
Electroactive 3D printable poly(3,4-ethylenedioxythiophene)-graft-poly(ε-caprolactone) copolymers as scaffolds for muscle cell alignment†
Antonio
Dominguez-Alfaro
ab,
Miryam
Criado-Gonzalez
 a,
Elena
Gabirondo
a,
Haizpea
Lasa-Fernández
b,
Jorge L.
Olmedo-Martínez
a,
Nerea
Casado
a,
Nuria
Alegret
bc,
Alejandro J.
Müller
ad,
Haritz
Sardon
a,
Ainara
Vallejo-Illarramendi
ce and
David
Mecerreyes
*ad
a,
Elena
Gabirondo
a,
Haizpea
Lasa-Fernández
b,
Jorge L.
Olmedo-Martínez
a,
Nerea
Casado
a,
Nuria
Alegret
bc,
Alejandro J.
Müller
ad,
Haritz
Sardon
a,
Ainara
Vallejo-Illarramendi
ce and
David
Mecerreyes
*ad
aPOLYMAT and Department of Polymers and Advanced Materials: Physics, Chemistry and Technology, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel de Lardizabal 3, 20018 Donostia-San Sebastián, Spain. E-mail: david.mecerreyes@ehu.es
bCarbon Bionanotechnology Group, Center for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), San Sebastian 20014, Spain
cIIS Biodonostia, Neurosciences Area, Group of Neuromuscular Diseases, Paseo Dr. Begiristain s/n, 20014 San Sebastian, Spain
dIkerbasque, Basque Foundation for Science, 48013 Bilbao, Spain
eGroup of Neuroscience, Department of Pediatrics, Faculty of Medicine and Nursing, UPV/EHU, Paseo Dr. Begiristain 105, 20014 San Sebastian, Spain
First published on 2nd December 2021
Abstract
The development of tailor-made polymers to build artificial three-dimensional scaffolds to repair damaged skin tissues is gaining increasing attention in the bioelectronics field. Poly(3,4-ethylene dioxythiophene) (PEDOT) is the gold standard conducting polymer for the bioelectronics field due to its high conductivity, thermal stability, and biocompatibility; however, it is insoluble and infusible, which limits its processability into three dimensional scaffolds. Here, poly(3,4-ethylendioxythiophene)-graft-poly(ε-caprolactone) copolymers, PEDOT-g-PCL, with different molecular weights and PEDOT compositions, were synthesized by chemical oxidative polymerization to enhance the processability of PEDOT. First, the chemical structure and composition of the copolymers were characterized by nuclear magnetic resonance, infrared spectroscopy, and thermogravimetric analysis. Then, the additive manufacturing of PEDOT-g-PCL copolymers by direct ink writing was evaluated by rheology and 3D printing assays. The morphology of the printed patterns was further characterized by scanning electron microscopy and the conductivity by the four-point probe. Finally, the employment of these printed patterns to induce muscle cells alignment was tested, proving the ability of PEDOT-g-PCL patterns to produce myotubes differentiation.
Introduction
Conducting polymers (CPs) with tunable electrical and mechanical functionalities are gaining increasing attention for the development of (bio)electronic devices such as electronic skin, biosensors, health monitoring electrodes or soft robotics.1–3 The additive manufacturing (AM) of CPs also plays a key role in the final properties of these electroactive objects, which makes it necessary to evolve from traditional two-dimensional (2D) thin films to shape-conformable three-dimensional (3D) structures.4,5 Besides, in muscle engineering applications, AM could improve the cell–material interface throughout the manufacturing of artificial patterned scaffolds that support unidirectional alignment of myotubes forming parallel arrays of muscle, mimicking real scenarios.6,7 In this regard, 3D printing of CPs has emerged in the last years as a versatile technique for the fabrication of electronic devices with structural defined shapes and functionalities at the micrometer scale.8 Therefore, the design of tailor-made conducting polymer materials that possess good conductivity and appropriate mechanical properties to be processed by 3D printing represents a fascinating approach.9,10Poly(3,4-ethylene dioxythiophene) (PEDOT) is one of the most used conducting polymers in the (bio)electronics field due to its commercial availability and inherent properties, i.e., high conductivity, thermal stability, and biocompatibility; however, it is insoluble and infusible, which limits its processability.11 This drawback can be overcome by stabilizing PEDOT with other polymers that provide the required properties to be processed by 3D printing techniques. PEDOT is usually stabilized with polystyrene sulfonate (PSS), leading to PEDOT:PSS dispersions that can be formulated with organic solvents or polymers, i.e., cellulose, alginate, gelatin, leading to viscous materials (102–103 Pa s) with shear-thinning properties to be processed by direct ink writing (DIW).12–15
Although simple blending is the easiest method to prepare processable PEDOT/polymer materials, miscibility issues can affect the structural and functional homogeneity of the manufactured material, which is undesirable and can be solved by covalent grafting of both homopolymers. This PEDOT functionalization depends on the molecular weight, functional groups, conformation, and side chains attached to the monomeric units to graft leading to copolymers that keep intact the conjugated PEDOT backbone, responsible of the conductive properties, while modulating the polymer physicochemical properties improving its processability.
Among different polymer materials that can be combined with PEDOT to synthesize processable and biocompatible copolymers, saturated polyesters such as polycaprolactone (PCL) or polylactide (PLA) are excellent candidates due to their biodegradability and biocompatibility.16,17 Very recently, we have shown that PEDOT-g-PLA copolymers can be processed by 3D direct ink writing leading to scaffolds for soft tissue engineering.18 Nevertheless, for hard tissue engineering applications, i.e., skeletal muscle tissues, materials with longer degradation times are searched, being PCL an excellent alternative.19 Moreover, PCL exhibits lower glass transition (Tg ≥ −60 °C) and melting (Tm > 60 °C) temperatures, and higher crystallization rate and flexibility than PLA,20,21 which favours the additive manufacturing by DIW. In this line, Stevens and co-workers synthesized PEDOT-co-PCL block copolymers by ring opening polymerization of ε-caprolactone with oligo-EDOT macromonomers functionalized with amino end-capped groups to fabricate fiber-made platforms through melt electrospinning writing for neural tissue engineering.22 Similar strategy was used to synthesize tetraaniline-b-PCL-b-tetraaniline copolymers to manufacture scaffolds for bone tissue engineering by DIW,23 and PPy-b-PCL scaffolds by electrohydrodynamic printing (EHD) for peripheral nerve injury repair.24 Apart from that, EDOT end-group functionalized PCL macromonomers, synthesized by ROP of hydrozymethyl-EDOT and ε-caprolactone in presence of Sn(Oct) catalyst, has shown their ability to self-assemble in selective organic solvents.25
In this work, we propose new PEDOT-g-PCL graft copolymers to manufacture electroactive scaffolds for muscle tissue engineering by DIW. These copolymers were synthesized through chemical oxidative polymerization of EDOT and new PCL macromonomers functionalized with an EDOT end group. The influence of the EDOT-PCL macromonomer molecular weight (Mn) on the copolymerization process, and its effect on the copolymer processability by extrusion printing and conductive properties were also evaluated. Finally, the biocompatibility of the PEDOT-g-PCL printed patterns was tested in contact with 8220 muscle cells, as well as their influence in the myoblast cell alignment for potential muscle tissue engineering applications.
Experimental section
Materials
ε-Caprolactone (97%) was supplied by Sigma-Aldrich and distilled before using it. Methanesulfonic acid (MSA), 4-dimethylaminopyridine (DMAP), 3,4-ethylenedioxythiophene (97%) (EDOT) and hydroxymethyl EDOT (95%) were provided by Sigma-Aldrich and used as received.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, PCL4k, PCL8k and PCL16k, were synthesized by ring-opening polymerization (ROP) of ε-caprolactone in bulk using EDOT-methanol as chain initiator and an organocatalyst, formed by a mixture of methanesulfonic acid and 4-dimethylaminopyridine (1MSA:1DMAP). First, the organocatalysts mixture was warmed up at 100 °C until forming a white salt. Then, the caprolactone and hydroxymethyl EDOT were added and allowed them to react at 130 °C with magnetic stirring and sealed under inert atmosphere for 5 days until 90% conversion is achieved. The resulting product, EDOT-PCL, was purified through precipitation in methanol, and subsequently vacuum-dried at room temperature overnight.
:5 PEDOT), the PCL macromonomers synthesized previously (1.5 g, 1.31 × 10−2 mol) and EDOT (0.078 g, 5.49 × 10−4 mol) were dissolved in 10 mL of CHCl3. Then, 0.135 g of FeCl3 (1.5 eq. respect EDOT monomer) was added to the mixture and let it react under magnetic stirring at room temperature overnight. The blue dispersion obtained was precipitated and washed with methanol until the iron residue was fully removed. Finally, the product was dried under vacuum at room temperature.
000 g mol−1, PCL4k, PCL8k and PCL16k, were synthesized by ring-opening polymerization (ROP) of ε-caprolactone in bulk using EDOT-methanol as chain initiator and an organocatalyst, formed by a mixture of methanesulfonic acid and 4-dimethylaminopyridine (1MSA:1DMAP). First, the organocatalysts mixture was warmed up at 100 °C until forming a white salt. Then, the caprolactone and hydroxymethyl EDOT were added and allowed them to react at 130 °C with magnetic stirring and sealed under inert atmosphere for 5 days until 90% conversion is achieved. The resulting product, EDOT-PCL, was purified through precipitation in methanol, and subsequently vacuum-dried at room temperature overnight.
:5 PEDOT), the PCL macromonomers synthesized previously (1.5 g, 1.31 × 10−2 mol) and EDOT (0.078 g, 5.49 × 10−4 mol) were dissolved in 10 mL of CHCl3. Then, 0.135 g of FeCl3 (1.5 eq. respect EDOT monomer) was added to the mixture and let it react under magnetic stirring at room temperature overnight. The blue dispersion obtained was precipitated and washed with methanol until the iron residue was fully removed. Finally, the product was dried under vacuum at room temperature.
Methods
800 Hz, and more than 10000 scans.
:2:1 (matrix:sample:salt). A total of 2000–3000 acquisitions were carried out in positive linear mode and registered by the software FlexControl 3.3 (Bruker Daltonics, Bremen, Germany).
:50 in PBS at room-temperature. Then, samples were washed with PBS three times, and they were mounted with ProLong Gold antifade reagent with DAPI (Life Technologies). Fluorescence images were acquired using a Zeiss LSM 510 confocal microscope with a 20× objective.
Results and discussion
Synthesis of α-EDOT-PCL macromonomers by ring-opening polymerization
Graft copolymers combining conducting polymers, i.e., PEDOT or polypyrrole, and aliphatic polyesters have been successfully synthesized by the macromonomer method.18,26–29 However, to the best of our knowledge, the graft copolymerization of PEDOT and PCL using α-EDOT-PCL macromonomers has not been reported yet. For such purpose, in a first step α-EDOT-PCL macromonomers, with molecular weights ranging from 4000 to 16000 g mol−1, were synthesized by ring-opening polymerization (ROP) using EDOT-methanol as chain initiator of ε-caprolactone polymerization (Scheme 1a). The ROP was carried out in bulk using an organocatalyst previously reported for the synthesis of poly(L-lactide) (PLA) using benzyl alcohol as initiator. This organocatalyst is formed by a mixture of methanesulfonic acid (MSA) and 4-dimethylaminopyridine (DMAP) in a ratio 1:1.30 A 90% conversion was reached after 5 days reaction at 130 °C as calculated by 1H-NMR (Fig. S1a, ESI†). Moreover, the molecular weight of the EDOT-PCL macromonomers was determined through the relative intensities of the 1H-NMR signals corresponding to the tiophene proton at 6.4 ppm and –CH2– aliphatic polyester chain at 4.3 ppm (Table S1, ESI†). The presence of the EDOT end group was also confirmed by 13C-NMR (Fig. S1b, ESI†). Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) spectroscopy (Fig. S1c, ESI†) also confirmed the presence of the end-group and a monomodal mass profile distribution of the polycaprolactone repeating unit (114 m/z).
 | ||
| Scheme 1 Chemical routes employed to synthesize: (a) EDOT-PCL macromonomers by ROP and (b) PEDOT-g-PCL copolymers by chemical oxidative polymerization. | ||
Synthesis and characterization of PEDOT-g-PCL copolymers
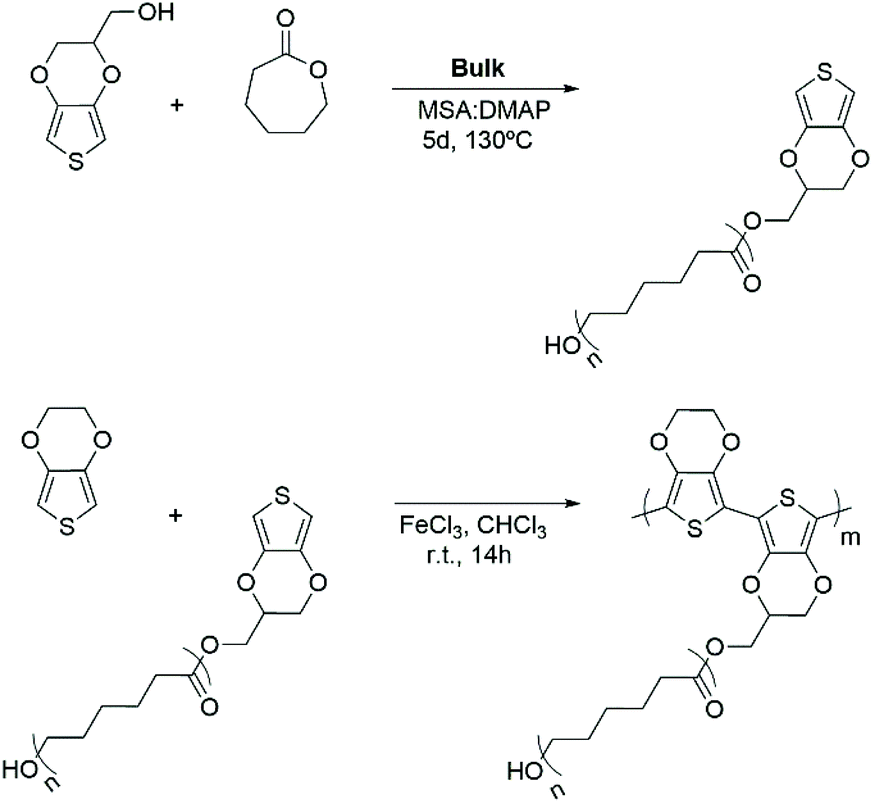
PEDOT-g-PCL copolymers were synthesized by chemical oxidative copolymerization of 3,4-ethylendioxythiophene and the previously synthesized PCL macromonomers (Scheme 1b). Thus, the EDOT monomer was oxidized with an excess of FeCl3 and grafted to the EDOT-PCL macromonomer in solution leading to a dark blue dispersion. It is expected that the conductive properties of the PEDOT-g-PCL copolymers increase as the PEDOT composition increases, whereas the printability properties will benefit from the PCL composition. For this reason, a balance between both components of the graft copolymer needs to be found. Therefore, copolymers with different PEDOT percentages were synthesized by changing the initial EDOT:PCL ratio employed in the reaction. As can be observed in Table 1, higher yields (>40%) are obtained when a low quantity of EDOT monomer is fed to the reaction (≤5 wt%), as well as the molecular weight of the PCL macromonomer increases. The presence of both components, PCL and PEDOT, in the copolymers was monitored by infrared spectroscopy (Fig. 1a). FT-IR spectra show the signals of the carbonyl group of PCL at 1747 cm−1 and the thiophene ring of PEDOT located at 985, 836 and 686 cm−1. Besides, the presence of PEDOT in the copolymers was also confirmed by UV-vis-NIR spectroscopy. All spectra show the polaron and bipolaron absorption bands in the range 800–1000 nm (Fig. S2, ESI†), which is characteristic of the PEDOT oxidized polymer.31 Regarding the PEDOT composition in the PEDOT-g-PCL copolymers, it was calculated by TGA using the relative thermal stability of the copolymers at 450 °C (Fig. 1b). As can be observed, all spectra present a proportional weight loss related to the PEDOT percentage in the copolymers. By comparing these results with the calibration curve obtained for different PEDOT/PCL blends (Fig. S3, ESI†), it is possible to determine the percentage of PEDOT present in the synthesized copolymers PEDOT-g-PCL, and results are collected in Table 1. The PEDOT composition in the copolymers was much higher than the one fed to the reaction, and as expected, the final percentage of PEDOT in the copolymers increased with the initial EDOT percentage used in the feed. These results are in agreement with previous works for the synthesis of other graft copolymers such as PEDOT-g-PLA and PPy-g-PCL and associated with the low reactivity of the macronomers.18,26,32 It was also observed an increase in the percentage of PEDOT incorporated in the copolymers as the molecular weight (Mn) of the PCL macromonomer decreased. This can be explained by the fact that more EDOT-end PCL chains are available to react with the EDOT monomer leading to PEDOT-g-PCL copolymers with a higher PEDOT percentage.
 | ||
| Fig. 1 (a) FT-IR and (b) TGA spectra of the copolymers PEDOT-g-PCL8k synthesized with different PEDOT compositions, 16, 26 and 68 wt%. | ||
| Code |
M
n PCLa (g mol−1) |
wt%EDOTb | Yield (wt%) | wt%PEDOTc |
|---|---|---|---|---|
| a M n EDOT-PCL macromonomers calculated by 1H-NMR. b Weight fraction of EDOT in the reaction feed. c Weight fraction of PEDOT in the copolymers calculated by TGA. | ||||
| 1 | 4000 | 10 | 12 | 40 |
| 2 | 7500 | 5 | 40 | 16 |
| 3 | 7500 | 10 | 26 | 26 |
| 4 | 7500 | 20 | 18 | 68 |
| 5 | 16000 |
5 | 50 | 10 |
| 6 | 16000 |
10 | 35 | 30 |
To evaluate the impact of the PEDOT presence in the copolymers to obtain homogeneous materials for additive manufacturing applications, the morphological structure was analysed by SEM (Fig. 2). The PCL macromonomer shows a continuous morphology that is substantially affected by the incorporation of PEDOT within the copolymer chain PEDOT-g-PCL. As long as PEDOT is incorporated in the graft copolymer, the film morphology shows the presence of particles throughout the whole surface, whose density considerably increases with the PEDOT percentage. In the case of the copolymers with the highest PEDOT percentage, 68% PEDOT-g-PCL, the morphology is granular, almost similar to the one of neat PEDOT, which could hinder the extrusion printing process. Even if the incorporation of PEDOT interrupts the flatten PCL morphology, each copolymer shows a homogeneous morphology from flat to rough particle surfaces for lower and higher PEDOT compositions respectively.
 | ||
| Fig. 2 SEM images of films obtained by drop casting of PCL, PEDOT and different PEDOT-g-PCL8k copolymer solutions in CHCl3. | ||
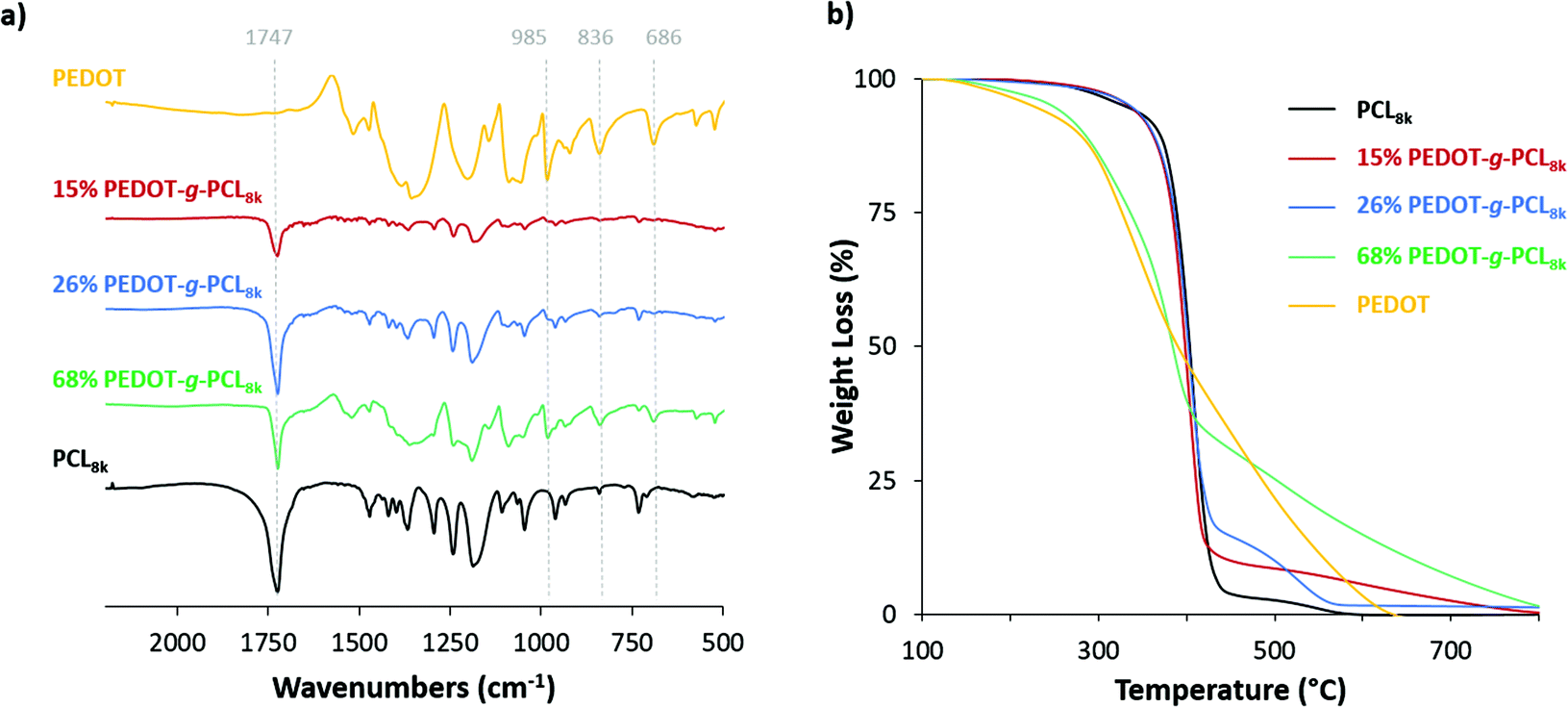
Prior to proceed with the additive manufacturing of these copolymers by direct ink writing in the melt state, the thermal properties need to be studied. DSC analysis was performed to evaluate the melting and crystallization processes of the graft copolymers under standard conditions. First, the sample was heated from room temperature up to 120 °C at a rate of 20 °C min−1 and then it was cooled down until −20 °C at the same rate. Fig. 3a exhibits the second DSC heating scans of PEDOT-g-PCL8k copolymers. A single endothermic peak is observed above 53 °C corresponding to the melting temperature (Tm) of the polyester (PCL). This melting temperature remains constant in the case of the copolymer with the highest PCL content, 16% PEDOT-g-PCL, while a shift to lower Tm is observed as the PEDOT percentage increases in the copolymers, Tm = 51 °C for 26% PEDOT-g-PCL and Tm = 50 °C for 68% PEDOT-g-PCL. As PEDOT is incorporated in the graft copolymer, the crystallisable linear PCL sequences are increasingly interrupted by the grafted chains, and thinner crystals are formed that melt at progressively lower temperatures. Fig. 3b shows the DSC cooling down scan with the presence of a single exothermic peak, which is attributed to the crystallization process from the melt. In that case, a shift to higher crystallization temperatures (Tc) is observed as long as PEDOT is incorporated in the copolymer, from Tc = 23 °C for PCL up to 27 °C and 29 °C for 16% PEDOT-g-PCL and 68% PEDOT-g-PCL, respectively. Similar behaviour was observed for the copolymers PEDOT-g-PCL16k, synthesized with higher Mn of the PCL macromonomer (Fig. S4, ESI†). The increase in crystallization temperature of the PCL component is probably due to a nucleation effect, which could be caused by PEDOT incorporation.
 | ||
| Fig. 3 DSC spectra of PCL8k and PEDOT-g-PCL8k copolymers, (a) 2nd heat scan at a temperature rate of 20 °C min−1 rate and (b) cooling down scan at 20 °C min−1. | ||
The effect of the thermal treatment on the nanoscale morphology of the PEDOT-g-PCL copolymers was examined by AFM. For such purpose, the copolymers were solubilized in chloroform, drop casted on a silicon substrate, and subjected to the thermal protocol specified in the Experimental section. AFM height images (Fig. 4) show the characteristic lamellar structure of the PCL. In the case of PEDOT-g-PCL copolymers, this lamellar structure is altered by round-shape areas that increase the average roughness (Ra) from 5 nm for PCL to 12 nm for 26% PEDOT-g-PCL, due to the presence of PEDOT as observed by SEM. After the thermal treatment, the bigger size lamellae remained unaltered, but some small darker areas appear in the interlamellar regions. These darker areas are attributed to the presence of amorphous PCL regions. During the thermal treatment, the samples are heated to temperatures above the melting point of the PCL phase, then they are cooled to room temperature only. During cooling just to room temperature (see Fig. 3b), it is possible that not all the previous thinner lamellae (that usually crystallize at lower temperatures) could be formed and the material where they were previously located is now constituting the amorphous interlamellar regions between the thickest lamellae (that formed at temperatures above 25 °C during cooling from the melt). A similar behaviour has also been observed for PCL-co-PLA diblock copolymers.33 These results allow us to conclude that the main structure and morphology of the PEDOT-g-PCL copolymers is not substantially affected by the heating process over the melting point (as they are able to reform as expected), which makes them ideal candidates to be manufactured by DIW in the melt state.
 | ||
| Fig. 4 AFM images of films obtained by drop casting of PCL and different PEDOT-g-PCL8k copolymer solutions in CHCl3 pre- (a) and after-thermal treatment (b). Blue arrows highlight darker regions (lower height) after thermal treatment. | ||
3D printing of PEDOT-g-PCL copolymers
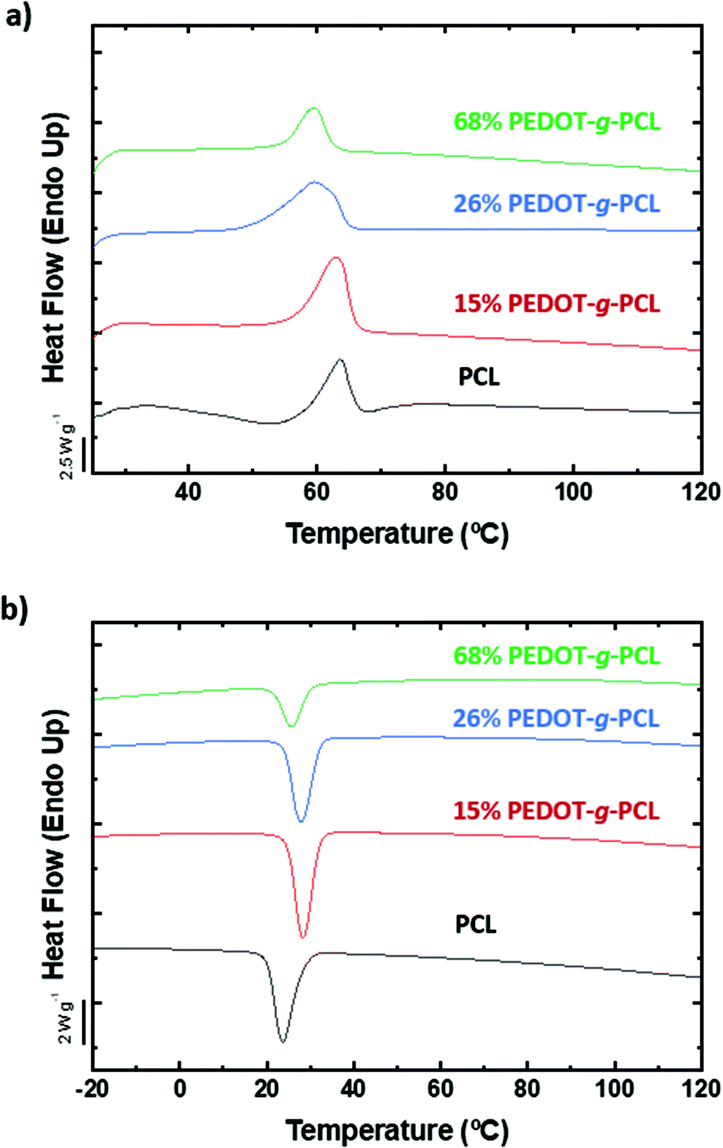
The additive manufacturing of PEDOT-g-PCL copolymers by 3D printing depends on their shear-thinning behaviour and ability to flow. Previously, we have reported that complex viscosities (η*) lower than 103 Pa s represent the most liquid-like compositions to be processed by 3D extrusion printing, whereas higher than 104 Pa s are characteristic of a solid-like behaviour. Complex viscosities between 103 Pa s < η* < 104 Pa s are indicative of solid to liquid transition with a high plasticity behaviour that hinders the polymer flow through the needle.18 For such purpose, frequency sweeps were performed at the melting temperature of the polyester, i.e., 65 °C, (Fig. 5 and S5, ESI†). In the case of the PCL macromonomer, a fluid-like behaviour is observed with complex viscosity values lower than 10 Pa s. The incorporation of PEDOT in the polymer chain gives rise to a huge increase in the complex viscosity reaching values of 400 and 5000 Pa s for 16% PEDOT-g-PCL8k and 26% PEDOT-g-PCL8k, respectively. This latter case is ranged in the transition-like area and could present limitations to flow. It was also observed a decrease of the complex viscosity in the copolymers when the molecular weight of the PCL macromonomer (Mn) increases, reaching a value of η* = 600 Pa s for 30% PEDOT-g-PCL16k. Nevertheless, all copolymers show a shear thinning behaviour with a high decrease of the complex viscosity with the frequency, which makes them accurate for 3D direct ink writing. | ||
| Fig. 5 Complex viscosity at 65 °C of (a) PCL8k and PEDOT-g-PCL8k copolymers, and (b) PCL16k and PEDOT-g-PCL16k copolymers. | ||
Then, 3D printing tests were performed by DIW of these copolymers in the melting state (Fig. 6a). In the case of the PCL macromonomers, they can be easily printed at the melting temperature (65 °C) with minimal pressure. However, the copolymers PEDOT-g-PCL require the combination of both properties, melting state and a physical pressure, to be printed.
 | ||
| Fig. 6 (a) Schematic representation of the printing process. (b) Pictures of the printed PEDOT-g-PCL patterns. Scale bar = 12 mm. (c) Topographical SEM images of the printed patterns corresponding to 16% PEDOT-g-PCL8k copolymers (top) and 26% PEDOT-g-PCL8k copolymers (bottom). (d) Zoom-in of the SEM pictures, where PEDOT particles are highlighted with yellow arrows. | ||
Besides, the copolymers with complex viscosity values in the solid to liquid transition zone, 103 Pa s < η* < 104 Pa s, present high plasticity, being necessary to combine the pneumatic pressure input by air with the mechanical action of a metallic piston to print them. Therefore, the copolymers PEDOT-g-PCL were inserted in a metallic extruder, warmed up at 65 °C, and subsequently printed by air/physical action. The material was deposited layer-by-layer, forming three-dimensional structures.
The PCL macromonomer and all the copolymers with PEDOT percentages between 10 and 30 wt% were successfully printed in a wide variety of complex shapes (Fig. 6b and S6, ESI†). The structural characterization by SEM shows a very high resolution of the printed structures retaining their shape after printing (Fig. 6c and S6, ESI†). It is worth mentioning that all the copolymers with PEDOT percentages between 10 and 30 wt% were successfully printed using the air/physical action. Then, the morphology of the printed copolymers was analysed in detail by zoom-in the printed area (Fig. 6d). Some particles can be distinguished all around the material, as marked by yellow arrows, and their density increases with the PEDOT percentage in the copolymers, accordingly with the results obtained for the drop casted films.
Electrical properties of the 3D printed scaffolds
The conductivity of the printed scaffolds was measured with a four-point probe. In the case of the copolymers PEDOT-g-PCL8k, there is an increase of the conductivity with the PEDOT percentage in the copolymers, from 1.17 × 10−4 S cm−1 for 16% PEDOT-g-PCL8k to 7.07 × 10−4 S cm−1 for 26% PEDOT-g-PCL8k. However, in the case of PEDOT-g-PCL16k copolymers, synthesized with the PCL macromonomer of high molecular weight, the conductivity substantially decreases up to values of 3.10 × 10−5 S cm−1 and 4.26 × 10−5 S cm−1 for 10 and 30% PEDOT-g-PCL16k, respectively. This conductivity is lower than the one obtained for highly conductive PEDOT:PSS; however, these values are in the same order as those obtained for other conducting graft copolymers such as PPy-b-PCL copolymers (2.8 × 10−4 S cm−1)24 and PCL/PANI blends (2.5 × 10−4 S cm−1),23 and higher than PCL/MWCNTs scaffolds (1.4 × 10−5 S cm−1).34 Taking into account that the conductive properties decrease for the copolymers with higher Mn of the PCL macromonomer, the copolymer 16% PEDOT-g-PCL8k was chosen for further studies with 8220 muscle cells.Electrochemical characterization was performed using cyclic voltammetry in 0.1 M NaCl aqueous solution. As can be observed in Fig. 7, the cyclic voltammogram (CV) of 16% PEDOT-g-PCL shows a capacitive behavior and broad anodic and cathodic peaks at 0.55 V and 0.4 V vs. Ag/AgCl, which are typical of PEDOT and demonstrates the electroactive behavior of the polymer. On the contrary, the CV of PCL is completely flat, indicating that is not electrochemically active. Besides, cyclic voltammetries at different scan rates were performed to analyze charge transfer processes. The CV at various scan rates of a 16% PEDOT-g-PCL film in 0.1 M NaCl aqueous solution are shown in Fig. S7.† The anodic and cathodic currents increase proportionally with the scan rate, indicating that the redox process is not limited by diffusion, and demonstrating that the whole PEDOT-g-PCL film is involved in the electrochemical reaction.
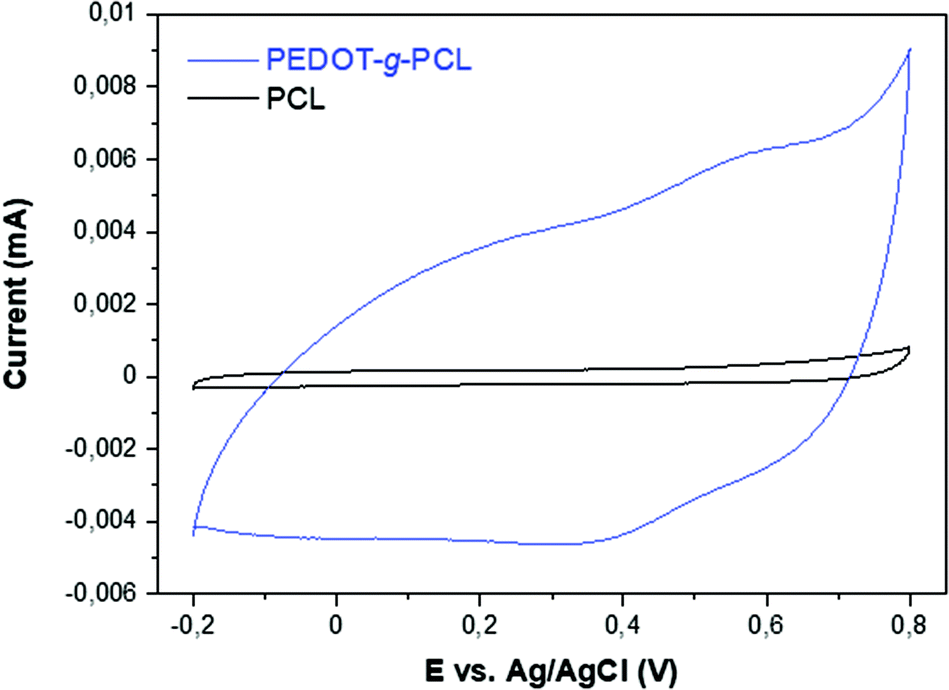 | ||
| Fig. 7 Comparison of the cyclic voltammograms of 16% PEDOT-g-PCL and PCL polymers in 0.1 M NaCl aqueous solution at 50 mV s−1. | ||
Muscle cells culture onto PCL and electroactive PEDOT-g-PCL patterns
As mentioned before, muscle tissues are formed by a skeletal muscle fiber arrangement of myotubes unidirectionally aligned.6 To that aim, patterns with different distances between strands, 460 μm for large width and 230 μm for small width, were printed to induce the muscle cells alignment (Fig. 8a). Firstly, the biocompatibility of these printed scaffolds was assessed. For such purpose, 8220 muscle cells were cultured on the printed scaffolds, and cell toxicity after 24 hours culture was calculated by a direct lactate dehydrogenase (LHD) assay measuring the absorbance at 490 nm (Fig. 8b). As can be observed, there are not significant differences in the absorbance for the PCL and PEDOT-g-PCL scaffolds as comparing with the gelatin-coated plate used as control (CTL), which means that the cell viability is not altered in contact with the PCL and PEDOT-g-PCL scaffolds proving the non-cytotoxic behaviour of the materials. Then, the effect of the composition and design of the printed scaffolds on the myoblasts cell alignment and differentiation was studied by immunofluorescent staining using myosin heavy chain (MyHC), a myotube maturation marker (Fig. 8c). A robust green fluorescence for MyHC is observed in the case of the patterns made with the copolymer PEDOT-g-PCL following the strand direction of the printed pattern. This is also corroborated by the blue fluorescence (DAPI) of the nuclei oriented along the channel between strands meaning that muscle cells are more fused tending to form myotubes. That is not observed in the case of the PCL scaffolds, where nuclei are randomly distributed all over the channel with not robust MyHC fluorescence intensity, meaning that myoblasts are not aligned. Cells are not either aligned in the case of CTL samples. This achievement was also corroborated by SEM characterization (Fig. 8d). Cells adhere on the PCL and PEDOT-g-PCL scaffolds surface; however, in the case of PCL scaffolds, nuclei are randomly distributed, whereas on PEDOT-g-PCL scaffolds, nuclei are clearly aligned, as indicated by the yellow arrows. Alignment of muscle cells depends on cell confluence, material composition, patterning, surface energy and wettability. PEDOT-g-PCL patterns are more hydrophilic (WCA = 49 ± 7°) than PCL ones (WCA = 70 ± 3°), as shown in Fig. S8,† which could favour the cell adhesion, a primary step for the later cell proliferation and differentiation.35 Apart from that, conductive materials can promote proliferation and differentiation when responsive cells are cultured on top or within them.36,37 In that sense, we have shown by the four-point probe and CV experiments that PEDOT-g-PCL scaffolds are electroactive, whereas PCL scaffolds do not exhibit that property. It means that PEDOT-g-PCL scaffolds are able to provide electrical stimulation per se to the cells in contact with them favouring the chemical exchanges and signal communications among the cells at the interface of cell membrane-material inducing their alignment. Therefore, the combination of different specific properties exhibited by PEDOT-g-PCL materials, including chemical composition, conductivity, redox activity and hydrophilicity changes, ensures differences in cell alignment and myogenesis. Indeed, the presence of MyHC marker could be indicative of myotubes differentiation in the case of the PEDOT-g-PCL, corroborating the hypothesis. Therefore, this type of 3D printed conducting scaffolds could mimic native skeletal muscle architecture for potential tissue engineering applications.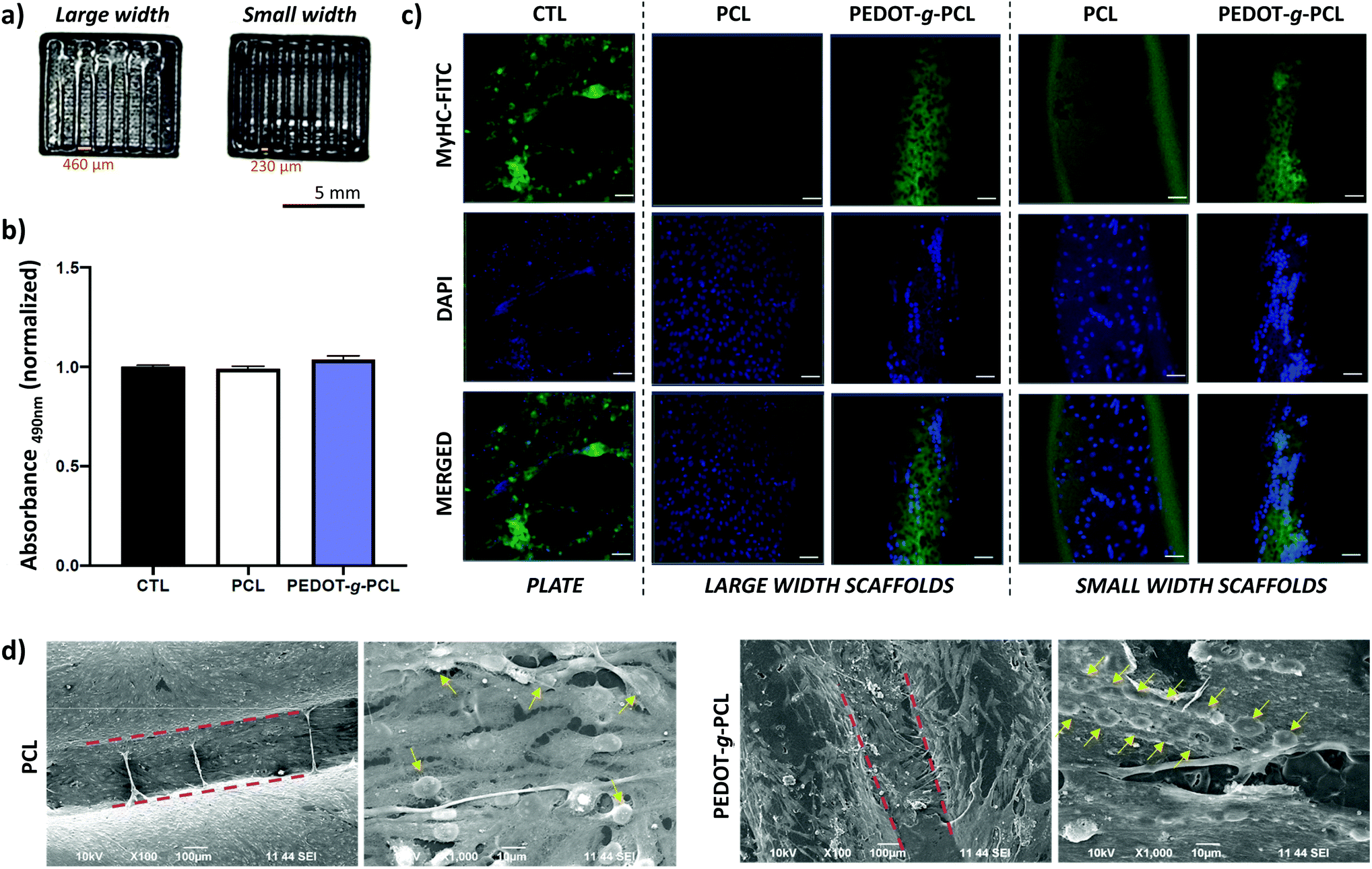 | ||
| Fig. 8 (a) Printed patterns employed for cellular tests. Large width patterns possess 460 μm width between strands and small width patterns 230 μm. (b) LDH test to determine the 8220 muscle cell toxicity after 24 h. Non-significant differences with **p ≤ 0.05. (c) Immunofluorescence images of 8220 muscle cells after 5 days culture on the plate (CTL) and on PCL or PEDOT-g-PCL scaffolds with large width and small width between strands. Cytoskeleton stained in green with MyHC-FITC (top), nuclei stained in blue with DAPI (center) and merged channels (bottom). Scale bar = 50 μm. (d) SEM images of 8220 muscle cells cultured on PCL or PEDOT-g-PCL small width scaffolds, where channels are delimited by dashed red lines, and zoom-in of the channel area between strands where nuclei are highlighted with yellow arrows. | ||
Conclusions
PEDOT-g-PCL copolymers with different molecular weights and compositions, were synthesized by chemical oxidative polymerization of EDOT and α-EDOT-PCL macromonomers, which were previously synthesized by ring-opening polymerization of εCL initiated by EDOT-methanol. The copolymerization process reached yields higher than 40% in the case of copolymers with lower PEDOT composition, 10% PEDOT-g-PCL16k, and 16% PEDOT-g-PCL8k. All the copolymers show a shear-thinning behaviour with a drastic decrease of the viscosity as the frequency increases. However, only the copolymers with lower PEDOT percentages show complex viscosity values lower than 103 Pa s to be easily processed by direct ink writing. The copolymers were successfully printed by DIW in different shapes. It was also determined that the melting process did not produce significant changes in the global morphology of the material, and larger size lamellar were able to reformed during cooling after the thermal treatment. Moreover, SEM images showed high resolution images of these printed patterns with conductivity values of 1.17 × 10−4 S cm−1 for 16% PEDOT-g-PCL8k and 7.07 × 10−4 S cm−1 for 26% PEDOT-g-PCL8k, as determined by the four-point probe. Finally, the biocompatibility properties of the printed scaffolds were proven in contact with 8220 muscle cells, as well as their ability to induce the cell alignment and myotubes differentiation mimicking the native muscle tissue for tissue engineering applications. These polymers show promising properties as artificial three-dimensional scaffolds mimicking the native skeletal muscle architecture for potential applications in bioelectronics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the spanish AEI-MICINN project PID2020-119026GB-I00 and Basque Government through grant IT1309-19. J. L. O.-M. thanks the Consejo Nacional de Ciencia y Tecnología (CONACyT, México) for the grant awarded no. 471837.References
- N. Alegret, A. Dominguez-Alfaro and D. Mecerreyes, Conductive Polymers Building 3D Scaffolds for Tissue Engineering, in Redox Polymers for Energy and Nanomedicine, The Royal Society of Chemistry, 2021, ch. 10, pp. 383–414 Search PubMed.
- J. C. Yang, J. Mun, S. Y. Kwon, S. Park, Z. Bao and S. Park, Electronic Skin: Recent Progress and Future Prospects for Skin-Attachable Devices for Health Monitoring, Robotics, and Prosthetics, Adv. Mater., 2019, 31(48), 1904765 CrossRef CAS.
- A. Maziz, E. Özgür, C. Bergaud and L. Uzun, Progress in conducting polymers for biointerfacing and biorecognition applications, Sens. Actuators Rep., 2021, 3, 100035 CrossRef.
- N. Alegret, A. Dominguez-Alfaro and D. Mecerreyes, 3D Scaffolds Based on Conductive Polymers for Biomedical Applications, Biomacromolecules, 2019, 20(1), 73–89 CrossRef CAS PubMed.
- Z. Qi, J. Ye, W. Chen, J. Biener, E. B. Duoss, C. M. Spadaccini, M. A. Worsley and C. Zhu, 3D-Printed, Superelastic Polypyrrole–Graphene Electrodes with Ultrahigh Areal Capacitance for Electrochemical Energy Storage, Adv. Mater. Technol., 2018, 3(7), 1800053 CrossRef.
- Y. Zhao, H. Zeng, J. Nam and S. Agarwal, Fabrication of skeletal muscle constructs by topographic activation of cell alignment, Biotechnol. Bioeng., 2009, 102(2), 624–631 CrossRef CAS PubMed.
- G.-W. Kim, G.-H. Nam, I.-S. Kim and S.-Y. Park, Xk-related protein 8 regulates myoblast differentiation and survival, FEBS J., 2017, 284(21), 3575–3588 CAS.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Polymers for 3D Printing and Customized Additive Manufacturing, Chem. Rev., 2017, 117(15), 10212–10290 CrossRef CAS.
- M. Criado-Gonzalez, A. Dominguez-Alfaro, N. Lopez-Larrea, N. Alegret and D. Mecerreyes, Additive Manufacturing of Conducting Polymers: Recent Advances, Challenges, and Opportunities, ACS Appl. Polym. Mater., 2021, 3(6), 2865–2883 CrossRef CAS.
- B. Narupai and A. Nelson, 100th Anniversary of Macromolecular Science Viewpoint: Macromolecular Materials for Additive Manufacturing, ACS Macro Lett., 2020, 9(5), 627–638 CrossRef CAS.
- D. Minudri, D. Mantione, A. Dominguez-Alfaro, S. Moya, E. Maza, C. Bellacanzone, M. R. Antognazza and D. Mecerreyes, Water Soluble Cationic Poly(3,4-Ethylenedioxythiophene) PEDOT-N as a Versatile Conducting Polymer for Bioelectronics, Adv. Electron. Mater., 2020, 6(10), 2000510 CrossRef CAS.
- H. Yuk, B. Lu, S. Lin, K. Qu, J. Xu, J. Luo and X. Zhao, 3D printing of conducting polymers, Nat. Commun., 2020, 11(1), 1604 CrossRef CAS PubMed.
- P. Bao, Y. Lu, P. Tao, B. Liu, J. Li and X. Cui, 3D printing PEDOT-CMC-based high areal capacity electrodes for Li-ion batteries, Ionics, 2021, 27(7), 2857–2865 CrossRef CAS.
- H. Françon, Z. Wang, A. Marais, K. Mystek, A. Piper, H. Granberg, A. Malti, P. Gatenholm, P. A. Larsson and L. Wågberg, Ambient-Dried, 3D-Printable and Electrically Conducting Cellulose Nanofiber Aerogels by Inclusion of Functional Polymers, Adv. Funct. Mater., 2020, 30(12), 1909383 CrossRef.
- A. R. Spencer, E. Shirzaei Sani, J. R. Soucy, C. C. Corbet, A. Primbetova, R. A. Koppes and N. Annabi, Bioprinting of a Cell-Laden Conductive Hydrogel Composite, ACS Appl. Mater. Interfaces, 2019, 11(34), 30518–30533 CrossRef CAS PubMed.
- I. Manavitehrani, A. Fathi, H. Badr, S. Daly, A. Negahi Shirazi and F. Dehghani, Biomedical Applications of Biodegradable Polyesters, Polymer, 2016, 8(1), 20 Search PubMed.
- F. A. M. M. Gonçalves, A. C. Fonseca, M. Domingos, A. Gloria, A. C. Serra and J. F. J. Coelho, The potential of unsaturated polyesters in biomedicine and tissue engineering: Synthesis, structure-properties relationships and additive manufacturing, Prog. Polym. Sci., 2017, 68, 1–34 CrossRef.
- A. Dominguez-Alfaro, E. Gabirondo, N. Alegret, C. M. De León-Almazán, R. Hernandez, A. Vallejo-Illarramendi, M. Prato and D. Mecerreyes, 3D Printable Conducting and Biocompatible PEDOT-graft-PLA Copolymers by Direct Ink Writing, Macromol. Rapid Commun., 2021, 42(12), 2100100 CrossRef CAS.
- I. Fortelny, A. Ujcic, L. Fambri and M. Slouf, Phase Structure, Compatibility, and Toughness of PLA/PCL Blends: A Review, Front. Mater., 2019, 6, 206 CrossRef.
- C. Zhang, Q. Lan, T. Zhai, S. Nie, J. Luo and W. Yan, Melt Crystallization Behavior and Crystalline Morphology of Polylactide/Poly(ε-caprolactone) Blends Compatibilized by Lactide-Caprolactone Copolymer, Polymer, 2018, 10(11), 1181 Search PubMed.
- S. E. Fenni, D. Cavallo and A. J. Müller, Nucleation and Crystallization in Bio-Based Immiscible Polyester Blends, in Thermal Properties of Bio-based Polymers, ed. M. L. Di Lorenzo and R. Androsch, Springer International Publishing, Cham, 2019, pp. 219–256 Search PubMed.
- K. I. Ritzau-Reid, C. D. Spicer, A. Gelmi, C. L. Grigsby, J. F. Ponder Jr., V. Bemmer, A. Creamer, R. Vilar, A. Serio and M. M. Stevens, An Electroactive Oligo-EDOT Platform for Neural Tissue Engineering, Adv. Funct. Mater., 2020, 30(42), 2003710 CrossRef CAS.
- A. Wibowo, C. Vyas, G. Cooper, F. Qulub, R. Suratman, A. I. Mahyuddin, T. Dirgantara and P. Bartolo, 3D Printing of Polycaprolactone–Polyaniline Electroactive Scaffolds for Bone Tissue Engineering, Materials, 2020, 13(3), 512 CrossRef CAS.
- S. Vijayavenkataraman, S. Kannan, T. Cao, J. Y. H. Fuh, G. Sriram and W. F. Lu, 3D-Printed PCL/PPy Conductive Scaffolds as Three-Dimensional Porous Nerve Guide Conduits (NGCs) for Peripheral Nerve Injury Repair, Front. Bioeng. Biotechnol., 2019, 7, 266 CrossRef PubMed.
- A.-D. Bendrea, L. Cianga, G.-L. Ailiesei, E.-L. Ursu, D. Göen Colak and I. Cianga, 3,4-Ethylenedioxythiophene (EDOT) End-Group Functionalized Poly-ε-caprolactone (PCL): Self-Assembly in Organic Solvents and Its Coincidentally Observed Peculiar Behavior in Thin Film and Protonated Media, Polymer, 2021, 13(16), 2720 CAS.
- D. Mecerreyes, R. Stevens, C. Nguyen, J. A. Pomposo, M. Bengoetxea and H. Grande, Synthesis and characterization of polypyrrole-graft-poly(ε-caprolactone) copolymers: new electrically conductive nanocomposites, Synth. Met., 2002, 126(2), 173–178 CrossRef CAS.
- A. C. da Silva, T. Augusto, L. H. Andrade and S. I. Córdoba de Torresi, One pot biocatalytic synthesis of a biodegradable electroactive macromonomer based on 3,4-ethylenedioxytiophene and poly(l-lactic acid), Mater. Sci. Eng., C, 2018, 83, 35–43 CrossRef CAS PubMed.
- A. C. da Silva, A. T. S. Semeano, A. H. B. Dourado, H. Ulrich and S. I. Cordoba de Torresi, Novel Conducting and Biodegradable Copolymers with Noncytotoxic Properties toward Embryonic Stem Cells, ACS Omega, 2018, 3(5), 5593–5604 CrossRef CAS.
- B. G. Molina, A. D. Bendrea, L. Cianga, E. Armelin, L. J. del Valle, I. Cianga and C. Alemán, The biocompatible polythiophene-g-polycaprolactone copolymer as an efficient dopamine sensor platform, Polym. Chem., 2017, 8(39), 6112–6122 RSC.
- A. Basterretxea, E. Gabirondo, C. Jehanno, H. Zhu, O. Coulembier, D. Mecerreyes and H. Sardon, Stereoretention in the Bulk ROP of l-Lactide Guided by a Thermally Stable Organocatalyst, Macromolecules, 2021, 54(13), 6214–6225 CrossRef CAS.
- J. Luo, D. Billep, T. Waechtler, T. Otto, M. Toader, O. Gordan, E. Sheremet, J. Martin, M. Hietschold, D. R. T. Zahn and T. Gessner, Enhancement of the thermoelectric properties of PEDOT:PSS thin films by post-treatment, J. Mater. Chem. A, 2013, 1(26), 7576–7583 RSC.
- S. Marina, D. Mantione, K. ManojKumar, V. Kari, J. Gutierrez, A. Tercjak, A. Sanchez-Sanchez and D. Mecerreyes, New electroactive macromonomers and multi-responsive PEDOT graft copolymers, Polym. Chem., 2018, 9(27), 3780–3790 RSC.
- J. K. Palacios, H. Zhang, B. Zhang, N. Hadjichristidis and A. J. Müller, Direct identification of three crystalline phases in PEO-b-PCL-b-PLLA triblock terpolymer by In situ hot-stage atomic force microscopy, Polymer, 2020, 205, 122863 CrossRef CAS.
- E. P. e Silva, B. Huang, J. V. Helaehil, P. R. L. Nalesso, L. Bagne, M. A. de Oliveira, G. C. C. Albiazetti, A. Aldalbahi, M. El-Newehy, M. Santamaria-Jr., F. A. S. Mendonça, P. Bártolo and G. F. Caetano, In vivo study of conductive 3D printed PCL/MWCNTs scaffolds with electrical stimulation for bone tissue engineering, Bio-Des. Manuf., 2021, 4(2), 190–202 CrossRef CAS.
- Y. Wan, X. Qu, J. Lu, C. Zhu, L. Wan, J. Yang, J. Bei and S. Wang, Characterization of surface property of poly(lactide-co-glycolide) after oxygen plasma treatment, Biomaterials, 2004, 25(19), 4777–4783 CrossRef CAS.
- S. Wang, C. Sun, S. Guan, W. Li, J. Xu, D. Ge, M. Zhuang, T. Liu and X. Ma, Chitosan/gelatin porous scaffolds assembled with conductive poly(3,4-ethylenedioxythiophene) nanoparticles for neural tissue engineering, J. Mater. Chem. B, 2017, 5(24), 4774–4788 RSC.
- A. G. Guex, J. L. Puetzer, A. Armgarth, E. Littmann, E. Stavrinidou, E. P. Giannelis, G. G. Malliaras and M. M. Stevens, Highly porous scaffolds of PEDOT:PSS for bone tissue engineering, Acta Biomater., 2017, 62, 91–101 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR, 13C-NMR and MALDI-TOF spectra of macromonomers, UV spectra copolymers, TGA calibration curve, DSC thermograms, frequency sweeps by rheology, SEM images of printed patterns. See DOI: 10.1039/d1py01185e |
| This journal is © The Royal Society of Chemistry 2022 |