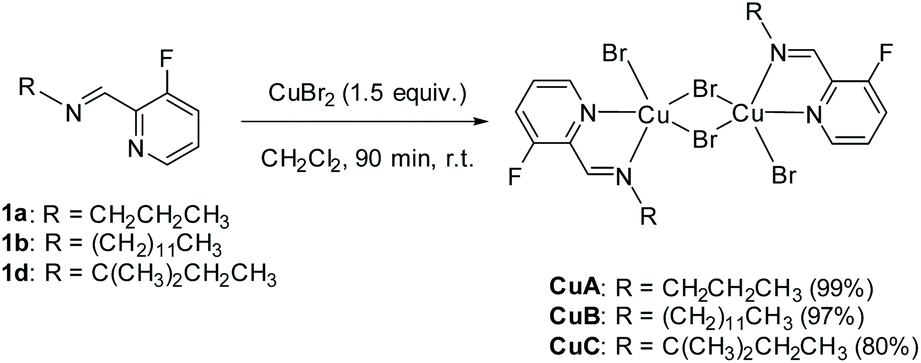 | ||
| Scheme 2 Synthesis of complexes CuA, CuB and CuC from aldimines 1a, 1b and 1d, respectively (isolated yields in brackets). | ||
CuA X-ray crystal structure shows the formation of a centrosymmetric bimetallic complex with two bromide anions bridging both metals (Fig. 3), in agreement with other related structures containing N,N-bidentated ligands: pyridyl-benzimidazole,27 pyridyl-oxime,28 and pyridyl-pyrazole.29 The asymmetric unit consists of a five-membered metallocycle featuring a bidentate N,N-coordination of the 1-(3-fluoropyridin-2-yl)-N-propylmethanimine ligand (bite angle N1Cu1N2 = 80.11(8)°) and two bromide anions in a slightly distorted square planar arrangement around the metal center (the distance between the N1N2Br1Br2 plane and Cu is 0.21 Å). Each Cu(II) center is pentacoordinated exhibiting a distorted square pyramidal arrangement constituted of a N1N2Br1Br2 plane and an apical bromide anion (Cu–Br(apical) distance = 2.781(1) Å); no metal–metal interaction is observed (Cu–Cu distance = 3.601(1) Å) (Fig. 3 and Fig. S1 in the ESI‡).
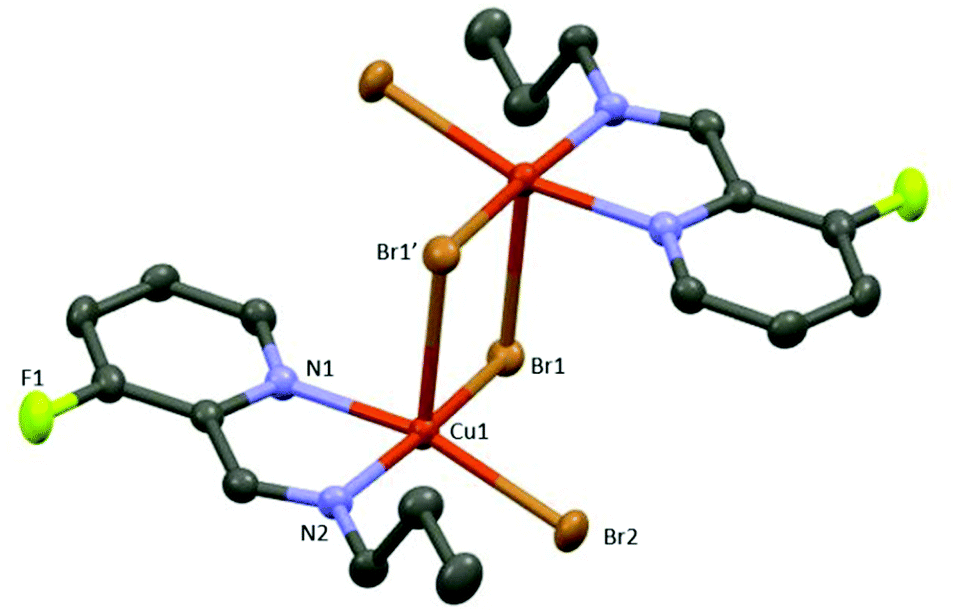 | ||
| Fig. 3 Molecular view of CuA. Hydrogen atoms have been omitted for clarity purposes (ellipsoids representing 50% probability). Selected distances (Å) and angles (°) [symmetry code (i): 1 − x, 2 − y, 1 − z]: Cu1–Br1′ = 2.781(1), Cu1–Br1 = 2.425(1), Cu1–Br2 = 2.386(1), Cu1–N1 = 2.065(2), Cu1–N2 = 2.039(2); N1–Cu1–N2 = 80.11(8); Br1–Cu1–Br2 = 91.24(1); N1–Cu1–Br1 = 92.33(6); N2–Cu1–Br2 = 94.48(6). | ||
Reactivity studies of CuA in the absence of base were carried out in CHCl3 and CH3CN, but no conversion was detected, pointing that this complex is not involved in the C–H bond activation, probably due to the formation of unsaturated reactive copper species (see Table S1 in the ESI‡). When Ac2O was used as solvent, only by-products coming from imine hydrolysis were observed. Even though the use of a solvent mixture of AcOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CHCl3 (1:4) rendered a very low conversion (5%), a new 3-alkyl-8-fluoroimidazo[1,5-a]pyridine product (2a) was identified albeit in low yield. Finding the right balance in terms of stability and reactivity of the Cu-based intermediates seems crucial to enable a selective transformation overriding hydrolysis pathways. To facilitate the C(sp3)–H amination, NH4OAc (1.5 equiv.) was used to perform the transformation under buffered conditions in a solvent mixture of AcOH:CHCl3 (1:4), but only 8-fluoro-3-(3-fluoropyridin-2-yl)imidazo[1,5-a]pyridine (3) and (2Z)-3-(3-fluoropyridin-2-yl)-2-methylprop-2-enenitrile (4; see the ESI‡ for single-crystal XRD structure) by-products were obtained (Table 1, entries 1 and 2) arising from deleterious hydrolytic pathways. The formation of 3 can be justified by imine transimination with ammonia followed by condensation with a second aldehyde in the absence of copper(II) species as described in the literature (Table 1, entry 2).30 This secondary reaction was also favored in glycerol (69% conversion, Table 1, entry 3). Concerning the formation of 4, a rational is proposed in the mechanistic discussion (see below). Transformation of the dimer CuA into the heterocycle 2a occurred in CH3CN and in the presence of NH4OAc, albeit in low yield (23%, Table 1, entry 4), whereas only traces of the product could be detected in CHCl3 (4% Table 1, entry 5).
:CHCl3 (1:4) rendered a very low conversion (5%), a new 3-alkyl-8-fluoroimidazo[1,5-a]pyridine product (2a) was identified albeit in low yield. Finding the right balance in terms of stability and reactivity of the Cu-based intermediates seems crucial to enable a selective transformation overriding hydrolysis pathways. To facilitate the C(sp3)–H amination, NH4OAc (1.5 equiv.) was used to perform the transformation under buffered conditions in a solvent mixture of AcOH:CHCl3 (1:4), but only 8-fluoro-3-(3-fluoropyridin-2-yl)imidazo[1,5-a]pyridine (3) and (2Z)-3-(3-fluoropyridin-2-yl)-2-methylprop-2-enenitrile (4; see the ESI‡ for single-crystal XRD structure) by-products were obtained (Table 1, entries 1 and 2) arising from deleterious hydrolytic pathways. The formation of 3 can be justified by imine transimination with ammonia followed by condensation with a second aldehyde in the absence of copper(II) species as described in the literature (Table 1, entry 2).30 This secondary reaction was also favored in glycerol (69% conversion, Table 1, entry 3). Concerning the formation of 4, a rational is proposed in the mechanistic discussion (see below). Transformation of the dimer CuA into the heterocycle 2a occurred in CH3CN and in the presence of NH4OAc, albeit in low yield (23%, Table 1, entry 4), whereas only traces of the product could be detected in CHCl3 (4% Table 1, entry 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Base | Conversionb (%) | Yieldb (%) | ||
| 2a | 3 | 4 | ||||
| a Reaction conditions: CuA (0.15 mmol, 117.0 mg), 0.45 mmol of base, 4 mL of the corresponding solvent, at 100 °C for 10 min under microwave irradiation (max. 200 W). b Conversions and yields were determined by 19F NMR with 4-fluorotoluene as standard. c 1a was used as starting material instead of CuA. d Complex mixture of degradation by-products. e Complex mixture composed of acetals and by-products. f n.d. for not detected. | ||||||
| 1 | AcOH:CHCl3 (1:4) |
NH4OAc | 76 | <1 | 49 | 15 |
| 2c | AcOH:CHCl3 (1:4) |
NH4OAc | 90d | 0 | 34 | 0 |
| 3 | Glycerol | NH4OAc | 69e | 5 | 54 | 4 |
| 4 | CH3CN | NH4OAc | 32 | 23 | 0 | 0 |
| 5 | CHCl3 | NH4OAc | 6 | 4 | 0 | 0 |
| 6 | CH3CN | NBu4OAc | 72 | 64 | n.d.f | n.d.f |
| 7 | CH3CN | KOPiv | 87 | 84 | n.d.f | n.d.f |
In CH3CN, other bases were tested (Table 1, entries 6–7 and Table S2 in the ESI‡). Notably, NBu4OAc furnished up to 72% conversion and good selectivity towards 2a (64% yield, Table 1, entry 6) and KOPiv, 87% conversion (84% yield, Table 1, entry 7). CsOAc or KOAc showed low conversions (8 and 26%, respectively, Table S2 in the ESI‡) albeit in excellent selectivity for the latter. Other salts were tested as bases, namely, KOH and KOtBu; KOH did not trigger any transformation and more basic KOtBu directed a nucleophilic attack to the imine moieties of the binuclear complex (see Table S2 in the ESI‡). These results point to a plausible anion exchange on the complex (bromide by carboxylate; encompassing an instantaneous color change of the reaction mixture from deep red to green) that enables the C–H bond functionalization.
Once the reactivity of the binuclear copper complex was optimized, we studied the in situ reaction of imines type-1 with CuBr2 (1.5 equiv.) in the presence of NBu4OAc (1.5 equiv.). Gratifyingly, under these conditions, the C(sp3)–H amination and cyclization furnished the desired imidazo[1,5-a]pyridine product (2a) in comparable yield and selectivity to the reaction carried out from the preformed complex CuA (52% vs. 64% yield, Table S3,‡ entry 1 vs.Table 1, entry 6, respectively).31
This result prompted us to examine the use of Cu(OAc)2 towards the C–H functionalization of the aldimine 1a (reaction conditions: 100 °C under microwave irradiation for 10 min in different solvents: EtOH, glycerol, AcOH, CF3COOH, CHCl3, EtOAc, CH3CN; see Table S4 in the ESI‡). Full decomposition was observed with superstoichiometric amounts of Cu(OAc)2 in both CF3COOH and EtOAc as solvents; moreover, major substrate hydrolysis to propylamine and 6-fluoropyridine-2-carboxaldehyde precursors was obtained for many of the tested conditions using protic solvents. When CH3CN and CHCl3 were used as solvents, 2a was obtained in low to moderate yields (48 and 17%, respectively, see Table S4 in the ESI,‡ entries 6 and 7). Interestingly, a cubane tetranuclear copper(II) complex (Cu4B) with the composition [Cu4OCl6(2b)4] was obtained by crystallization of the crude reaction mixture in CHCl3 (Fig. 4). Its single-crystal X-ray analysis showed a central oxygen atom coordinated to four copper atoms in a tetrahedral geometry (Cu–O–Cu angles are 110.57(5)° and 108.92(2)°), each of which connected to the three other Cu atoms via bridging chlorine atoms (placed at the tetrahedron edges), analogously to related structures already reported in the literature.32 Each copper center shows a distorted trigonal bipyramid geometry with chlorine atoms occupying the three equatorial positions and the central oxygen atom and imine nitrogen of the heterocycle 2b in axial positions (Fig. 4).
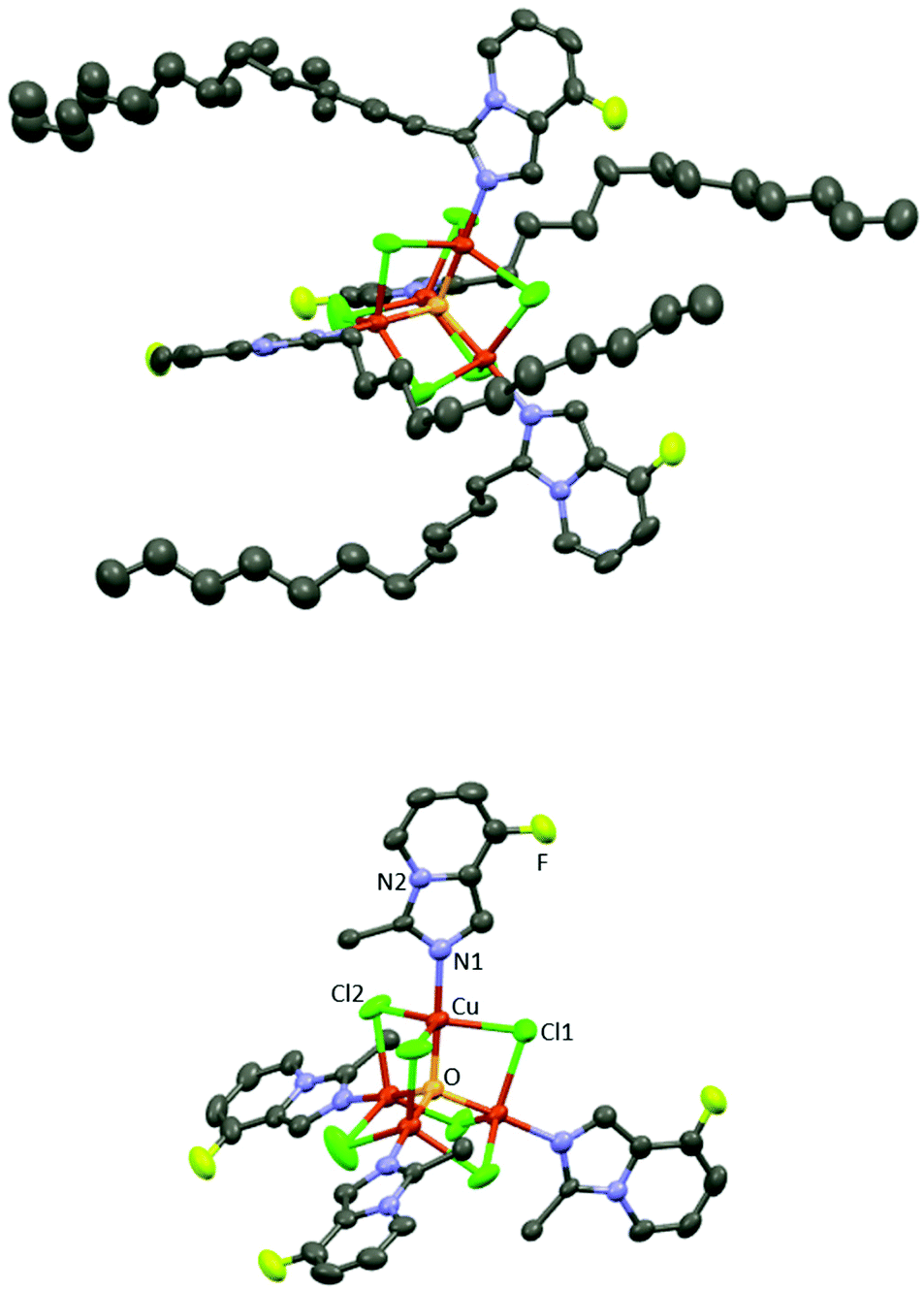 | ||
| Fig. 4 Top: Molecular view of a cubane tetranuclear copper(II) complex Cu4B with the composition [Cu4OCl6(2b)4] (ellipsoids representing 50% probability) where hydrogen atoms have been omitted for clarity purposes and only the major position of the disordered alkyl chain is shown (for atom labelling see bottom view). Bottom: Cu4B molecular view where hydrogen and carbon atoms of the alkyl chains have been omitted for clarity purposes. Selected distances (Å) and angles (°): Cu–N1 = 1.967(6); Cu–Cl = 2.374(2), 2.407(3) and 2.424(2); Cu–O = 1.909(1); Cl–Cu–Cl = 114.04(6), 119.85(4) and 123.60(6); Cl–Cu–N1 = 93.26(16), 94.50(15) and 97.99(18); Cl–Cu–O 84.03(6), 84.36(5) and 85.75(5); N1–Cu–O = 177.36(17). | ||
The isolation of this copper cluster encouraged us to assess its role in the reaction. Thus, its plausible precursor [Cu4OCl6(CH3CN)4] (5) was prepared.33 However, the reaction of 5 with aldimine 1b, aiming at the in situ generation of Cu4Bvia the displacement of the CH3CN ligands by 1b, did not furnish any traces of the desired product 2b under our reaction conditions, suggesting that the copper cluster 5 is inactive towards C(sp3)–H activation (see Scheme S1 in the ESI‡).
With the aim of obtaining Cu(II) carboxylate complexes, we carried out a number of attempts to prepare the corresponding 1a and 1b aldimine complexes with Cu(OPiv)2 (due to its better reactivity; see below); unfortunately, no suitable monocrystals for XRD analyses were obtained. However, MS analyses revealed the complexation of 1a affording bis-pivalate [Cu(OPiv)2(1a)] species ([M + H], m/z 432.1), together with the presence of the mono-pivalate peak, [Cu(OPiv)(1a)]+ (m/z 330.1; see Fig. S2 in the ESI‡).
To assess the determinant role of copper carboxylates in facilitating the C–H amination, several copper salts (namely formate, acetate, benzoate and pivalate) were tested (see Table S3 in the ESI,‡ entries 3–5). Despite the poor results obtained for both copper formate and copper benzoate (2a obtained in 55 and 14% yields, respectively), these results point to the key assistance of carboxylate groups in the C–H activation event. Among the copper(II) carboxylates used, Cu(OPiv)2 exhibited the best conversion and selectivity (95% conversion and 83% yield; Scheme 3 and see Table S3 in the ESI,‡ entry 5).
 | ||
| Scheme 3 Optimized reaction conditions for the synthesis of imidazo[1,5-a]pyridines type-2 with Cu(OPiv)2. | ||
In order to translate the stoichiometric reaction into its catalytic version, several oxidizing reagents were studied under microwave irradiation (10–20 mol% catalyst loadings of Cu(OPiv)2 in CH3CN at 100 °C; see Table 2 and Table S5 in the ESI‡). Among the oxidizing agents tested, only stoichiometric amounts (related to copper salt) of the desired product were obtained in the presence of pivalic anhydride (1.5 equiv.), benzoquinone (1.5 equiv.) and a mixture of K3[Fe(CN)6] (1.5 equiv.) and dibenzo-18-crown-6 (for further details, see Table S5 in the ESI‡). It is noteworthy to mention that a new 8-fluoroimidazo[1,5-a]pyridine-1-carbonitrile product (6a) was obtained when 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) was used (Table 2, entry 1; see the ESI‡ for single-crystal XRD of 6a). Taking into account literature reports on the role of CH3CN as a cyanating reagent with copper catalysts,34 we reasoned that the nitrile group could come from the solvent. However, this hypothesis was discarded as the reaction in CpOMe did also provide 6a together with by-products (Table 2, entry 2), whereas the reaction in the absence of DDQ did not provide any traces of 6a (Table 2, entry 3). These results indicate that DDQ acts as cyanating agent, in agreement with copper-catalyzed cyanation of arylboronic acids described by Cheng and co-workers.35 Optimization of the reaction was carried out (Table 2, entries 4–6), finding the following optimal conditions: Cu(OPiv)2 (20 mol%), DDQ (1 equiv.) in CH3CN at 100 °C for 10 min under microwave irradiation (Table 2, entry 4). This new copper-mediated C–H cyanation reaction brings a new entry to non-hazardous cyanide sources.36
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst loading (mol%) | DDQ (equiv.) | Solvent | Conversiona (%) |
6aa (%) |
| a Conversions and yields were determined by 19F NMR with 4-fluorotoluene as standard. b Complex mixture of 6-fluoropyridine-2-carboxaldehyde and by-products. c 2a was obtained in the absence of DDQ. d Two non-identified by-products were obtained (in 15% each), m/z 218 and 230. | |||||
| 1 | 20 | 1.5 | CH3CN | 100 | 87 |
| 2 | 20 | 1.5 | CpOMe | 100 | 25b |
| 3 | 20 | 0 | CH3CN | 18 | 0c |
| 4 | 20 | 1 | CH3CN | 100 | 89 |
| 5 | 10 | 1 | CH3CN | 100 | 68d |
| 6 | 0 | 1 | CH3CN | 100 | 0b |
With the optimized reaction conditions both in stoichiometric and catalytic versions in hand, we next investigated the substrate scope. A variety of aldimines gave the corresponding 3-substituted 8-fluoroimidazo[1,5-a]pyridines and 8-fluoroimidazo[1,5-a]pyridine-1-carbonitrile derivatives in good to excellent yields (Scheme 4). Substrates 1a and 1b including linear alkyl chains (Cn, n = 3, 12; Scheme 4) showed excellent regioselectivity for the geminal C(sp3)–H functionalization α′ to the aldimine, providing 2a–b and 6a–b in good yields. To test the feasibility of remote functionalization in β′ or γ′ positions, α′-branched substrates (isopropyl 1c, neopentyl 1d and tert-butyl 1e) were tested without success. Gratifyingly, when an aldimine substrate bearing a benzyl group was employed, the desired heterocycles arylated in position 3 were obtained (2h in 52%, and 6h 92% yield) in high regioselectivity, precluding C(sp2)–H activation on the aryl ring. The ester group is a well-tolerated functionality and gives the desired product 6i (55% yield).
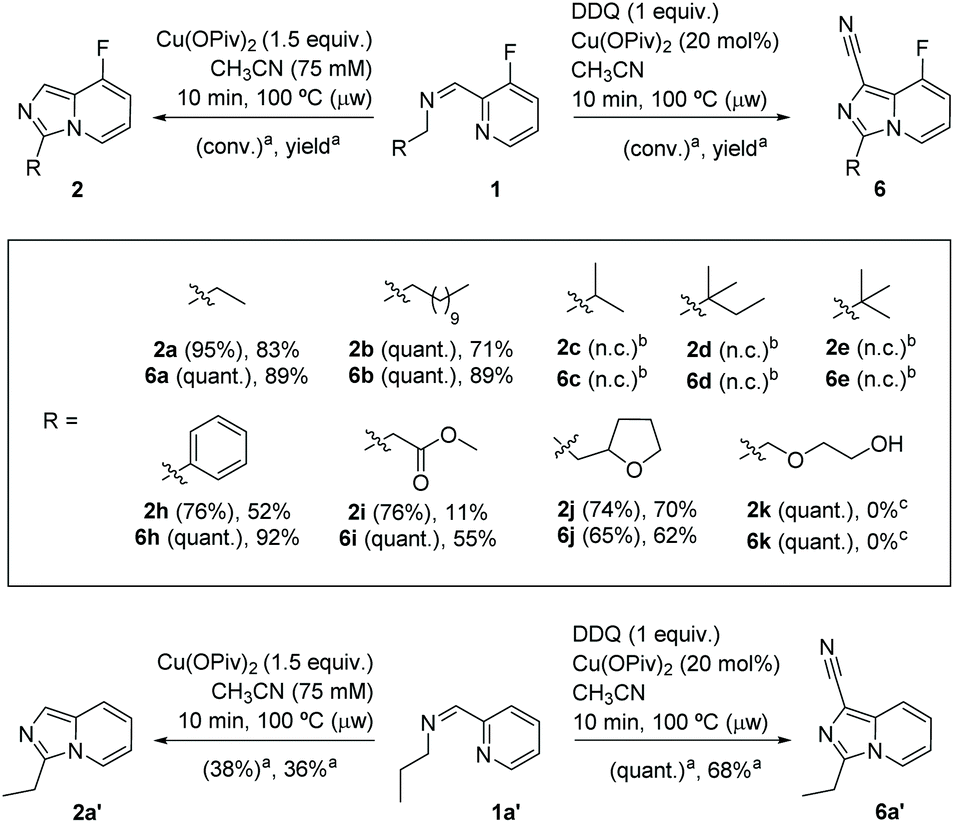 | ||
| Scheme 4 Top: Reaction scope for the synthesis of imidazo[1,5-a]pyridines type-2 and type-6 in both stoichiometric and catalytic versions. Bottom: Reactivity of 1a′ towards 2a′ and 6a′. aConversions and yields were determined by 19F NMR with 4-fluorotoluene as internal standard. b No conversion (n.c.). c Complex mixture. | ||
In the search of applying this methodology to other substrates incorporating heterocyclic units, we found that the placement of a tetrahydrofurfuryl moiety provides the C–H cyclization products 2j and 6j in good yields (70% and 62%, respectively, Scheme 4), despite the fact that free alcohol functional groups were not compatible with these reaction conditions (only decomposition by-products were obtained from 1k).
The generality of both methods has been assessed with a non-fluorinated substrate 1a′, leading to 2a′ (38% conv., 36% yield) and 6a′ (quant. conv., 68% yield), respectively (Scheme 4). Notably, the bidentate N,N′-coordination of the substrate poises 1a′ to selectively undergo C(sp3)–H activation and cyclization (no C(sp2)–H functionalization by-products on the pyridine ring were detected).
With the aim of studying a plausible surface reactivity, preformed zero-valent copper nanoparticles (CuNPs, see the synthesis protocol in the ESI†) were used as catalyst for the synthesis of 2a and 6a; the reactivity observed was similar to that using Cu(OPiv)2 (see Fig. S3 and S4 in the ESI‡). Considering that only copper agglomerates were detected after reaction (TEM analysis), it can be assumed that the C–H functionalization is merely promoted by molecular species, due to copper leaching from CuNPs.37 Taking into account that imidazo[1,5-a]pyridines are only formed in the presence of copper(II) carboxylates together with the coordination ability of aldimines to give five-coordinated Cu(II) complexes ([I], Fig. 5), a concerted metalation–deprotonation mechanism can be proposed for the α′-C(sp3)–H activation via the formation of a six-membered cupracycle [II], analogously to what has been proposed for Pd-promoted C–H activation.38,39 A reductive elimination then delivers 2a and Cu(0), both observed for the stoichiometric reaction [Cu(0) evidenced by powder X-ray diffraction; see Fig. S5 in the ESI‡]. To gain an insight on the metal intermediates, monitoring studies by cyclic voltammetry of the stoichiometric reaction [Cu(OPiv)2 and 1a in CH3CN] revealed the exclusive presence of Cu(II) species in solution, together with a peak intensity decrease over time, due to the formation of insoluble Cu(0) (see Fig. S6 in the ESI‡).
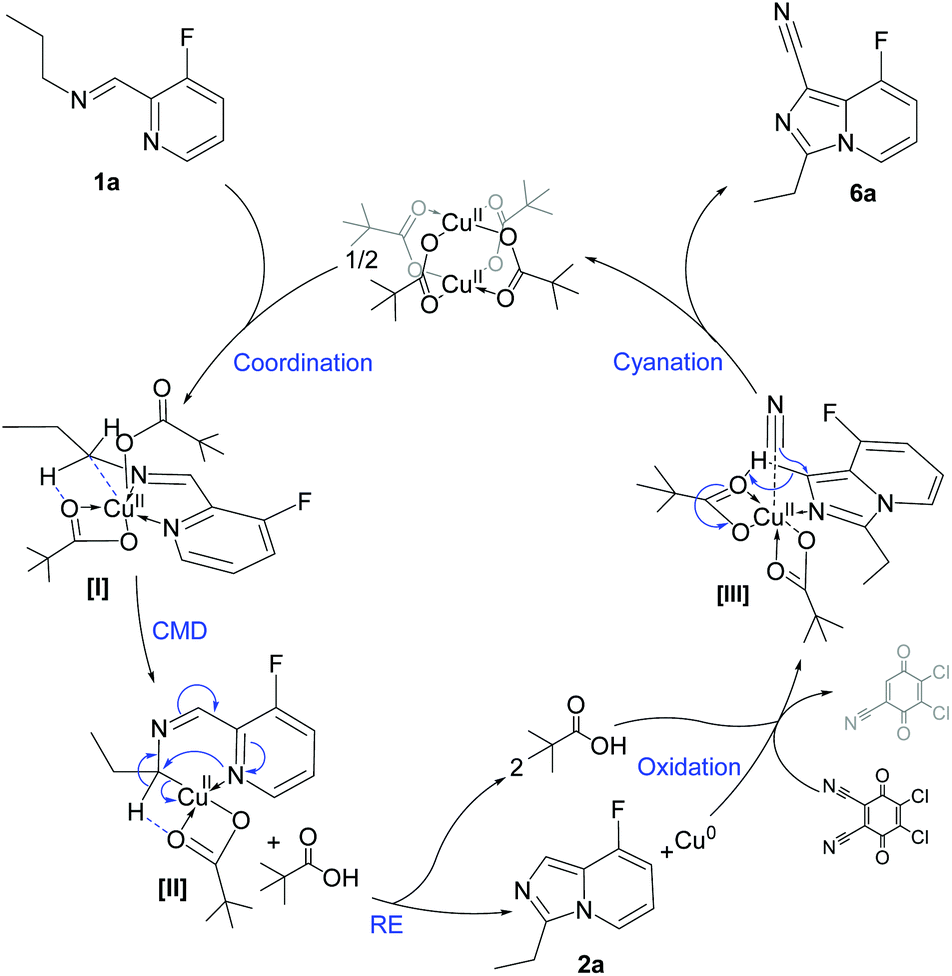 | ||
| Fig. 5 A plausible reaction mechanism for the synthesis of imidazo[1,5-a]pyridines via Cu(II) C–H bond functionalization reactions. For simplicity purposes, 1a was used as substrate. | ||
Under catalytic conditions, DDQ acts both as oxidant and cyanating agent, taking advantage of the fairly acidic proton in the imidazole ring and leading to the formation of 6a through a plausible intermediate [III]. According to control experiments, copper(II) cyanide species were detected by FTIR when DDQ was reacted with Cu(0) in the presence of potassium pivalate (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching bands at ca. 2160 cm−1; see Fig. S7 in the ESI‡). The cyanation step was demonstrated by the addition of DDQ (1 equiv.) to 2a, furnishing 6a (62% yield; see Scheme S2 in the ESI‡).
N stretching bands at ca. 2160 cm−1; see Fig. S7 in the ESI‡). The cyanation step was demonstrated by the addition of DDQ (1 equiv.) to 2a, furnishing 6a (62% yield; see Scheme S2 in the ESI‡).
Reaction manifolds involving single electron transfer processes and Cu(I) species could be ruled out (see Scheme S3 in the ESI‡). Thus, the presence of the radical scavenger 2,6-di-tert-butyl-4-methylphenol (1 equiv.) did not inhibit the catalytic transformation (53% yield of 2a). The reaction of 1a with CuI (1.5 equiv.) and potassium pivalate (1.5 equiv.) only resulted in traces of 2a (9% yield).40 These experiments evidence the crucial role of both electrophilic Cu(II) species and carboxylates to promote aldimine α′-C(sp3)–H functionalization. Furthermore, the isolation of an acrylonitrile derivative (4) under harsher reaction conditions, indicates that successive C–H deprotonation events may be feasible (see Scheme S4 in the ESI‡).
Conclusion
This work focuses on the study of N,N′-bidentate substrates that act as directing groups towards multiple C–H bond functionalization reactions via coordination to Cu(II) carboxylate salts. We have developed a Cu(II)-mediated C(sp3)–H amination/cyclization of 1-(2-pyridinyl)methanimines to afford imidazo[1,5-a]pyridine heterocycles via a Cu(II)/Cu(0) mechanism, under microwave irradiation. In addition, a telescoped Cu-catalyzed C(sp3)–H amination/cyclization followed by C(sp2)–H cyanation process could be optimized towards the synthesis of imidazo[1,5-a]pyridine-1-carbonitrile derivatives through a Cu(II)/Cu(0) mechanism. Notably, the N,N′-bidentate coordination renders this process selective towards C(sp3)–H functionalization, precluding side C(sp2)–H activation reactions on the pyridine ring. This synthetic platform offers great potential for the straightforward syntheses of bioisosteres of purine alkaloids and analogues thereof, providing novel means to explore the vast chemical space of non-natural alkaloids. Transition-metal promoted C–H functionalization strategies confer powerful and versatile tools towards late stage molecular edition via the installation of functional groups in a selective manner. Thus, orthogonal methods for the synthesis of imidazo[1,5-a]pyridine and imidazo[1,5-a]pyridine-1-carbonitrile derivatives were successfully achieved.These methods highlight the emerging value of unactivated C(sp3)–H bonds as functional groups in organic synthesis. A fine tuning of the carboxylate basicity resulted a determinant factor, suggesting a concerted metalation–deprotonation mechanism. Moreover, this contribution enables the development of sustainable processes based on simple copper salts.
Experimental section
General procedure for the synthesis of aldimines 1a–1l
Amine a–l (8.00 mmol) was added to a suspension of 3-fluoro-2-pyridinecarboxaldehyde (1.00 g, 8.00 mmol) and sodium sulfate (6.81 g, 47.99 mmol, 6 equiv.) in 2-methyltetrahydrofuran (40 mL), and the resulting mixture was stirred at r.t. for 16 h. The solid was then filtered out and the filtrate was evaporated under reduced pressure to furnish aldimine 1a–1l (yields 89 to 99%, see the ESI‡ for characterization details).General procedure for the preparation of CuBr2–aldimine complexes CuA, CuB and CuC
CuBr2 (402 mg, 1.80 mmol) was added in one solid portion to a solution of aldimine 1a (200 mg, 1.20 mmol) in CH2Cl2 (10 mL). The initial green solution was stirred for 90 min at rt, turning rapidly to a dark red color. The reaction mixture was then filtered through a 0.2 μm PTFE filter and the solvent was removed under reduced pressure, furnishing the desired complex CuA (459 mg, 99%) as a dark solid that was used without further purification. Suitable monocrystals for XRD analysis of CuA were obtained by vapor diffusion crystallization of pentane in a CH2Cl2 solution.
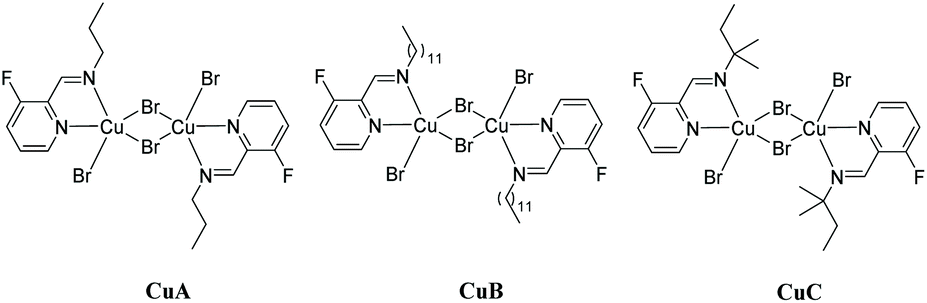
In an analogous manner and starting from 1b (351 mg, 1.20 mmol) or 1d (233.1 mg, 1.20 mmol), CuB and CuC complexes were obtained [548 mg, 89% (CuB), and 401 mg, 80% (CuC)], respectively (see the ESI‡ for characterization details).
Stoichiometric C–H bond functionalization reactions
Author contributions
M. C. prepared the substrates, optimized the C–H bond functionalization reactions, studied the scope and mechanistic studies. I. F. carried out the cyclic voltammetry experiments and proofread the experimental part and main article. S. M.-L. carried out the X-ray crystallographic studies and the refinement of the structures. D. P. identified the heterocycle via C–H bond functionalization, revised the experimental part, designed a research plan and co-wrote the manuscript. M. G. supervised the work, co-wrote the manuscript and coordinated the overall project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Centre National de la Recherche Scientifique (CNRS) and the Université Toulouse 3 – Paul Sabatier are gratefully acknowledged for their financial support. M. C. thanks the Ministère de l'Enseignement Supérieur et de la Recherche of the French government for a PhD scholarship. Dr Gyorgy Szaloki is acknowledged for his valuable help in the cyclic voltammetry studies. The authors thank Dr Nathalie Saffon-Merceron of the X-ray diffraction service of the Institut de Chimie de Toulouse ICT-UAR 2599 (http://www.ict.ups-tlse.fr) for her valuable help in single crystal analysis.Notes and references
- (a) X. Ribas, Catalytic C–H/C–X Bond Functionalization, Royal Society of Chemistry, Cambridge, 2013 RSC; (b) A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129–133 CrossRef CAS; (c) S. Korwar, K. Brinkley, A. R. Siamaki, B. F. Gupton and K. C. Ellis, Org. Lett., 2015, 17, 1782–1785 CrossRef CAS PubMed; (d) X. Jia, L. I. Frye, W. Zhu, S. Gu and T. B. Gunnoe, J. Am. Chem. Soc., 2020, 142, 10534–10543 CrossRef CAS PubMed; (e) T. M. Gentle and E. L. Muetterties, J. Phys. Chem., 1983, 87, 2469–2472 CrossRef CAS; (f) K. Funaki, H. Kawai, T. Sato and S. Oi, Chem. Lett., 2011, 40, 1050–1052 CrossRef CAS; (g) X.-P. Fu, Q.-Q. Xuan, L. Liu, D. Wang, Y.-J. Chen and C.-J. Li, Tetrahedron, 2013, 69, 4436–4444 CrossRef CAS; (h) R. Feng, J. Yao, Z. Liang, Z. Liu and Y. Zhang, J. Org. Chem., 2013, 78, 3688–3696 CrossRef CAS; (i) I. J. S. Fairlamb and A. F. Lee, Fundamental Pd(0)/Pd(II) redox steps in cross-coupling reactions: homogeneous, hybrid homogeneous–heterogeneous to heterogeneous mechanistic pathways for C–C couplings, in Catalytic C–H/C–X Bond Functionalization, RSC Publishing Cambridge, 2013, pp. 88–123 Search PubMed; (j) H. Duan, M. Li, G. Zhang, J. R. Gallagher, Z. Huang, Y. Sun, Z. Luo, H. Chen, J. T. Miller, R. Zou, A. Lei and Y. Zhao, ACS Catal., 2015, 5, 3752–3759 CrossRef CAS; (k) J. Dong, Y. Huang, X. Qin, Y. Cheng, J. Hao, D. Wan, W. Li, X. Liu and J. You, Chem. – Eur. J., 2012, 18, 6158–6162 CrossRef CAS; (l) A. M. Contreras, M. Montano, S. J. Kweskin, M. M. Koebel, K. Bratlie, K. Becraft and G. A. Somorjai, Top. Catal., 2006, 40, 19–24 CrossRef CAS; (m) Y.-H. Chin, M. García-Diéguez and E. Iglesia, J. Phys. Chem. C, 2016, 120, 1446–1460 CrossRef CAS; (n) W. Chen, M. Wang, P. Li and L. Wang, Tetrahedron, 2011, 67, 5913–5919 CrossRef CAS; (o) S. Bhowmik, G. Pandey and S. Batra, Chem. – Eur. J., 2013, 19, 10487–10491 CrossRef CAS PubMed; (p) C. G. Baumann, S. De Ornellas, J. P. Reeds, T. E. Storr, T. J. Williams and I. J. S. Fairlamb, Tetrahedron, 2014, 70, 6174–6187 CrossRef CAS; (q) M. Al-Amin, M. Arisawa, S. Shuto, Y. Ano, M. Tobisu and N. Chatani, Adv. Synth. Catal., 2014, 356, 1631–1637 CrossRef CAS; (r) L. Adak, S. Bhadra and B. C. Ranu, Tetrahedron Lett., 2010, 51, 3811–3814 CrossRef CAS; (s) C. Taglang, L. M. Martínez-Prieto, I. del Rosal, L. Maron, R. Poteau, K. Philippot, B. Chaudret, S. Perato, A. Sam Lone, C. Puente, C. Dugave, B. Rousseau and G. Pieters, Angew. Chem., Int. Ed., 2015, 54, 10474–10477 CrossRef CAS.
- (a) P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495–2495 CrossRef CAS; (b) J. J. Mousseau and A. B. Charette, Acc. Chem. Res., 2013, 46, 412–424 CrossRef CAS; (c) K. Hattori, K. Yamaguchi, J. Yamaguchi and K. Itami, Tetrahedron, 2012, 68, 7605–7612 CrossRef CAS; (d) M. Harari, F. Couly, C. Fruit and T. Besson, Org. Lett., 2016, 18, 3282–3285 CrossRef CAS; (e) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS; (f) F. Valentini, G. Brufani, L. Latterini and L. Vaccaro, Advanced Heterogeneous Catalysts Volume 1: Applications at the Nano-Scale, American Chemical Society, 2020, vol. 1359, ch. 17, pp. 513–543 Search PubMed; (g) K. Hirano and M. Miura, Yuki Gosei Kagaku Kyokaishi, 2011, 69, 252–265 CrossRef CAS; (h) M. Camats, D. Pla and M. Gómez, in Recent Advances in Nanoparticle Catalysis, ed. P. W. N. M. van Leeuwen and C. Claver, Springer International Publishing, 2020, pp. 249–280 Search PubMed; (i) G. Danoun, A. Tlili, F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2012, 51, 12815–12819 CrossRef CAS PubMed; (j) P. Gandeepan, J. Koeller and L. Ackermann, ACS Catal., 2017, 7, 1030–1034 CrossRef CAS; (k) J. Li, S. De Sarkar and L. Ackermann, in C-H Bond Activation and Catalytic Functionalization, ed. P. H. Dixneuf and H. Doucet, Springer International Publishing, Cham, 2016, pp. 217–257 Search PubMed; (l) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS; (m) F. Pesciaioli, U. Dhawa, J. C. A. Oliveira, R. Yin, M. John and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 15425–15429 CrossRef CAS PubMed; (n) C. Zhu, J. C. A. Oliveira, Z. Shen, H. Huang and L. Ackermann, ACS Catal., 2018, 8, 4402–4407 CrossRef CAS; (o) B. Nie, W. Wu, Y. Zhang, H. Jiang and J. Zhang, Org. Chem. Front., 2020, 7, 3067–3099 RSC.
- (a) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790–6791 CrossRef CAS PubMed; (b) Y. Zhang, B. Nie and J. Zhang, Youji Huaxue, 2015, 35, 2067–2085 CrossRef CAS; (c) H. Yu, S. Su, C. Zhang and Z. Dang, Youji Huaxue, 2013, 33, 1628–1646 CrossRef CAS; (d) Y. Yamamoto, in Copper-Mediated Cross-Coupling Reactions, ed. G. Evano and N. Blanchard, John Wiley & Sons, Inc., 2014, ch. 10, pp. 335–399 Search PubMed; (e) R.-J. Song and J.-H. Li, in Copper-Mediated Cross-Coupling Reactions, ed. G. Evano and N. Blanchard, John Wiley & Sons, Inc., 2014, ch. 11, pp. 401–453 Search PubMed; (f) G. K. Reen, A. Kumar and P. Sharma, Beilstein J. Org. Chem., 2019, 15, 1612–1704 CrossRef CAS; (g) M. Matsumura, Yakugaku Zasshi, 2020, 140, 1101–1106 CrossRef CAS; (h) Q. Liao and C. Xi, Youji Huaxue, 2012, 32, 986–993 CrossRef CAS; (i) A. P. Jadhav, D. Ray, V. U. B. Rao and R. P. Singh, Eur. J. Org. Chem., 2016, 2369–2382 CrossRef CAS; (j) K. Hirano and M. Miura, Chem. Lett., 2015, 44, 868–873 CrossRef CAS; (k) M. C. Henry, M. A. B. Mostafa and A. Sutherland, Synthesis, 2017, 4586–4598 CAS; (l) S. Cacchi, G. Fabrizi and A. Goggiamani, in Innovative Catalysis in Organic Synthesis: Oxidation, Hydrogenation, and C–X Bond Forming Reactions, ed. P. G. Andersson, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, ch. 10, pp. 211–232 Search PubMed; (m) A. A. Almasalma and E. Mejia, Synthesis, 2020, 2613–2622 CAS.
- (a) J. Liu, G. Chen and Z. Tan, Adv. Synth. Catal., 2016, 358, 1174–1194 CrossRef CAS; (b) M. Corbet and F. De Campo, Angew. Chem., Int. Ed., 2013, 52, 9896–9898 CrossRef CAS; (c) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed.
- Y. Taniguchi, S. Horie, K. Takaki and Y. Fujiwara, J. Organomet. Chem., 1995, 504, 137–141 CrossRef CAS.
- (a) Z. Li, D. S. Bohle and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8928–8933 CrossRef CAS; (b) F.-F. Wang, C.-P. Luo, G. Deng and L. Yang, Green Chem., 2014, 16, 2428–2431 RSC; (c) D. Talukdar, S. Borah and M. K. Chaudhuri, Tetrahedron Lett., 2015, 56, 2555–2558 CrossRef CAS; (d) V. R. Regalla, R. R. Addada and A. Chatterjee, Tetrahedron Lett., 2018, 59, 4161–4164 CrossRef CAS; (e) G.-J. Cheng, L.-J. Song, Y.-F. Yang, X. Zhang, O. Wiest and Y.-D. Wu, ChemPlusChem, 2013, 78, 943–951 CrossRef CAS PubMed; (f) S. K. Rout, S. Guin, W. Ali, A. Gogoi and B. K. Patel, Org. Lett., 2014, 16, 3086–3089 CrossRef CAS; (g) T. Wang, M. Schrempp, A. Berndhaeuser, O. Schiemann and D. Menche, Org. Lett., 2015, 17, 3982–3985 CrossRef CAS PubMed.
- Y. Li, K. Dong, F. Zhu, Z. Wang and X.-F. Wu, Angew. Chem., Int. Ed., 2016, 55, 7227–7230 CrossRef CAS.
- (a) Z. Cui, X. Shang, X.-F. Shao and Z.-Q. Liu, Chem. Sci., 2012, 3, 2853–2858 RSC; (b) A. Gogoi, S. Guin, S. K. Rout and B. K. Patel, Org. Lett., 2013, 15, 1802–1805 CrossRef CAS PubMed.
- (a) F. Shi and X. Cui, in Catalytic Amination for N-Alkyl Amine Synthesis, ed. F. Shi and X. Cui, Academic Press, 2018, ch. 6, pp. 149–180 Search PubMed; (b) T.-S. Zhang, W.-J. Hao, N.-N. Wang, G. Li, D.-F. Jiang, S.-J. Tu and B. Jiang, Org. Lett., 2016, 18, 3078–3081 CrossRef CAS PubMed; (c) H. Wang, W. Xu, Z. Wang, L. Yu and K. Xu, J. Org. Chem., 2015, 80, 2431–2435 CrossRef CAS; (d) J. M. Munoz-Molina, T. R. Belderrain and P. J. Perez, Synthesis, 2021, 51–64 CrossRef CAS; (e) T. Truong, K. D. Nguyen, S. H. Doan and N. T. S. Phan, Appl. Catal., A, 2016, 510, 27–33 CrossRef; (f) J. Long, L. Le, T. Iwasaki, R. Qiu and N. Kambe, J. Org. Chem., 2020, 85, 482–492 CrossRef PubMed; (g) G. Zhang, Y. Zhao and H. Ge, Angew. Chem., Int. Ed., 2013, 52, 2559–2563 CrossRef PubMed.
- (a) C. Wang, Y. Yang, D. Qin, Z. He and J. You, J. Org. Chem., 2015, 80, 8424–8429 CrossRef PubMed; (b) K. Nozawa-Kumada, S. Saga, Y. Matsuzawa, M. Hayashi, M. Shigeno and Y. Kondo, Chem. – Eur. J., 2020, 26, 4496–4499 CrossRef PubMed; (c) P.-C. Huang, P. Gandeepan and C.-H. Cheng, Chem. Commun., 2013, 49, 8540–8542 RSC; (d) A. Ilangovan and G. Satish, Org. Lett., 2013, 15, 5726–5729 CrossRef PubMed.
- (a) N. Takemura, Y. Kuninobu and M. Kanai, Org. Biomol. Chem., 2014, 12, 2528–2532 RSC; (b) A. Modak, U. Dutta, R. Kancherla, S. Maity, M. Bhadra, S. M. Mobin and D. Maiti, Org. Lett., 2014, 16, 2602–2605 CrossRef CAS.
- X. Wu, Y. Zhao and H. Ge, Chem. – Asian J., 2014, 9, 2736–2739 CrossRef PubMed.
- S. Shi, P. Zhang, C. Luo, S. Zhuo, Y. Zhang, G. Tang and Y. Zhao, Org. Lett., 2020, 22, 1760–1764 CrossRef PubMed.
- (a) M. L. Kantam, C. Gadipelly, G. Deshmukh, K. R. Reddy and S. Bhargava, Chem. Rec., 2019, 19, 1302–1318 CrossRef PubMed; (b) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef PubMed; (c) K. Hirano and M. Miura, Chem. Commun., 2012, 48, 10704–10714 RSC; (d) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef.
- (a) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC; (b) X. Wu, Y. Zhao, G. Zhang and H. Ge, Angew. Chem., Int. Ed., 2014, 53, 3706–3710 CrossRef CAS.
- (a) F. Roudesly, J. Oble and G. Poli, J. Mol. Catal. A: Chem., 2017, 426, 275–296 CrossRef CAS; (b) A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797–9804 CrossRef CAS.
- M. Li, Y. Xie, Y. Ye, Y. Zou, H. Jiang and W. Zeng, Org. Lett., 2014, 16, 6232–6235 CrossRef CAS PubMed.
- H. Wang, W. Xu, L. Xin, W. Liu, Z. Wang and K. Xu, J. Org. Chem., 2016, 81, 3681–3687 CrossRef CAS.
- D. S. Tan, Nat. Chem. Biol., 2005, 1, 74–84 CrossRef CAS.
- (a) S. Sharma, S. Mehndiratta, S. Kumar, J. Singh, P. M. S. Bedi and K. Nepali, Recent Pat. Anti-Cancer Drug Discovery, 2015, 10, 308–341 CrossRef CAS; (b) F. Li, H. Maag and T. Alfredson, J. Pharm. Sci., 2008, 97, 1109–1134 CrossRef CAS PubMed.
- N. S. M. Ismail, G. M. E. Ali, D. A. Ibrahim and A. M. Elmetwali, Future J. Pharm. Sci., 2016, 2, 60–70 CrossRef.
- (a) D. Pla, D. S. Tan and D. Y. Gin, Chem. Sci., 2014, 5, 2407–2415 RSC; (b) J. Tang, B. Wang, T. Wu, J. Wan, Z. Tu, M. Njire, B. Wan, S. G. Franzblauc, T. Zhang, X. Lu and K. Ding, ACS Med. Chem. Lett., 2015, 6, 814–818 CrossRef CAS PubMed.
- (a) D. J. Paymode, F. S. P. Cardoso, T. Agrawal, J. W. Tomlin, D. W. Cook, J. M. Burns, R. W. Stringham, J. D. Sieber, B. F. Gupton and D. R. Snead, Org. Lett., 2020, 22, 7656–7661 CrossRef CAS PubMed; (b) R. T. Eastman, J. S. Roth, K. R. Brimacombe, A. Simeonov, M. Shen, S. Patnaik and M. D. Hall, ACS Cent. Sci., 2020, 6, 672–683 CrossRef CAS.
- G. Volpi and R. Rabezzana, New J. Chem., 2021, 45, 5737–5743 RSC.
- P. M. Cowley, M. A. Mcgowan, T. J. Brown, Y. Han, K. Liu, Q. Pu, A. Wise, H. Zhang and H. Zhou, Novel substituted imidazopyridine compounds as inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan-2,3-dioxygenase. Int. Pat. ApplWO2017189386, 2020 Search PubMed.
- X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed.
- J. Hu, Y. Guo, J. a. Zhao and J. Zhang, Bioorg. Med. Chem., 2017, 25, 5733–5742 CrossRef CAS.
- B. L. Westcott, G. Crundwell, M. Remesic, K. Knopf, K. Chandler, J. McMaster and E. S. Davies, Inorg. Chem. Commun., 2016, 74, 79–81 CrossRef CAS.
- J. Salinas Uber, Y. Vogels, D. van den Helder, I. Mutikainen, U. Turpeinen, W. T. Fu, O. Roubeau, P. Gamez and J. Reedijk, Eur. J. Inorg. Chem., 2007, 2007, 4197–4206 CrossRef.
- G. A. Ardizzoia, G. Colombo, B. Therrien and S. Brenna, Eur. J. Inorg. Chem., 2019, 2019, 1825–1831 CrossRef CAS.
- One-pot reaction attempts aiming at in situ condensation of 1-propylamine and 3-fluoro-2-pyridinecarboxaldehyde preclude C–H bond functionalization probably due to the strong coordination of copper with primary amines (see Table S3‡, entry 7).
- (a) S. Löw, J. Becker, C. Würtele, A. Miska, C. Kleeberg, U. Behrens, O. Walter and S. Schindler, Chem. – Eur. J., 2013, 19, 5342–5351 CrossRef; (b) A. El-Toukhy, G. Z. Cai, G. Davies, T. R. Gilbert, K. D. Onan and M. Veidis, J. Am. Chem. Soc., 1984, 106, 4596–4605 CrossRef CAS; (c) F. S. Keij, J. G. Haasnoot, A. J. Oosterling, J. Reedijk, C. J. O'Connor, J. H. Zhang and A. L. Spek, Inorg. Chim. Acta, 1991, 181, 185–193 CrossRef CAS; (d) A. M. Atria, A. Vega, M. Contreras, J. Valenzuela and E. Spodine, Inorg. Chem., 1999, 38, 5681–5685 CrossRef CAS; (e) S. V. Voitekhovich, P. N. Gaponik, A. S. Lyakhov, J. V. Filipova, A. G. Sukhanova, G. T. Sukhanov and O. A. Ivashkevich, Tetrahedron Lett., 2009, 50, 2577–2579 CrossRef CAS.
- S. Löw, J. Becker, C. Würtele, A. Miska, C. Kleeberg, U. Behrens, O. Walter and S. Schindler, Chem. – Eur. J., 2013, 19, 5342–5351 CrossRef PubMed.
- Y. Zhu, M. Zhao, W. Lu, L. Li and Z. Shen, Org. Lett., 2015, 17, 2602–2605 CrossRef CAS PubMed.
- G. Zhang, S. Chen, H. Fei, J. Cheng and F. Chen, Synlett, 2012, 2247–2250 CAS.
- Q. Wen, J. Jin, L. Zhang, Y. Luo, P. Lu and Y. Wang, Tetrahedron Lett., 2014, 55, 1271–1280 CrossRef CAS.
- ICP analyses of the organic extracts coming from the reactions under stoichiometric and catalytic conditions revealed 0.3 mg and 0.8 mg of Cu, respectively.
- (a) J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2572 CrossRef CAS; (b) F. Tjosaas and A. Fiksdahl, J. Organomet. Chem., 2007, 692, 5429–5439 CrossRef CAS; (c) T. Yang, C. Kong, S. Yang, Z. Yang, S. Yang and M. Ehara, Chem. Sci., 2020, 11, 113–125 RSC; (d) S. Rousseaux, B. Liégault and K. Fagnou, Chem. Sci., 2012, 3, 244–248 RSC; (e) G. Aullón, R. Chat, I. Favier, M. Font-Bardia, M. Gómez, J. Granell, M. Martínez and X. Solans, Dalton Trans., 2009, 8292–8300 RSC; (f) M. Gómez, J. Granell and M. Martinez, J. Chem. Soc., Dalton Trans., 1998, 37–44 RSC.
- Given that the cleavage of the α′-C(sp3)–H via the direct formation of copper hydride species has not been detected (Table S1 in the ESI,‡ entries 1 and 2), and the fact that the selectively formation of 2a can only be achieved by addition of carboxylate salts (Table 1), both experimental results point to the assistance of the carboxylate anion in triggering C(sp3)–H bond functionalization through a concerted metalation–deprotonation pathway.
- A partial oxidation of copper(I) salts by air can be feasible as microwave reactions are carried out under aerobic conditions.
- The instability of 2a and 2b upon purification precluded their isolation in pure form (see Fig. S8 in the ESI‡ for 2b).
Footnotes |
| † Dedicated to the memory of Prof. David Y. Gin and his contributions in the synthesis of bioactive alkaloids. |
| ‡ Electronic supplementary information (ESI) available: Instrumentation, materials, experimental procedures, Tables S1–S5, Fig. S1–S8, Schemes S1–S4, synthesis and characterization of compounds type-1, type-2, 4 and 6a, NMR spectra of type-1, type-2, 4 and 6a compounds, and single-crystal X-ray diffraction data for CuA, Cu4B, 4 and 6a (Tables S6–S9) (PDF). CCDC 2077016 (CuA), 2077017 (Cu4B), 2077019 (4) and 2077018 (6a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob02118d |
| This journal is © The Royal Society of Chemistry 2022 |