
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A disulfide-based linker for thiol–norbornene conjugation: formation and cleavage of hydrogels by the use of light†
Markus
Lunzer‡
 ab,
Boris
Maryasin
cd,
Tommaso
Zandrini
b,
Stefan
Baudis
a,
Aleksandr
Ovsianikov
b and
Robert
Liska
*a
ab,
Boris
Maryasin
cd,
Tommaso
Zandrini
b,
Stefan
Baudis
a,
Aleksandr
Ovsianikov
b and
Robert
Liska
*a
aInstitute of Applied Synthetic Chemistry, Technische Universität Wien, Getreidemarkt 9/E163, 1060 Vienna, Austria. E-mail: robert.liska@tuwien.ac.at
bInstitute of Materials Science and Technology, Technische Universität Wien, Getreidemarkt 9/E308, 1060 Vienna, Austria
cInstitute of Organic Chemistry, University of Vienna, Währinger Strasse 38, 1090 Vienna, Austria
dInstitute of Theoretical Chemistry, University of Vienna, Währinger Strasse 17, 1090 Vienna, Austria
First published on 10th January 2022
Abstract
Photolabile groups are the key components of photo-responsive polymers, dynamically tunable materials with multiple applications in materials and life sciences. They usually consist of a chromophore and a labile bond and are inherently light sensitive. An exception are disulfides, simple reversible linkages, which become photocleavable upon addition of a photoinitiator. Despite their practical features, disulfides are rarely utilized due to their impractical formation. Here, we report a disulfide-based linker series bearing norbornene terminals for facile crosslinking of thiol-functionalized macromers via light-triggered thiol–ene conjugation (TEC). Besides finding a highly reactive lead compound, we also identify an unexpected TEC-retardation, strongly dependent on the molecular linker structure and affecting hydrogel stability. Finally, we present a useful method for localized disulfide cleavage by two-photon irradiation permitting micropatterning of disulfide-crosslinked networks.
Introduction
Photo-responsive polymers are highly useful “smart” materials, as their properties can be locally tuned by irradiation with light. Applications include tissue engineering and regenerative medicine,1 advanced cell culture2 and drug delivery3 as well as technical utilizations in self-healing materials,4 recycling,5 thin films,6 and as potential key components for sensors or hydrogel machines.7 Hydrogels are especially often modified with photosensitive groups, due to the high transparency of water for light in the visible to near-infra red (VIS-NIR) region.8–10 Longer wavelengths allow for deeper penetration and are generally less harmful for living tissue. Light-induced mechanisms include stiffness modifications such as secondary photo-crosslinking,11,12 reversible softening13–16 or complete degradation17–20 of the hydrogel backbone, as well as reversible patterning of biomolecules and proteins21–24 or activation of biological tethers by uncaging.25 By now, a considerable tool box of light-controllable chemical reactions has been developed.3,8,26–28 One of the most commonly used and studied photocleavable triggers is the o-nitrobenzyl (oNB) group.19,29–31 Alternatives are coumarin-derivatives,32,33o-nitrobiphenylpropyl-derivatives25,34 or ruthenium-complexes.35,36 All of these phototriggers consist of chromophores as light harvesting units and labile bonds, which can be photocleaved by irradiation with UV-VIS or multiphoton excitation using NIR-light. Due to the inherent light sensitivity of photocleavable groups, handling under light protection is required during all experimental steps including synthesis, analysis and application. Furthermore, the preparation of such chromophores can be quite laborious and usually involves multistep synthetic modifications with often moderate yields.An interesting but rarely utilized alternative are disulfide linkages. These molecularly quite simple building blocks can be reversibly formed and cleaved by various triggers, but are not photosensitive at ambient light conditions.37 Generally, cleavage and remodeling of disulfide-networks relies on the thiol-disulfide metathesis reaction, in which a sulfur-species attacks a disulfide linkage by forming a new disulfide bond and releasing another sulfur-species. The metathesis can be induced chemically via a nucleophilic attack of a thiol anion38 or photochemically in a radical-mediated reaction.39 Due to their reversible character, disulfide networks are dynamic and exhibit self-healing properties.39–42 For the mild chemical reduction of disulfides, usually an excess of a small molecule thiol or dithiol reagent is used.43–45 Other common reducing agents for disulfides are TCEP46 and NaBH4.47
Photoscission of disulfides in polymers has been directly induced by irradiation with high intensity UV light, resulting in rearrangement and self-healing of materials.42,48 Apart from such harsh conditions, disulfide networks are stable at ambient light. Yet, in presence of a photoinitiator, disulfide linkages in hydrogels can be cleaved using UV-VIS light at low intensities through a radical-mediated fragmentation reaction.39 Radicals, formed from the scission of the photoinitiator, attack and cleave disulfides, liberating thiyl radicals, which again undergo fragmentation and exchange reactions. Depending on the ratio of radicals to disulfides either photodeformation, photowelding, or photodegradation can be induced. This approach is quite practical, as the weak bond is already present in the network, while the photosensitive trigger is added when required.
Disulfide-based networks are usually directly formed from thiol-terminated precursors by oxidative coupling of thiols to disulfides mediated by oxygen,49 hydrogen peroxide,39,50 or enzymatically.51 However, in oxidative coupling the gelation rate depends on the concentrations of thiol and redox partner, making handling challenging and procedures have to be optimized for each formulation to get controllable results. On the contrary, horseradish peroxidase-mediated crosslinking proceeds slowly.51 Hence, it is advantageous to introduce disulfide linkages via simple linkers into networks. Disulfide-containing linkers have been studied bearing acrylate or acrylamide terminals, and were either incorporated into polymer networks via thiol-Michael addition with thiol-terminated macromers45 or by free radical copolymerization with acrylates.52–54 However, formulations based on Michael-acceptors and thiols have to be handled very skilled and quickly, as gelation sets in fast upon combining of the reactive components,18 which can be especially challenging when it comes to delicate procedures such as cell encapsulation or molding of complex geometries. Besides, acrylates are cytotoxic.55
For the ease of handling, an externally triggered gelation-reaction such as photo-induced step growth polymerization would be highly beneficial. As disulfides can be cleaved by increased concentrations of radicals,39 a highly efficient coupling reaction is required. The thiol–norbornene addition is exceptionally reactive, but yet well tolerated in cell encapsulation.56–58 Hence, to facilitate the fast and mild formation of disulfide-based hydrogels and the general application of this useful linkage, we developed a water-soluble disulfide-containing crosslinker for the light-induced formation of such networks from thiol-terminated precursors by thiol–ene coupling (TEC) reaction. To keep the approach simple and convenient, we aimed at designing the linker to be synthesized from cheap and readily available starting materials.
Results and discussion
Design and synthesis of disulfide crosslinkers
The first synthetic approach to such a disulfide-containing thiol–ene-crosslinker was based on the esterification of the readily available chemicals 2-hydroxyethyl disulfide and carbic anhydride, leading to the simple symmetric target molecule Nor-O-SS (Scheme 1), bearing carboxylate groups to facilitate water-solubility. A non-cleavable aliphatic reference linker Nor-O-CC was synthesized as well. However, Nor-O-SS appeared to be self-immolating and thus could not be purified from its decay products (Fig. S20–S22†). The synthesis of the corresponding amide from cystamine seemed unpromising as well.59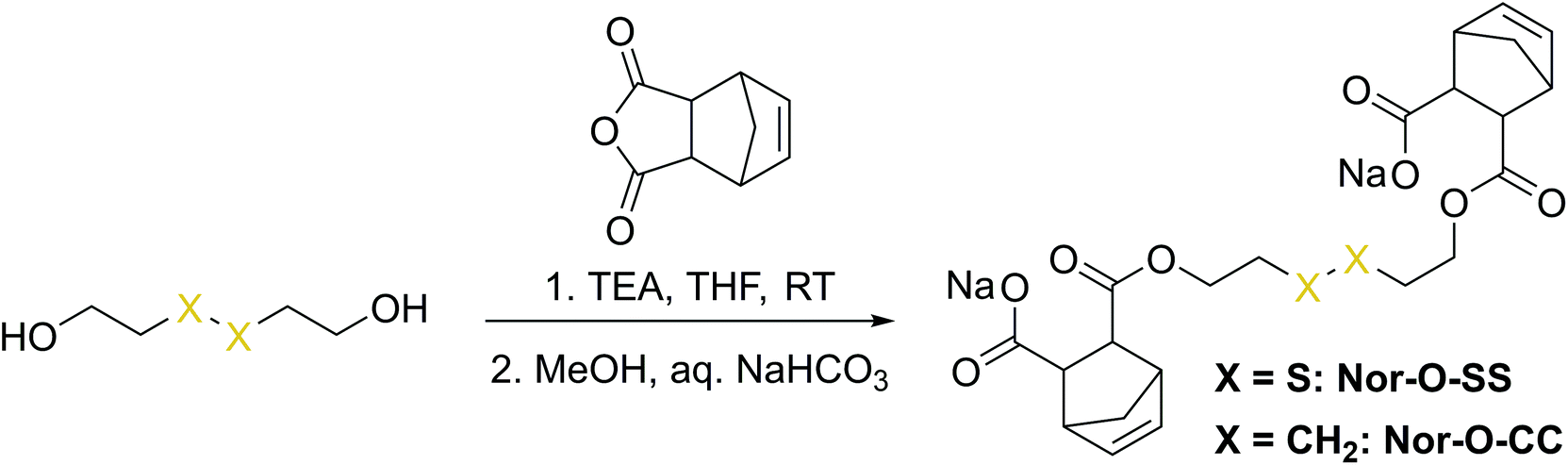 | ||
| Scheme 1 Synthesis of Nor-O-SS and the analog non-cleavable reference compound Nor-O-CC. | ||
In a second approach, the photo-crosslinker was synthesised from L-cystine, the readily available dimer of the amino acid L-cysteine, by coupling with 5-norbornene-carboxylic acid chloride in a Schotten–Baumann-type reaction (Scheme 2).52 However, in using cystine as core unit the complexity of the stereo-chemistry increases, since two more stereocenters are integrated into the molecule. For practical and economic reasons, the acid chloride was synthesized from 5-norbornene-2-carboxylic acid in its most common commercial form, a racemic mixture of exo- and endo-isomers with predominantly endo-molecules. Thus, the linker Nor-L-Cys (Scheme 2, Fig. S23†) was obtained as a mixture of diastereomers, which was used as received without applying complex purification protocols.
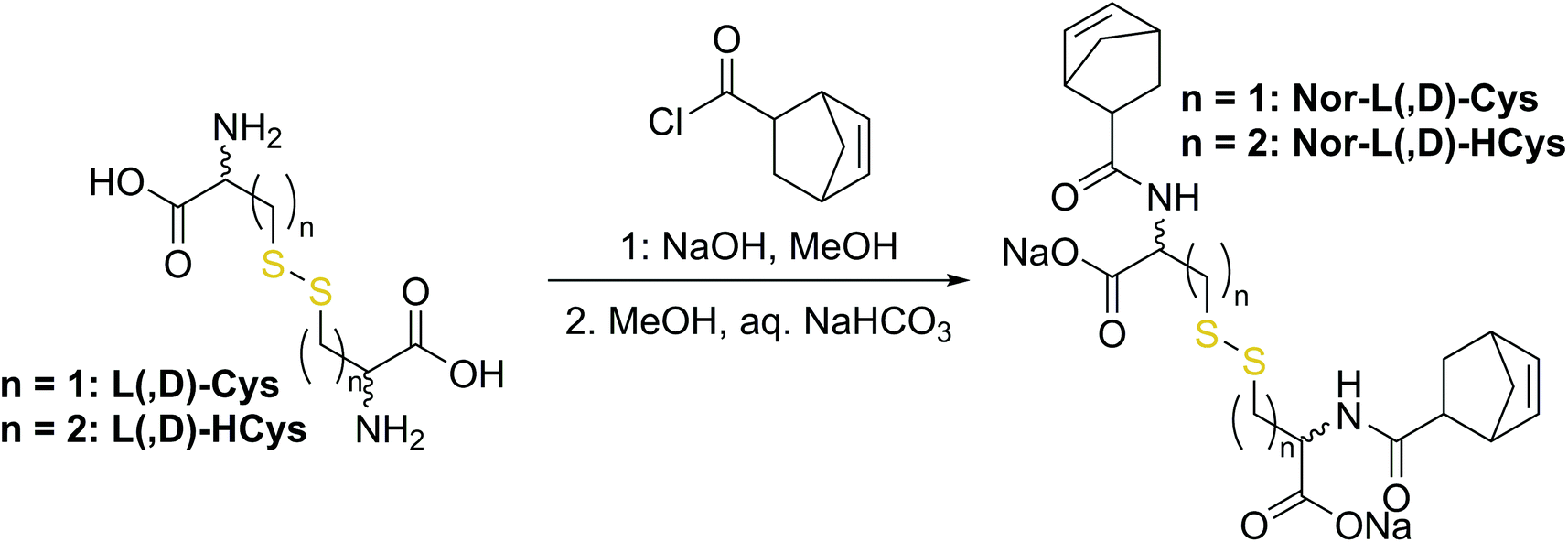 | ||
| Scheme 2 Synthesis of cystine- and homocysteine-based linkers. | ||
Next, a preliminary 1H-NMR conversion test with 2-mercaptoethanol was performed to test the reactivity of Nor-L-Cys (Fig. S22b). Whereas the double bonds of Nor-O-SS were readily consumed within 5 min of irradiation, full conversion of Nor-L-Cys was surprisingly retarded. Even with a SH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ene ration of 2:1, full conversion of Nor-L-Cys could not be observed within 10 min.
:ene ration of 2:1, full conversion of Nor-L-Cys could not be observed within 10 min.
The retarded reactivity of Nor-L-Cys is probably based on steric interactions, which could be either caused by the molecular configuration of norbornene or the core subunit. Hence, reasonable molecular modification strategies to improve the reactivity towards TEC include both the (I) norbornene group as well as the (II) disulfide-based core unit.
(I) Nor-L-Cys was synthesized from a mixture of exo- and endo-5-norbornene-carboxylic acid. Since the ene in exo-5-norbornenecarboxylic acid faces away from the carboxylic group, the exo-derivative might be sterically less hindered than the corresponding endo-based analogue. Thus, to test the influence of the stereoisomerism of the norbornene group, the exo-only linker exo-Nor-L-Cys (Fig. S23†) was synthesized from L-cystine and exo-5-norbornene carboxylic acid.
(II) In contrast to 2-hydroxyethyl disulfide, cystine is a chiral compound containing two stereocenters. Due to the close proximity of the functional groups, their relative orientation might affect the reactivity of the norbornenes by intramolecular interactions. Additionally, disulfide dihedral angles are usually in the range of 90°,60 adding up to the sterical effects based on the configuration of functional groups. The core unit can be simply modified by changing the enantiopure L-cystine to the racemic D,L-cystine giving the linker Nor-D,L-Cys (Fig. S23,†Scheme 2). A further strategy to reduce steric hindrance between the two branches of the molecule is elongation of the core by exchanging cystine for homocystine, containing an additional methylene group per subunit (Nor-L-HCys, Nor-D,L-HCys, Fig. 1, Scheme 2). Finally, as the water solubility of the molecule is facilitated by carboxylate anions, also hydratization effects could impact the reactivity of the ene. Hence, the reactivity of the linker in aprotic medium was tested as well. Hence, the dimethylated linker Nor-L-CysMe was synthesised (Fig. S23c†).
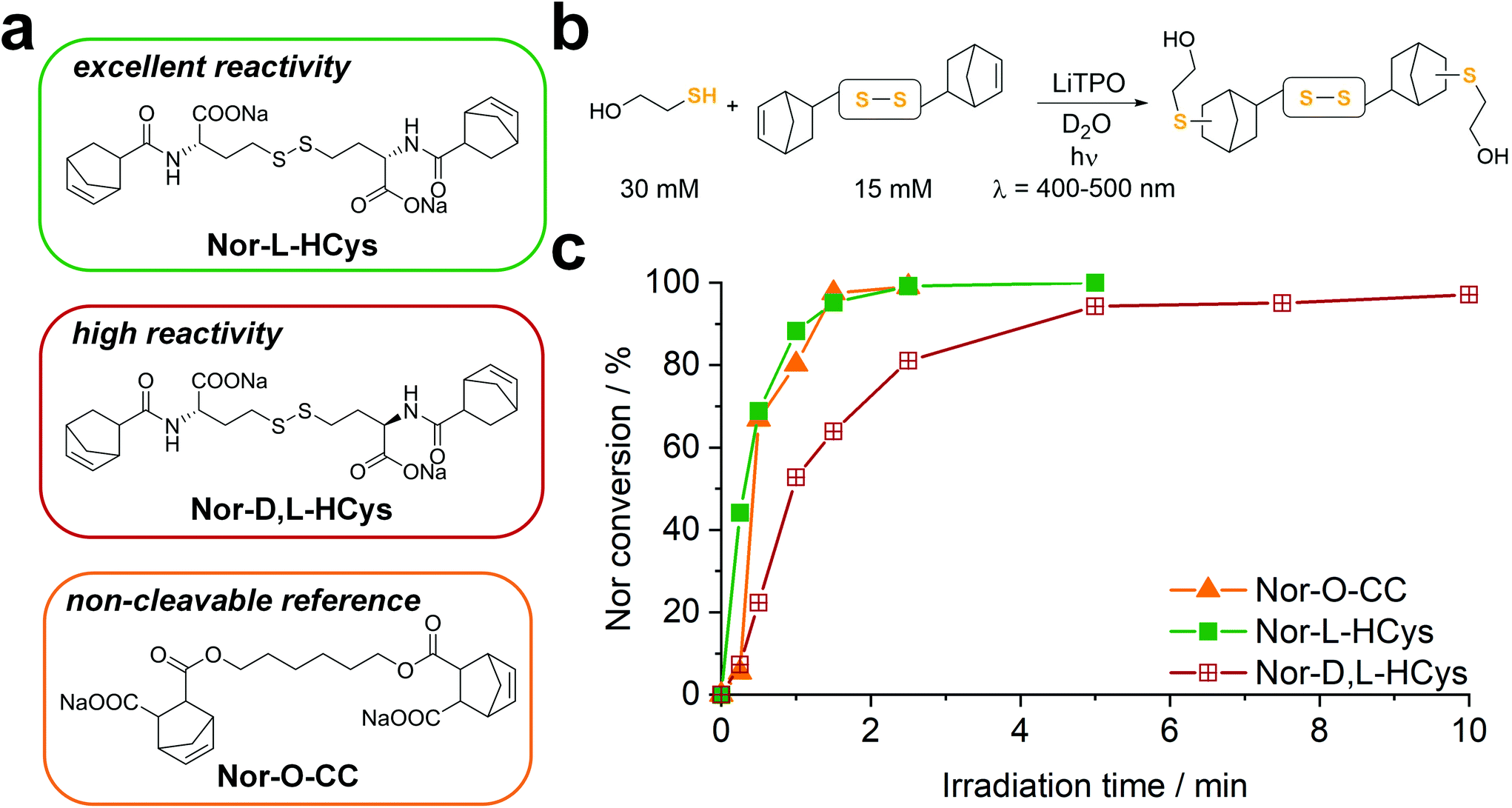 | ||
| Fig. 1 (a) The water-soluble disulfide linkers Nor-L-HCys and Nor-D,L-HCys for thiol–norbornene conjugation (TEC) were synthesized from L- and D,L-homocysteine. The aliphatic linker Nor-O-CC served as non-cleavable reference. (b) The linker reactivity was investigated in an 1H-NMR study. Linkers (15 mM) were reacted with 2-mercaptoethanol (30 mM) in D2O using LiTPO as photoinitiator (0.6 mM, λ = 400–500 nm, 20 mW cm−2). (c) Both Nor-O-CC and Nor-L-HCys reached full conversion within 2 min of reaction time. In contrast, Nor-D,L-HCys reacted slower and reached high (96%) but not complete ene-conversion after 10 min. | ||
Reactivity estimation by 1H-NMR
To examine the reactivity of the linkers towards TEC and evaluate the efficiency of this reaction in presence of disulfides, a small molecule 1H-NMR study was performed. The respective linker (15 mM) was combined with 2-mercaptoethanol (30 mM) in an equimolar thiol–ene ratio in D2O and irradiated (400–500 nm, 20 mW cm−2) in presence of the photoinitiator LiTPO61,62 for different time periods (Fig. 1b, Fig. S23b†). A low concentration of LiTPO (0.6 mM, 2 mol% of SH) was used to prevent interaction of excess radicals with disulfides.39 The evolution of double bond signals (∼6.0–6.4 ppm, HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH) was analysed. The signal of the proton at the α-carbon (∼4.2–4.5 ppm, CH–NH) served as internal reference. Surprisingly, the experiment revealed a strong dependence of the ene-conversion from the molecular linker design (Fig. 1b, Fig. S23†). Although all linkers were expected to efficiently react with a thiol in a thiol–ene “click”-reaction,56,63 the results showed that homocysteine-based linkers are highly reactive (Fig. 1b), while the TEC reaction of cystine-based derivatives is strongly retarded (Fig. S23†). Nevertheless, a distinct difference in reactivity among the homocysteine linkers could be found. Nor-L-HCys reacted much quicker than Nor-D,L-HCys and showed full conversion of norbornenes within 2.5 min of irradiation. With Nor-D,L-HCys high (∼94%) but not quantitative ene-conversion was reached within 10 min. Thus, the TEC reaction of Nor-D,L-HCys seems to be slightly retarded. For comparison, also the non-cleavable linker Nor-O-CC was tested (Fig. 1a and c and Scheme 1). As with Nor-L-HCys, full conversion was achieved within 2.5 min of irradiation. Since Nor-O-CC contains a non-rigid aliphatic spacer without a distinct configuration of side groups, minimal intramolecular interactions can be assumed. As Nor-L-HCys shows equal reactivity, minimal intramolecular interactions can be expected as well. Based on these findings, the reactivity of the linkers regarding TEC reaction can be graded in the following order: Nor-O-CC ∼ Nor-L-HCys > Nor-D,L-HCys ≫ Nor-L-Cys ∼ Nor-D,L-Cys > exo- Nor-L-Cys.
CH) was analysed. The signal of the proton at the α-carbon (∼4.2–4.5 ppm, CH–NH) served as internal reference. Surprisingly, the experiment revealed a strong dependence of the ene-conversion from the molecular linker design (Fig. 1b, Fig. S23†). Although all linkers were expected to efficiently react with a thiol in a thiol–ene “click”-reaction,56,63 the results showed that homocysteine-based linkers are highly reactive (Fig. 1b), while the TEC reaction of cystine-based derivatives is strongly retarded (Fig. S23†). Nevertheless, a distinct difference in reactivity among the homocysteine linkers could be found. Nor-L-HCys reacted much quicker than Nor-D,L-HCys and showed full conversion of norbornenes within 2.5 min of irradiation. With Nor-D,L-HCys high (∼94%) but not quantitative ene-conversion was reached within 10 min. Thus, the TEC reaction of Nor-D,L-HCys seems to be slightly retarded. For comparison, also the non-cleavable linker Nor-O-CC was tested (Fig. 1a and c and Scheme 1). As with Nor-L-HCys, full conversion was achieved within 2.5 min of irradiation. Since Nor-O-CC contains a non-rigid aliphatic spacer without a distinct configuration of side groups, minimal intramolecular interactions can be assumed. As Nor-L-HCys shows equal reactivity, minimal intramolecular interactions can be expected as well. Based on these findings, the reactivity of the linkers regarding TEC reaction can be graded in the following order: Nor-O-CC ∼ Nor-L-HCys > Nor-D,L-HCys ≫ Nor-L-Cys ∼ Nor-D,L-Cys > exo- Nor-L-Cys.
This simple 1H-NMR study demonstrates significant differences in TEC reactivity depending on the molecular design, as recently suggested.64 While the radical-mediated TEC reaction has often been considered an archetype of “click”-reactions,63 recently several studies pointed out its limitations.64–67 It was shown that the efficiency of TEC reactions can be reduced by the nature of the thiol or the ene, the photoinitiator used or the chemical reaction environment. Hence, these studies highlight the importance of carefully optimizing the molecular design in order to achieve high conversion. Yet, no ultimate design guidelines for highly efficient TEC systems could be established yet. Obviously in the current case, both the length of the core as well as its configuration affect the ene-conversion. Hence, intramolecular interactions are expected to influence the reactivity. As the length of the core is critical, since homocystine-based linkers are much more reactive than their cystine counterparts, such interactions most probably occur between functional groups of opposing linker-subunits.
Computational study
To get a better insight into the cause of the TEC retardation and further evaluate the structure–property relationship, the reaction of the retarded cystine-based linker Nor-L-Cys (system A) and the elongated, highly-reactive homocystine derivative Nor-L-HCys (system B) with a 2-mercaptoethanol thiyl radical was computationally modelled (ESI†). System A is more rigid compared to the elongated, and therefore, more flexible system B. As a consequence, system B has a higher degree of freedom for the formation of stabilizing intermolecular interactions and gives the more stabilized thermodynamically product. The thermodynamic driving force of the computationally modeled event is 2.5 times larger for system B than for system A. This agrees well with the experimentally observed reactivity difference for Nor-L-Cys (A) (Fig. S23†) and Nor-L-HCys (B) (Fig. 1).Development of the disulfide-based hydrogel platform
Next, hydrogel formation of the highly reactive homocystine-based linkers Nor-L-HCys and Nor-D,L-HCys was investigated by photorheology. Linkers were reacted with 10 wt% of thiol-terminated 8-armed PEG (8armPEG20k-SH, Mw ∼20 kDa) by irradiation with an LED (385 nm, ∼6 mW cm−2). First, the lowest required concentration of LiTPO was established, as an excess of photoinitiator is known to induce degradation of disulfide cross-linked hydrogels.39 Stock solutions of 8armPEG20k-SH, Nor-D,L-HCys (190 mM) and LiTPO (18 mM) in PBS were combined to give formulations with final concentrations of 10 wt% 8armPEG20k-SH, 22 mM Nor-D,L-HCys (SH:ene = 1:1) and a variable LiTPO concentration (0.2 mM, 0.4 mM, 0.6 mM, 15 mM). After 1 min of equilibration, the prepolymer solutions were irradiated for 5 min (Fig. 2a).
 | ||
| Fig. 2 (a) In situ oscillatory photorheology of hydrogel formulations containing 10 wt% 8armPEG20k-SH, Nor-D,L-HCys (22.2 mM, SH:ene = 1:1) and variable concentrations of LiTPO (0.2 mM, 0.4 mM, 0.6 mM, 15 mM). The prepolymer solutions were exposed to 385 nm light (6 mW cm−2) for 5 min continuously, or (b) light was shuttered with 5 s on-time and 15 s off-time followed by 3 min of continuous exposure. Periods of irradiation are highlighted by a purple background. | ||
At low LiTPO concentrations (0.2 mM, 0.4 mM, 0.6 mM), a delay of polymerization was observed, which decreases with an increasing amount of photoinitiator, while the final storage modulus G′ is increasing from 17.6 ± 1.7 kPa (0.2 mM), over 20.2 ± 2.2 kPa (0.4 mM) to 22.9 ± 3.9 kPa (0.6 mM). Similar behavior has been reported before, when a TEC reaction had been sensitized using different concentrations of Eosin Y.68 Anyhow, G′ is unaffected by further irradiation after a plateau is reached, showing that crosslinking via TEC occurs quicker than degradation of disulfides and LiTPO concentrations of 0.6 mM and below are sufficient for gel formation without provoking significant network degradation.
In contrast, when a high concentration of LiTPO (15 mM) is used, G′ rises very rapidly within 5 s to a maximal value of 21.5 ± 0.3 kPa, before declining again. This result illustrates initial very rapid step-growth polymerization followed by network degradation via excess photoinitator, again demonstrating that the TEC reaction is preferred over radical-mediated disulfide-cleavage. Here, also the loss modulus G′′ is significantly increased during irradiation. As 15 mM LiTPO is not sufficient for complete reverse gelation of a hydrogel based on 22 mM Nor-D,L-HCys, G′ reaches a plateau after 5 min of irradiation due to still intact disulfide-crosslinks. When the light is dynamically shuttered (5 s on-time, 15 s off-time, 3 min continuous irradiation, 15 mM, Fig. 2b, purple shading) the partial reversibility of the radical induced disulfide cleavage becomes apparent.39 While during irradiation G′ decreases and G′′ increases, the inverse behavior is observed when irradiation is interrupted, indicating rapid reformation of disulfide bonds by thiyl radical recombination. However, with every irradiation circle G′ is gradually reduced, as thiyl-radicals are consumed by photoinitiator fragments, impeding full restauration of the initial crosslinking density and G′max. After establishing the impact of the photoinitiator concentration on the hydrogel, further characterizations were performed with a hydrogel formulation based on 10 wt% 8armPEG20k-SH, Nor-(D,)L-HCys (22 mM, SH:ene = 1:1) and 0.4 mM LiTPO, which will herein be referred to as 8arm-(D,)L-HCys 100 hydrogel.
Thermo- and mechanostability of 8arm-D,L-HCys 100 hydrogel
To investigate the thermostability, a sample of 8arm-D,L-HCys 100 hydrogel was photopolymerized at 20 °C on the rheometer, before the temperature was continuously increased up to 90 °C at a heating rate of 1 °C min−1 (ESI Fig. S24†). Only at temperatures above 60 °C a sharp decrease of G′ could be observed, demonstrating thermal stability of the hydrogel at ambient and physiological conditions. The mechanostability of 8arm-(D,)L-HCys 100 hydrogel was investigated in a strain sweep experiment, where the strain was logarithmically increased from 0.1% to 1000% at a constant frequency of 1 Hz (ESI Fig. S25†) or dynamically cycled between 1% and 350% (ESI Fig. S26†). Up to a strain of 100% the moduli of the hydrogel remained unaltered. At higher strains rupture of the gel occurred.Stabilization of 8arm-D,L-HCys 100 hydrogel
When immersed in PBS at room temperature, 8arm-D,L-HCys 100 started swelling heavily, leading to first deformation and then dissolution within a few days. In the course of deformation, cylindrical samples turned into spherical caps (Fig. 3a). This behavior indicates relaxation of surface tension, which is most likely occurring via thiol-disulfide metathesis involving free thiols (Fig. 3b). It is in accordance with the observation that partly degraded disulfide networks adapted to an applied strain to minimize the system's free energy.39 This is explicable, as the 1H-NMR reactivity estimation revealed incomplete ene-conversion of Nor-D,L-HCys (Fig. 1c). Upon swelling and relaxation by thiol-disulfide metathesis the hydrogel expands. Since the linkers contain carboxylate anions to facilitate solubility, further swelling of water into the hydrogel volume is provoked by the increased ion concentration, finally leading to dissolution of the hydrogel.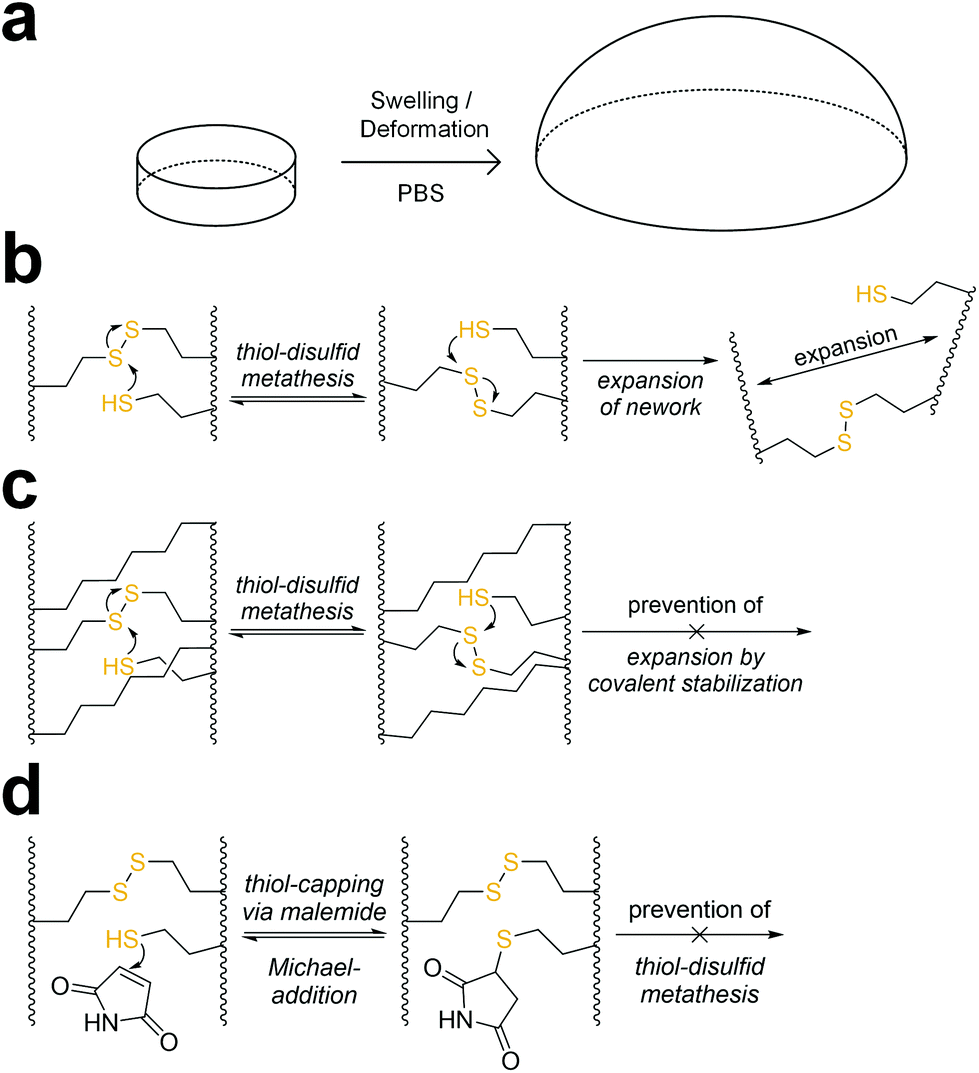 | ||
| Fig. 3 (a) In the course of swelling in PBS, 8arm-D,L-HCys 100 hydrogel disks lost their shape by turning into spherical caps before finally dissolving within a few days. (b) A possible reason for this behavior is that unreacted thiols can undergo thiol-disulfide metathesis with crosslinks leading to relaxation of internal stress and resulting in expansion of the network. The network could be stabilized by (c) a non-cleavable background network or (d) capping of free thiols by maleimide or an excess of linker. | ||
Interestingly, when swollen in 10x PBS for the first 20 h after formation, 8arm-D,L-HCys 100 hydrogel remained stable for weeks thereafter (Fig. S27†). Due to the increased ion concentration of the swelling medium in the initial period, the samples were losing mass via osmosis first. After the medium was changed to 1× PBS, samples began swelling and gained mass, but remained their shape and did not dissolve. This behavior can be explained by formation of additional crosslinks by unreacted thiols upon condensing of the network due to initial negative swelling.
A strategy to prevent shape-relaxation and expansion of 8arm-D,L-HCys 100 hydrogel involves the inclusion of non-cleavable aliphatic crosslinks by addition of Nor-O-CC (Fig. 1 and 3c). In order to stabilize a hydrogel based on an 8armPEG20k-SH backbone, at least two arms of the macromer have to be connected to a non-cleavable unit to meet the percolation threshold. Hence, 25% of Nor-D,L-HCys must be substituted by Nor-O-CC to form a non-cleavable background network, in which every macromer is theoretically connected to two units of Nor-O-CC. This hypothesis was confirmed by comparing the swelling behavior of different formulations based on variable ratios of Nor-D,L-HCys to Nor-O-CC. Whereas both non-stabilized 8arm-D,L-HCys 100 and 8arm-SS-CC (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) hydrogel (12.5% Nor-O-CC) were disintegrating within a few days, a mixture of 75% Nor-D,L-HCys and 25% Nor-O-CC gave the stable hydrogel 8arm-SS-CC (3:1). Further details are given in the ESI (Fig. S28†). This experiment demonstrates that unrestricted expansion of the hydrogel is a reason for dissolution of 8arm-D,L-HCys 100. The most probable cause for this expansion is the dynamic reformation of linkages induced by free thiols.
:1) hydrogel (12.5% Nor-O-CC) were disintegrating within a few days, a mixture of 75% Nor-D,L-HCys and 25% Nor-O-CC gave the stable hydrogel 8arm-SS-CC (3:1). Further details are given in the ESI (Fig. S28†). This experiment demonstrates that unrestricted expansion of the hydrogel is a reason for dissolution of 8arm-D,L-HCys 100. The most probable cause for this expansion is the dynamic reformation of linkages induced by free thiols.
The presence of pendant thiols can be indirectly proven by capping and thus disabling their participation in thiol-disulfide metathesis. As free thiols readily react in a thiol-Michael addition, they can simply be consumed by a small molecule Michael-acceptor (Fig. 3d).69 Hence, freshly polymerized samples of 8arm-D,L-HCys 100 were immersed in a solution of maleimide (4 mM) for 1 h and thereafter swollen in PBS (Fig. S29†). Indeed, this treatment led to a stabilization of the hydrogel, indicating(I) the presence of unreacted thiols as well as (ii) their involvement in the instability of this hydrogel.
Another simple yet convenient method to prevent the presences of unreacted thiols is the use of an excess of linker. However, this ene-excess has to be optimized precisely, since it also reduces the overall connectivity of the network, leading to pending norbornenes. Hence, three formulations with increasing ene-excess (15, 20 and 23.5 mol%) were tested by photorheology. Further details can be found in the ESI Fig. S30.† This study could well demonstrate, that (i) with Nor-D,L-HCys ene-conversion is incomplete when reacted at an 1:1 ratio of functional groups. Moreover, (ii) the crosslinking density and stiffness can even be increased by using an excess of linker, whereby an optimum excess of ene could be established at 20 mol% (8arm-D,L-HCys 120, Fig. 4a and b, ESI Fig. S30†).
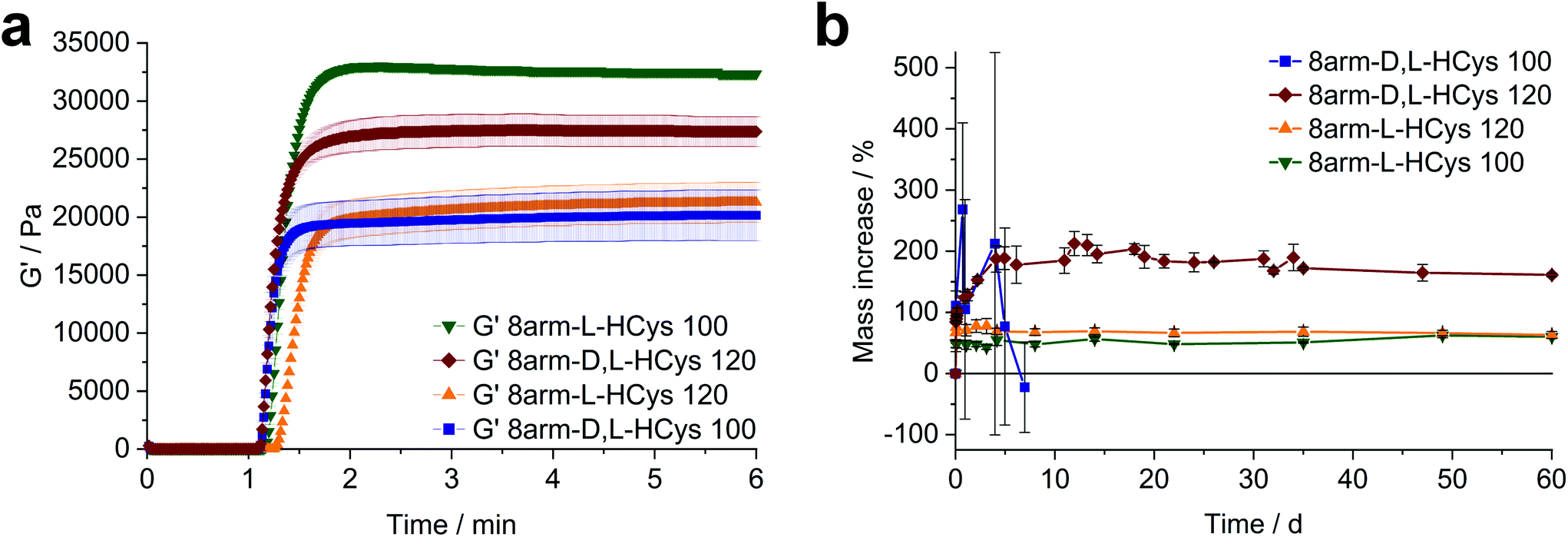 | ||
| Fig. 4 The effect of the configuration of homocysteine-based linkers on hydrogel behavior. (a) Comparison of the photopolymerization of formulations based on 10 wt% 8armPEG20k SH, 0.4 mM LiTPO and linkers with a homocysteine core. When using Nor-D,L-HCys a 20 mol% excess of ene (8arm-D,L-HCys 120) gives a higher storage modulus G′ than in the equimolar case. In contrast, with Nor-L-HCys the equimolar formulation 8arm-L-HCys 100 exhibits the highest G′ of the series, whereas an excess of Nor-L-HCys results in a reduced storage modulus. (b) Swelling behavior of the studied hydrogels in PBS. Nor-L-HCys gives stable hydrogels at equimolar conditions and when an excess of linker is used. In contrast, with Nor-D,L-HCys and excess of ene is required to yield a stable hydrogel. The data is depicted as mean values of at least triplicates and error bars illustrating the standard deviation. | ||
Nor-L-HCys based hydrogels
Nor- L -HCys showed the highest reactivity of all investigated linkers and exhibited quantitative conversion of ene in the model reaction with 2-mercaptoethanol within 2.5 min of irradiation, whereas Nor-D,L-HCys reached ∼96% conversion only after 10 min of irradiation (Fig. 1c). This leads to remarkable behavior in corresponding rheological and swelling experiments, when comparing hydrogel formulations based on these two linkers (Fig. 4a). While formulations with Nor-L-HCys react slightly slower, they reach higher G′ values than their Nor-D,L-HCys counterparts. More importantly, whereas with Nor-D,L-HCys a 20 mol% excess of ene is required to reach maximum G′ (8arm-D,L-HCys 120, 27.4 ± 1.3 kPa) and form a stable hydrogel (Fig. 4b), with Nor-L-HCys the equimolar formulation exhibits the highest G′ of the series (8arm-L-HCys 100, 32.4 ± 0.3 kPa). In contrast, using a 20% excess of ene leads to a lower stiffness when Nor-L-HCys is used (8arm-L-HCys 120, 21.3 ± 1.7 kPa). It lies in the range of the equimolar formulation of Nor-D,L-HCys (8arm-D,L-HCys 100, 20.2 ± 2.2 kPa).These results indirectly confirm the very high to quantitative ene-conversion of Nor-L-HCys observed in the 1H-NMR reactivity estimation. The equimolar formulation of Nor-L-HCys reaches a higher storage modulus than when a 20 mol% excess of ene is used. In contrast, with Nor-D,L-HCys, an excess of ene leads to maximum G′. This is also reflected in the swelling behavior, since hydrogels of both Nor-L-HCys formulations remain stable, whereby the equimolar formulation exhibits the lowest swelling of the series, again indicating high network connectivity (Fig. 4b).
In the case of the linker Nor-D,L-HCys, the final storage modulus is reached within less than 60 s of irradiation (Fig. 4a and Fig. S30†), while in the NMR experiments it takes 2.5 min to reach a norbornene conversion of 80%, and only after 10 min a high ene-conversion of 96% is obtained. We hence reason that this lower reactivity of Nor-D,L-HCys compared to Nor-L-HCys is the cause why an excess of linker is required here to form a stable hydrogel.
Two-photon micropatterning of disulfide-crosslinked hydrogels
Disulfide crosslinked hydrogels can be degraded by UV-light in presence of a cleavable photoinitiator via a radical-mediated disulfide fragmentation.39 Here, we demonstrate that this concept also works in the two-photon irradiation regime, permitting 3D-micropatterning of such hydrogels. For this purpose, the cleavable, water-soluble two-photon initiator DAS was used (Fig. 5f).70,71 Two-photon induced hydrogel cleavage was studied by microchannel formation and subsequent visualization of the channels via confocal microscopy, after swelling with high molecular weight fluorescent dextran.18 Micropatterning was tested on 8arm-D,L-HCys 120 and 8arm-L-HCys 100 hydrogels. Samples were formed in molds by irradiation at 365 nm, and first swollen in PBS to reach equilibrium swelling and subsequently submersed in solutions of DAS at different concentrations (0.5 mM, 1.0 mM, 2.0 mM), to permit diffusion of the chromophore into the network (Fig. 5a–c). Thereafter, the samples were micropatterned at a scanning speed of 200 mm s−1 using a fs-pulsed laser (720 nm) focused through a water immersion objective (Fig. 5d). Parallel channels were eroded into the hydrogel at laser powers ranging from 10–120 mW. Individual x,y-planes were either scanned once or twice. For examination of the channels by LSM, the samples were swollen with high molecular weight fluorescent dextran (2000 kDa, FITC2000). FITC2000 only infiltrates open micro-channels but not the bulk hydrogel (Fig. 5e). When 8arm-D,L-HCys 120 hydrogel was micropatterned using 2.0 mM DAS, open channels formed after a single scan at laser powers starting from 20 mW (Fig. 6a, ESI Fig. S31†). At lower concentrations it was necessary to scan each x,y-plane twice to produce open microchannels. At a concentration of 1 mM DAS bright fluorescence was visible in regions scanned at 30 mW and above, while at 0.5 mM DAS at least 40 mM were required to produce channels. When no DAS was used, no fluorescence could be observed in scanned regions. This experiment demonstrates that the two-photon cleavage requires the use of a two-photon active initiator and that the threshold for this cleavage is concentration dependent.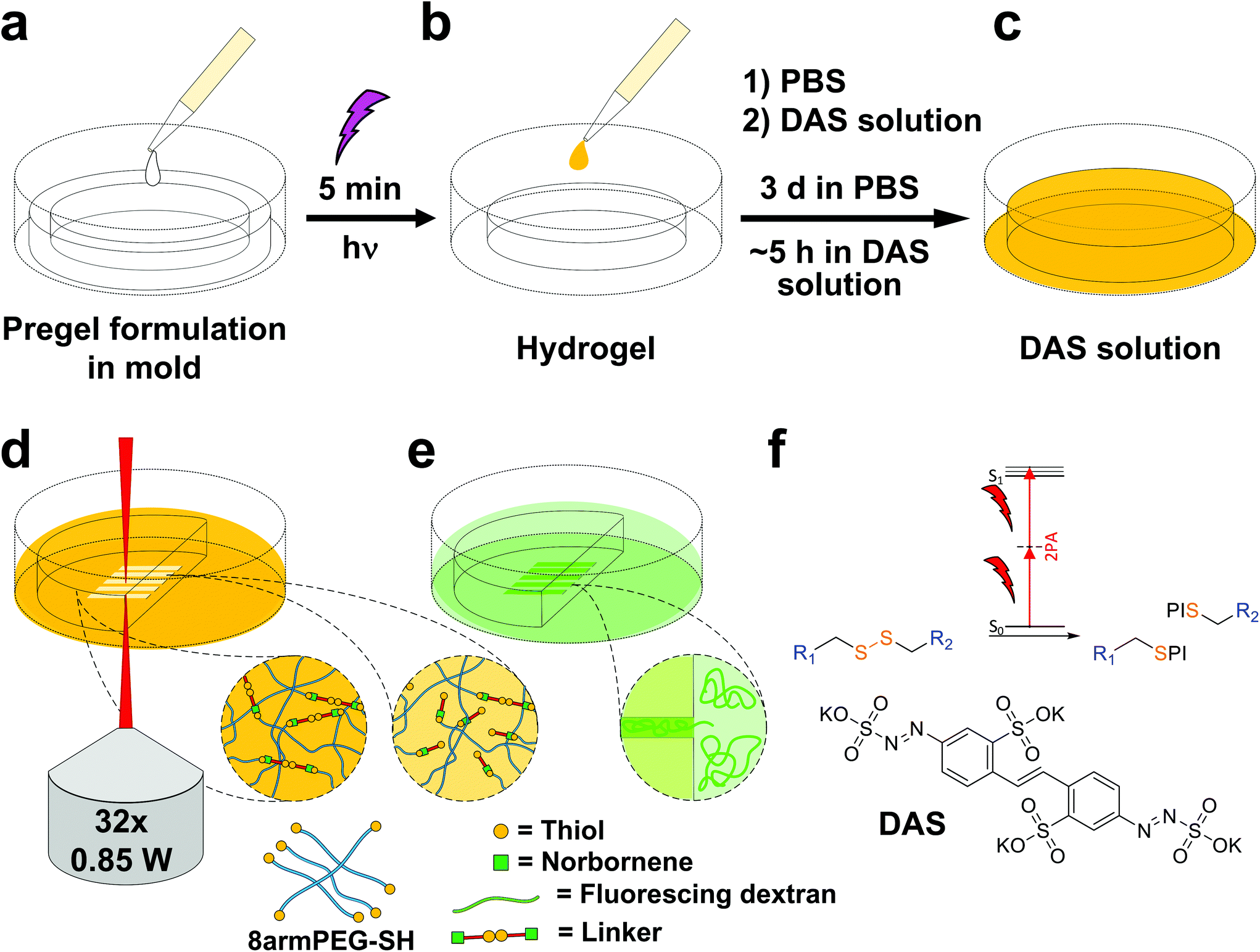 | ||
| Fig. 5 Workflow of hydrogel formation via UV-light and treatment with DAS prior to two-photon micropatterning and post-treatment for visualization. (a) Disulfide-crosslinked PEG hydrogels were formed by photoinduced TEC upon irradiation with UV-light. (b) Thereafter, hydrogels were swollen in PBS, (c) before being soaked in solutions of DAS. (d) The activated hydrogels were then two-photon micropatterned using a fs-pulsed laser. (e) Microchannels were visualized by confocal microscopy, after swelling in fluorescing dextran. (f) The presence of the two-photon active DAS is crucial for effective cleavage of disulfide linkages by two-photon irradiation. PI denotes a photoinitiator fragment. | ||
 | ||
| Fig. 6 Microchannels were fabricated by two-photon degradation of disulfide-based hydrogels in presence of two-photon initiator DAS (0.5–2.0 mM). Individual x,y-planes were either scanned once or twice. Thereafter, hydrogels were soaked in a solution of high molecular weight fluorescent dextran (FITC2000) and microchannels were visualized by confocal microscopy. (a) 8arm-D,L-HCys 120 could be fully degraded in presence of 2.0 mM DAS in a single scan. At lower concentration double scanning was required for complete erosion. (b) Microchannel fabrication in stiffer 8arm-L-HCys 100 hydrogel required double scanning in presence of 2.0 mM DAS for complete erosion. When 1 mM DAS was used, channels could only be visualized using lower molecular weight dextran (FITC500), indicating localized softening of the material. Images display orthogonal cross sections (y,z-plane) of micro channels. Scale bar 20 μm. | ||
The scanning parameters for complete degradation can be optimized in multiple ways, as magnification, laser power, scanning speed, hatch and z-layer spacing influence the applied light dose. Here, a hatch distance of 0.1 μm and a z-layer spacing of 0.5 μm was used. At DAS concentrations below 2 mM, x,y-planes had to be scanned twice. In contrast, for photodegradation of a hydrogel based on an oNB ester moiety, which undergoes defined molecular photolysis in response to pulsed light (λ = 740 nm), 5–7 frame repeat scans using 1.9 W laser power with a pixel dwell time of 2 μs were reported to be required recently.17 However, no additional oNB-cleavage-promoting two-photon sensitizer18 was used in these experiments.
When using the same scanning parameters as above on the stiffer 8arm-L-HCys 100 hydrogel, double scanning in presence of 2.0 mM DAS was required to fabricate micro-channels at laser powers of 40 mW and above, which could be infiltrated by FITC2000 (Fig. 6b, ESI Fig. S32†). With lower molecular weight FITC-dextran (500 kDa, FITC500) channels fabricated at 30 mW were visible as well. When only 1.0 mM DAS was used in 8arm-L-HCys 100, microchannels produced by double scanning at laser powers of 50 mW and above required swelling in FITC500 for visualization, indicating local softening but no complete degradation at these scanning parameters. In contrast, when channels were only scanned once in presence of 2.0 mM DAS, hydrogel degradation was not sufficient for FITC-dextran to enter the irradiated areas and no fluorescence could be observed (Fig. S33†).
With these experiments, two-photon cleavage of disulfides in presence of DAS could be demonstrated. Based on the scanning parameters used either complete degradation or localized softening of hydrogels can be achieved, depending on the application of choice. Moreover, with the developed linker photodegradable hydrogels can be produced directly from thiol-terminated macromers (thiol-gelatin,72 thiol-HA,49 thiol-PEG,39 thiol-PVA73) without the use of linkers containing photolabile groups, significantly simplifying precursor preparation and easing handling of hydrogels, as no inherent light sensitivity is given prior to the addition of a photoinitiator.
Noteworthy, others have developed a photoresist based on thiol-terminated macromers and a phenacyl sulfide linker for the direct formation of disulfide networks upon two-photon irradiation at 700 nm. In this system the phenacyl sulfide releases a photo-caged thioaldehyde species, which reacts with a thiol to form a disulfide bond.74 Hence, our two-photon induced cleavage of disulfide networks also expands the methodology for localized 3D-printing and degradation of polymer networks by means of two-photon irradiation, as recently shown by Batchelor et al.75
Conclusion
A disulfide-crosslinker for the redox-free, photochemical formation of cleavable hydrogels from thiol-terminated precursors via TEC reaction was developed. Interestingly, the reactivity of the TEC reaction was found to be strongly dependent on the molecular design of the linker. Whereas linkers based on homocysteine were highly reactive, those with a cystine core exhibited hindered thiol–norbornene addition. However, full consumption of free thiols is crucial. It could be shown that pendant thiol moieties lead to an unstable network and hence have to be avoided. In the absence of free thiols, disulfide-based hydrogels formed from a thiol-terminated macromer were stable for months in PBS. Furthermore, a method for two-photon induced degradation of disulfide-based hydrogels was demonstrated by microchannel fabrication. The presented crosslinker extends the scope of photolabile functional groups, as it is easily accessible and allows the mild formation of various disulfide-based cleavable networks in a controllable way by the use of light. Due to the reversible nature of the disulfide bond, it provides various opportunities for the design of tunable materials systems with tailored properties, allowing to engineer materials for numerous applications from tissue engineering to drug delivery, microfluidics and materials science.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank Simon Liedl, Sophia Thiele, Simon Scherrer and Daniel Glöcklhofer (all TU Wien) for their assistance in synthesis and photorheology experiments. Calculations were partially performed at the Vienna Scientific Cluster (VSC). We thank Prof. Leticia González and Prof. Nuno Maulide for generous support, resources and fruitful discussions. This work was financially supported by TU Wien “Innovative Projects” Research Funds (M. L.) and the Austrian Science Fund (FWF) project I2444N28.References
- T. E. Brown and K. S. Anseth, Chem. Soc. Rev., 2017, 46, 6532–6552 RSC.
- A. M. Rosales and K. S. Anseth, Nat. Rev. Mater., 2016, 1, 15012 CrossRef CAS PubMed.
- E. R. Ruskowitz and C. A. DeForest, Nat. Rev. Mater., 2018, 3, 17087 CrossRef CAS.
- D. Habault, H. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244–7256 RSC.
- S. Radl, M. Kreimer, J. Manhart, T. Griesser, A. Moser, G. Pinter, G. Kalinka, W. Kern and S. Schlögl, Polymer, 2015, 69, 159–168 CrossRef CAS.
- A. Romano, I. Roppolo, E. Rossegger, S. Schlögl and M. Sangermano, Materials, 2020, 13(12), 2777 CrossRef CAS PubMed.
- X. Liu, J. Liu, S. Lin and X. Zhao, Mater. Today, 2020, 36, 102–124 CrossRef CAS.
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 1807333 CrossRef PubMed.
- T. L. Rapp and C. A. DeForest, Adv. Healthcare Mater., 2020, 9, 1901553 CrossRef CAS PubMed.
- A. Alabugin, Photochem. Photobiol., 2019, 95, 722–732 CrossRef CAS PubMed.
- T. E. Brown, J. S. Silver, B. T. Worrell, I. A. Marozas, F. M. Yavitt, K. A. Günay, C. N. Bowman and K. S. Anseth, J. Am. Chem. Soc., 2018, 140, 11585–11588 CrossRef CAS PubMed.
- C. B. Highley, C. B. Rodell and J. A. Burdick, Adv. Mater., 2015, 27, 5075–5079 CrossRef CAS PubMed.
- A. M. Rosales, S. L. Vega, F. W. DelRio, J. A. Burdick and K. S. Anseth, Angew. Chem., Int. Ed., 2017, 56, 12132–12136 CrossRef CAS PubMed.
- L. Liu, J. A. Shadish, C. K. Arakawa, K. Shi, J. Davis and C. A. DeForest, Adv. Biosyst., 2018, 2, 1800240 CrossRef PubMed.
- M. Hörner, K. Raute, B. Hummel, J. Madl, G. Creusen, O. S. Thomas, E. H. Christen, N. Hotz, R. J. Gübeli, R. Engesser, B. Rebmann, J. Lauer, B. Rolauffs, J. Timmer, W. W. A. Schamel, J. Pruszak, W. Römer, M. D. Zurbriggen, C. Friedrich, A. Walther, S. Minguet, R. Sawarkar and W. Weber, Adv. Mater., 2019, 31, 1806727 CrossRef PubMed.
- A. M. Rosales, K. M. Mabry, E. M. Nehls and K. S. Anseth, Biomacromolecules, 2015, 16, 798–806 CrossRef CAS PubMed.
- C. K. Arakawa, B. A. Badeau, Y. Zheng and C. A. DeForest, Adv. Mater., 2017, 29, 1703156 CrossRef PubMed.
- M. Lunzer, L. Shi, O. G. Andriotis, P. Gruber, M. Markovic, P. J. Thurner, D. Ossipov, R. Liska and A. Ovsianikov, Angew. Chem., Int. Ed., 2018, 57, 15122–15127 CrossRef CAS PubMed.
- D. R. Griffin and A. M. Kasko, J. Am. Chem. Soc., 2012, 134, 13103–13107 CrossRef CAS PubMed.
- T. E. Brown, I. A. Marozas and K. S. Anseth, Adv. Mater., 2017, 29, 1605001 CrossRef PubMed.
- C. A. DeForest and K. S. Anseth, Angew. Chem., Int. Ed., 2012, 51, 1816–1819 CrossRef CAS PubMed.
- C. A. DeForest and D. A. Tirrell, Nat. Mater., 2015, 14, 523–531 CrossRef CAS PubMed.
- J. C. Grim, T. E. Brown, B. A. Aguado, D. A. Chapnick, A. L. Viert, X. Liu and K. S. Anseth, ACS Cent. Sci., 2018, 4, 909–916 CrossRef CAS PubMed.
- J. A. Shadish, G. M. Benuska and C. A. DeForest, Nat. Mater., 2019, 18, 1005–1014 CrossRef CAS PubMed.
- A. Farrukh, J. I. Paez and A. del Campo, Adv. Funct. Mater., 2019, 29, 1807734 CrossRef.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS PubMed.
- M. Fernandez-Villamarin, L. Brooks and P. M. Mendes, Adv. Opt. Mater., 2019, 7, 1900215 CrossRef.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- N. Kretschy, A.-K. Holik, V. Somoza, K.-P. Stengele and M. M. Somoza, Angew. Chem., Int. Ed., 2015, 54, 8555–8559 CrossRef CAS PubMed.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723–1736 CrossRef CAS.
- P. J. LeValley, R. Neelarapu, B. P. Sutherland, S. Dasgupta, C. J. Kloxin and A. M. Kloxin, J. Am. Chem. Soc., 2020, 142, 4671–4679 CrossRef CAS PubMed.
- M. A. Azagarsamy, D. D. McKinnon, D. L. Alge and K. S. Anseth, ACS Macro Lett., 2014, 3, 515–519 CrossRef CAS.
- M. S. Lee and J.-C. Kim, J. Appl. Polym. Sci., 2012, 124, 4339–4345 CrossRef CAS.
- Y. Zheng, A. Farrukh and A. del Campo, Langmuir, 2018, 34, 14459–14471 CrossRef CAS PubMed.
- S. Theis, A. Iturmendi, C. Gorsche, M. Orthofer, M. Lunzer, S. Baudis, A. Ovsianikov, R. Liska, U. Monkowius and I. Teasdale, Angew. Chem., Int. Ed., 2017, 56, 15857–15860 CrossRef CAS PubMed.
- T. Rapp, C. Highley, B. Manor, J. Burdick and I. J. Dmochowski, Chem. – Eur. J., 2018, 24, 2328–2333 CrossRef CAS PubMed.
- E.-K. Bang, M. Lista, G. Sforazzini, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1752–1763 RSC.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS PubMed.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142–149 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- H. Otsuka, S. Nagano, Y. Kobashi, T. Maeda and A. Takahara, Chem. Commun., 2010, 46, 1150–1152 RSC.
- J. C. Lukesh, M. J. Palte and R. T. Raines, J. Am. Chem. Soc., 2012, 134, 4057–4059 CrossRef CAS PubMed.
- R. Singh and G. M. Whitesides, in Techniques in Protein Chemistry, ed. W. C. John, Academic Press, 1995, vol. 6, pp. 259–266 Search PubMed.
- J. Zhang, A. Skardal and G. D. Prestwich, Biomaterials, 2008, 29, 4521–4531 CrossRef CAS PubMed.
- J. A. Burns, J. C. Butler, J. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648–2650 CrossRef CAS.
- Y. Chujo, K. Sada, A. Naka, R. Nomura and T. Saegusa, Macromolecules, 1993, 26, 883–887 CrossRef CAS.
- H. Xiang, J. Yin, G. Lin, X. Liu, M. Rong and M. Zhang, Chem. Eng. J., 2019, 358, 878–890 CrossRef CAS.
- X. Z. Shu, Y. Liu, Y. Luo, M. C. Roberts and G. D. Prestwich, Biomacromolecules, 2002, 3, 1304–1311 CrossRef CAS PubMed.
- X. Z. Shu, Y. Liu, F. Palumbo and G. D. Prestwich, Biomaterials, 2003, 24, 3825–3834 CrossRef CAS PubMed.
- S. Singh, F. Topuz, K. Hahn, K. Albrecht and J. Groll, Angew. Chem., Int. Ed., 2013, 52, 3000–3003 CrossRef CAS PubMed.
- M. Mackiewicz, K. Kaniewska, J. Romanski, E. Augustin, Z. Stojek and M. Karbarz, J. Mater. Chem. B, 2015, 3, 7262–7270 RSC.
- K. Raghupathi, V. Kumar, U. Sridhar, A. E. Ribbe, H. He and S. Thayumanavan, Langmuir, 2019, 35, 7929–7936 CrossRef CAS PubMed.
- W. Xu, J. Qian, Y. Zhang, A. Suo, N. Cui, J. Wang, Y. Yao and H. Wang, J. Mater. Chem. B, 2016, 4, 3339–3350 RSC.
- B. Husár and R. Liska, Chem. Soc. Rev., 2012, 41, 2395–2405 RSC.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS PubMed.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- S. R. Caliari, S. L. Vega, M. Kwon, E. M. Soulas and J. A. Burdick, Biomaterials, 2016, 103, 314–323 CrossRef CAS PubMed.
- L. J. Alcock, K. D. Farrell, M. T. Akol, G. H. Jones, M. M. Tierney, H. B. Kramer, T. L. Pukala, G. J. L. Bernardes, M. V. Perkins and J. M. Chalker, Tetrahedron, 2018, 74, 1220–1228 CrossRef CAS.
- R. J. Boyd, J. S. Perkyns and R. Ramani, Can. J. Chem., 1983, 61, 1082–1085 CrossRef CAS.
- S. Benedikt, J. Wang, M. Markovic, N. Moszner, K. Dietliker, A. Ovsianikov, H. Grützmacher and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2015, 473–479, DOI:10.1002/pola.27903.
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Biomaterials, 2009, 30, 6702–6707 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- B. Colak, J. C. S. Da Silva, T. A. Soares and J. E. Gautrot, Bioconjugate Chem., 2016, 27, 2111–2123 CrossRef CAS PubMed.
- B. D. Fairbanks, D. M. Love and C. N. Bowman, Macromol. Chem. Phys., 2017, 218, 1700073 CrossRef.
- D. M. Love, K. Kim, J. T. Goodrich, B. D. Fairbanks, B. T. Worrell, M. P. Stoykovich, C. B. Musgrave and C. N. Bowman, J. Org. Chem., 2018, 83, 2912–2919 CrossRef CAS PubMed.
- D. Love, B. Fairbanks and C. Bowman, ACS Macro Lett., 2020, 9, 174–179 CrossRef CAS.
- H. Shih, A. K. Fraser and C.-C. Lin, ACS Appl. Mater. Interfaces, 2013, 5, 1673–1680 CrossRef CAS PubMed.
- G. A. Barcan, X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2015, 137, 5650–5653 CrossRef CAS PubMed.
- M. Tromayer, A. Dobos, P. Gruber, A. Ajami, R. Dedic, A. Ovsianikov and R. Liska, Polym. Chem., 2018, 9, 3108–3117 RSC.
- W. Steiger, P. Gruber, D. Theiner, A. Dobos, M. Lunzer, J. Van Hoorick, S. Van Vlierberghe, R. Liska and A. Ovsianikov, Opt. Mater. Express, 2019, 9, 3567–3581 CrossRef CAS.
- S. V. Vlierberghe, E. Schacht and P. Dubruel, Eur. Polym. J., 2011, 47, 1039–1047 CrossRef.
- S. Baudis, D. Bomze, M. Markovic, P. Gruber, A. Ovsianikov and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2060–2070 CrossRef CAS.
- M. M. Zieger, P. Mueller, A. S. Quick, M. Wegener and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2017, 56, 5625–5629 CrossRef CAS PubMed.
- R. Batchelor, T. Messer, M. Hippler, M. Wegener, C. Barner-Kowollik and E. Blasco, Adv. Mater., 2019, 31, 1904085 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Instrumentation, computational study, synthesis of linkers, detailed rheological procedures and supporting experiments, 1H-NMR and 13C-NMR information, and additional confocal microscopy images. See DOI: 10.1039/d1py00914a |
| ‡ Present Addresses: UpNano GmbH, Modecenterstrasse 22/D36, 1030 Vienna, Austria. |
| This journal is © The Royal Society of Chemistry 2022 |