
Amide-derived lysine analogues as substrates and inhibitors of histone lysine methyltransferases and acetyltransferases†
Jordi C. J.
Hintzen‡
 a,
Jona
Merx‡
b,
Marijn N.
Maas
a,
Sabine G. H. A.
Langens
b,
Paul B.
White
b,
Thomas J.
Boltje
*b and
Jasmin
Mecinović
*a
a,
Jona
Merx‡
b,
Marijn N.
Maas
a,
Sabine G. H. A.
Langens
b,
Paul B.
White
b,
Thomas J.
Boltje
*b and
Jasmin
Mecinović
*a
aDepartment of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense, Denmark. E-mail: mecinovic@sdu.dk; Tel: +45 6550 3603
bInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525AJ, Nijmegen, The Netherlands. E-mail: thomas.boltje@ru.nl; Tel: +31 24 3652331
First published on 29th November 2021
Abstract
Histone lysine methyltransferases and acetyltransferases are two classes of epigenetic enzymes that play pivotal roles in human gene regulation. Although they both recognise and posttranslationally modify lysine residues in histone proteins, their difference in histone peptide-based substrates and inhibitors remains to be firmly established. Here, we have synthesised lysine mimics that posses an amide bond linker in the side chain, incorporated them into histone H3 tail peptides, and examined synthetic histone peptides as substrates and inhibitors for human lysine methyltransferases and acetyltransferases. This work demonstrates that histone lysine methyltransferases G9a and GLP do catalyse methylation of the most similar lysine mimic, whereas they typically do not tolerate more sterically demanding side chains. In contrast, histone lysine acetyltransferases GCN5 and PCAF do not catalyse acetylation of the same panel of lysine analogues. Our results also identify potent H3-based inhibitors of GLP methyltransferase, providing a basis for development of peptidomimetics for targeting KMT enzymes.
Introduction
Histones are nuclear proteins that are crucial for compaction of DNA and transcriptional regulation in eukaryotes. Protruding from the core histone proteins are flexible, unstructured N-terminal tails, which are the site of heavy post-translational modifications (PTMs). Through histone PTMs, many proteins and enzymes interact with the histone tails and thereby regulate which areas of the genome are actively transcribed and which ones are silenced.1,2 Among the vast landscape of histone PTMs, lysine methylation and acetylation are some of the most well-known and abundant modifications (Fig. 1a). Methylation of histone's lysine residues is associated both with gene activation and repression, depending on the histone site and methylation state.3 Lysine can be found in mono- (Kme1), di- (Kme2) and trimethylated (Kme3) forms.4,5 The methylation reactions are catalysed by histone lysine methyltransferases (KMTs), which use S-adenosylmethionine (SAM) as the cosubstrate (Fig. 1b).6 Lysine acetylation on histones is, on the other hand, generally associated with gene activation.7,8 Upon the installation of the acetyl group on lysine (Kac), the positive charge of lysine is permanently neutralised (Fig. 1a), causing the histone protein to associate weaker with the negatively charged DNA, thus leading to a more accessible chromatin structure.9 Acetylation of histones is catalysed by histone lysine acetyltransferases (KATs), which use acetyl coenzyme A (AcCoA) as the cosubstrate (Fig. 1c).10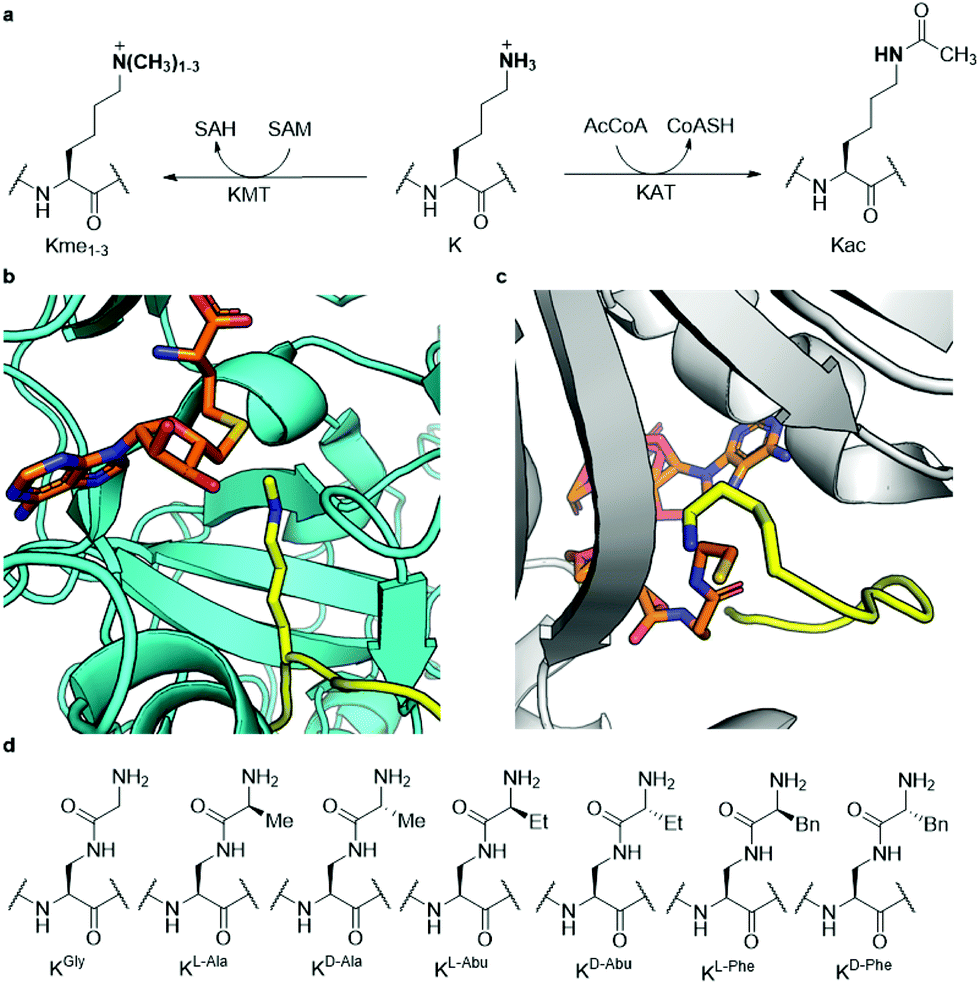 | ||
| Fig. 1 Histone lysine methylation and acetylation. (a) KMT-catalysed lysine methylation in the presence of SAM cosubstrate and KAT-catalysed lysine acetylation in the presence of AcCoA cosubstrate; (b) view from a crystal structure of GLP (cyan) complexed with H3K9me1 (yellow) and S-adenosylhomocysteine (SAH, orange) (PDB ID: 3HNA); (c) view from a crystal structure of GCN5 (gray) complexed with H3K14 (yellow) and coenzyme A (CoASH, orange) (PDB ID: 1Q2C); (d) the panel of lysine analogues incorporated in the H3 peptide for examination of KMT and KAT catalysis and inhibition. | ||
Since both classes of epigenetic enzymes are of crucial importance to the function of the cell and show a specificity towards the lysine site that undergoes PTM, gaining a deeper understanding of their mode of action and substrate scope is highly desirable. Furthermore, both KMTs and KATs are known to be involved in many disease phenotypes, including cancer.11,12 Previously, both classes of enzymes have been successfully subjected to a wide variety of lysine analogues to assess which chemical changes are tolerated by the two families of enzymes and potentially leading to the discovery of novel inhibitors.13–25 KATs were shown to be more promiscuous than KMTs towards the lysine's side chain length,16,24 while γ-difluorolysine was accepted as an excellent substrate for both families of enzymes.14 Adding to this body of work, here we have developed an expanded set of lysine analogues, containing an amide bond in the side chain, which introduce some degree of steric bulk and rigidity into lysine's δ-position (Fig. 1d). A set of seven lysine analogues was chemically synthesised, incorporated into histone 3 tail peptides and subsequently evaluated as substrates for methyltransferases G9a and GLP, as well as the acetyltransferases GCN5 and PCAF, which catalyse the methylation and acetylation of lysine K9 of histone H3, respectively.
Results and discussion
As functionalisation of lysine's aliphatic side chain is synthetically challenging, we aimed to synthesise a panel of easily accessible lysine analogues starting from diamino propionic acid and employing amide bond formation to generate lysine analogues using amino acids with varying side chains. Fmoc-protected lysine analogues were synthesised starting from commercially available N-Cbz protected diamino propionic acid (Dap). Facile protection of the acid in methanolic HCl allows functionalisation of the side chain's amine in Dap with commercially available N-Boc protected amino acids under agency of HATU. Deprotection of the methyl ester and subsequent one-pot exchange of the amine protecting group, by hydrogenolysis of the Cbz and treatment with Fmoc-Cl, yielded the Fmoc-protected lysine analogues in moderate yields (Fig. 2).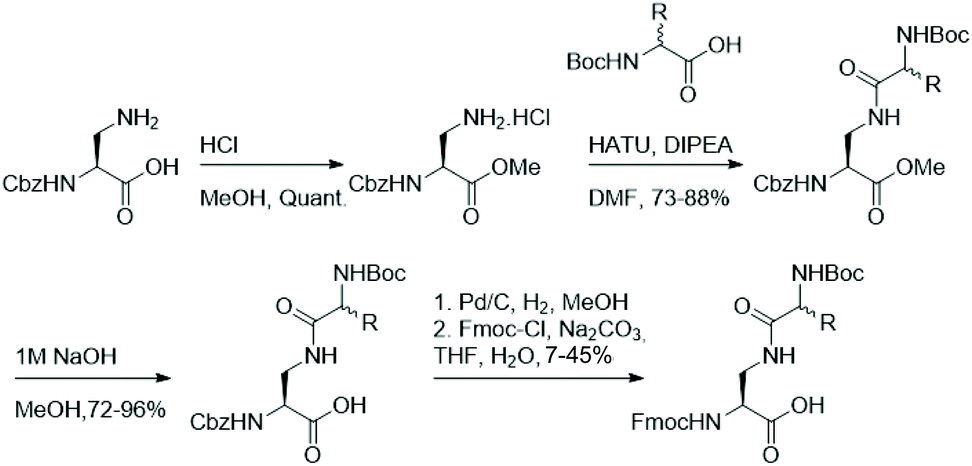 | ||
| Fig. 2 Synthetic pathway for the preparation of amide-derived lysine analogues. | ||
The set of synthesised Fmoc/Boc-protected lysine analogues, including natural lysine as a control, was subsequently incorporated into histone H3 tail peptides (residues 1–13, ARTKQTAR![[K with combining low line]](https://www.rsc.org/images/entities/b_char_004b_0332.gif) 9STGG) by standard Fmoc-SPPS. The synthetic peptides were then purified by reverse-phase HPLC and their purity was confirmed by MALDI-TOF MS and analytical HPLC (Fig. S1–S8†). The potential of the histone peptides possessing lysine analogues being substrates for KMTs and KATs was evaluated by carrying out enzymatic assays and evaluation by MALDI-TOF MS.
9STGG) by standard Fmoc-SPPS. The synthetic peptides were then purified by reverse-phase HPLC and their purity was confirmed by MALDI-TOF MS and analytical HPLC (Fig. S1–S8†). The potential of the histone peptides possessing lysine analogues being substrates for KMTs and KATs was evaluated by carrying out enzymatic assays and evaluation by MALDI-TOF MS.
H3 peptides (100 μM) were incubated with the recombinantly expressed human GLP or G9a (2 μM) in the presence of SAM as the cosubstrate (500 μM) for 3 hours at 37 °C, while conversion was also checked after 1 hour, according to previously reported standard conditions.14 This revealed predominant dimethylation and trimethylation of the H3K9 peptide already after 1 hour for both KMTs, while the reaction was also found to be dependent on both the presence of the enzyme and SAM (Fig. S9†). The H3K9 peptide was found to be fully trimethylated after 3 hours (Fig. 3a). Among the panel of lysine analogues, only the H3 peptide containing KGly (H3KGly9) was found to be accepted as a GLP/G9a substrate (Fig. 3b and S10†).
 | ||
| Fig. 3 MALDI-TOF MS data showing methylation of H3 peptides (100 μM) in presence of GLP (2 μM) and SAM (500 μM) after 3 hours at 37 °C at pH 8.0. Control reactions without GLP present are shown in black, GLP-catalysed reactions are shown in red. (a) H3K9, (b) H3KGly9, (c) H3KL-Ala9, (d) H3KD-Ala9, (e) H3KL-Abu9, (f) H3KD-Abu9, (g) H3KL-Phe9, (h) H3KD-Phe9. | ||
Interestingly, only dimethylation was observed for the analogue under standard conditions, reaching completion after 3 hours. This finding might indicate that while the analogue is well accommodated in the KMTs binding pocket, an extra barrier exists to reach the final trimethylated product, as found for lysine. This can be attributed to the limited flexibility of lysine's side chain in the case of H3KGly9 due to introduction of the amide functionality and the increased steric demand of the two Nε-methyl groups present. Previously, it was shown that other analogues also only reach the dimethyl stage when reacted with GLP and G9a.15,18,19 To assess this finding, H3KGly9 (20 μM) was incubated with GLP (2 μM) and SAM (500 μM), to obtain a lower ratio between peptide and substrate. Under these conditions, dimethylation was readily observed and traces of trimethylation were detected (Fig. S11†). Then, H3KGly9 was incubated with a higher concentration of GLP (10 μM) and cosubstrate (1 mM). This showed that at higher enzyme and cosubstrate concentration, GLP does have the ability to predominantly catalyse trimethylation of H3KGly9 (Fig. S12†). This result further suggests the extra barrier for trimethylation, indicating that a higher enzyme concentration plays a role in pushing the reaction to completion. Meanwhile, all other analogues, derived from amino acids that contain some degree of steric bulk in their side chains, were not methylated, within the limits of detection (Fig. 3c–h and Fig. S10†). Seeing that higher methylation states can be reached for H3KGly9 under optimised conditions, all other lysine analogues were incubated under these conditions (Fig. S11 and S12†). It was found that for all analogues, traces of monomethylation and dimethylation could be observed in varying degrees, with high enzyme concentration (10 μM) resulting in slightly higher conversion, as already observed for H3KGly9. Specifically, peptides H3KL-Ala9, H3KL-Abu9 and H3KD-Abu9 were found to be dimethylated to a minor degree. These results strongly indicate that while the presence of the amide bond in the side chain itself is tolerated, introduction of additional steric bulk at the ε-position leads to a loss of enzymatic methylation activity. To evaluate and compare the progression of the methylation of H3KGly9 (100 μM) at 2 and 10 μM of GLP, a timecourse experiment was performed under previously stated conditions, taking aliquots of the enzymatic reactions at several time points, and plotting the methylated species over time (Fig. S13†). A clear increase of higher methylation states was observed over time, at both 2 μM and 10 μM GLP, with a trimethylated product only evident in the latter case (Fig. S13†).
Similarly, to evaluate KAT-catalysed acetylation, H3 peptides (100 μM) were incubated with human GCN5 or PCAF (2 μM) and AcCoA as the cosubstrate (300 μM) for 3 hours at 37 °C.14 Full conversion of H3K9 to H3K9ac was reached already after 1 hour, with the dependence on the enzyme and cosubstrate proven in controls (Fig. 4a and S9†). Interestingly, none of the H3 peptides bearing lysine analogues was accepted as a substrate for KATs within the detection limit of our assay, suggesting that introduction of the amide functionality in the side chain is not tolerated by these representative KATs (Fig. 4 and S14†). Increasing the concentration of GCN5 and PCAF to 10 μM resulted in a trace of acetylation for H3KGly9 (Fig. S15†), whereas other lysine analogues were not acetylated within the limit of detection.
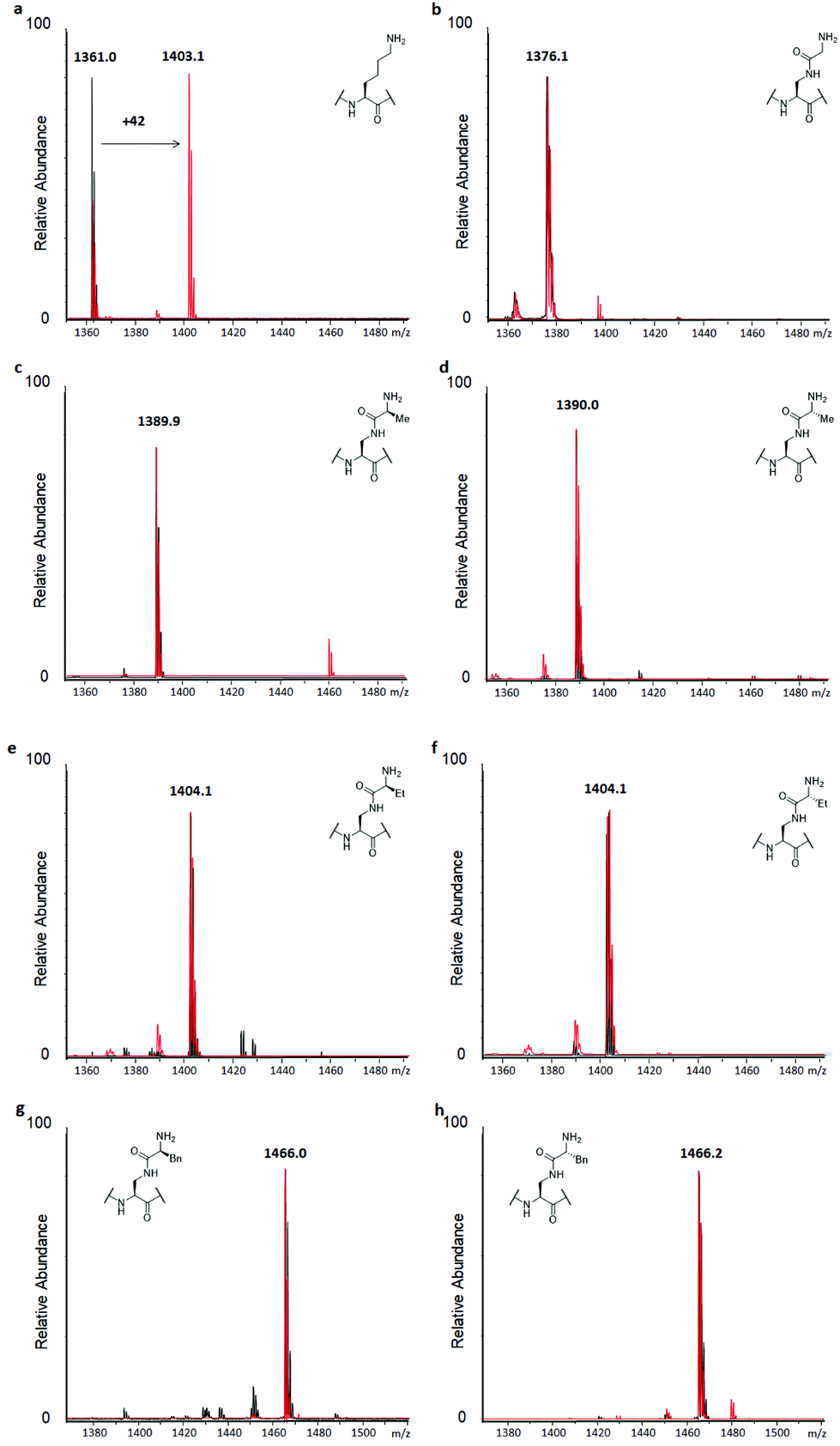 | ||
| Fig. 4 MALDI-TOF MS data showing acetylation of H3 peptides (100 μM) in the presence of GCN5 (2 μM) and AcCoA (300 μM) after 3 hours at 37 °C at pH 8.0. Control reactions without GCN5 present are shown in black, GCN5-catalysed reactions are shown in red. (a) H3K9, (b) H3KGly9, (c) H3KL-Ala9, (d) H3KD-Ala9, (e) H3KL-Abu9, (f) H3KD-Abu9, (g) H3KL-Phe9, (h) H3KD-Phe9. | ||
Having established H3KGly9 as a substrate for both GLP and G9a, we set out to evaluate the kinetic parameters of H3KGly9 in comparison to the H3K9 peptide using MALDI-TOF MS assays. To obtain an accurate representation of the kinetic profile of substrate methylation, it must be ensured that the lysine and its mimic are only monomethylated during the enzymatic reaction. Therefore, previously reported conditions were employed, using GLP/G9a (100 nM), SAM (5 μM) and a varying concentration of the H3K9 peptide between 5 and 200 μM.14 All reactions were carried out for 5 minutes at 37 °C. However, under these conditions, methylation of the H3KGly9 peptide proved to be too low for accurate determination of conversion, and therefore the enzyme concentration was increased to 400 nM. This revealed that both H3 peptides behaved similarly for both G9a and GLP (Fig. 5, Table 1). Furthermore, H3KGly9 was found to have about 4- to 5-fold decrease in enzyme efficiency (kcat/KM) in comparison to lysine. The loss in activity can be attributed to both an increased KM and a decreased kcat, unsurprisingly, the same pattern is observed for both enzymes.
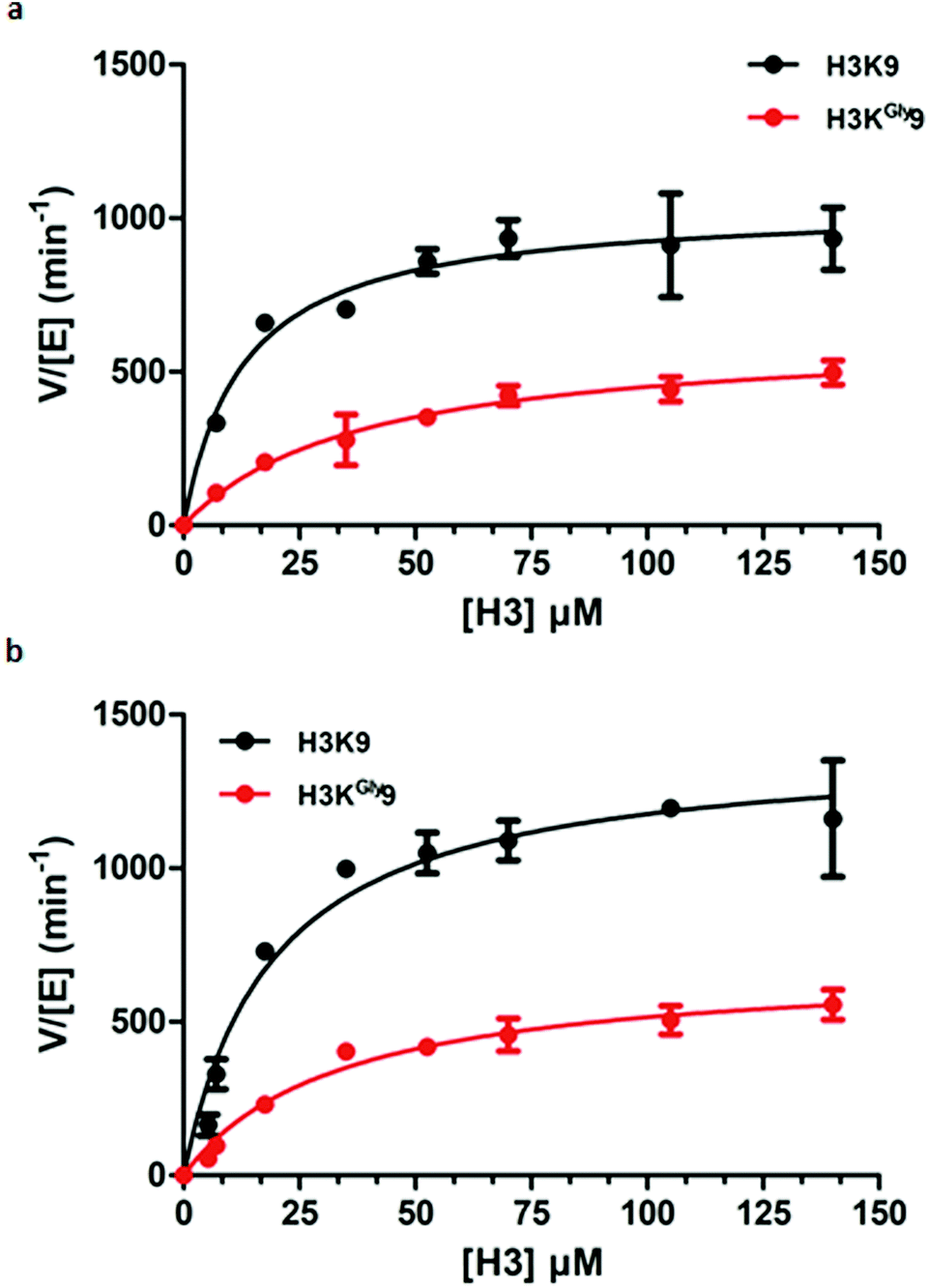 | ||
| Fig. 5 Kinetic plots of (a) G9a-catalysed methylation of H3K9 (black) and H3KGly9 (red) and (b) GLP-catalysed methylation of H3K9 (black) and H3KGly9 (red). | ||
| k cat (min−1) | K M (μM) | k cat/KM (μM−1 min−1) | |
|---|---|---|---|
| GLP | |||
| H3K9 | 1401 ± 79 | 19.1 ± 3.8 | 73.4 |
| H3KGly9 | 687 ± 44 | 33.7 ± 6.1 | 20.4 |
| G9a | |||
| H3K9 | 1042 ± 58 | 12.7 ± 3.1 | 82.0 |
| H3KGly9 | 624 ± 55 | 38.5 ± 9.2 | 16.2 |
To investigate the methylation of H3KGly9 by GLP and further validating it as a substrate, we examined the enzymatic reactions by NMR spectroscopy (Fig. 6). The H3KGly9 peptide (400 μM) or unmodified H3K9 (400 μM) as a control were incubated with GLP (8 μM) and SAM (2 mM) in Tris-d11 buffer (pD = 8.0) for 1 h at 37 °C. The samples were then transferred to an NMR tube and subjected to 1H and 1H–13C HSQC NMR experiments. The conversion of the H3K9 control resulted in an appearance of a resonance at 3.04 ppm (NMe3) corresponding to H3K9me3 with a corresponding resonance of 52.6 ppm for 13C (NMe3), in line with previous work (Fig. 6a and c).14 For the H3KGly9 peptide, under the employed conditions, a resonance appeared at 3.23 ppm (NMe3) with a corresponding 13C resonance of 54.6 ppm (NMe3), indicating trimethylation (Fig. 6b and d). The conversion of SAM to SAH, with the appearance of a triplet at 2.61 ppm (SAH-CH2γ), was observed in both reactions (Fig. 6a and b). The relative higher enzyme concentration used in the NMR experiments and the long measurement time (22 h) can result in the complete formation of H3KGly9me3.
 | ||
| Fig. 6 NMR analysis of GLP-catalysed methylation of H3K9 and H3KGly9. (a) 1H NMR spectrum showing GLP-catalysed methylation of H3K9, (b) 1H NMR spectrum showing GLP-catalysed methylation of H3KGly9, (c) 1H–13C HSQC (red = CH/CH3, blue = CH2) spectrum showing GLP-catalysed methylation of H3K9, (d) 1H–13C HSQC (red = CH/CH3, blue = CH2) spectrum showing GLP-catalysed methylation of H3KGly9. | ||
Peptides that were not found to be accepted as enzyme substrates, were then evaluated for potential inhibition of both lysine methylation and acetylation reactions. A single point screen was carried out where the analogue (10 or 100 μM) was preincubated with GLP (200 nM) and SAM (100 μM) for 5 minutes, after which H3K9 (10 μM) was added and reacted for the additional 30 minutes (Fig. 7 and Fig. S16†). Interestingly, peptides H3KD-Abu9, H3KL-Phe9 and H3KD-Phe9 showed residual enzyme activity well below 50% at 10 μM of inhibitor already, while H3KL-Abu9 showed a residual activity around 50%, suggesting strong inhibition by these amino acid side chains (Fig. 7). Out of these peptides, the former three in particular showed good inhibition at this concentration, suggesting that KMTs can accommodate the bulkier side chain containing lysine analogues, but are prevented from an efficient methyl transfer. To gain a more detailed insight into their inhibitory properties, IC50 values were determined for H3KL-Abu9, H3KD-Abu9, H3KL-Phe9 and H3KD-Phe9 and were found to be between 21.6 and 1.29 μM (Table 2 and Fig. S17†). Previously, an H3 peptide bearing a meta-aminophenylalanine residue at position 9 was found to have an IC50 of 26 μM.18 H3KL-Abu9 was found to have a similar IC50 value, but our most potent inhibitor, H3KD-Abu9, has a 20-fold stronger inhibition (IC50 = 1.29 μM), pushing the limits of peptide-based inhibitors of GLP.
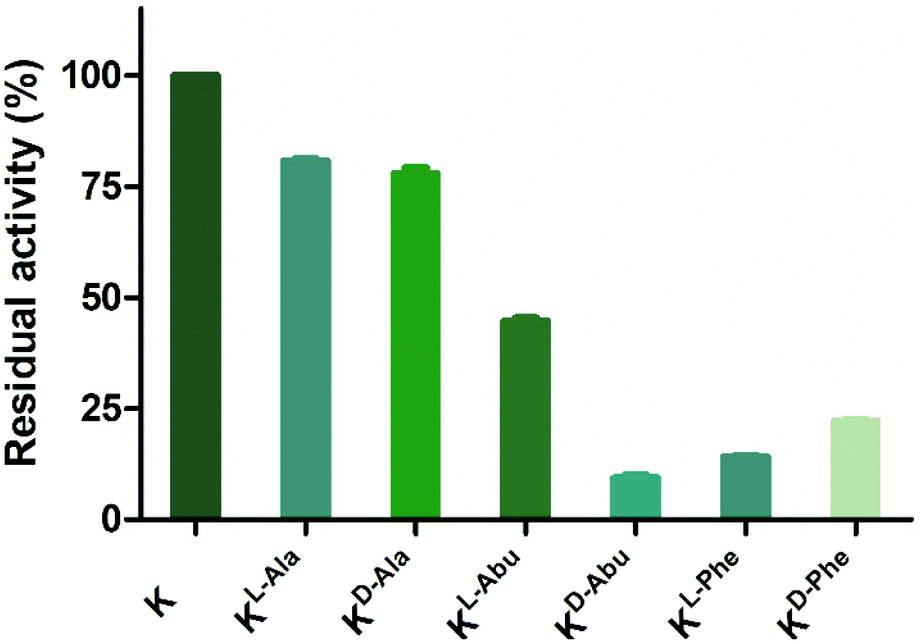 | ||
| Fig. 7 Single point inhibition of GLP (200 nM) by H3K9 peptides (10 μM). Error bars reported as standard error (SE), experiments carried out in duplicate. | ||
| Peptide | IC50 (μM) | Peptide | IC50 (μM) |
|---|---|---|---|
| H3KL-Abu9 | 21.6 | H3KL-Phe9 | 1.74 |
| H3KD-Abu9 | 1.29 | H3KD-Phe9 | 7.17 |
A single point screen was also carried out to assess GCN5-catalysed lysine acetylation by preincubating a competing histone peptide (10 or 100 μM) with GCN5 (500 nM) in the presence of AcCoA (100 μM) for 10 minutes. Then the H3K9 substrate (10 μM) was added to the enzymatic mixture and left for 30 minutes (Fig. S18†). Peptides H3KL-Abu9 and H3KD-Abu9 were not evaluated for acetylation inhibition due to overlapping signals in the MS analysis. These results revealed that even at 100 μM of H3 peptides present, no reduced acetylation activity was observed (Fig. S18†). This result confirms that H3 peptides possessing lysine analogues not only are non-substrates, but even do not seem to bind to the active site of GCN5 to be able to compete with H3K9 binding. These results are in line with recent examinations of other lysine analogues as substrates and inhibitors of KATs, as reflected by high KM values and poor inhibitory potency.16,17,20
Computational analysis was then performed to gain insight into potential binding conformations of the lysine analogue-containing H3 peptides and to shed light upon the observed selectivity for GLP-mediated methylation of H3KGly9 (Fig. 8 and Fig. S19†). Dockings of H3K9 peptides using the X-ray structure of GLP-H3K9me2-SAH ternary complex (PDB-ID: 2RFI26) with mildly restrained peptide backbones illustrate that the modified H3KGly9 side chain adopts a similar binding pose to substrate H3K9 (Fig. 8a). The remaining analogues were similarly observed to position their side chains in conformations comparable to H3K9 (Fig. 8b). Unlike H3KGly9, however, none of the other lysine analogues seem to position its ε-amino group towards the S atom of SAH (the product of SAM-mediated methylation reaction), with the introduced aliphatic side chains of Ala, Abu and Phe being positioned towards SAH instead (Fig. S19†). Interestingly, the introduction of an amide to the analogue side chains potentially allows for the formation of additional hydrogen bonding interactions with the enzyme's L1143 backbone in a β-sheet like manner, while for both H3KL-Phe9 and H3KD-Phe9 binding is enhanced through π–π stacking with Y1221 of GLP, presumably contributing to high potency of both peptides (Fig. 8b and Fig. S19†). Similarly, the amine group in H3KL-Abu9 points away from the active site Y1221, while the amine in H3KD-Abu9 directly faces this residue, indicating that the protonated Nε-amino group can interact with the aromatic ring of Y1221 via favourable cation–π interactions. This difference possibly gives rise to a stronger binding interaction with the enzyme and the improved IC50 observed for H3KD-Abu9 (Fig. S20†).
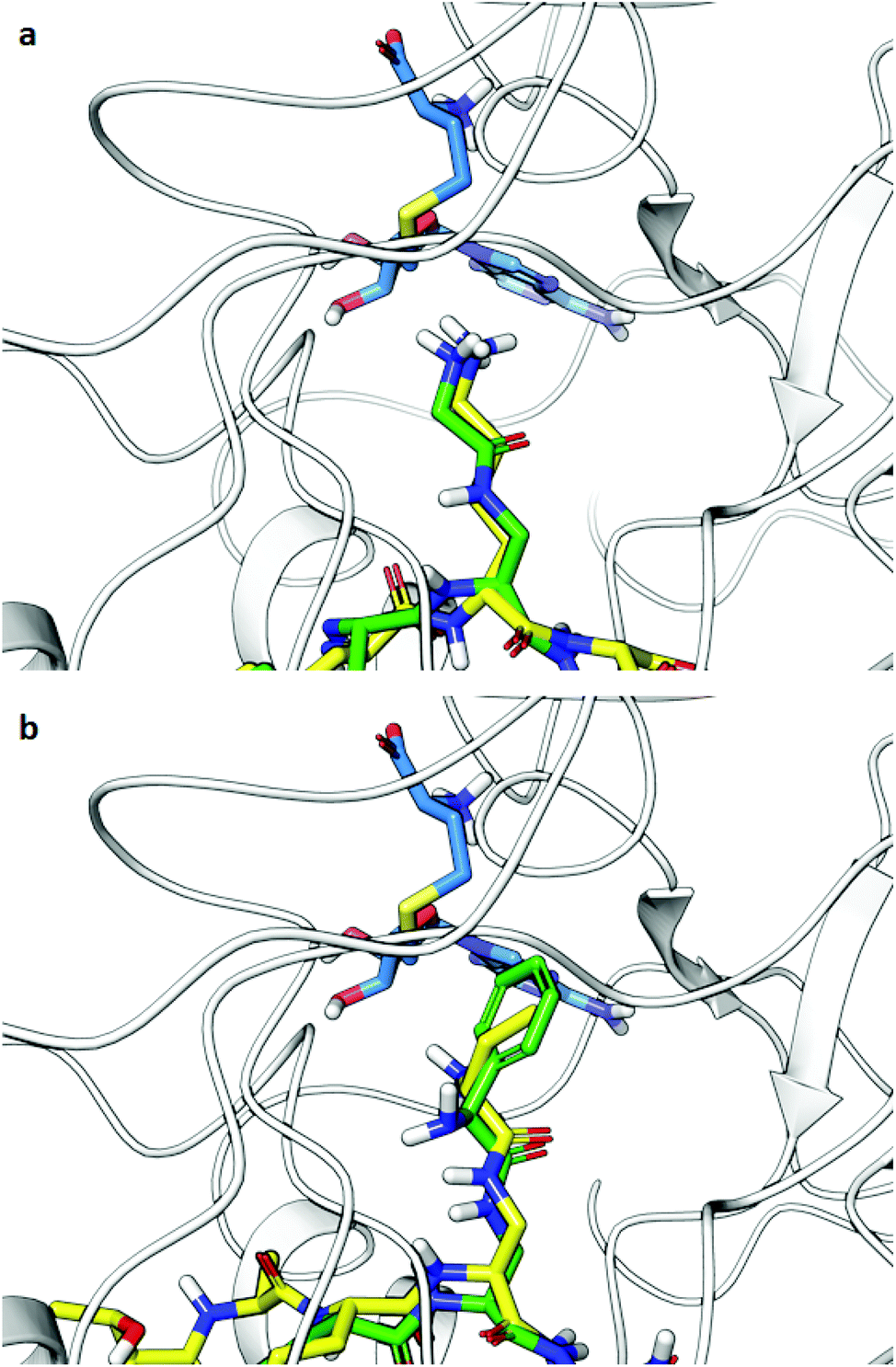 | ||
| Fig. 8 Docking poses of H3 peptides possessing lysine analogues (residues 6–11) in GLP (PDB-ID: 2RFI): (a) overlay of substrates H3K9 (yellow) and H3KGly9 (green), (b) overlay of inhibitors H3KD-Abu9 (yellow) and H3KL-Phe9 (green). SAH is shown in blue. | ||
Conclusions
In conclusion, we have synthesised seven novel Fmoc-protected lysine analogues by functionalising the amine side chain amine of Dap with N-Boc protected amino acids. The analogues were subsequently incorporated into histone H3 peptides and assayed with human histone lysine methyltransferases and acetyltransferases. Our results demonstrate that methyltransferases appear to be more promiscuous than acetyltransferases towards these analogues, with KGly being accepted as a substrate and its sterically more demanding mimics found as potent GLP inhibitors. Acetyltransferases did not catalyse acetylation of the H3 peptides possessing amide-derived lysine analogues, nor were they inhibited by such peptides. Furthermore, we have confirmed enzymatic methylation of the histone peptide bearing KGly by elegant NMR studies. The analogues used in this work further exemplify different substrate specificities of two biomedically important enzyme classes, the KMTs and KATs, allowing to gain a deeper understanding of their molecular requirements for catalysis.Experimental
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 water:MeCN, v/v). The compounds were lyophilized to obtain compounds 9–15 as white solids which were used in the SPPS.
:4 water:MeCN, v/v). The compounds were lyophilized to obtain compounds 9–15 as white solids which were used in the SPPS.
Synthesis and purification of histone peptides
The H3 13-mer peptides (ARTKQTAR![[X with combining low line]](https://www.rsc.org/images/entities/b_char_0058_0332.gif) 9STGG) were chain assembled on Rink amide resin using manual solid phase peptide synthesis. Amino acid couplings were carried out with the equivalent ratio of [5]:[5]:[7.5] of [Fmoc-protected amino acid]:[HATU]:[DIPEA] at room temperature for 30 minutes. Analogues of lysine were coupled by manual SPPS with the equivalent ratio of [5]:[5]:[7.5] of [Fmoc-protected amino acid]:[HATU]:[DIPEA] overnight. Full conversion was checked by Kaiser test and further amino acids were coupled manually with the equivalent ratio of [5]:[5]:[7.5] of [Fmoc-protected amino acid]:[HATU]:[DIPEA] for 1 hour. The peptide proceeded to standard cleavage from resin using 0.5% TIPS, 0.5% H2O in conc. TFA for 3 hours. TFA was blown off using N2 and the resultant residue suspended in cold Et2O. After suspension it was subjected to centrifugation for 5 minutes at 5000 rpm in an Eppendorf 5804R centrifuge (Eppendorf, Hamburg, Germany) after which the supernatant was decanted into the waste. The remaining white to yellow solid was washed twice by cold Et2O and subjected to centrifugation after which the crude peptide was dissolved in a mixture of ACN in H2O and purified using preparative reverse-phase HPLC (RP-HPLC) using a gradient of H2O + 0.1% TFA and ACN + 0.1% TFA from 5% ACN to 30% ACN over 30 minutes at 4 ml min−1 using a Gemini 10 μm NX-C18 110 Å LC column (Phenomenex, Torrance, CA, USA). Analytical RP-HPLC was carried out on a Gemini 5 μm C18 110 Å LC column (Phenomenex) at a flow rate of 1 mL min−1. Analytical injections were monitored at 215 nm. A gradient was used of 0% ACN to 3% ACN over 10 minutes and 3% ACN to 100% ACN in 20 minutes. The mass of the peptide was confirmed by mixing with a 1:2 with α-cyano-4-hydroxycinnamic acid (CHCCA) matrix and loaded onto an MTP 384 polished steel target to be analysed by a MALDI-TOF UltrafleXtreme-II tandem mass spectrometer (Bruker).
9STGG) were chain assembled on Rink amide resin using manual solid phase peptide synthesis. Amino acid couplings were carried out with the equivalent ratio of [5]:[5]:[7.5] of [Fmoc-protected amino acid]:[HATU]:[DIPEA] at room temperature for 30 minutes. Analogues of lysine were coupled by manual SPPS with the equivalent ratio of [5]:[5]:[7.5] of [Fmoc-protected amino acid]:[HATU]:[DIPEA] overnight. Full conversion was checked by Kaiser test and further amino acids were coupled manually with the equivalent ratio of [5]:[5]:[7.5] of [Fmoc-protected amino acid]:[HATU]:[DIPEA] for 1 hour. The peptide proceeded to standard cleavage from resin using 0.5% TIPS, 0.5% H2O in conc. TFA for 3 hours. TFA was blown off using N2 and the resultant residue suspended in cold Et2O. After suspension it was subjected to centrifugation for 5 minutes at 5000 rpm in an Eppendorf 5804R centrifuge (Eppendorf, Hamburg, Germany) after which the supernatant was decanted into the waste. The remaining white to yellow solid was washed twice by cold Et2O and subjected to centrifugation after which the crude peptide was dissolved in a mixture of ACN in H2O and purified using preparative reverse-phase HPLC (RP-HPLC) using a gradient of H2O + 0.1% TFA and ACN + 0.1% TFA from 5% ACN to 30% ACN over 30 minutes at 4 ml min−1 using a Gemini 10 μm NX-C18 110 Å LC column (Phenomenex, Torrance, CA, USA). Analytical RP-HPLC was carried out on a Gemini 5 μm C18 110 Å LC column (Phenomenex) at a flow rate of 1 mL min−1. Analytical injections were monitored at 215 nm. A gradient was used of 0% ACN to 3% ACN over 10 minutes and 3% ACN to 100% ACN in 20 minutes. The mass of the peptide was confirmed by mixing with a 1:2 with α-cyano-4-hydroxycinnamic acid (CHCCA) matrix and loaded onto an MTP 384 polished steel target to be analysed by a MALDI-TOF UltrafleXtreme-II tandem mass spectrometer (Bruker).
Expression and purification of lysine methyltransferases and acetyltransferases
Human KMT enzymes were expressed and purified as previously described.27 The GLP (residues 951–1235) and G9a (residues 913–1193) plasmids were transformed into E. coli Rosetta BL21 (DE3)pLysS cells. Purification of the N-terminally His-tagged GLP and G9a was performed using Ni–NTA affinity chromatography column, which was prewashed with lysis buffer. GLP and G9a enzyme was then eluted using a linear gradient concentration of imidazole. The enzymes were further purified by gel filtration on a Superdex 75 column (GE Healthcare) on an AKTA system. Protein concentrations were determined using the Nanodrop DeNovix DS-11 spectrophotometer and the purity was monitored by SDS-PAGE on a 4–15% gradient polyacrylamide gel (Bio-Rad).Plasmid carrying recombinant His-tagged Human GCN5 catalytic domain (residues 497–662 in pET28a-LIC vector) was purchased from Addgene (25482). The protein was expressed and purified as previously described.28 Plasmid carrying recombinant SNAP-His-tagged Human PCAF catalytic domain (residues 498–658 in pET16b vector), kindly provided by Professor Milan Mrksich (Northwestern University), was expressed and purified as previously described.17 Purity of the eluted proteins was assessed with SDS-PAGE, and was separated in aliquots, rapidly flash-frozen and stored at −80 °C.
Enzymatic assays
Histone methyltransferase assays were carried out as described in 30 μL final total volume.24 In brief, purified GLP/G9a (2 or 10 μM) was incubated with 20 or 100 μM of purified histone peptides in the methyltransferase assay buffer Tris HCl (pH 8.0) and methyl donor SAM (500 μM or 1 mM) at 37 °C. Histone acetyltransferase assays were carried out by incubating peptides under standard conditions (2 or 10 μM GCN5/PCAF, 100 μM peptide, 300 or 500 μM AcCoA) in the reaction buffer (50 mM HEPES, 0.1 mM EDTA, 1 mM DTT, pH = 8.0). The reactions were carried out in a final volume of 50 μL. All reactions were shaken in a Thermomixer C at 750 rpm, at 37 °C. All reactions were quenched by the addition of 10% TFA in Milli-Q water at different time points. All reactions were aliquoted and mixed 1:2 with a solution of α-cyano-4-hydroxycinnamic acid (CHCCA) in 1:1 MQ and ACN (0.1% TFA) and loaded onto an MTP 384 polished steel target to be analysed by a UltrafleXtreme-II tandem mass spectrometer (Bruker, Billerica, MA, USA).
Histone lysine methyltransferase kinetics studies were performed as described.24 A solution of histone peptide (0–200 μM), was added to a solution of SAM (5 μM) in assay buffer (50 mM Tris, pH 8.0) at room temperature (final volume of 30 μL). The reaction was then initiated by the addition of GLP or G9a (100 nM) and shaken for 5 min. Reactions were incubated at 37 °C, shaken at 750 rpm and quenched with TFA 10% in Milli-Q water at different time points, within linear production of monomethylated peptides extrapolated from timecourse plots. All reactions were aliquoted and mixed 1:2 with a solution of α-cyano-4-hydroxycinnamic acid (CHCCA) in 1:1 MQ and ACN (0.1% TFA) and loaded onto a MTP 384 polished steel target to be analysed by UltrafeXtreme-II tandem mass spectrometer (Bruker). The amount of methylated peptide was calculated by integration of the product peak area and divided it by the amount of unmodified peptide, taking in account all the ionic species, at any concentration points using the FlexAnalysis software. Kinetic values were extrapolated by fitting V0 values (μM of produced peptide per minutes) and histone peptide concentrations to the Michaelis–Menten equation using the GraphPad Prism 5 software. Kinetic experiments were carried out in duplicates and final values are reported as value ± SD.
Inhibition assays
GLP inhibition assays were performed by preincubating a mixture of histone peptide (0–250 μM) and GLP (100 nM final concentration) for 5 minutes at 37 °C in 25 mM Tris–HCl buffer pH 8. Then 2 μL of a pre-mixture of SAM (50 μM) and H3K9 peptide (100 μM) was added and the enzymatic reaction was incubated for an additional 10 minutes before quenching with 10% TFA in Milli-Q water. GCN5 inhibition assays were performed by preincubating a mixture of histone peptide (10 or 100 μM) and GCN5 (200 nM) for 10 minutes at 37 °C in 25 mM Tris–HCl buffer pH 8. Then AcCoA and H3K9 peptide were added to a final concentration of 300 μM and 100 μM, respectively, and the enzymatic reaction was incubated for an additional 20 minutes before quenching with 10% TFA in Milli-Q water. The samples were then aliquoted and mixed 1:1 with CHCCA matrix and loaded onto a MTP 384 polished steel target to be analysed by UltrafeXtreme-II tandem mass spectrometer (Bruker). Residual enzymatic activity was determined by calculating the relative integral of the methylated/acetylated peptide to a control reaction in absence of potential inhibitory peptides. Experiments were carried out in replicate.
Docking
The crystal structure of GLP in complex with a H3K9me2 peptide (PDB ID: 2RFI, X-ray crystal structure resolution 1.59 Å) was obtained from the Protein Data Bank. 2RFI as imported into Schrödinger Suite's Maestro module.292RFI was pre-processed by adding missing hydrogen atoms, removing water molecules, and assigning bond orders. The PROPKA tool was utilized to determine the protonation states at pH = 7.0, and restrained minimization of the hydrogen atoms was executed using the OPLS_2005 force field to alleviate steric contacts.30 H3 tail sequences containing lysine analogues were imported into the Maestro module and minimized tautomers were generated using the LigPrep tool by applying force field minimization (OPLS_2005) and ionization at pH 7.0 (Epik).31,32 A receptor grid was generated of the 2RFI structure using Glide with initial van der Waals scaling of 1 Å. The grid box was defined using H3K9 as the centroid. Unmodified 6-mer H3 (TARKST) peptide was docked into 2RFI using the generated receptor grid with Glide using the Extra Precision (XP) mode with ligand flexibility.33 The resulting predicted H3–2RFI complex was subsequently used for Induced Fit Docking (IFD) of the histone tail hexapeptides using the XP-docking mode.34 IFD was executed by using initial van der Waals scaling of 0.5 for the receptor and ligand, ligand backbone restrainment within 2 Å of the docked H3 (TARKST) backbone, and with refinement of receptor residues within 5 Å of the docking ligands.NMR assays
Histone lysine methyltransferase NMR studies were carried out in a total volume of 500 μL. GLP (8 μM) was added to a solution of histone peptide (400 μM) and SAM (2 mM) in 50 mM Tris-D11 buffer pD 8.0. After shaking at 750 rpm for 1 hour at 37 °C in Eppendorf, the reaction mixture was transferred to an NMR tube. NMR spectroscopy was conducted on a Bruker 500 MHz Advance III equipped with a Prodigy cryoprobe.Author contributions
J. Mx. and S. G. H. A. L. synthesised lysine analogues. M. N. M. and J. C. J. H. synthesised and purified histone peptides. J. C. J. H. carried out enzymatic, kinetic and inhibition assays. M. N. M. performed docking. J. Mx. and P. B. W. carried out NMR experiments. J. C. J. H., J. Mx., M. N. M. and J. Mc. wrote the original draft. T. J. B. and J. Mc. supervised the project. All authors edited and agreed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the European Research Council (ERC Starting Grant to J. M., ChemEpigen-715691, and ERC Starting Grant to T. J. B., GlycoEdit-758913).References
- A. J. Bannister and T. Kouzarides, Cell Res., 2011, 21, 381–395 CrossRef CAS PubMed.
- B. D. Strahl and C. D. Allis, Nature, 2000, 403, 41–45 CrossRef CAS PubMed.
- K. Hyun, J. Jeon, K. Park and J. Kim, Exp. Mol. Med., 2017, 49, e324 CrossRef CAS.
- J. C. Black, C. van Rechem and J. R. Whetstine, Mol. Cell, 2012, 48, 491–507 CrossRef CAS PubMed.
- C. Martin and Y. Zhang, Mol. Cell Biol., 2005, 6, 838–849 CAS.
- M. Luo, Chem. Rev., 2018, 118, 6656–6705 CrossRef CAS.
- C. Choudhary, B. T. Weinert, Y. Nishida, E. Verdin and M. Mann, Nat. Rev. Mol. Cell Biol., 2014, 15, 536–550 CrossRef CAS PubMed.
- R. Marmorstein and M.-M. Zhou, CSH Perspect. Biol., 2014, 6, a018762 Search PubMed.
- A. J. Bannister and E. A. Miska, Cell. Mol. Life Sci., 2000, 57, 1184–1192 CrossRef CAS PubMed.
- A. Drazic, L. M. Myklebust, R. Ree and T. Arnesen, BBA – Proteins Proteom., 2016, 1864, 1372–1401 CrossRef CAS.
- D. Husmann and O. Gozani, Nat. Struct. Mol. Biol., 2019, 26, 880–889 CrossRef CAS.
- N. Avvakumov and J. Cote, Oncogene, 2007, 26, 5395–5407 CrossRef CAS PubMed.
- M. N. Maas, J. C. J. Hintzen, M. R. B. Porzberg and J. Mecinovic, Int. J. Mol. Sci., 2020, 21, 9451 CrossRef CAS.
- J. C. J. Hintzen, Y. Luo, M. R. B. Porzberg, P. B. White, J. Jian, G. Proietti and J. Mecinovic, Chem. Commun., 2021, 57, 6788–6791 RSC.
- A. H. K. Al Temimi, J. Merx, C. J. van Noortwijk, G. Proietti, R. Buijs, P. B. White, F. P. J. T. Rutjes, T. J. Boltje and J. Mecinovic, Sci. Rep., 2020, 10, 21574 CrossRef CAS PubMed.
- G. Proietti, Y. Wang, G. Rainone and J. Mecinovic, Sci. Rep., 2020, 10, 13046 CrossRef CAS PubMed.
- G. Proietti, G. Rainone, J. C. J. Hintzen and J. Mecinovic, Bioconjugate Chem., 2020, 31, 844–851 CrossRef CAS.
- A. H. K. Al Temimi, V. Tran, R. S. Teeuwen, A. J. Altunc, H. I. V. Amatdjais-Groenen, P. B. White, D. C. Lenstra, G. Proietti, Y. Wang, A. Wegert, R. H. Blaauw, P. Qian, W. Ren, H. Guo and J. Mecinovic, Sci. Rep., 2020, 10, 3671 CrossRef CAS.
- A. H. K. Al Temimi, H. I. V. Amatdjais-Groenen, Y. V. Reddy, R. H. Blaauw, H. Guo, P. Qian and J. Mecinovic, Commun. Chem., 2019, 2, 112 CrossRef.
- G. Proietti, Y. Wang, C. Punzo and J. Mecinovic, Int. J. Mol. Sci., 2021, 22, 846 CrossRef CAS.
- A. H. K. Al Temimi, P. B. White, M. J. M. Mulders, N. G. A. van der Linden, R. H. Blaauw, A. Wegert, F. P. J. T. Rutjes and J. Mecinovic, Chem. Commun., 2020, 56, 3039–3042 RSC.
- A. H. K. Al Temimi, R. van der Wekken-de Bruijne, G. Proietti, H. Guo, P. Qian and J. Mecinovic, Bioconjugate Chem., 2019, 30, 1798–1804 CrossRef CAS.
- T. Yang, X.-M. Li, X. Bao, Y. M. E. Fung and X. D. Li, Nat. Chem. Biol., 2016, 12, 70–72 CrossRef CAS.
- A. H. K. Al Temimi, Y. V. Reddy, P. B. White, H. Guo, P. Qian and J. Mecinovic, Sci. Rep., 2017, 7, 16148 CrossRef PubMed.
- R. Belle, A. H. K. Al Temimi, K. Kumar, B. J. G. E. Pieters, A. Tumber, J. E. Dunford, C. Johansson, U. Oppermann, T. Brown, C. J. Schofield, R. J. Hopkinson, R. S. Paton, A. Kawamura and J. Mecinovic, Chem. Commun., 2017, 53, 13264–13267 RSC.
- H. Wu, J. Min, V. V. Lunin, T. Antoshenko, L. Dombrovski, H. Zeng, A. Allali-Hassani, V. Campagna-Slater, M. Vedadi and C. H. Arrowsmith, PLoS One, 2010, 5, e8570 CrossRef.
- Y. Shinkai and M. Tachibana, Genes Dev., 2011, 25, 781–788 CrossRef CAS.
- A. Schütz, G. Bernstein, A. Dong, T. Antoshenko, H. Wu, P. Loppnau, A. Bochkarev and A. N. Plotnikov, Proteins, 2007, 68, 403–407 CrossRef PubMed.
- Schrödinger Release 2021-1: Maestro, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
- Schrödinger Release 2021-1: Protein Preparation Wizard: Epik, Schrödinger, LLC, New York, NY, 2021 Search PubMed; Impact, Schrödinger, LLC, New York, NY, 2021 Search PubMed; Prime, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
- Schrödinger Release 2021-1: LigPrep, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
- Schrödinger Release 2021-1: Epik, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
- Schrödinger Release 2021-1: Glide, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
- Schrödinger Release 2021-1: Induced Fit Docking protocol; Glide, Schrödinger, LLC, New York, NY, 2021 Search PubMed; Prime, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, peptide synthesis, MALDI-MS enzyme assays, NMR spectra, docking. See DOI: 10.1039/d1ob02191e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |