
Ag(I)-Promoted homo-dimerization of 2-(alk-2-yn-1-onyl)-1-alkynylbenzenes via a [4 + 2] cycloaddition of benzopyrylium ions: access to structurally unique naphthalenes†
Beeraiah
Baire
 * and
Jampani
Santhi
* and
Jampani
Santhi
Department of Chemistry, Indian Institute of Technology Madras, Chennai, Tamil Nadu 600036, India. E-mail: beeru@iitm.ac.in
First published on 2nd December 2021
Abstract
Herein we report a Ag(I)-promoted homo-dimerization of 2-(alk-2-yn-1-onyl)-1-alkynylbenzenes for the synthesis of structurally novel and functionalized naphthalene derivatives. This transformation exhibits a broad scope for the alkyl as well as aryl groups present on alkynes. Observations made from control experiments suggest the possible mechanism as (i) the homo-dimerization of the in situ generated benzopyrylium ion intermediates through a head–tail [4 + 2] cycloaddition, followed by (ii) the competitive ring-opening vs. decarbonylative aromatization of the adduct to give formylated and deformylated naphthalenes, respectively.
Introduction
The chemistry of 2-alkynyl-1-acylbenzenes 1 through a benzopyrylium ion intermediate 2 is well known in the literature as a building block for the development of new reactions and for the synthesis of polycyclic frameworks.1 Additionally, employing the benzopyrylium ion 2 as a 4π-component in a [4 + 2] cycloaddition reaction has also been well developed (Scheme 1).2 On the other hand, the reactions of benzopyrylium ions 2′ generated from 2-(alk-2-yn-1-onyl)-1-alkynylbenzenes 1′ (R = alkyne in 1) with external nucleophiles are scarce. For example, the nucleophilic addition of H2O and ROH to the benzopyrylium ions 2′ in the presence of Ag(I) and Au(I) has been reported.3 | ||
| Scheme 1 Divergent reactivity of benzopyrylium ions generated from 2-alkynyl-1-acylbenzenes. | ||
Results and discussion
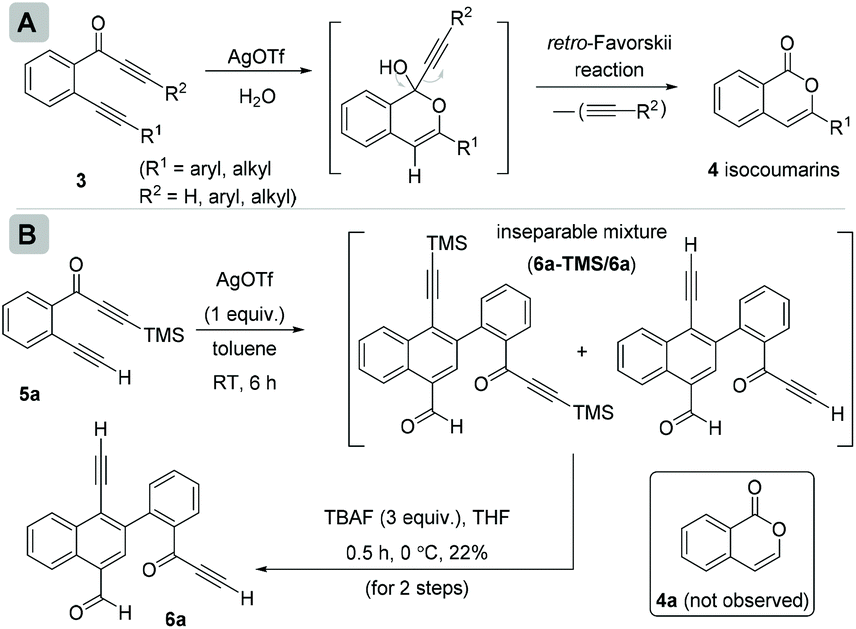
In continuation of our interest in exploring the reactivity of 2-(alk-2-yn-1-onyl)-1-alkynylbenzenes,4 recently, we reported a retro-Favorskii reaction like fragmentation of 2-(alk-2-yn-1-onyl)-1-alkynylbenzenes 3 in the presence of Ag(I)5 for the synthesis of isocoumarins 4 (Scheme 2A).4c During this study, when we employed the substrate (R1 = H, R2 = TMS) 5a, an inseparable mixture of two new compounds 6a-TMS/6a (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 ratio; one of the compounds with the TMS group, observed from the NMR spectrum) was isolated instead of isocoumarin 4a (Scheme 2B). To achieve the chromatographic separation of the two compounds, we treated the mixture with TBAF in anhydrous THF at 0 °C. Surprisingly, a single product, naphthalene 6a, was isolated in 22% overall yield in two steps. Compound 6a was found to be the same as the compound without TMS that was present in the mixture obtained earlier. The structure of 6a was confirmed by spectroscopic analysis.
:1.1 ratio; one of the compounds with the TMS group, observed from the NMR spectrum) was isolated instead of isocoumarin 4a (Scheme 2B). To achieve the chromatographic separation of the two compounds, we treated the mixture with TBAF in anhydrous THF at 0 °C. Surprisingly, a single product, naphthalene 6a, was isolated in 22% overall yield in two steps. Compound 6a was found to be the same as the compound without TMS that was present in the mixture obtained earlier. The structure of 6a was confirmed by spectroscopic analysis.
 | ||
| Scheme 2 (A) Our earlier work on 2-(alk-2-yn-1-onyl)-1-alkynylbenzenes for isocoumarins and (B) discovery of homodimerization of 2-(alk-2-yn-1-onyl)-1-alkynylbenzenes with naphthalenes. | ||
Delighted by this unexpected formation of structurally novel naphthalene, we next performed an optimization study to find out the suitable reaction conditions for the efficient and exclusive formation of these naphthalene derivatives. Accordingly, we chose 2-(alk-2-yn-1-onyl)-1-alkynylbenzene 5b as the model substrate, which possesses a terminal alkyne (R1 = H). Initially we treated 5b with AgOTf (0.5 equiv.) at 0 °C (entry 1, Table 1). To our delight, the reaction exclusively formed a separable mixture of two naphthalenes 6b (with CHO) and 7b (without CHO) in 50% and 13% respective yields, after 19 h. Next, performing the reaction at RT (entry 2) gave improved yields of products 6b (65%) and 7b (21%). Decreasing the amount of AgOTf to 0.25 equiv. at RT gave 65% and 23% yields for 6b and 7b, respectively (entry 3).
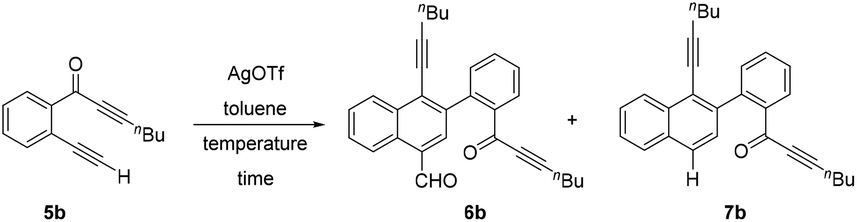
Changing the temperature to 0 °C and 50 °C with 0.25 equiv. of AgOTf (entries 4 and 5) did not improve the efficiency of the process. Similarly, performing the reaction with 0.1 equiv. of AgOTf at 0 °C and RT (entries 6 and 7) also failed to improve the yields of the products. Other metal catalysts like AgSbF6, Cu(OTf)2 and AgNO3 were found to be inefficient to promote this dimerization even when used in higher equivalents. Therefore, we chose 0.25 equiv. of AgOTf in toluene at RT (entry 3) as the suitable conditions for the exclusive and efficient formation of the two naphthalene derivatives 6b and 7b.
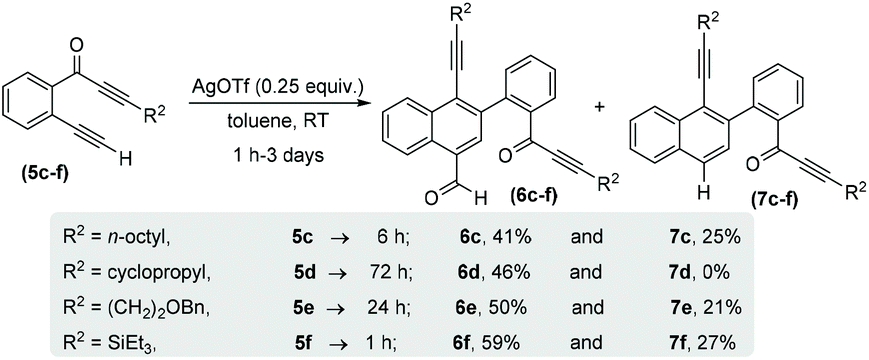
With the optimized reaction conditions in hand, we next performed a scope study to verify the generality of this process (Scheme 3). Initially, we extended this process to alkyl substituted alkynones (R2 = alkyl) 5c–f and generated structurally novel naphthalene derivatives 6c–f and 7c–f. In the case of cyclopropylalkynone 5d, no decarbonylated naphthalene 7d was observed and carbonylated naphthalene 6d was afforded as the sole product in 46% yield after 3 days. The triethylsilylalkynone 5f also afforded the corresponding carbonylated and decarbonylated naphthalenes 6f and 7f in 59% and 27% respective yields.
 | ||
| Scheme 3 Scope for aliphatic alkynones. | ||
Subsequently, diversely substituted arene attached alkynones 8a–l were also employed under the standard reaction conditions (entry 3, Table 1). When an arene attached alkynone 8a′ (R1 = H) was employed under the standard reaction conditions, the formation of the expected naphthalene derivatives 9a and 10a along with an inseparable complex mixture of by-products was observed. On the other hand, using 2-(arylalkynone)trimethylsilyl alkynylbenzene 8a, which possesses the TMS alkyne (R1 = TMS instead of H), resulted in a clean and efficient formation of the naphthalene derivatives 9a (45%) and 10a (25%) without the formation of any complex mixture (entry 1, Scheme 4). Hence, various 2-(3-aryl-2-alkyn-3-onyl)-1-alkynylbenzenes 8a–l with the TMS alkyne (R1 = TMS, R2 = Ar) and diversely functionalized arenes were synthesized and employed as substrates for the generation of a divergent library of naphthalenes 9a–l and 10a–l (Scheme 4). All these substrates 8a–l underwent the dimerization process very smoothly to give the respective naphthalene derivatives 9a–l (44–68%) and 10a–l (5–30%) in good yields. In the case of 2-(arylalkynone)alkynylbenzene 8l6 with –SiEt3 (in place of TMS, entry 12, Scheme 4) naphthalenes 9l and 10l were isolated in 50% and 23% yields, along with 14% of isocoumarin 4a.4c
 | ||
| Scheme 4 Scope study of arylalkynones. | ||
To unambiguously confirm the presence of carbonylated naphthalene and decarbonylated naphthalene frameworks, single crystal X-ray diffraction analyses were performed for compounds 9a and 10c. The ORTEP diagrams of compounds 9a and 10c are shown in Fig. 1.7a,b
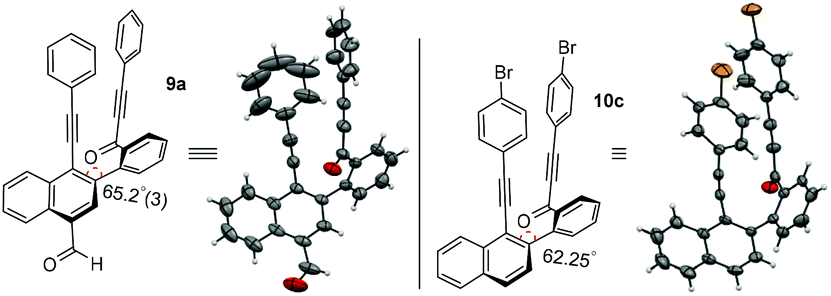 | ||
| Fig. 1 ORTEP diagram for compounds 9a and 10c. | ||
Furthermore, when we employed arylalkynones 8m–o, possessing electron-rich arenes (Scheme 5), only the corresponding formyl-napthalenes 9m–o were obtained but in relatively poor yields (compare with Scheme 4). The o-anisyl as well as 3,5-dimethoxyphenyl substrates 8m and 8n gave the corresponding formyl-napthalenes 9m and 9n in 39% and 45% respective yields (Scheme 5A). However, the p-anisyl derivative 8o gave a new product dicarbonyl 11 in 60% yield along with only 10% of naphthalene 9o after 24 h. The formation of 11 can be explained via the hydration of the benzopyrylium ion intermediate 12 followed by ring opening of the in situ generated tetrahedral intermediate 13 (Scheme 5B).4a In order to verify the intermediacy of this dicarbonyl 11 during the formation of the naphthalenes, we subjected it to the standard reaction conditions for 24 h. Surprisingly, α-naphthol 14 was isolated as the exclusive product in 87% yield possibly via an intramolecular Michael addition. There was no formation of either naphthalenes 9o or 10o even in trace amounts. This observation ruled out the possible intermediacy of dicarbonyl 11 for naphthalenes.
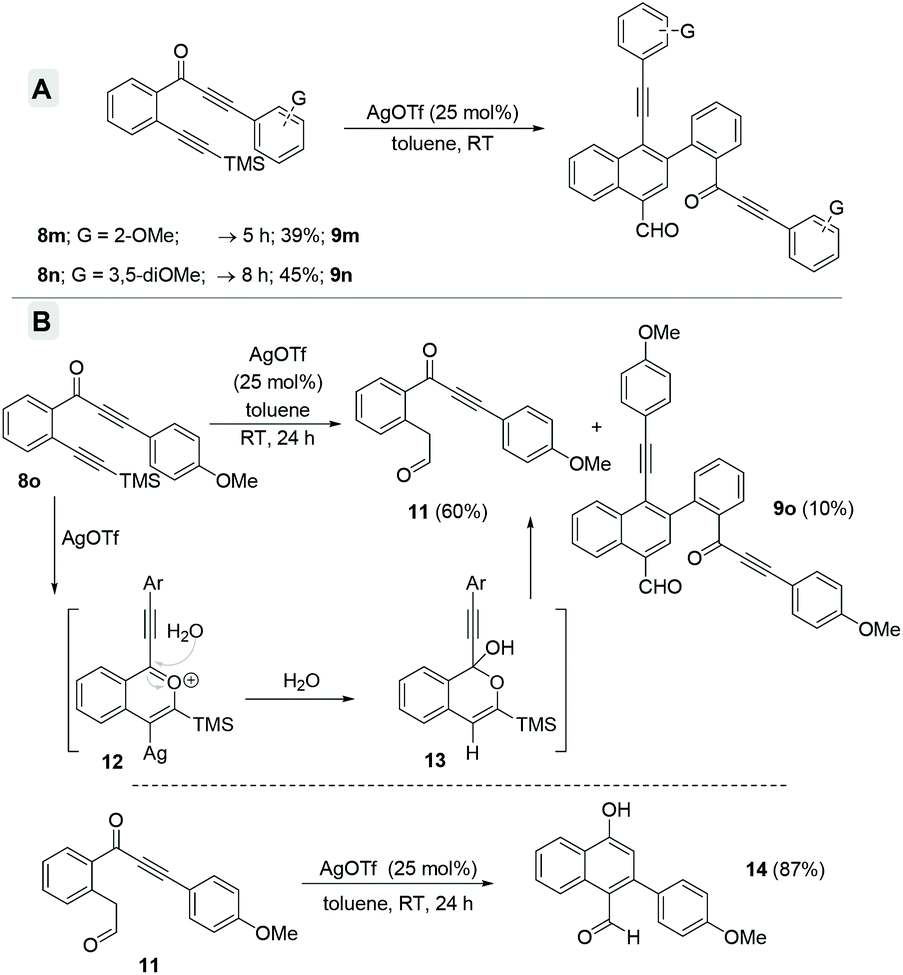 | ||
| Scheme 5 (A) Scope study of electron-rich arylalkynones and (B) employing p-anisylalkynones. | ||
After verifying the generality of this homodimerization process, next we performed a series of control experiments to understand the mechanistic details (Scheme 6). Initially, to verify the possibility of the formation of decarbonylated naphthalenes from carbonylated naphthalenes via decarbonylation, we subjected naphthaldehyde 6b to the standard reaction conditions (Scheme 6A). No traces of 7b were detected even after 14 h. This observation supported the competitive formation of both the carbonylated naphthalenes as well as decarbonylated naphthalenes under the reaction conditions.
 | ||
| Scheme 6 Control experiments: (A) failed conversion of carbonylated naphthalene to decarbonylated naphthalene; (B) attempted cross-over [4 + 2] cycloaddition of the benzopyrylium ion with alkynes; and (C) the ruled-out competitive cross [4 + 2] cycloaddition reaction between the benzopyrylium ion and alkyne. | ||
According to the literature, the Diels–Alder reactions using the benzopyrylium ion 15 as the 4π component and an alkyne or an olefin as the 2π component are known to give acylated-naphthalenes and deacylated-naphthalenes. To verify the possibility for this cross [4 + 2] cycloaddition, we performed a cross-over experiment with 5b and external 2π-components such as diphenylacetylene and diethylacetylene dicarboxylate under the standard conditions (Scheme 6B). In both cases, the cross-products 16a and 16b were not identified; instead, the regular naphthalenes 6b and 7b were isolated in comparable yields. This observation ruled out the possible cross [4 + 2] cycloaddition. Furthermore, this also suggested the possibility of a homo-dimerization via head–tail [4 + 2] cycloaddition between two molecules of the in situ generated benzopyrylium ion 15.
Based on the above observations from control experiments, a possible mechanism has been proposed (Scheme 7) for the formation of both naphthalenes 17 and 18 from 2-(alk-2-yn-1-onyl)-1-alkynylbenzenes 19. Initially, a benzopyrylium ion 20 is generated via the Ag(I)-mediated intramolecular nucleophilic attack from the carbonyl group of 19. The in situ formed benzopyrylium ion 20 undergoes a head–tail [4 + 2] cycloaddition to give the cyclic-oxonium ion 21.8 This 21 undergoes aromatization to afford the formylated-naphthalene 17. Next, 21 also undergoes a competitive decarbonylative aromatization process to give the deformylated-naphthalene 18. The whole process represents a novel homodimerization of benzopyrylium ions generated from 2-(alk-2-yn-1-onyl)-1-alkynylbenzenes.
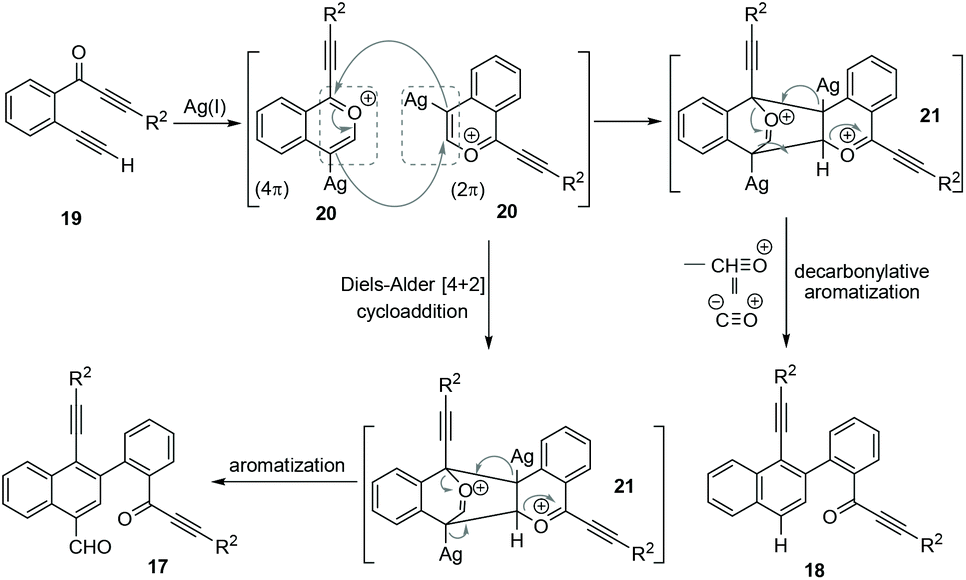 | ||
| Scheme 7 Proposed mechanism through the head–tail [4 + 2] cycloaddition of two benzopyrylium ions. | ||
Finally, the synthetic utility of the synthesized naphthalenes has also been described (Scheme 8). Heating a 1,2-DCB solution of naphthalenes 6b and 6c at 150 °C, respectively, for 21 h and 28 h gave the corresponding chrysene derivatives 22 (65%) and 23 (48%) via a thermal alkyne–carbonyl metathesis reaction.9 Single crystal X-ray diffraction analysis was also performed for compound 22 to unambiguously confirm the chrysene framework.10 The ORTEP diagram is shown in Scheme 8.
 | ||
| Scheme 8 Conversion of naphthalenes to chrysenes via a thermal alkyne–carbonyl metathesis reaction and the ORTEP diagram of 22. | ||
Conclusions
In conclusion we have developed a homodimerization of benzopyrylium ions (generated from 2-(alk-2-yn-1-onyl)-1-alkynylbenzenes) via a head–tail [4 + 2] cycloaddition process for the synthesis of the functionalized naphthalene derivatives in good to excellent yields. The observations made from several control experiments suggested and supported the proposed mechanism. This homodimerization exhibits a very broad substrate scope for aliphatic and aromatic 2-(alk-2-yn-1-onyl)-1-alkynylbenzenes. The generated naphthalenes were utilized for the synthesis of substituted chrysenes via a thermal alkyne–carbonyl metathesis reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Indian Institute of Technology Madras, Chennai, for infrastructural facilities and SERB-India for financial support through the CRG/2019/000988 grant. J. S. thanks IIT Madras for the HTRA fellowship. We thank Mr Ramkumar for the single crystal X-ray diffraction analysis.Notes and references
- Pyrylium ion reactivity: (a) A. Das, H.-H. Liao and R. S. Liu, J. Org. Chem., 2007, 72, 9214 CrossRef CAS PubMed; (b) N. Asao, T. Kasahara and Y. Yamamoto, Angew. Chem., Int. Ed., 2003, 42, 3504 CrossRef CAS PubMed; (c) N. Asao, H. Aikawa and Y. Yamamoto, J. Am. Chem. Soc., 2004, 126, 7458 CrossRef CAS PubMed; (d) B. D. Mokar, D. B. Huple and R.-S. Liu, Angew. Chem., Int. Ed., 2016, 55, 11892 CrossRef CAS PubMed; (e) F. Ye, M. Haddad, V. Michelet and V. R. Vidal, Org. Lett., 2016, 18, 5612 CrossRef CAS PubMed; (f) F. Ye, C. Tran, L. Jullien, T. L. Saux, M. Haddad, V. Michelet and V. R. Vidal, Org. Lett., 2018, 20, 4950 CrossRef CAS PubMed; (g) D. Pena, D. Perez, E. Guitian and L. Castedo, Eur. J. Org. Chem., 2003, 1238 CrossRef CAS; (h) N. T. Patil, N. K. Pahadi and Y. Yamamoto, J. Org. Chem., 2005, 70, 10096 CrossRef CAS PubMed; (i) J.-M. Tang, T.-A. Liu and R.-S. Liu, J. Org. Chem., 2008, 73, 8479 CrossRef CAS PubMed; (j) J. Wang, H.-T. Zhu, Y.-X. Li, L.-J. Wang, Y.-F. Qiu, Z.-H. Qiu, M.-J. Zhong, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2014, 16, 2236 CrossRef CAS PubMed; (k) M. E. Domaradzki, Y. Long, Z. She, X. Liu, G. Zhang and Y. Chen, J. Org. Chem., 2015, 80, 11360 CrossRef CAS PubMed; (l) H. Wang, Y. Kuang and J. Wu, Asian J. Org. Chem., 2012, 1, 302 CrossRef CAS; (m) B. Paroi, S. P. Sancheti and N. T. Patil, Chem. Rec., 2021, 21, 1 CrossRef; (n) J.-M. Tang, T.-A. Liu and R.-S. Liu, J. Org. Chem., 2008, 73, 8479 CrossRef CAS PubMed; (o) B. Guo, Y. Zhou, L. Zhang and R. Hua, J. Org. Chem., 2015, 80, 7635 CrossRef CAS PubMed; (p) M. Mandal, S. Sakthivel and R. Balamurugan, J. Org. Chem., 2021, 86, 333 CrossRef CAS PubMed.
- Pyrylium ion-based cycloadditions: (a) A. Das, H.-H. Liao and R. S. Liu, J. Org. Chem., 2007, 72, 9214 CrossRef CAS PubMed; (b) B. Guo, Y. Zhou, L. Zhag and R. Hua, J. Org. Chem., 2015, 80, 7635 CrossRef CAS PubMed; (c) L. Li, D. Huang, C. Shi and G. Yana, Adv. Synth. Catal., 2019, 361, 1958 CrossRef CAS; (d) S. Bhunia, K.-C. Wang and R.-S. Liu, Angew. Chem., 2008, 120, 5141 CrossRef; (e) L. Chen, K. Chen and S. Zhu, Chem, 2018, 4, 1208 CrossRef CAS; (f) B. Guo, L. Zheng, L. Yang and R. Hua, J. Org. Chem., 2014, 79, 4352 CrossRef CAS PubMed; (g) L. Chen, Z. Liu and S. Zhu, Org. Biomol. Chem., 2018, 16, 8884 RSC; (h) C. Zhang, L. Chen, K. Chen, H. Jiang and S. Zhu, Org. Chem. Front., 2018, 5, 1028 RSC; (i) S. Kotha, S. Misra and S. Halder, Tetrahedron, 2008, 64, 10775 CrossRef CAS; (j) N. Asao, Synlett, 2006, 1645 CrossRef CAS; (k) N. T. Patil and Y. Yamamoto, ARKIVOC, 2007, 6 Search PubMed; (l) N. Asao and H. Aikawa, J. Org. Chem., 2006, 71, 5249 CrossRef CAS PubMed; (m) N. Asao, T. Nogami, S. Lee and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 10921 CrossRef CAS PubMed.
- Pyrylium ion reactions with water and alcohols: (a) N. T. Patil, N. K. Pahadi and Y. Yamamoto, J. Org. Chem., 2005, 70, 10096 CrossRef CAS PubMed; (b) J.-M. Tang, T.-A. Liu and R.-S. Liu, J. Org. Chem., 2008, 73, 8479 CrossRef CAS PubMed.
- (a) J. Santhi and B. Baire, ChemistrySelect, 2017, 2, 4338 CrossRef CAS; (b) J. Santhi and B. Baire, ChemistrySelect, 2020, 5, 8151 CrossRef CAS; (c) J. Santhi and B. Baire, Chem. – Asian J., 2019, 14, 3161 CrossRef CAS PubMed.
- For reviews on Ag-assisted reactions, see: (a) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS PubMed; (b) M. Alevarez-Corral, M. Munoz-Dorado and I. Rodriguez-Garcia, Chem. Rev., 2008, 108, 3174 CrossRef PubMed; (c) G. Fang and X. Bi, Chem. Soc. Rev., 2015, 44, 8124 RSC.
- J. Santhi and B. Baire, Adv. Synth. Catal., 2016, 358, 3817 CrossRef CAS.
- (a) CCDC 1878982† contains the supplementary crystallographic data for compound 9a. The ORTEP diagram and further details are given in the ESI (Table 1†); ; (b) CCDC 1965357† contains the supplementary crystallographic data for compound 10c. The ORTEP diagram and further details are given in the ESI (Table 2†).
- For a simultaneous double-site activation by two metals, see: P. N. Bagle, M. V. Mane, K. Vanka, D. R. Shinde, S. R. Shaikh, R. G. Gonnade and N. T. Patil, Chem. Commun., 2016, 52, 14462 RSC.
- For alkyne–carbonyl metathesis, see: (a) T. Jin, F. Yang, C. Liu and Y. Yamamoto, Chem. Commun., 2009, 3533 RSC; (b) K. Murai, K. Teteishi and A. Saito, Org. Biomol. Chem., 2016, 14, 10352 RSC; (c) M. Nayak and I. Kim, Org. Biomol. Chem., 2015, 13, 9697 RSC; (d) S. Jalal, K. Bera, S. Sarkar, K. Paul and U. Jana, Org. Biomol. Chem., 2014, 12, 1759 RSC; (e) K. Bera, S. Jalal, S. Sankar and U. Jana, Org. Biomol. Chem., 2014, 12, 57 RSC; (f) C. Ao, X. Yang, S. Jia, X. Xu, Y. Yan, D. Zhang and W. Hu, J. Org. Chem., 2019, 84, 15331 CrossRef CAS PubMed; (g) S. Parpart, S. Boldt, P. Ehlers and P. Langer, Org. Lett., 2018, 20, 122 CrossRef CAS PubMed; (h) M.-N. Lin, S.-H. Wu and M.-C. P. Yeh, Adv. Synth. Catal., 2011, 353, 3290 CrossRef CAS.
- CCDC 1878983† contains the supplementary crystallographic data for compound 22. The ORTEP diagram and further details are given in the ESI (Table 3†).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1878982, 1878983 and 1965357. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob02229f |
| This journal is © The Royal Society of Chemistry 2022 |