
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSyntheses of polylactides by means of tin catalysts
Hans R.
Kricheldorf
*a and
Steffen M.
Weidner
 b
b
aUniversität Hamburg, Institut für Technische und Makromolekulare Chemie, Bundesstr. 45, D-20146 Hamburg, Germany. E-mail: hrkricheldorf@aol.de
bBundesanstalt für Materialforschung und -prüfung – BAM, Richard Willstätter Str. 11, D-12489 Berlin, Germany
First published on 3rd March 2022
Abstract
This article reviews the usefulness of tin(II) and tin(IV) salts and compounds as catalysts for the polymerization of lactides. The text is subdivided into nine parts mainly reflecting different polymerization strategies, such as ring-opening polymerization (ROP), ring-expansion polymerization (REP), ROP combined with simultaneous polycondensation (ROPPOC), various catalysts with unknown polymerization mechanisms, and polycondensation of lactic acid. Since the toxicity of tin salts and compounds is a matter of concern and frequently mentioned in numerous publications, the first section deals with facts instead of myths about the toxicity of tin salts and compounds.
1. Introduction
This review deals with the role of tin catalysts in ring-opening polymerizations (ROP), ring-expansion polymerization (REP), and ring-opening polymerization combined with simultaneous polycondensation (ROPPOC) of lactides, but also with polycondensation of lactic acid. Just now, and even more in the future, polylactides play an important role as biodegradable materials for a broad variety of applications with the additional benefit that the monomers (L-lactic acid, L-lactide (LLA), meso-lactide (mesoLA), rac-lactide (racLA)) can be produced from natural resources. Almost all commercial polylactides are produced by ring-opening polymerization of the cyclic dimers, the lactides using tin(II) 2-ethylhexanoate (SnOct2) as the standard catalyst for their technical production. Therefore, tin catalyzed polymerizations have stimulated numerous scientific studies and the present review makes an attempt to summarize the results of these studies. Despite many review articles about ROPs of lactides and lactones,1–10 a review focused on tin-catalyzed syntheses of polylactides has never been published to the best knowledge of the authors.The performance of catalyst is usually defined and determined in terms of rate of polymerization, the extent of final conversion and the maximum molar mass that can be achieved. In case of biodegradable polymers, and above all, when part of these polymers should be used for medical and pharmaceutical applications, a fourth parameter plays an important role, namely toxicity. In this regard tin-based catalysts have a negative image and therefore, this aspect needs to be discussed in detail. Hence, the present review also tries to present facts about the toxicity of tin salts and compounds in comparison with toxicity data of other catalysts and drugs.
In addition to a section dealing with toxicity (part 2), this review is subdivided into seven parts (3–9) according to the polymerization methods: ROP, REP, ROPPOC and polycondensation of lactic acid. Furthermore, chemical reactions in solid poly(L-lactide)s (PLLA), and their consequences for the physical properties are reviewed.
Finally, the origin of tin-catalyzed polymerizations of lactides should be mentioned here. In 1959 Kleine and Kleine reported on SnCl4-catalyzed polymerizations of L-lactide in bulk.11 At that time SEC was not available and those authors listed inherent viscosities as crude measure of molar mass. From those data it may be roughly estimated that only low molar mass polylactides (weight average molar masses (Mw) < 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were obtained. However, the work of Kleine et al. has the important merit that it reported for the first time on the extent of racemization caused by different catalysts. Those authors found that the risk of racemization is particularly low for SnCl4 and higher for the less toxic catalysts based on zinc or magnesium salts. This finding was later confirmed by the first author of this review in a detailed study including a broader variety of Sn, Zn and Mg salts or complexes.12 Since crystallinity, mechanical properties and rate of hydrolytic degradation depend very much on the optical purity of pol(L-lactide) the results of Kleine et al. were and are still of great importance.
000) were obtained. However, the work of Kleine et al. has the important merit that it reported for the first time on the extent of racemization caused by different catalysts. Those authors found that the risk of racemization is particularly low for SnCl4 and higher for the less toxic catalysts based on zinc or magnesium salts. This finding was later confirmed by the first author of this review in a detailed study including a broader variety of Sn, Zn and Mg salts or complexes.12 Since crystallinity, mechanical properties and rate of hydrolytic degradation depend very much on the optical purity of pol(L-lactide) the results of Kleine et al. were and are still of great importance.
2. Toxicity – rumours and facts
First, it is necessary to distinguish between catalysts based on tin(II) ions and organotin(IV) compounds having alkyl or aryl residues covalently attached to the tin atom. In the case of tin(II) ions the toxicity has been intensively studied over the past 40 years and found to be rather low for humans for the following reasons.Both the upper middle and upper class of the European population have used tin dishes, such as plates, cups, mugs, jars, pots, tankards, and bottles, over a period of more than 300 years, and fatalities or chronic diseases resulting from intoxication with tin have not been clearly evidenced (in contrast to intoxication with lead). More precise data about the toxicity of tin(II) ions in food has been elaborated from extensive studies of canned food. Over the past 120 years the mankind has produced many billions of cans made from steel having the interior walls plated with tin to protect the steel walls against corrosion. Thus, in British English tin became a synonym for can. The low toxicity of tin(II) ions for humans found in those studies in combination with its high toxicity against any kind of microbes and fungi has prompted the American FDA to accept tin(II) 2-ethylhexanoate and other tin(II) salts, above all SnCl2, as food stabilizer. For example, SnCl2 is the standard stabilizer for any kind of tomato products such as soups, sauces and juice, and it is well known that hundreds of thousands of litres of tomato juice are consumed e.g. in airplanes every year. A concrete figure, the authors have found for SnCl2, says that up to 200 mg kg−1 of human body is harmless.13–15 SnCl2 is preferentially used as food stabilizer, because it much less expensive than tin(II) acetate, less expensive and slightly less toxic than SnOct2, since the chloride ions belong to the human metabolism in contrast to 2-ethyl hexanoate ions.
It is interesting to compare this ratio with what is known about pyridine (Table 1). For that amine the LD50 amounts to 400 mg kg−1, when orally applied by humans (see footnote (b) in Table 1). This figure means that 50% of humans who swallow 500 mg kg−1 pyridine will die, whereas the other 50% will be more or less damaged by intoxication. Hence, it is obvious that pyridine is considerably more toxic than SnCl2. The data in Table 1 also indicate that N,N-dimethyl-4-aminopyridine is 5–10 times more toxic than pyridine itself. This comparison is of interest, because in a first paper claiming that metal-free organo-catalysts may be advantageous over metal-based catalysts Nederberg et al. used N,N-dimetly-4-aminopyridine (and 4-pyrrolidinopyridine) as catalysts.16 Such pyridine derivatives were also reported in papers of other authors.17–19 Furthermore, the toxicity data compiled in Table 1 suggest that trialkyl amines are more toxic than pyridine.
Nonetheless, several research groups used bases such as diazabicycloundecene (DBU) or triazabicyclodecene (TBD) as catalysts, although their basicity and nucleophilic reactivity are several orders of magnitude higher than those of triethylamine.20–23 We were not able to find toxicity data, but a higher toxicity than trimethylamine or guanidine is predictable, in as much as these amines are registered as dangerous, highly caustic, corrosive liquids. Nonetheless, the following statement can be found in a recent review: “Such organo-catalysts present lower toxicity than their organometallic counterparts, and can for the most part, be safely used in biomedical and electronic applications and can be easily removed from the final PLA material”.23 This statement turns the known facts about toxicity upside down, apparently because of lack of any knowledge about toxicity of the catalysts they have reviewed (toxicity data were not mentioned). However, two publications of Coulembier and co-workers appeared recently, which report on studies of cytotoxicity of several organocatalysts including thioureas and tertiary amines.24,25 The toxicities covered a broad range and confirm that it is not a priori justified to attribute a negligible toxicity to organocatalysts.
Finally, it is worth mentioning that most widely used drugs are more toxic than SnCl2, when the LD50 oral doses for rats are compared (Table 2). Those drugs are consumed in much larger quantities than SnCl2 or other tin(II) salts. It is certainly true that tin ions are not desirable in polyesters used for medical and pharmaceutical applications, but the quantities needed for such applications are small, not to say negligible, compared to what is needed for other applications for example as films and foils in agriculture, for packaging or for 3-D printing and other technical applications.26 This amount will increase in the future, when more and more ‘immortal’ polymers will be substituted by biodegradable ones. For all those large-scale applications the low toxicity of tin(II) ions represents no serious problem. Dangerous accumulation in nature causing long-term problems to the environment is probably not a concern, because tin(II) is a reducing agent which is oxidized by oxygen and Fe(III) ions (ubiquitous on the surface of Earth). The oxidation product SnO2 is an extremely insoluble and harmless chemical known and used by the humankind for more than 5000 years as a mineral named Cassiterite.
| Pharmaceutical name | LD50 [mg kg−1] oral (rat) | Pharmaceutical name | LD50 [mg kg−1] oral (rat) |
|---|---|---|---|
| a Retrieved from Wikipedia. | |||
| Paracetamol | 338 | Diclofenac | 245 |
| Dexamethasone | 50 | Ibuprofen | 636/740 (mouse) |
| Resorcin | 330 | Phenobarbital | 162 |
| Lidocaine | 314 | Diazepam | 249 |
| Codeine | 427 | Methamphetamine | —/34 (mouse) |
Alkyltin compounds are in general considerably more toxic than Sn(II) salts. Their toxicity decreases with increasing length and decreasing number of the alkyl groups. Due to a moderate toxicity, dibutyltin oxide has found commercial applications, mainly as transesterification catalyst in the production of insulating lacquers. The less toxic dioctyltinoxide has found a much broader application for example as catalyst and additive in insulating lacquers, paints, coatings, fresheners or in machine wash liquids. Its production amounts to approximately 10000 tons per year in Europe. Nonetheless, catalyst based on dibutyltin(IV) compounds (see below) are mainly of interest as catalyst for academic research.
3. Ring-opening polymerization (ROP) catalyzed by tin alkoxides
3.1 Homopolymerization and the coordination–insertion (C–I) mechanism
For more than three decades ROPs of cyclic esters were believed to proceed either by a cationic or an anionic mechanism.27 However, the existence of ionic polymerization mechanisms catalyzed by tin salts has never been evidenced, so that all polymerization methods reviewed in the following sections are based on the C–I mechanism.The concept of a mechanism exclusively based on a rearrangement of covalent bonds, later called coordination–insertion mechanism, grew up step by step beginning with papers of Teyssié and co-workers in 1976/1977.28,29 That group studied ROPs of ε-caprolactone catalyzed by Al-isopropoxide and by a bimetallic complex based on Al and Zn, and formulated for the first time a non-ionic insertion mechanism which included the insertion of the lactone into a covalent Al–O bond (Scheme 1).
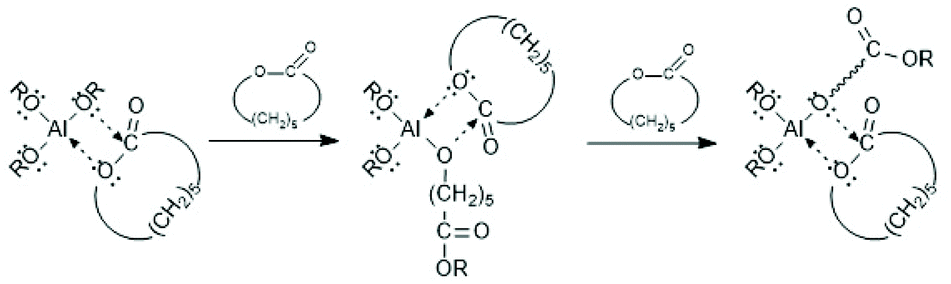 | ||
| Scheme 1 C–I mechanism formulated for εCL by Ouhadi et al.29 (with permission from Wiley). | ||
In 1991 these studies were extended to ROPs of lactide.30 Meanwhile (1985), Kricheldorf et al. reported on copolymerizations of glycolide with L-lactide (or lactones) using various tin alkoxides such as Bu3SnOMe, Bu2Sn(OMe)2, BuSn(OMe)3 and BuSn(OPr)3.31 Random 1/1 copolyesters were obtained at 150 °C in bulk and by a comparison with the different results obtained by ionic ROP it was concluded, that the C–I mechanism illustrated in Scheme 2 was operating.
 | ||
| Scheme 2 C–I mechanism formulated for glycolide by Kricheldorf et al.31 (reproduced with permission of Wiley). | ||
Characteristic for this mechanism are two steps. At first the carbonyl oxygen interacts with a free orbital of the Sn atom with the consequence that the carbonyl C turns more electrophilic and the Sn–O bond more nucleophilic. The second step consist of a rearrangement of covalent bonds, so that an insertion of the cyclic monomer into the Sn–O bond occurs. In an ensuing publication (1988) Kricheldorf et al. studied homopolymerizations of L-lactide and various lactones with several covalent metal alkoxides including Bu3SnOMe and Bu2Sn(OMe)2.32 For all monomer-catalyst combinations it was found that at temperatures >50 °C all alkoxide groups initiated a chain, so that the average degree of polymerization (DP) paralleled the monomer/alkoxide ratio. It was concluded that all covalent metal alkoxides initiate a C–I mechanism for ROPs of all cyclic esters regardless of their ring size. From this time on the C–I mechanism became an established mechanism, which is in the case of lactides for two reasons of paramount importance. Firstly, anionic and zwitterionic ROPs of L-lactide cause racemization and cationic polymerization only proceed with super acids, such as fluorosulfonic acid or trifluoromethane sulfonic acid, as catalysts in solution.33,34 Therefore, the C–I mechanism is underlaying all the polymerization methods discussed in the following sections.
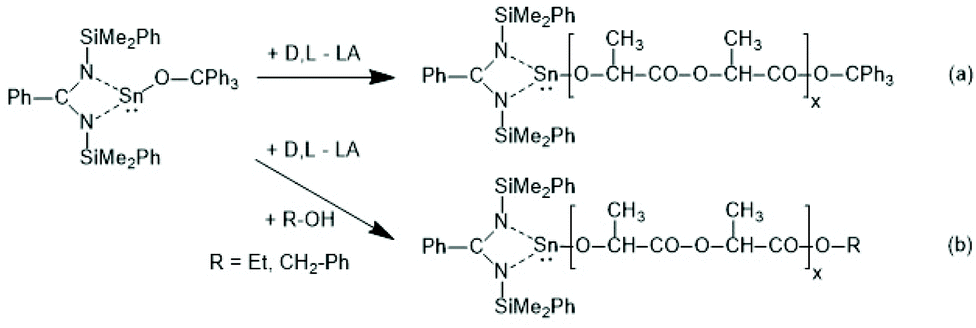
In the following years covalent tin alkoxides were rarely used as catalysts/initiators of ROPs of lactides because most research groups working on ROPs of lactide focused their interest on SnOct2-catalyzed polymerizations. When the Penczek group started to investigate the mechanism of alcohol-initiated, SnOct2 catalyzed ROPs of L-lactide, they used Sn(OBu)2 for comparative studies.35–37 It was found that this tin(II) alkoxide is more reactive that the afore-mentioned butyl tin(IV) alkoxides and rapid ROPs became feasible at 80 °C in THF. Living ROPs resulting in polymers with narrow dispersities (Đ) were observed. NMR spectra revealed that both butoxide groups initiated chains growth. By means of MALDI TOF mass spectrometry it was found that the Sn atom was attached to the O–CH(CH3) end group. In this way, the Sn(OBu)2-initiated ROPs contributed an important mosaic piece to the elucidation of the alcohol-initiated ROPs of lactides, which are discussed in more detail in the next section.
In the years 2001–2009 Gibson and co-workers performed detailed studies on synthesis and properties of Sn(II) single site catalysts containing ancillary ligands (Schemes 3–6).38–42 Those studies were mainly focused on syntheses and characterization of those complexes by means of NMR spectroscopy and X-ray methods. All polymerization experiments were conducted in toluene at 60 °C with racLA as monomer. They were found to behave 1st order for racLA, whereas the reaction order of the catalyst depended on whether it was monomeric or dimerized in toluene. A living character was attested to those ROPs with low dispersities (Đ). In most cases dispersities even below 1.1 (down to 1.04) were found at low conversions along with a steady increase at higher conversions. Quite similar results were obtained by Aubrecht et al.43 The single-site catalysts outlined in Scheme 7 were synthesized and characterized. Again, ROPs of racLA were performed in toluene (at 80 °C). The catalysts did aggregate under these conditions, but the rate of propagation was 1st order with regard to the monomer, and low dispersities (<1.1) increasing with the conversion were found.
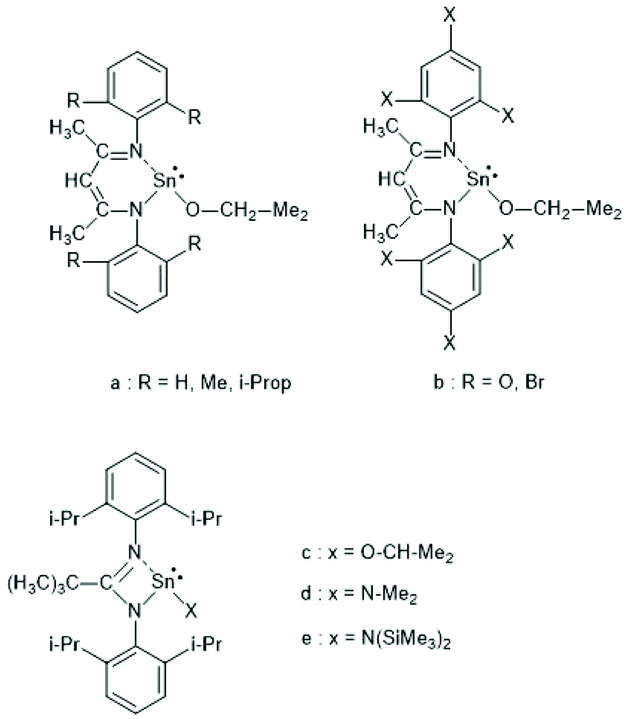 | ||
| Scheme 3 Sn(II) complexes used by Gibson et al. as catalysts for ROPs of racLA (reproduced with permission from ref. 39 Copyright 2006 American Chemical Society). | ||
 | ||
| Scheme 4 Sn(II) complexes used by Gibson et al. for ROPs of racLA (reproduced with permission from ref. 41 Copyright 2006 American Chemical Society). | ||
 | ||
| Scheme 5 Sn(II) complexes used by Gibson et al. for ROPs of racLA (reproduced with permission from ref. 41 Copyright 2006 American Chemical Society). | ||
 | ||
| Scheme 6 ROP of racLA formulated by Gibson et al. indicating an intramolecular DA-interaction of the LA unit directly attached to the Sn-atom.42 (with permission from RSC). | ||
 | ||
| Scheme 7 Sn(II) complex used by Aubrecht et al. for ROPs of racLA without or with addition of an alcohol as initiator (reproduced with permission from ref. 43 Copyright 2002 American Chemical Society). | ||
However, poly(D,L-lactide) was more soluble than PLLA in any solvents, so that precipitation into a non-solvent may entail significant fractionation. This is also true for methanol and for heptane, and the removal of oligomers enhances Mn and reduces the dispersities. The influence of fractionation increases with decreasing molar mass of the polylactide, and thus, the extremely low dispersities reported by the Gibson and Aubrecht were at least partly a consequence of fractionation.38–43
From the preparative point of view, the complexes outlined in Schemes 3–7 have two disadvantages. First, they will racemize L-lactide at higher temperatures due to the basicity of the nitrogen atoms. Second, only uncorrected Mw's below 50000 were obtained. However, such low molar mass PLLAs can easily be prepared by a variety of catalysts including catalysts based on metal ions of low toxicity such as zinc, magnesium, or bismuth. Furthermore, the toxicity of the tin complexes is unknown, and it should be considered that organic complexes of tin(II) may have a much higher toxicity than the free Sn2+ ion, in analogy to other organotin compounds (see part 2).
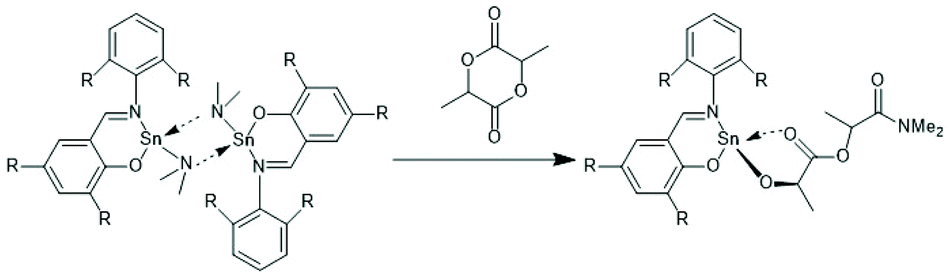
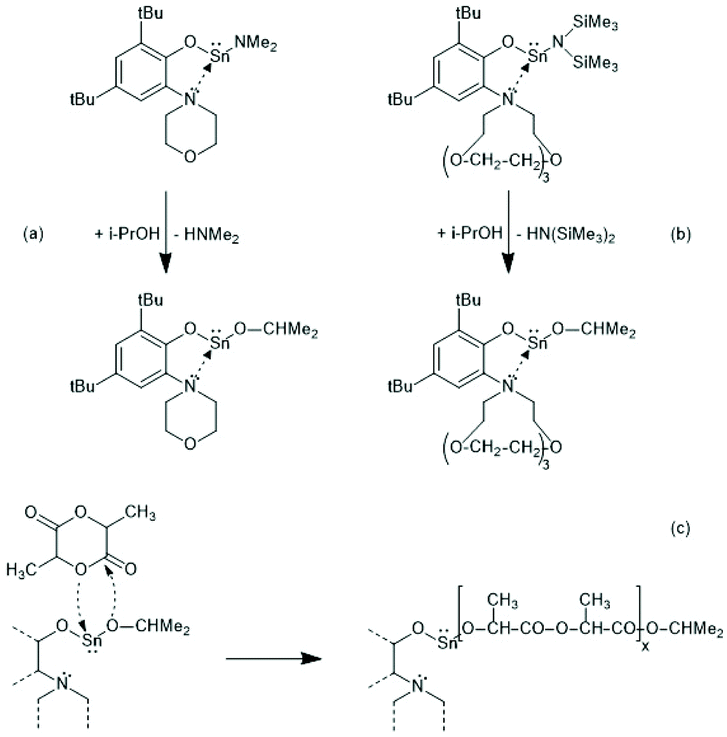
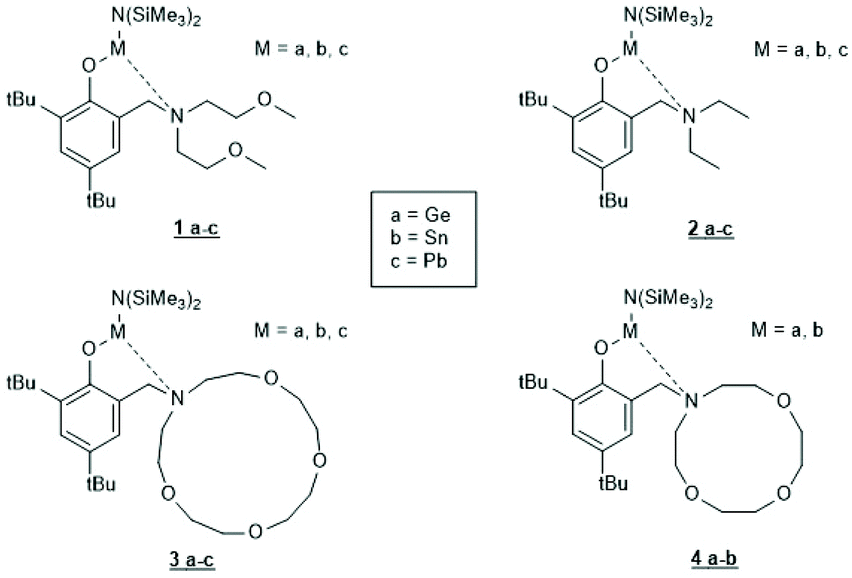
Tin(II) complexes with nitrogen-containing ancillary ligands were also studied by the group of Carpentier (Schemes 8 and 9).44–48 Again a 1st order polymerization mechanism regarding the monomer with living character and low dispersities were observed. The incorporation of isopropoxide groups as dead chain ends was confirmed by 1H NMR spectroscopy. LLA was used as monomer, but despite the strongly basic tertiary amino group in the catalyst, no information on racemization was disclosed. In the paper of Wang et al. Sn-bis-isopropoxide was also synthesized by means of a large excess of isopropanol and used as polymerization catalyst.46 In agreement with the results Penczek et al.35–37 had found for Sn-bis-n-butoxide, those authors observed that both isopropoxide groups initiated a PLLA chain. The consequences of the excess alcohol are discussed in part 4.3. In their last paper dealing with Sn(II) complexes Carpentier et al. studied the synthesis and catalytic activity of the Ge, Sn and Pb complexes (1a–c)–(4a–c) in Scheme 9.48 It was found that the reactivity strongly increased with increasing radius of the metal ion. All the papers of Gibson, Tolman or Carpentier and co-workers have in common that their kinetic results agree with a C–I mechanism, which was proposed analogous to that of Scheme 2 (see Scheme 8).40,45
 | ||
| Scheme 8 Sn(II) complexes used by Carpentier et al. for ROPs of rac.LA or LLA44 and CI mechanism formulated in ref. 45 (with permission from Wiley). | ||
 | ||
| Scheme 9 Ge, Sn and Pb complexes used by Carpentier and cow. for ROPs of racLA46 (with permission from RSC). | ||
Quite recently Punyodom et al. published three studies using Bu3Sn butoxide or tin(II) n-alkoxides as initiators for ROPs of LA in bulk.49–51 Kinetic studies using 1H NMR spectroscopy or DSC measurements performed with variation of temperature and time were used to determine activation energies and to compare the efficiency of their catalysts with those of SnOct2–alcohol combinations.50 Sn(OnBu)2 was also used to prepare graft copolymers having PLLA side arms and antimicrobial coated medical grade LLA/εCl copolymers.51,52 However, the statement of those authors that Sn(nBu)2 is a novel initiator is not quite correct, because it was already used before by the Penczek group.35–37
A new dimension of preparative work based on cyclic dibutyltin bisalkoxides was opened by the Kricheldorf group in 1994, but since those polymerizations fall under the definition of ring-expansion polymerization (REP) they will be discussed in part 5.
3.2 Stereocopolymerization
Copolymerizations of lactides with other cyclic esters, such as glycolide or ε-caprolactone (εCL) have been studied by numerous research groups, but either alcohols served as initiators (see part 4) or catalysts based on other metals (e.g. Al, Zn) were used. However, covalent Sn-catalysts played a role for early studies of stereo-copolymers of D- and L-lactyl units.Since lactide contains two chiral carbons, this dimeric monomer exists in the form of four different isomers: L,L-lactide (LLA), D,D-lactide (DLA), D,D,L,L-lactide (racLA) and D,L-lactide (mesoLA). It was predictable and confirmed in the early days of lactide research that the percentage of D-units in PLLA has strong influence on all physical properties.53 Therefore, it was a major success of the early work of Kleine and Kleine that they could demonstrate that Sn-based catalyst do not cause racemization of lactide even at 160 °C.11 Racemization is typically a consequence of a reversible, temporary proton abstraction from an α-carbon of the lactide (Scheme 10). Since the temporarily formed partial C–O double bond hinders rotation around this C–O bond, its formation is favoured in the cyclic monomer over a formation in the polylactide backbone. The basicity of Sn–O, Sn–S and Sn–Cl bonds is low enough to avoid racemization up to temperatures of 180 °C when the reaction time is short (1–4 h depending on the structure and concentration of the catalyst). The influence of small amounts of D-units on Tm and other physical properties have been studied by several research groups.53–57 It was found that Tm decreases from 176–178 °C to 138–139 °C when the D-content increases from 0–10.5%.53 When the D-content approaches 15% in a random sequence crystallinity vanishes completely. The sequence of D- and L-units plays of course a decisive role for the influence the D-units have on the properties of poly(L-lactide). Two-block copolymers can form so-called stereocomplexes that have high Tm values (up to 230 °C), whereas random stereosequences favour an amorphous character of polylactides.58,59
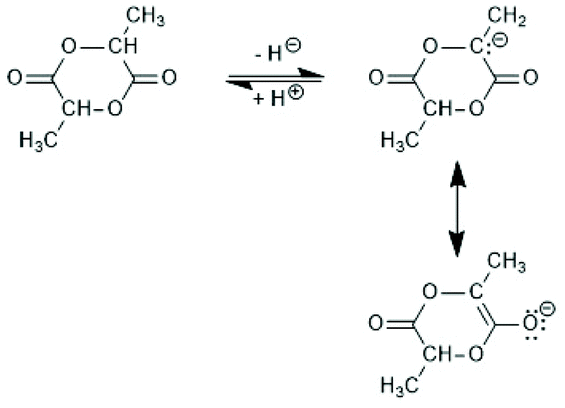 | ||
| Scheme 10 Mechanism of the base-catalyzed racemization of LLA. | ||
Stereosequences, in principle, can be analysed by high resolution 1H and 13C NMR spectroscopy. The first NMR spectroscopic stereosequence analyses of polylactides prepared by ROP of racLA were reported by Lillie and Schulz in 1975, but those authors used Zn and Al catalysts.60 One year later Schindler and Harper polymerized racLA in bulk at 130 °C for 260 h with neat SnOct2 as catalyst and characterized the resulting stereo-co-PLLA by means of 100 MHz 1H NMR spectra.61 Despite the long polymerization time they assumed that transesterification reactions were negligible. From NMR spectra, they concluded that the copolymerization obeyed a Bernoullian statistics. This means that the D–D and L–L bonds preformed in the monomers were not involved in transesterification, so that syndiotactic sequences (mrmr) were not formed. The same authors later studied the ROP of mesoLA at 140 °C/42 h catalyzed by neat SnOct2.62 On the basis of a 250 MHz 1H NMR spectrum now syndiotactic tetrads (mrm and rmr) were prevailing, whereas isotactic tetrads (mmm) were absent.
A broader study of stereo-copolylactides was performed by Kricheldorf et al. in 1992. These samples were analysed by means of 360 MHz homo-decoupled 1H NMR spectroscopy (to eliminate the influence of vicinal coupling) and 90 MHz 13C NMR spectroscopy.63 Around 50 ROPs of racLA and 40 ROPs of mesoLA were conducted either in bulk or in xylene. Bu3SnOMe and SnOct2 served as catalysts. The ROPs in bulk were conducted at 20, 150 and 180 °C and the time was varied from 2 to 24 h. The stereosequences were characterized by an average length of the isotactic blocks, Li, meaning the number of directly connected D or L units. With Bu3SnOMe as catalyst a maximum Li of 3.8 was found for racLA and 1.4 for mesoLA (indicating a predominance of syndiotactic dyads and tetrads), at 120 °C/2 h. When temperature and time were enhanced, the Li value of racLA decreased, whereas that of mesoLA increased. Finally, at 180 °C/24 h random stereosequences were obtained from both monomers characterized by an average isotactic block length of 2.0. An analogous series of ROPs performed with SnOct2 showed the same trends. However, a random stereo-copolymer was not obtained, because at 180 °C total degradation of the polylactides occurred when polymerization time was extended beyond 8 h. Polymerizations in xylene at 90 and 120 °C were unsuccessful when Bu3SnOMe served as catalyst and mesoLA as monomer. For racLA a maximum Li-value of 4.2 was found. With SnOct2 low yields and molar masses were achieved with both monomers, but the stereospecificity of the ROPS were quite similar to those observed for polymerizations in bulk at 120 °C. In this connection, a recent study of Bu3Sn butoxide-initiated ROPS of racLA also conducted in bulk should be mentioned.64
ROPs of racLA and mesoLA followed by NMR spectroscopic sequence analyses were also performed later by other research groups, but those groups used Zn or Al-based catalysts, so that a discussion of their results was outside the scope of this review.
4. Alcohol-initiated polymerizations
4.1 Mechanistic aspects of SnOct2-catalyzed ROPs
Since tin(II)2-etylhexanoate plays an extraordinary role for research and technical production of polylactides, the authors have attempted to trace back the history of this catalyst. In a patent of Ethicon Inc. filed by A K. Schneider in 1971, most polymerizations of L-lactide were performed with diethylzinc, but SnOct2 was used for a copolymerization of lactide and glycolide.65 Homo- and copolymerizations of LLA were described in 1972 by Sinclair and Gynn in a report of the Battelle Columbus Laboratories (Ohio),66 but Schneider and Sinclair used neat ScOct2 as catalyst. In the following years, a rapidly increasing number of research groups began to use SnOct2 for various preparative purposes either as neat reagent (discussed in part 6) or in combination with an alcohol as initiator. Medical or pharmaceutical applications, on the one hand, and exploration of the physical properties of polylactides, on the other hand, were in the focus of those research groups. The usefulness of alcohols as cocatalysts/initiators was apparently for the first time explored in a publication of Schindler et al. who used εCL and not lactides as monomer.67 Those authors demonstrated that each alcohol molecule initiates one chain, allowing for a control of the average molar mass of the resulting polylactones. They also demonstrated that relatively narrow dispersities may be obtained, and above all, a significant acceleration of the polymerization was found. This report prompted numerous research groups to explore the preparative potential of this catalyst system. Particularly important was its implementation as the standard process for the technical production of polylactides. In numerous academic studies the usefulness of mono- or multifunctional alcohols was explored mainly with the purpose to expand medical and pharmaceutical applications of homo- and copolylactides.The reaction mechanism of alcohol-initiated and SnOct2-catalyzed ROPs of lactides has been intensively studied by several research groups and even today (2021) there are still open questions. A first formulation of a mechanistic concept was published in 1994 by the McAuley group.68 Those authors found that the molar mass depended on the LA/alcohol ratio and accelerated the ROP, whereas addition of carboxylic acids reduced the rate of polymerization. They postulated that initially a rapid equilibration between alcohol and SnOct2 occurs generating a Sn–alkoxide bond (eqn (a) in Scheme 11), which was considered to be the genuine initiator due to its higher nucleophilicity relative to the tin carboxylate groups in SnOct2. For initiation and propagation, they proposed the C–I mechanism published by Kricheldorf et al. several years before on the basis of dibutyl and tributyl tin alkoxides (eqn (b) and (c) Scheme 11).31,32 Those authors also discussed an alternative mechanism, which proposed a reaction of alcohol with LLA both associated to the tin(II) atom via sp3d2 orbitals without formation of a covalent Sn–alkoxide bond.69 Yet, on the basis of further experiments conducted in bulk with NMR spectroscopic identification of end groups, a little later Kricheldorf et al. abandoned this mechanism in favour of the mechanism outlined in Scheme 11.70 The mechanism of Scheme 11 was also confirmed at the same time by detailed kinetic studies of the Penczek group, which used butanol as initiator in THF at 80 °C.35,37 Those authors demonstrated that under these conditions neat SnOct2 is inactive and a rapid ROP with narrow dispersities is only possible upon addition of an alcohol. According to the definition of a living polymerization the DP parallels the LA/In ratio and the conversion.37 A later study of Prud'Homme et al. conducted in toluene at 70 °C confirmed these conclusions.71
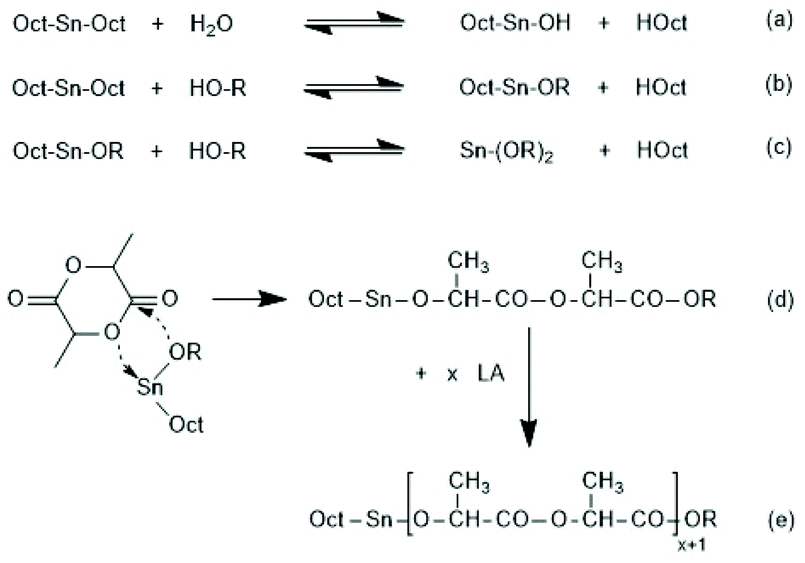 | ||
| Scheme 11 Mechanism of the alcohol-initiated and SnOct2-catalyzed polymerization of lactides first formulated by Zhang et al.68 (with permission of Wiley) and Kricheldorf et al.32 (with permission Copyright 1988 American Chemical Society). | ||
However, this picture has one important shortcoming. The reaction conditions are quite different from those used for the technical production, which is conducted in bulk at temperatures increasing from 130 to 180 °C to keep the PLLA in the molten state. Extrusion from the reactor even requires temperatures around 200 °C for a few minutes. Prior to extrusion a catalyst poison is added to the melt to suppress the rapid degradation of the initially formed high molar mass PLLA, a typical consequence of SnOct2 observed first by Jamshidi et al.72 Experimental studies of the authors at 130, 160 and 180 °C revealed the following results. ROPs in bulk above 120 °C differ from those described for temperatures below 100 °C in solution in several important aspects.
First, at temperatures >120 °C at least four transesterification mechanism occur: three intermolecular reactions (Schemes 12–14) and the intramolecular formation of cycles by “back-biting” (Scheme 15, see “Third” below).73,74 The intermolecular transesterifications occur at temperatures around or below 120 °C, and thus, are more efficient than ring-chain equilibration by “back-biting”.75 Particularly interesting is the existence of a transesterification mechanism, which converts the initially formed even-membered chains into odd-numbered ones without broadening of the MWD. Apparently a single lactyl unit is exchanged as illustrated by the hypothetical mechanism outlined in Scheme 14. This mechanism assumes that the last lactyl unit fixed to the Sn atom is activated by formation of a five membered ring with donor–acceptor (DA) interaction between the carbonyl oxygen and Sn. Evidence for the existence of this intramolecular DA interaction was contributed by three research groups by X-ray structure analyses of isolated complexes,39 by 13C NMR spectroscopy45 and by computer modelling.45,76 However, a definite prove for the single unit exchange (SUE) mechanism is still lacking.
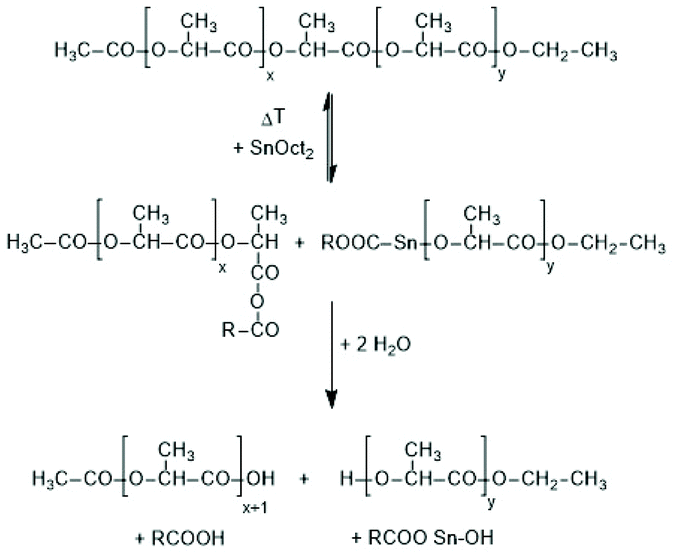 | ||
| Scheme 12 Intermolecular transesterification via cleavage of polylactide chains by SnOct2.73 | ||
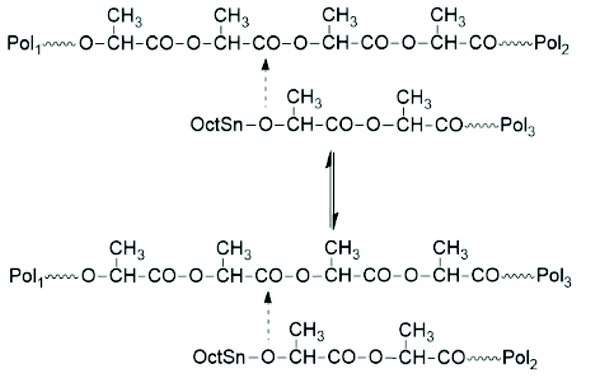 | ||
| Scheme 13 Intermolecular transesterification via reaction of active chains ends with the polylactide backbone (chain transfer equilibration).73 | ||
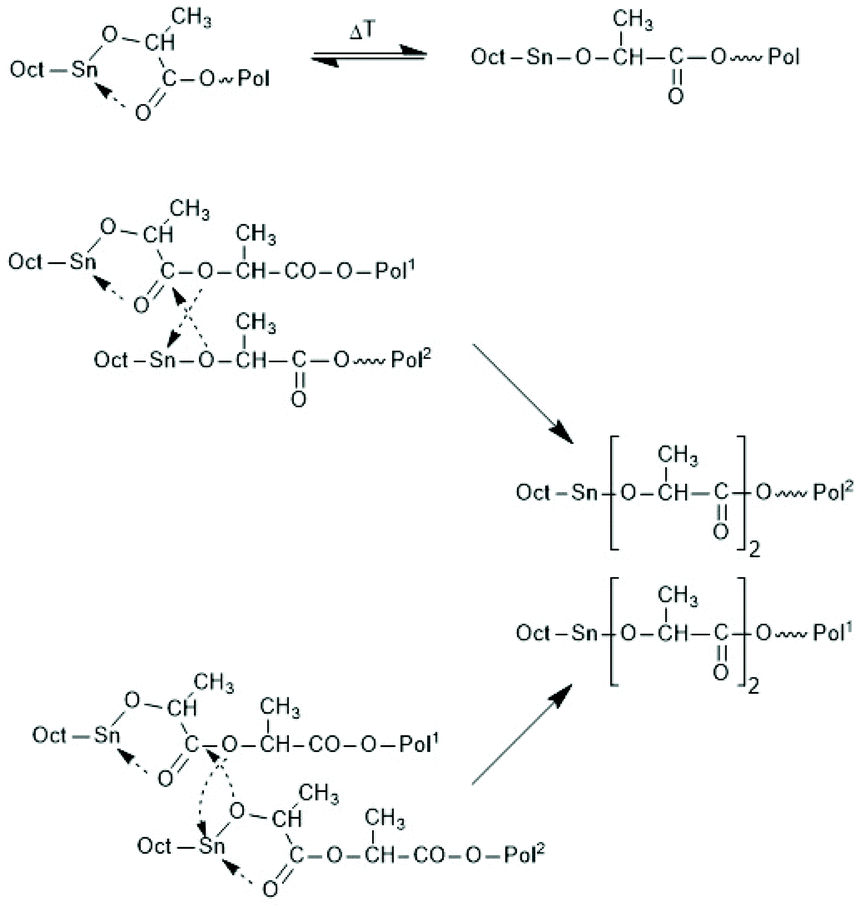 | ||
| Scheme 14 Two versions of the hypothetical single unit transfer mechanism. | ||
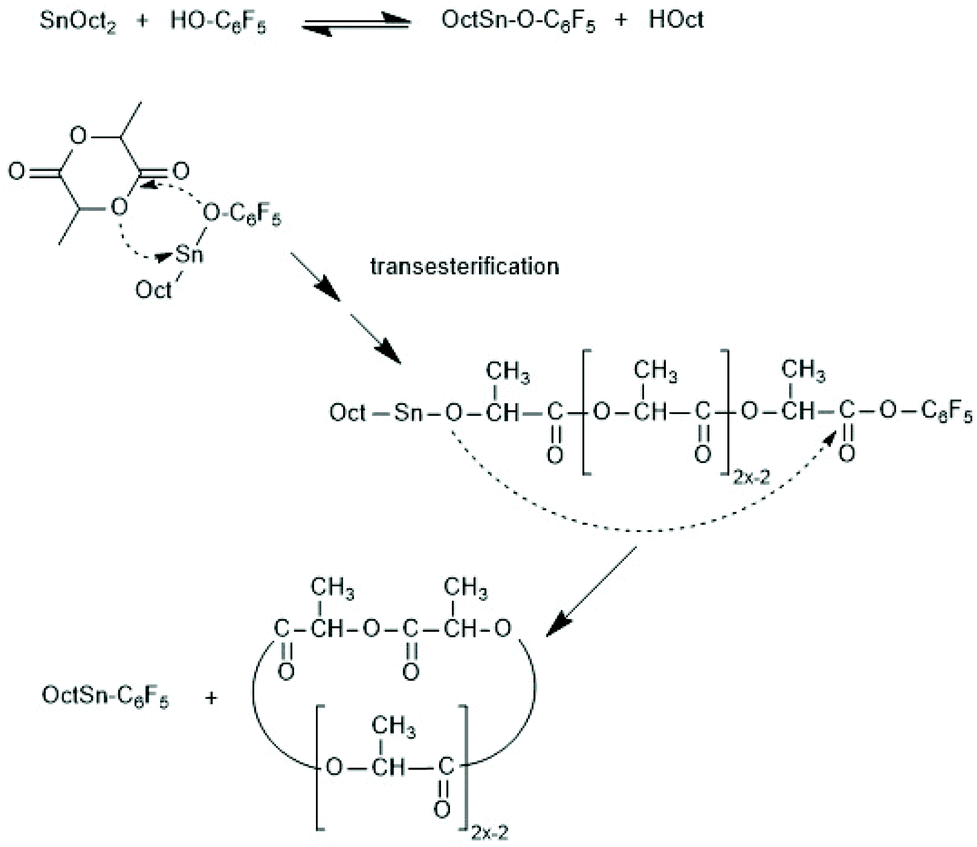 | ||
| Scheme 15 Formation of cycles in SnOct2-catalyzed ROPs initiated with acidic phenols. | ||
Second, it was recently observed by MALDI TOF mass spectrometry that the nature of the alcohol used as initiator has a significant influence on the efficiency of transesterification reactions above all on the efficiency of the even–odd equilibration.77 Two extreme cases are illustrated in Fig. 1. Somehow the initiators interact with the SnOct2 in a way, which has not been clarified yet. A comparison of three different alkanols by Pholharn et al. did not include mass spectrometric studies, and an influence of different alcohols on the course of low temperature ROPs in solution was not reported.78 When acidic OH-compounds such as cyclohexanone oxime, 4-chloro-, 4-cyanao-, 4-nitrophenol or pentafluoro phenol are used as initiators polylactides having activated ester end groups are formed in a clean ROP at temperatures around 130 °C, whereas a considerable fraction of cycles is forms at 160 °C (and above). The fraction of cycles increase with higher reactivity (electrophilicity) of the ester end group, what indicates that under these conditions most cycles are formed by end-to-end cyclization (Scheme 15).79
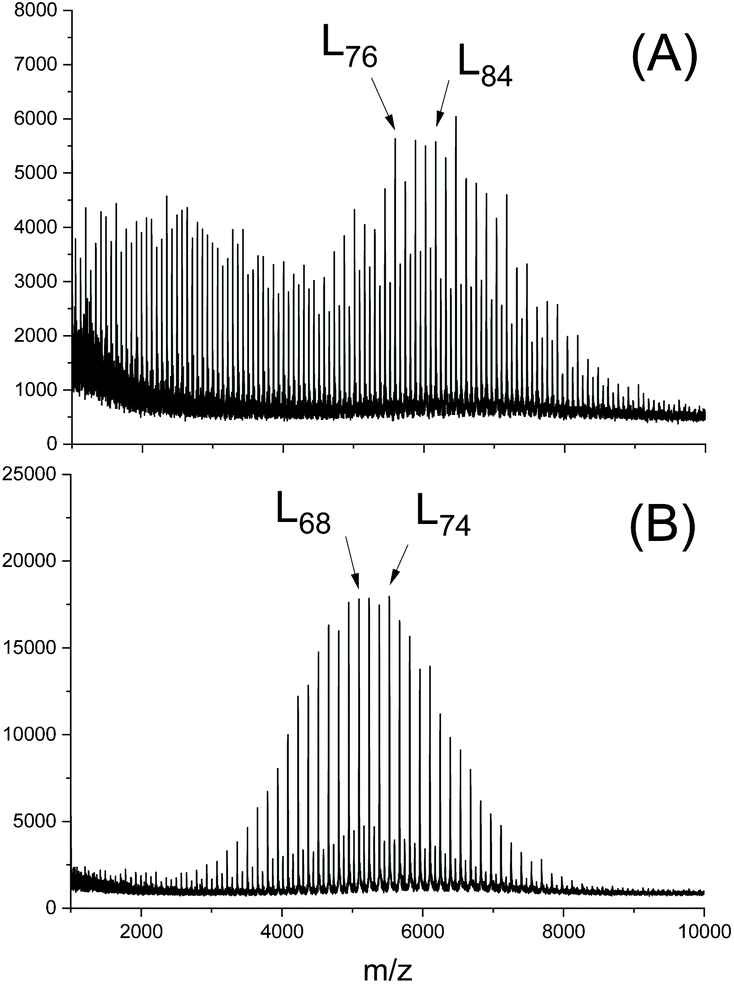 | ||
| Fig. 1 MALDI TOF mass spectra of PLLAs prepared in bulk at 130 °C with SnOct2 (LLA/Cat = 1000/1) and (LLA/In = 50/1): (A) initiated with n-butanol, (B) initiated with 4-nitrobenzyl alcohol (reproduced from ref. 77 with permission from Elsevier). | ||
Third, the ROPs of LA around or above 130 °C include formation of cycles. Two different mechanisms account for this fact. The reactivity of neat SnOct2 increases dramatically and allows for rapid polymerization of lactides at 160 °C or above even in the absence of alcohols.73 At LLA/Cat ratios >1000/1 typically used for the technical production, the resulting PLLA almost completely consists of cycles, because in the absence of an initiator a ROPPOC mechanism is operating as described in part 6 in more detail.73 The formation of cycles by neat SnOct2 has the consequence that the ROPPOC mechanism and the alcohol initiated ROP compete, and the content of cycles depends on the In/Cat ratio. Low LA/In ratios combined with high In/Cat ratios suppress the formation of cycles, while low In/Cat ratios (and thus, high LA/In ratios) favour formation of cycles via end-to-end (ete) cyclization, because the ROPPOC mechanism is favoured. However, ete-cyclization is not the only source of cycles. The second source of cycles is ring-chain equilibration via “back-biting”. This type of transesterification reaction (Scheme 16) is less effective than intermolecular transesterification reactions formulated in Schemes 12–14, but is favoured at temperatures >130 °C and longer reaction times. Regardless how the cycles are formed, their formation is unavoidable, when high molar mass polylactides are prepared in bulk >130 °C. Therefore, commercial PLLAs usually contain 1–3 weight% of cycles corresponding to 2–8 mol% of cycles, that represent the predominant species in mass spectra in the mass range below 6000 Da.
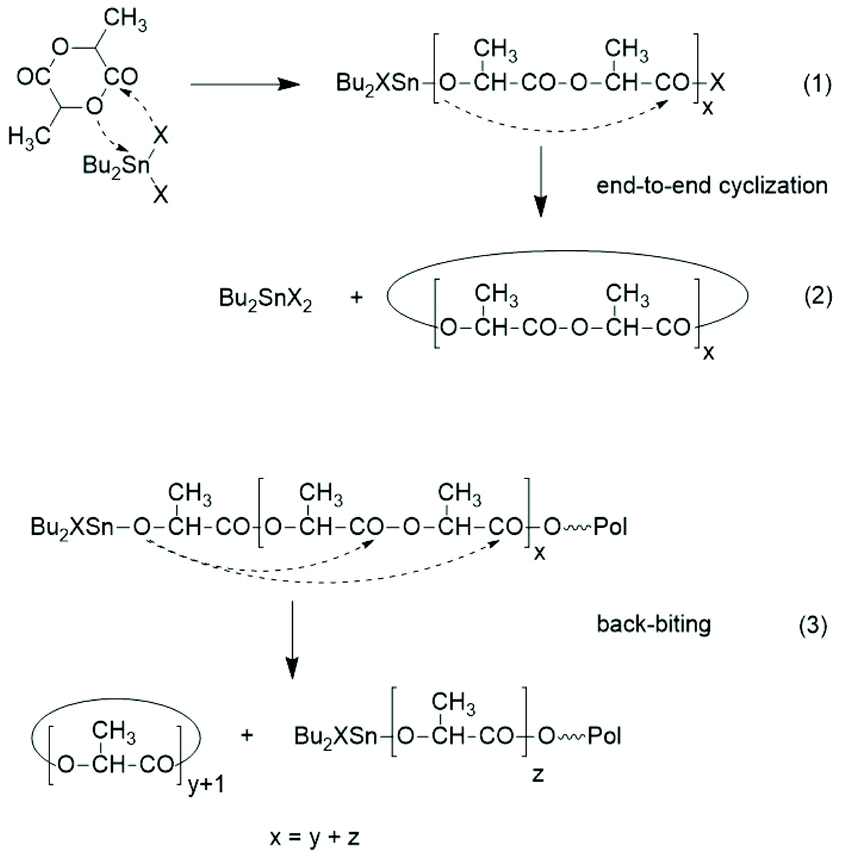 | ||
| Scheme 16 Ring-chain equilibration via “back-biting”. | ||
The formation of cycles has two consequences. First, the number average molar masses (Mn's) do not parallel the LA/In ratio but lag behind. This is an effect, which is particular remarkable at LA/In ratios >200.80 Nonetheless, even for ROPs conducted in bulk at higher temperatures the molar masses can be roughly controlled via the LA/In ratio. The second important effect of cyclization reactions and of intermolecular transesterification reactions is a significant broadening of the MWDs. Dispersities (Đ) > 2.0 and even >3.0 are quite normal.
Over the past twenty years, polymerizations proceeding according to the “living pattern” with full control of molar mass and dispersities < 1.2 have become a kind of academic fetish, and polymerizations that do not satisfy these properties are considered to be in lower quality and less useful. However, this view is not only irrelevant for the technical production of PLLA and other polymers, but also misleading. Whenever, commercial applications with thermal processing come into play (e.g. injection moulding, extrusion, bottle blowing, fibre spinning) a broad MWD is desirable, because it includes a better balance between moderate melt viscosity and good mechanical properties. Hence, the characteristic properties of LLA polymerizations in bulk at higher temperatures are in fact favourable for almost all commercial applications based on thermal processing.
In connection with the afore-mentioned mechanistic studies of alcohol-initiated ROPs catalyzed by SnOct2 the following publication merits mentioning. Kowalski et al. have demonstrated that primary amines used as initiators yield almost the same kinetic pattern as primary alcohols.81 The crude LA isolated from fermentation processes contains numerous impurities including amino acids, ethanol, glucose and sodium carbonate. Itzinger et al. have recently studied to what extent these impurities can influence the properties of PLLA when the polymerization is performed at 180 °C for 1 h.82 Whereas the influence on glass transition (Tg) and melting temperature (Tm) was negligible, the influence on molar mass and dispersity was remarkable. Not unexpectedly, the strongest effect was found for sodium carbonate, which can not only cause racemization, but also favour cleavage of ester groups via β-elimination (yielding acrylate and carboxylic end groups).
Extensive computer modelling studies of alcohol-initiated and SnOct2-catalyzed ROPs at 130 °C performed by Morbidelli and co-workers should be mentioned.83–85 More recently, extensive kinetic studies concerning the synthesis of linear and star-shaped PLLAs were reported by Karidi et al.86
4.2 Preparative aspects of alcohol-initiated, SnOct2-catalyzed ROPs
The usefulness of the alcohol/SnOct2 catalyst system for various preparative purposes is based on the fact that the structure of the alcohols used as initiators can be varied over a broad range, and this variation entails, in turn, a broad variation of structure, architecture and properties of the resulting polylactides. The following variants of their architecture were studied.(A) Linear homo polylactides and random copolylactides initiated with a monofunctional alcohol.
(B) Block copolymers initiated with a diol including oligomeric and polymeric α,ω-diols. The use of polymeric α,ω-diols as initiators had usually the purpose to prepare ABA triblock-copolymers with A segments consisting of crystalline PLLA.
(C) Star-shaped polylactides initiated by a multifunctional alcohol.
(D) Comb-shaped polylactides initiated by a polymer having pendant OH groups.
(E) Branched and hyperbranched polylactides.
The references cited regarding A–E are not exhaustive but focused on those publications that document the earliest application of the mentioned initiators and later review articles. A more detailed comment follows here.
(A) The technical production of linear poly(L-lactide)s is based on initiation by a monofunctional alcohol. The commercial polylactides vary with regard to their molar masses (uncorrected Mw's are typically in the range of 100–200 kDa), but they also vary with regard to their optical purity. Small amounts of D-lactyl units are purposely incorporated by addition of meso-lactide or racemic lactide to reduce the melting temperature (Tm) to values around or below 170 °C. Such a reduction eases thermal processing (e.g. injection moulding or extrusion) at temperatures < 200 °C, thereby lowering the risk of thermal degradation.4 Linear homopolymers of LLA or DLA were prepared by means of lauryl alcohol and SnOct2 to study the formation of stereocomplexes (racemates of PLLA and PDLA). These studies began in 1987 based on the work of Ikada and Tsuji and entailed numerous publications and review articles.87–95
Copolymers of L-lactide and other cyclic monomers such as Glycolide (GL) or εCL having a more or less random sequence were prepared by numerous research groups mainly for medical and pharmaceutical applications. A first study of alcohol-initiated copolymerization of LA and GL was reported by Gilding and Reed in 1979.96 Furthermore, a n-butanol-initiated copolymerization of LA and εCL in supercritical CO2 should be mentioned.97 A complete enumeration of all publications and patents of copolymers of racLA or LLA and their medical and pharmacological applications is beyond the scope of this article, and the reader is referred to pertinent reviews.5,98–102
(B) Diblock copolymers can be prepared by alcohol-initiated sequential copolymerization of racLA or LLA and another monomer such as GL or εCL. A first example was contributed by In't Veld et al. who performed an ethanol-initiated ROP of εCL in bulk at 110 °C and used the resulting PCL as macroinitiator for LLA under the same conditions.103 Interestingly, those authors found that the inverse sequence of copolymerization results in random copolyesters. Pensec et al. used the same approach.104 They prepared PCL-PLLA and PCL-PDLA block copolymers and studied the formation and properties of the corresponding stereocomplexes. Further diblock copolymers were prepared by using commercial poly(ethylene glycol) monomethyl ethers as macro initiators and either LLA or DLA as monomer. Jeong et al. examined the usefulness of their diblock copolymers as injectable drug-delivery systems.105 The formation and characterization of stereocomplexes were in the focus of the Kimura group.106,107 Jeong et al. also used their diblock copolymers for the preparation of A–B–A triblock copolymers by coupling of the diblocks with 1,6-hexamethylene-diisocyanate.105 Diblock copolymers based on poly(ethylene glycol) methyl ether and LLA/εCL copolyester were prepared and characterized by Min et al.108 The LA/εCL ratio was varied over a broad range. However, almost all research groups synthesized A–B–A triblock copolymers by means of a polymeric diol as telechelic macroinitiator for ROPs of LLA or DLA. Only two groups used telechelic PCL as initiator (Scheme 17), whereas most research groups used PEGs.109,110
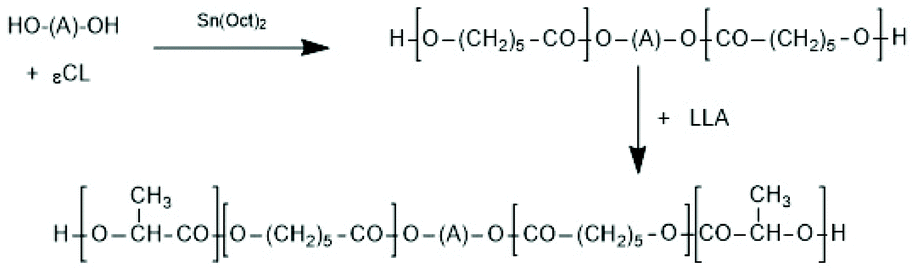 | ||
| Scheme 17 Synthesis of A–B–A triblock copolylactides via ROP with telechelic PεCL109,110 (with permission Copyright 2009 American Chemical Society). | ||
Starting out from commercial PEGs, Zhu et al. prepared A–B–A triblocks with amorphous poly(D,L-lactide) A-blocks and studied to what extent the crystallization of the PEG blocks depended on their length.111–113 Other examples of such amorphous-amorphous triblocks were contributed by Deng et al.114 Triblock copolyesters based on a PEG B-block and crystalline PLLA A-blocks were first synthesized by Kricheldorf et al.115,116 A special working field which emerged around 1995 concerned the formation and characterization of stereocomplexes of PEG-based A–B–A block copolymers having PLLA or PDLA A-blocks.110,116–120 Kubies et al. studied the microdomain structure of such triblock copolymers.121
ROPs initiated with a small diol such as ethanediol, 1,3-propanediol or 1,4-butanediol may be useful to prepare telechelic PLLAs, which may serve as crystalline central blocks in A–B–A triblock copolymers. However, this approach to triblock copolymers was rarely used, because almost all A–B–A triblock copolymers studied so far, were synthesized with a soft segment as B-block and polylactides as A-Blocks. One example was published by Tsuji et al.122
Quite recently, triblock copolymers containing a central poly(propylene glycol) should be mentioned.123 Their synthesis was achieved in bulk by coupling of two 1-dodecanol-initiated PLLA chains with a poly(propylene glycol) end capped with two epoxide groups.
(C) The first synthesis of star-shaped polylactides was reported by the Pennings group starting out from myo-inositol Scheme 18 as a hexafunctional star center.124 The resulting star-shaped polylactides were crosslinked by means of a diisocyanate derived from the ethyl ester of L-lysine. The resulting biodegradable polyurethane networks were designed to serve as coatings or scaffolds for medical applications, because all components of these networks were compatible with the human metabolism after hydrolytic degradation. A few years later Kissel et al. applied the same strategy and used glucose and sugar related polyols such as mannitol, sorbitol and xylitol (Scheme 19) as multifunctional initiators.125 The purpose of that work was elaboration of parenteral depot-systems for various drugs. Bruin et al. also used pentaerythritol as initiator,124 but four armed stars based on pentaerythritol were first prepared by Zhu et al.113 The arms of the star consisted of PLLA and PEO blocks, which were considered to be useful as “super microcapsules” for drugs. Kim et al. used triethanolamine in addition to pentaerythritol as initiators for SnOct2-catalyzed ROPs of LLA.126–129 However, tertiary amines have the disadvantage to cause rapid racemization of LLA at elevated temperatures. Pentaerythritol and dipentaerythritol were also used by numerous other research groups as star centers.130–141 In this connection the work of Biela et al. is noteworthy, because those authors modified pentaerythritol and dipentaerythritol by selective introduction of protecting groups in such a way, that they finally obtained di-, tri-, tetra-, penta- and hexa-functional star centres of similar structure.132–135
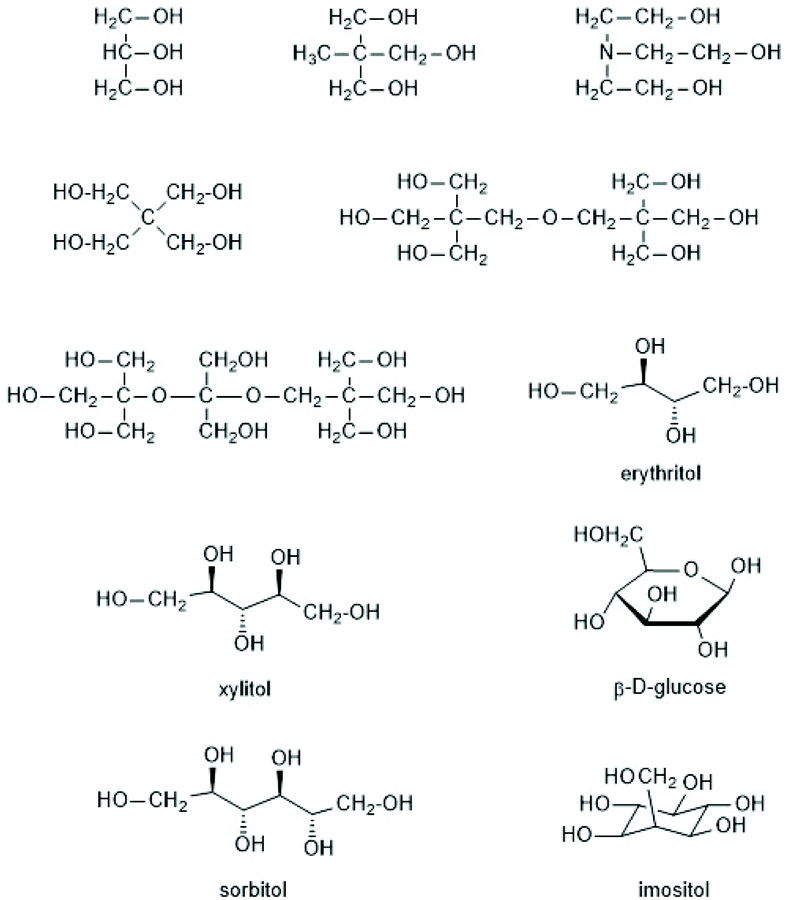 | ||
| Scheme 18 Multifunctional alcohols used as initiators for syntheses of star-shaped polylactides134 (with permission from Wiley). | ||
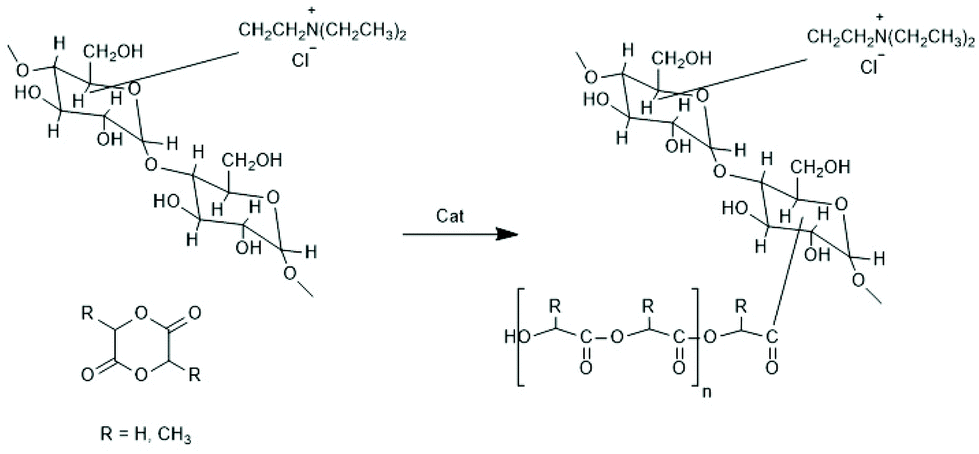 | ||
| Scheme 19 Grafting of racLA and glycolide from hydrophilic dextranes modified by attached ammonium groups144 (with permission from Elsevier). | ||
Several research groups were interested in the formation and properties of stereocomplexes of star-shaped polylactides. In this context two papers of Shaver et al. should be mentioned in which the hexafunctional dipentaerythritol and hexa(hydroxymethyl) benzene (Scheme 19) served as star center.138,139 Using tin and aluminium catalysts the sequence of L and D-units was systematically varied and the influence of the stereosequences on the physical properties of the resulting star-shaped polylactides was explored. More recently three papers of the Tsuji group were published, who prepared four- and six-armed stars from LLA and DLA and also synthesized star-shaped stereo diblock copolymers by sequential copolymerization of LLA and DLA.122,140,141 Star-shaped diblock copolymers consisting of poly(εCL) A-blocks and PLLA as B-blocks were prepared by Choochottiros et al. and examined with regard to their mechanical properties.142,143
(D) Comb-like polylactides were prepared by several research groups by grafting LLA from polymers such as (modified) polysaccharides, polyvinyl alcohol or poly(2-hydroxymethacrylate). Research in this field began with the work of the Kissel et al. who were interested in hydrolytic degradation profiles of these copolymers with regard to their usefulness as drug-release systems.125,144,145 Those authors used partly acetylated dextran, hydroxypropyl cellulose or polyvinyl alcohol as macroinitiators. In the case of dextran, the authors not only used partial acetylation to improve the solubility in organic solvents, but they also modified the dextran with an electric charge to improve its wettability. Either the sodium salt of sulphated dextran or dextranes containing ammonium salts were used (see Scheme 19).
Other groups used partially silylated dextranes prepared by silylation with hexamethyl disilazane in DMSO.146,147 The degree of substitution varied with the reaction conditions, but typically the least reactive OH group at C-3 remained free and served as initiator for the ROP of racLA. Grafting of racLA onto hydroxyethyl chitosan was achieved by B. Luo et al. by means of microwave irradiation.148 Two groups studied grafting of LA >on polyhydroxy ethyl methacrylates.149,150 In the work of Lim et al.150 oligomeric side chains either consisting of LLA or DLA were synthesized, and the formation and properties of stereo complexes were explored, whereas Zhao et al.149 studied crystallization and thermal properties of comb polymers having long PLLA side chains. Two different methods for the preparation of co-polyacrylates having polylactide side chains were described by Kobayashi and co-workers (Schemes 20 and 21).151 Either, itaconic acid terminated polylactides were synthesized first, and then copolymerized with acrylates/methacrylates (Scheme 21) or vinyl copolymers of itaconic anhydride were prepared first and OH-terminated polylactides were grafted onto these copolymers via esterification with the pendant anhydride group (Scheme 22).
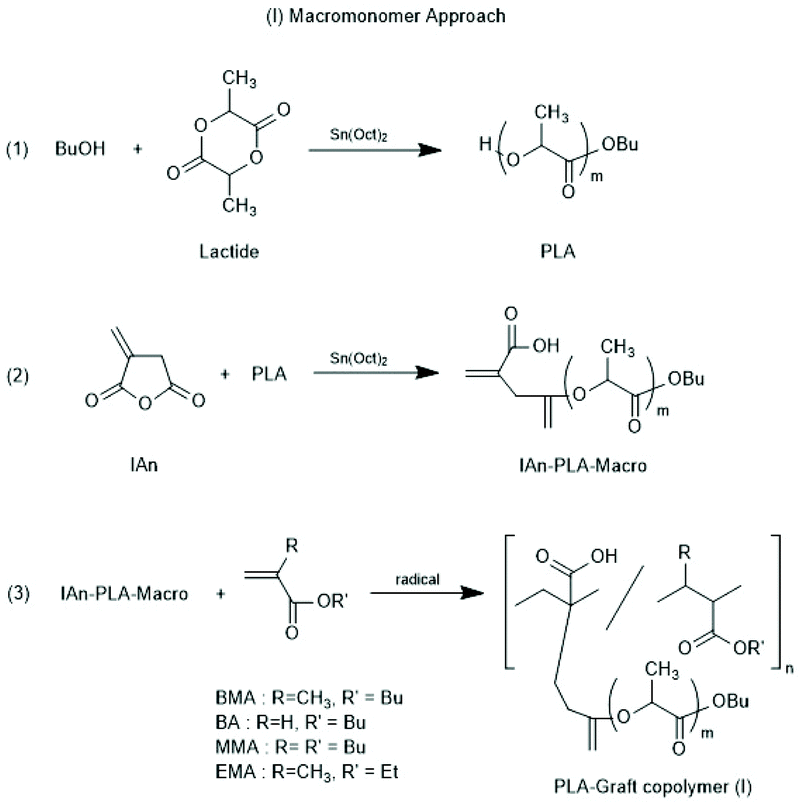 | ||
| Scheme 20 Whole reaction processes via macromonomer approach (reproduced from ref. 151 Copyright 2012 American Chemical Society). | ||
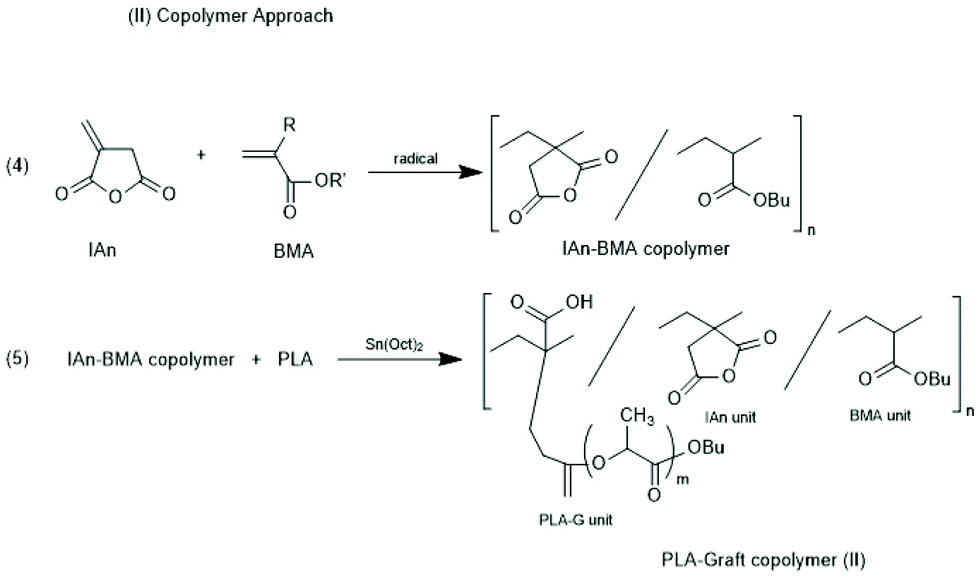 | ||
| Scheme 21 Whole reaction processes via copolymer approach (reproduced from ref. 151 Copyright 2012 American Chemical Society). | ||
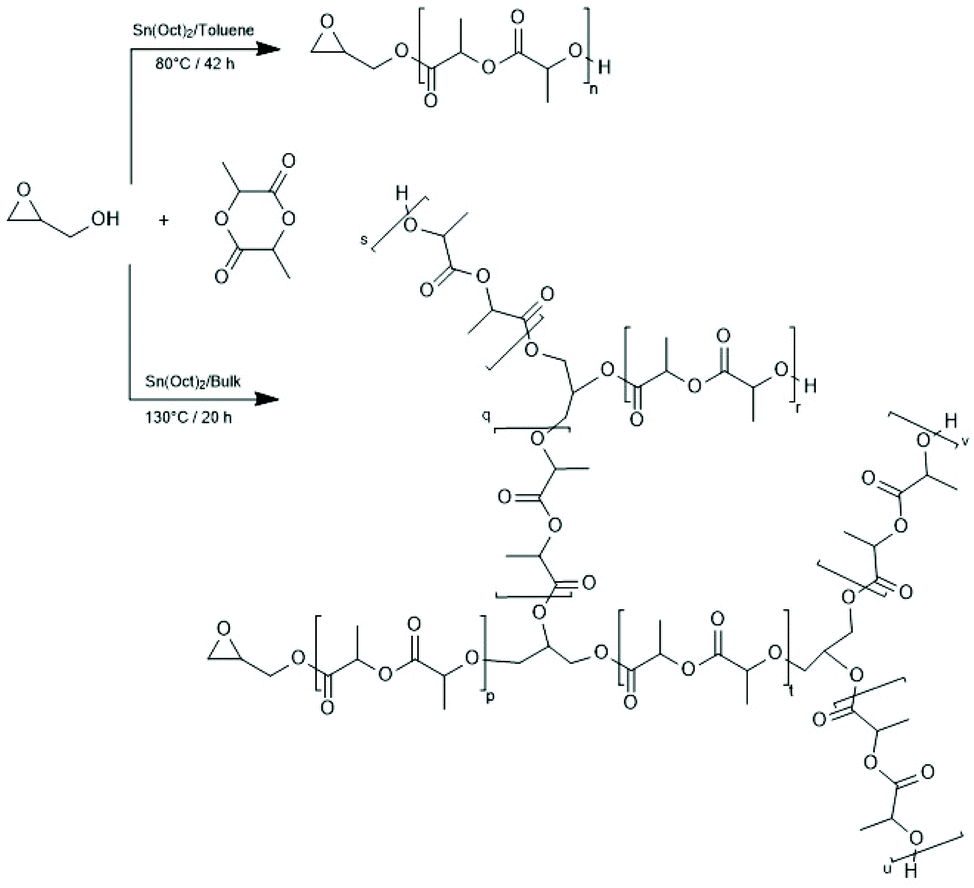 | ||
| Scheme 22 Synthesis of glycidol-lactide copolymers under different conditions (reproduced from ref. 152 Copyright 2007 American Chemical Society). | ||
(E) Compared to block- or star-shaped polylactides only few publications deal with syntheses of branches, including hyperbranched polymers and studies in that direction appeared relatively late. Apparently, the first paper in this field was published by Pitet et al. who initiated a SnOct2-catalyzed ROP of LLA with glycidol.152 At 80 °C in toluene a normal ROP occurred so that linear PLLA end-capped with a glycidol group was formed. However, at 130 °C in bulk the epoxide group also polymerized and yielded a branched polyether structure (Scheme 22). A similar approach was explored by Wolf and Frey (Scheme 23).153
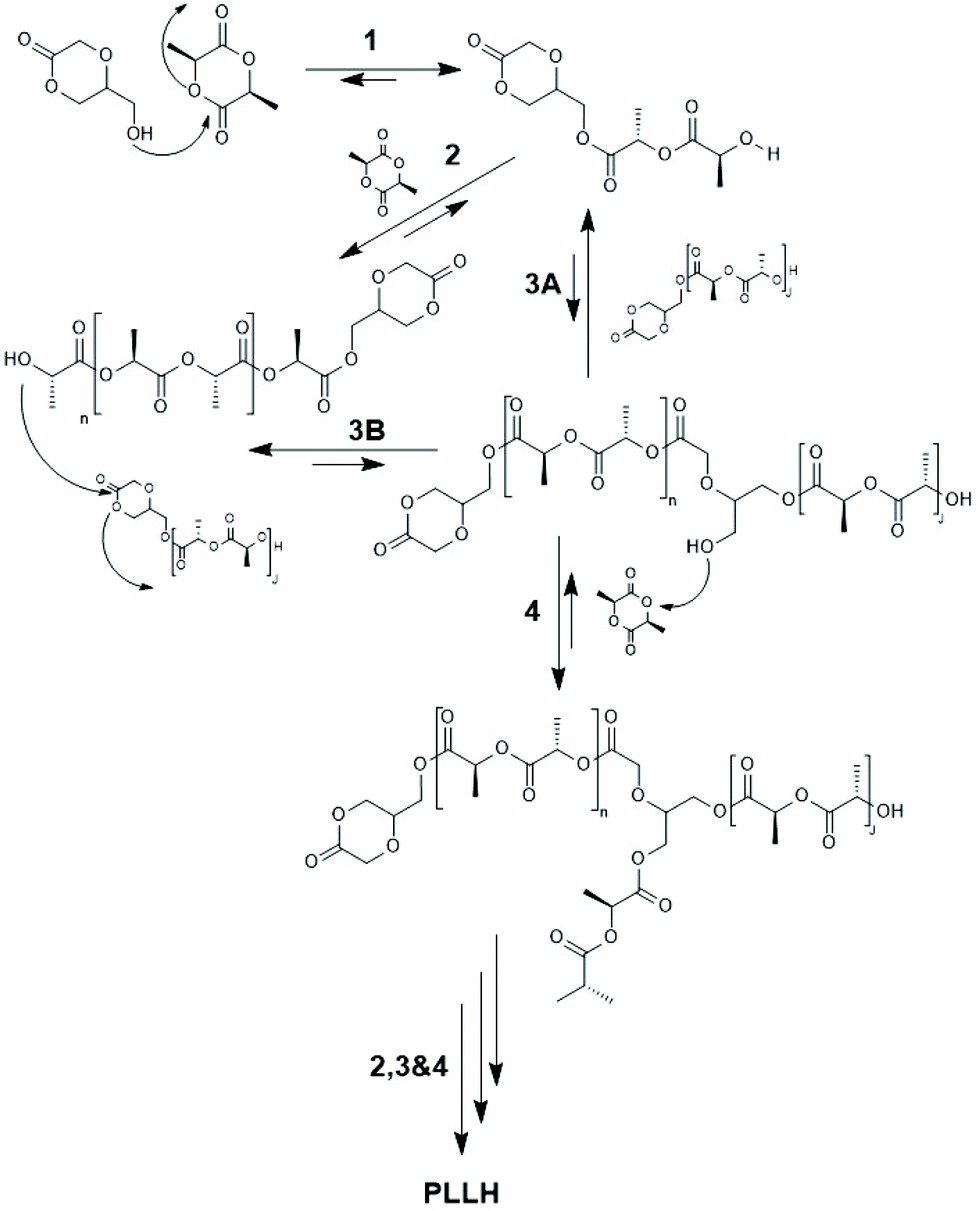 | ||
| Scheme 23 Quantitative reaction scheme for ROMBP (ring opening multibranching polymerization) of 5-HDON with L-lactide derived from kinetic measurements (reproduced from ref. 153 Copyright 2009 American Chemical Society). | ||
They used 5-hydroxymethyl-1.4-dioxanone as comonomer and obtained a branched copolyester, but only low molar masses were achieved (Mn < 1300, Mw < 5000). Later, a totally different approach was elaborated by the same group starting out from 2,2-bishydroxymethyl propionic acid as initiator and comonomer of LLA (Scheme 24).154 The first step of this approach consisted in a normal ROP, whereupon the two hydroxymethyl groups served as initiators. The second step consisted in a polycondensation involving the carboxylic groups activated with dicyclohexyl carbodiimide and 4-dimethylamino pyridine. A typical strategy for the preparation of biodegradable networks based on PLLA segments was described by Ivirico et al.155 Starting from 1,6-hexanediol as initiator telechelic PLLAs were prepared, and the OH end groups were end-capped with methacrylate groups. Their radical polymerization yielded networks with tailored water sorption.
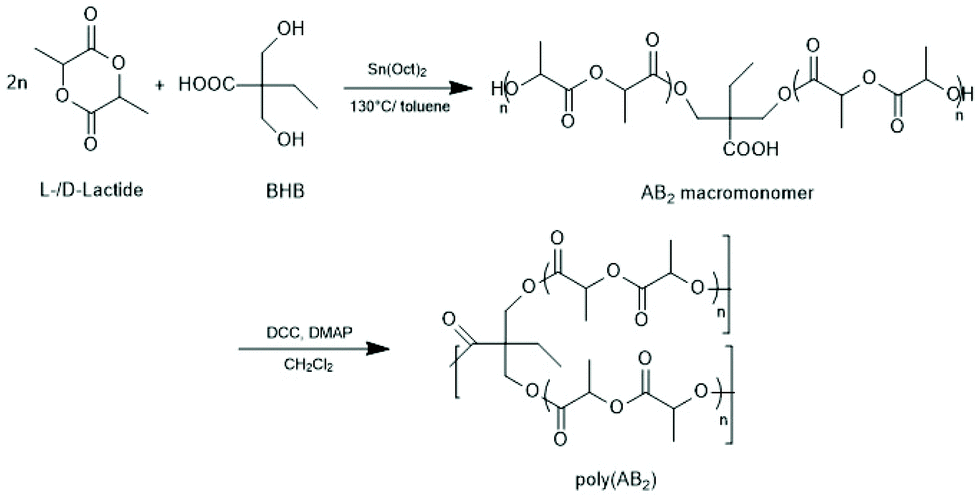 | ||
| Scheme 24 Synthetic route to long chain branched poly D,L-lactide by SnOct2 catalyzed ROP and subsequent condensation with DCC DMAP (reproduced from ref. 154 with permission from Wiley). | ||
4.3 Alcohol-initiated ROPs catalyzed by various tin compounds
In addition to SnOct2 various tin salts and compounds can be used as catalysts for alcohol initiated ROPs of lactides. At first, ROPs with two commercial tin(II) salts, namely SnCl2 and tin acetate (SnAc2), should be discussed.Although these tin salts were commercially available for many decades and their toxicity is low or lower than that of SnOct2 (see part 2, chloride and acetate belong to the human metabolism), systematic exploration of their usefulness as catalysts for ROPs of lactide and ε-caprolactone was only reported quite recently.79,156 With SnAc2 and temperatures < 160 °C a full control of the molar mass is feasible up to a LA/In ratio of 100. The MALDI the mass spectra do not display formation of cycles in close analogies to ROPs catalyzed by SnOct2 under identical conditions.156 With SnCl2 side reactions occur and the control of the molar mass was less perfect.79 Surprisingly, even cyclic dibutyltin catalysts, such as DSTL,157 BuSnBuca,158 or BuSnNaph159 (see Scheme 25) which, when used as neat compounds, catalyse formation of cyclic polylactides by the REP mechanism (see part 4), also catalyse formation of linear chains, when an alcohol was added as initiator.
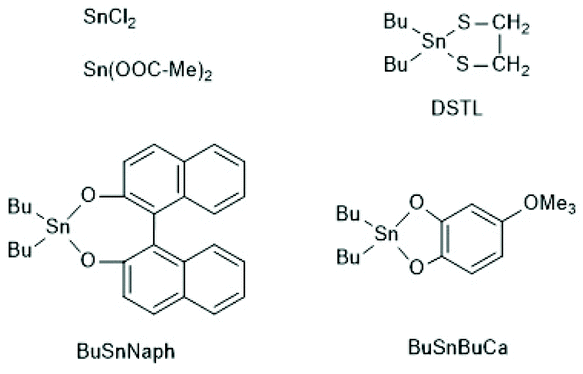 | ||
| Scheme 25 Various tin compounds used as catalysts for alcohol initiated ROPs of LLA ref. 159 (with permission from Elsevier). | ||
Full control was again feasible at temperatures around or below 160 °C and LLA/In ratios up to 100/1. All these alcohol-initiated ROPs have in common that the even-numbered chains predominate over the odd-numbered ones. However, detailed studies about the influence of different alcohols, different cyclic catalysts, In/Cat ratio and addition of solvents on the extent of even-/odd-equilibration and other transesterification reactions are missing.
Finally, a recent publication of Novák et al. discussing the usefulness of the cationic Sn(II) complexes (Schemes 26–28) as catalysts for ROPs of LLA should be mentioned.160 In the title and in the section “Discussion” those authors claim that these cationic catalysts initiate an activated monomer mechanism. However, MALDI TOF mass spectra and NMR spectroscopic end group analyses were missing. Furthermore, the authors have ignored that, Kricheldorf et al.161,162 and Bourissou et al.34 demonstrated long ago that only super acids such as fluorosulfonic acid and trifluoromethane sulfonic acid catalyse a cationic activated monomer mechanism of lactides, whereas any other kind of cationic species does not.
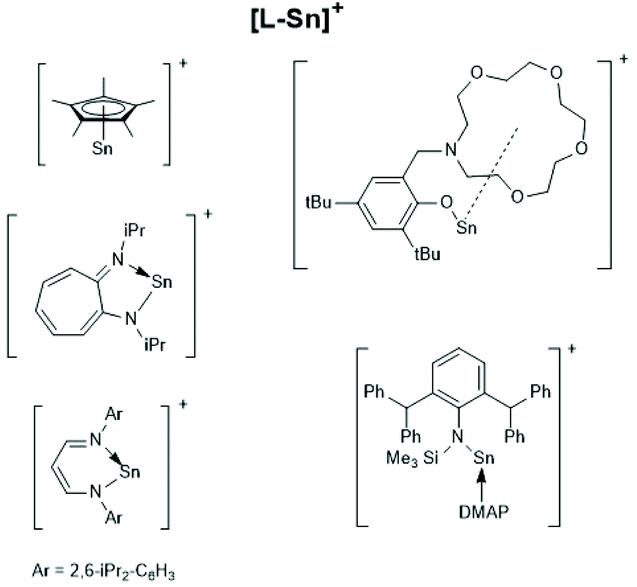 | ||
| Scheme 26 [L-Sn]+ type tin cations (modified from ref. 160, copyright 2021 American Chemical Society). | ||
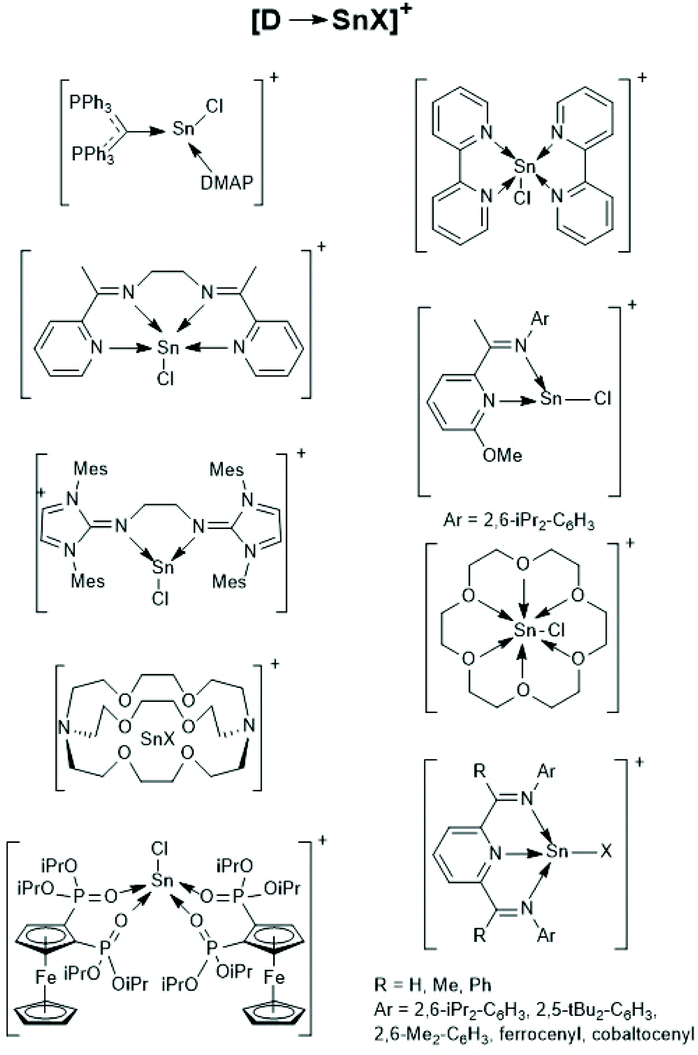 | ||
| Scheme 27 [D-SnX]+ type tin cations (modified from ref. 160, copyright 2021 American Chemical Society). | ||
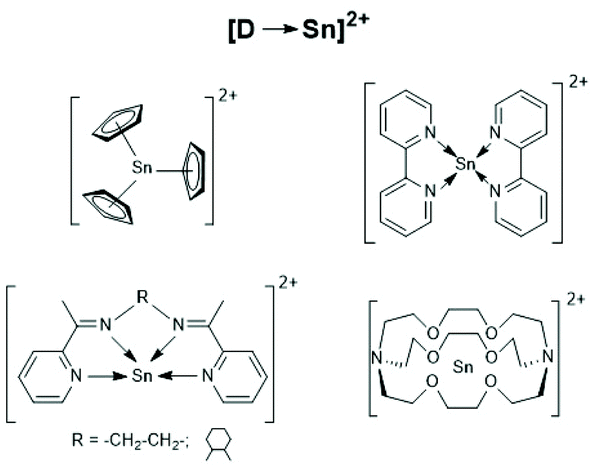 | ||
| Scheme 28 [D-Sn]2+ type tin cations (modified from ref. 160, copyright 2021 American Chemical Society). | ||
5. Ring-expansion polymerization (REP)
5.1. Cyclic dibutyl tin(IV) alkoxides as catalysts
The first successful experiments of REP were reported by Kricheldorf and Lee in 1994 and 1995.163,164 β-Butyrolactone and ε-caprolactone were used as monomers and the cyclic dibutyltin alkoxide (Scheme 29) served as catalyst. The structures and formula numbers of the cyclic tin alkoxides used later as catalysts for REPs of lactides and lactones are compiled in Scheme 30. The three standard methods used for their preparation are outlined in Scheme 31. The most convenient method is method (b) but requires the preparation of dibutyltin bis-methoxide. Although it was a commercial chemical in previous decades, it is hard to find a commercial source around 2021 (when this manuscript was written) due to its toxicity. Method (a) is simpler and less expensive but limited to a catalyst soluble in the reaction medium. Toluene or xylene are typically used for this purpose to remove the liberated water by azeotropic distillation. However, the five and six-membered cycles 1a + b, 2, 3 and 7 (Scheme 31) form dimers or oligomers in the solid state combined with a 3D network based on donor–acceptor interactions (DA) between oxygen and tin atoms. The consequence is insolubility in all inert solvents. The same is true for dibutyltin oxide, and so it may happen that part of the reaction product begins to precipitate before all dibutyltin oxide went into solution. Therefore, most REP experiments were performed with the soluble liquid catalyst 4. In the case of the ethylene glycol derivatives 6, an intracircular DA interaction, confirmed by 118Sn NMR spectroscopy, stabilizes the monomeric character.165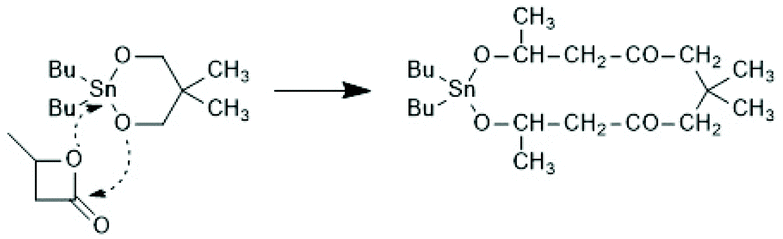 | ||
| Scheme 29 First REP of a cyclic ester as reported by Kricheldorf and Lee (reproduced with permission from ref. 163 Copyright 1994 American Chemical Society). | ||
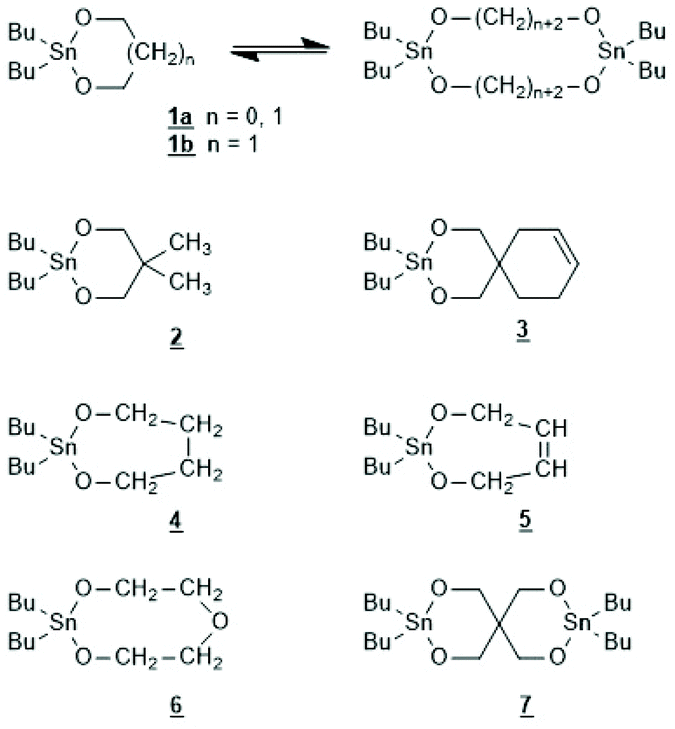 | ||
| Scheme 30 Structures of cyclic dibutyltin bis alkylene oxides used for REPs of LLA and several lactones. | ||
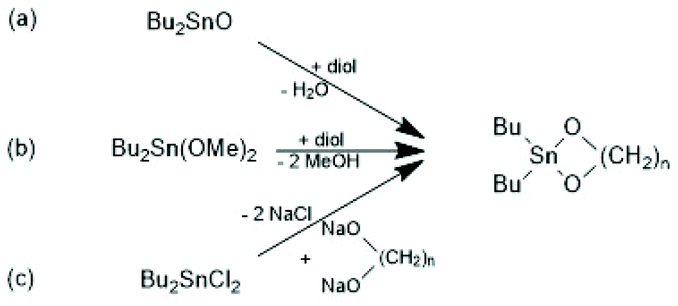 | ||
| Scheme 31 Three synthetic methods useful the preparation of cyclic dibutyltin bisalkyleneoxides. | ||
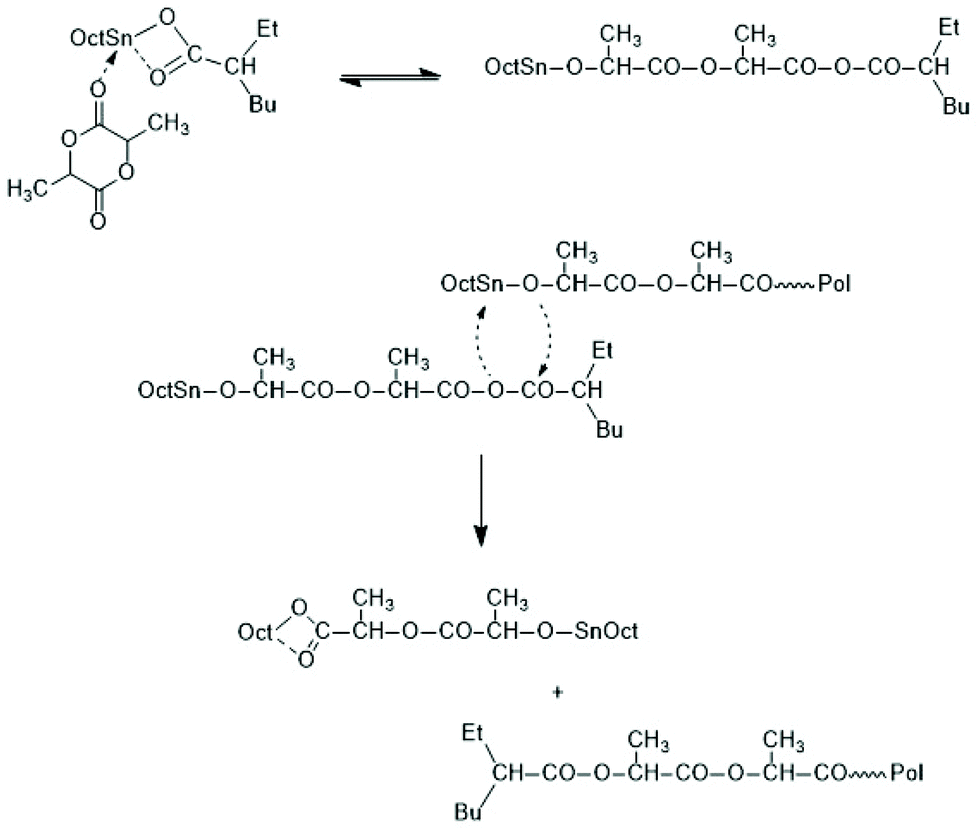
Regardless of monomer and catalyst, the REPs proceed via the normal C–I mechanism discussed above for non-cyclic thin alkoxides (see Scheme 30). The catalyst, which is in fact an initiator, remains covalently fixed in the chain with a negative and several positive consequences. The negative consequence is that cyclic polylactides free of initiator groups cannot be prepared by direct polymerization. Furthermore, the Sn–O–CH bonds are sensitive to hydrolysis and alcoholysis, so that these cyclic polylactides (and corresponding polylactones) are useless for any application on air. On the other hand, three positive aspects may be mentioned. First, the average ring size may be controlled via the LLA/Cat ratio. Second, the hydrolytically sensitive Sn–O bonds can be replaced in a second reaction step, as illustrated by reactions (1) and (2) in Scheme 32. The third approach consists of a second step which keeps the ring closed for example by incorporation of γ-thiobutyrolactone. Incorporation of thiobutyrolactone is a stoichiometric reaction, because this lactone is for thermodynamic reasons not polymerizable.166 The resulting Sn–S bond is more stable against hydrolysis than the ester groups of lactide units. Further ring closing reactions were reported for REPs with εCL.166–168
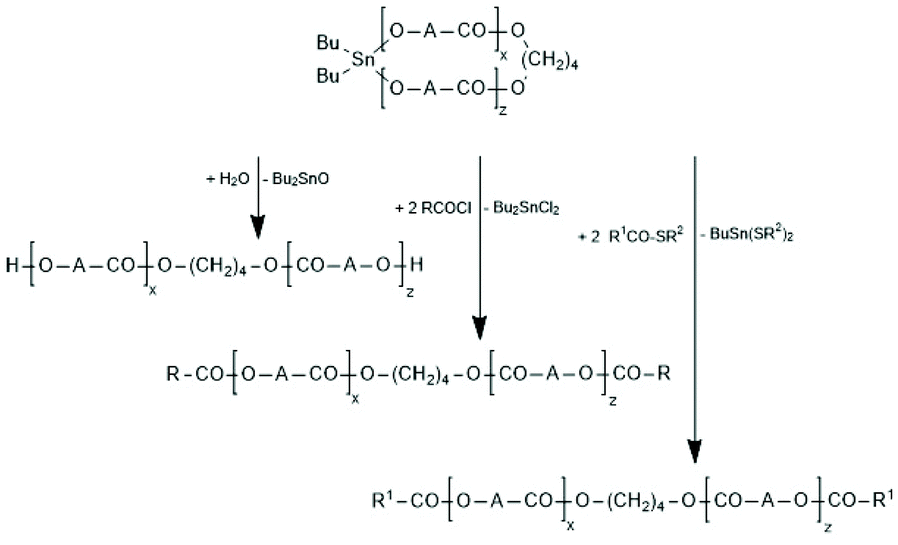 | ||
| Scheme 32 Transformation of cyclic Bu2Sn-containing polylactides into telechelic linear polyesters. | ||
The high reactivity of the Sn–O bond allows for a quantitative ring-opening with numerous reagents such as water, alcohols, thioesters, acid anhydrides and acid chlorides, so that a broad variety of telechelic polyesters can be prepared in this way (Scheme 32). Star-shaped polyesters and biodegradable networks (useful as matrices for drug delivery) can also be prepared in his way. Most of these preparative applications were explored with lactones as monomers and thus, are not discussed here in detail, but they were summarized and discussed in a previous review article.169 However, two methods used for preparation of telechelic A–B–A triblock copolymers having crystalline poly(L-lactide) blocks are illustrated in Scheme 33170 and Scheme 34.171,172
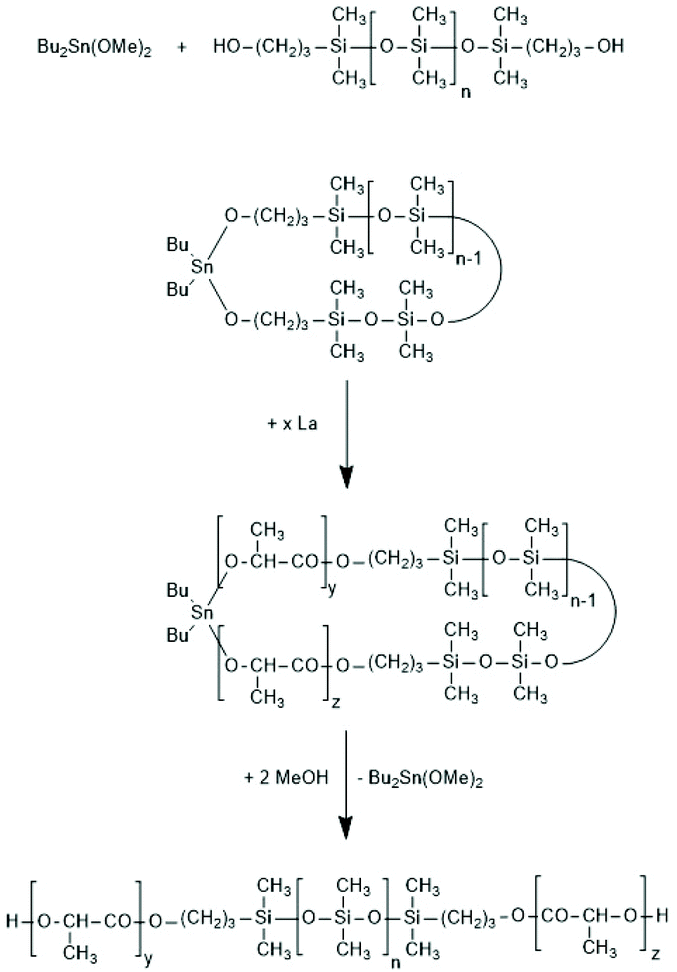 | ||
| Scheme 33 Synthesis of A–B–A triblock copolymers via a cyclic polysiloxane initiator (adapted from ref. 170 with permission from Wiley). | ||
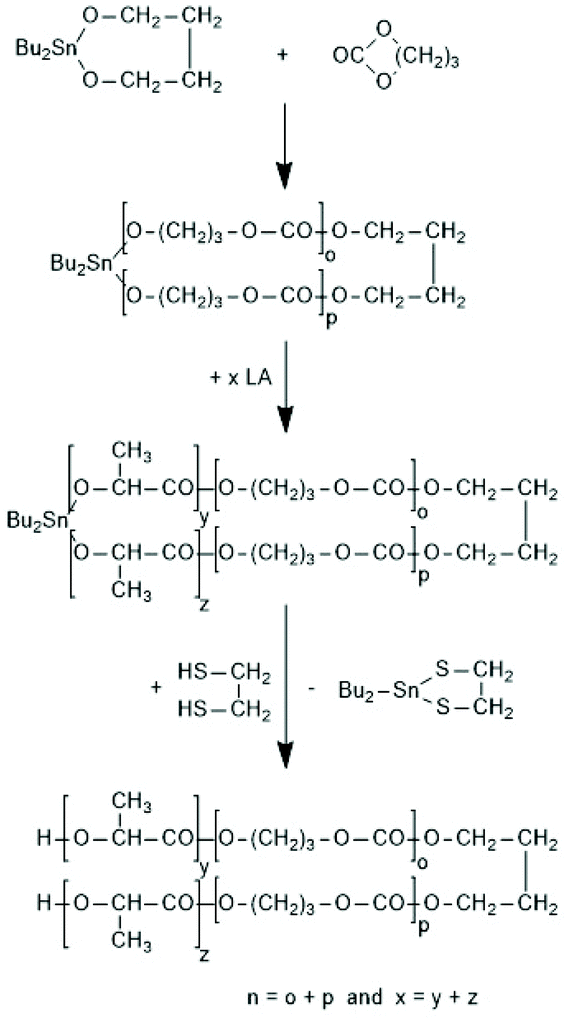 | ||
| Scheme 34 Synthesis of A–B–A triblock copolymers via REP with 2,2-dibutyltin-2-stanna-1,3-dioxaalkanes171 (with permission from Wiley). | ||
Important for the success both synthetic approaches is the optimization of the reaction conditions, so that formation of large amounts of cyclics via intracircular back-biting is avoided. In the case of the second approach (Scheme 34), intermolecular transesterification between both polyester blocks needs also to be avoided. Furthermore, the preparation of a network starting out from the REP of racLA followed by crosslinking with glycerol triglutarate trichloride should be mentioned.173 This gel exclusively consists of biocompatible components and may serve as drug delivery system.
5.2 Cyclic tin mercaptides and bisphenoxides as catalysts
A new dimension of REP catalyzed by cyclic tin compounds was opened when the reactivity of DSTL (2,2-dibutyl-2-stanna-1,3-dithiolane) (Scheme 35) was reinvestigated.157 Originally, it was reported that DSTL is inactive as catalyst for REP of lactide, for two reasons.174 First, the Sn–S bond is thermodynamically more stable than the Sn–O bond formed in the (hypothetical) initiation step. Second, in an attempt to polymerize L-lactide in bulk with a high concentration of catalyst (LLA/Cat ratio of 100/1) no significant polymerization was observed after 8 h at 120 °C. This negative result was more recently confirmed, but in a re-investigation with longer reaction times (e.g. 24 h) at 120 °C or with shorter times (>3 h) at 160 °C nearly complete conversions were achieved.157 Both, 1H NMR spectra and MALDI-TOF mass spectra revealed that the reaction products almost completely consisted of cyclic polylactides free of initiator fragments. A confirmation of this interpretation by comparison of the intrinsic viscosities with those of commercial linear polylactides was not reported at that time but was achieved later. A typical example of such Mark–Houwink–Sakurada (MHS) measurement is presented in Fig. 2. A simplified scheme of the polymerization mechanism, which is based on the normal coordination/insertion mechanism, is presented in Scheme 36. The formation of odd-numbered cycles indicated that the kinetically controlled polymerization reaction yielding even-numbered chains was followed by rapid intracircular back-biting reactions, which finally result in thermodynamic control of the entire REP (Scheme 37).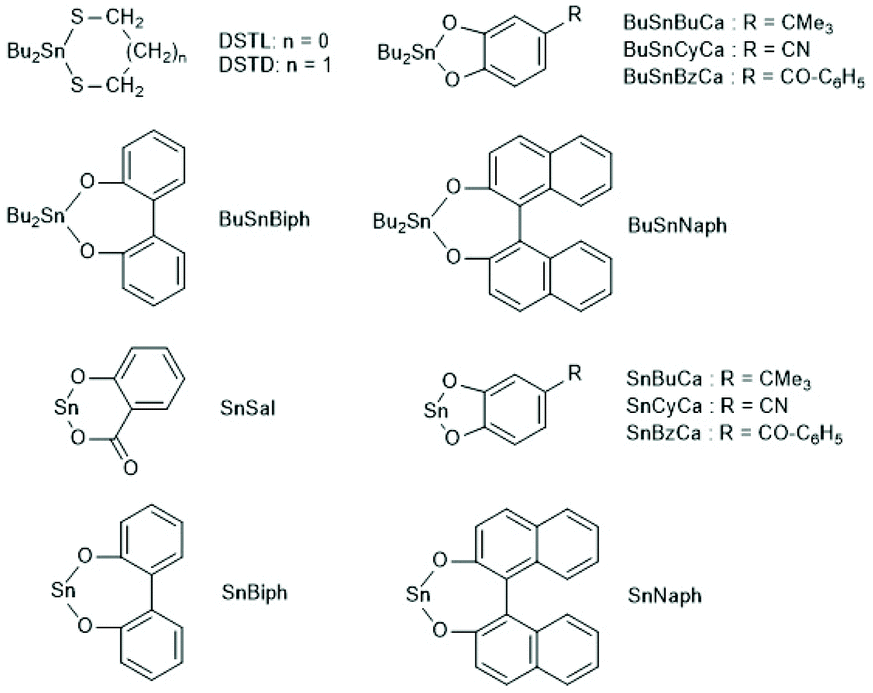 | ||
| Scheme 35 Cyclic tin catalysts used by the Kricheldorf group for REP of LLA.157,175–177 | ||
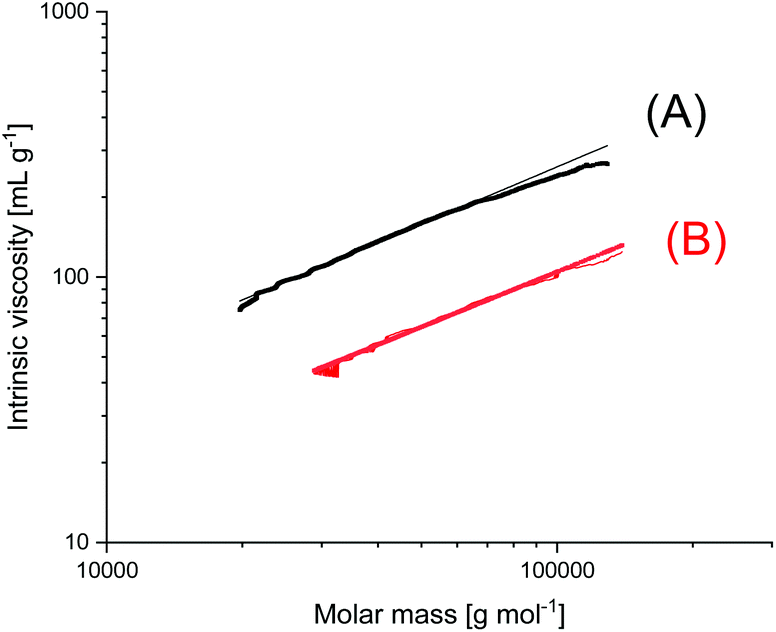 | ||
| Fig. 2 MHS plots of (A) commercial linear Purapol L105, (B) cyclic PLLA prepared with DSTL in bulk at 160 °C (unpublished measurements, sample (B) was prepared in ref. 178). | ||
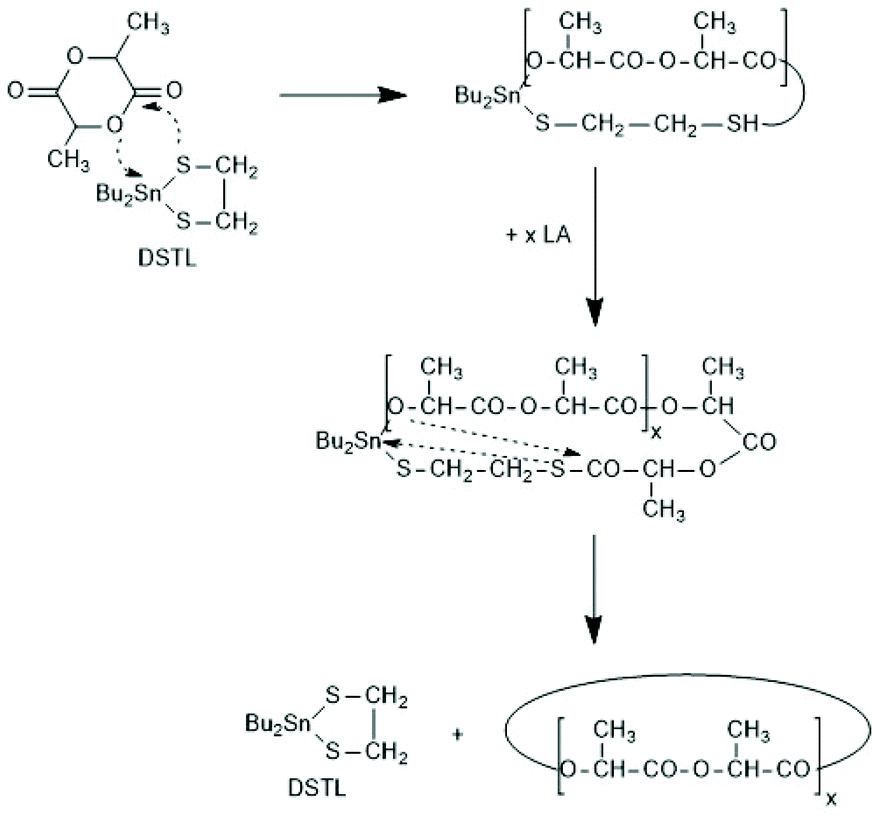 | ||
| Scheme 36 Simplified mechanism of the REP of LLA catalyzed by DSTL. | ||
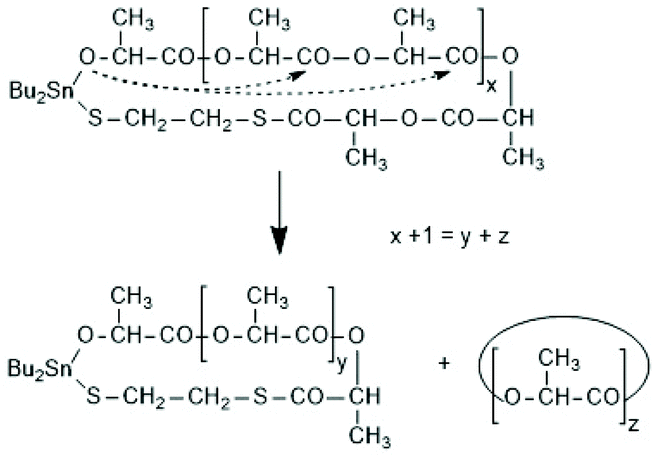 | ||
| Scheme 37 Formation of cyclic oligolactides via intracircular “back-biting”. | ||
In agreement with the existence of equilibration reactions, dispersities above 2.5 were found and Mw's up to 140000 were obtained without racemization. Only few polymerizations were conducted with the 2,2-dibutyl-2-stanna-1,3-dithiane (DSDT, Scheme 35), which indicated that this six-membered ring is slightly less reactive than DSTL.158
Following the successful synthesis of cyclic polylactides bare of initiator fragments with DSTL, sulphur-free cyclic tin catalysts derived from suitable diphenols such as catechols, 1,1′-diphenol, 1,1′dinaphthol and salicylic acid were synthesized (Scheme 35) and their catalytic potential was explored in much detail. All these catalyzed are easily accessible from inexpensive, commercially available starting materials in three ways. The dibutyltin derivatives are best prepared from dibutyltin oxide in hot toluene with azeotropic removal of water. The cyclic Sn(II) catalysts can be prepared by heating of the diphenol with SnOct2 in toluene, whereupon the desired catalyst crystalizes after cooling and drives the equilibrium into the desired direction. Higher yield can be achieved by using silylated diphenols or salicylic acid, a method which can be performed in a one-pot procedure.
REPs with Bu2Sn-catalyst derived from catechols were studied first revealed that with BuSnBuCa cycles almost free of linear by-products were obtained.158 Polymerizations in bulk at 160 °C with variation of LA/Cat ratio and time yielded (uncorrected) Mw's in the range of 70000 to 100000 with high dispersities. Characteristic for the BuSnBuCa catalyst, is a predominance of even numbered cycles in the reaction product. However, when BuSnCyCa or BuSnBzCa were used as catalysts at temperatures below 100 °C, the odd-numbered cycles were large prevalent.175 A mechanistic explanation for this surprising result was discussed. With the seven-membered cyclic dibutyltin compounds BuSnBiph and BuSnNaph higher polymerization rates and higher molar masses were achieved.159 REPs performed in bulk at 160 °C were completed within 1 h and yielded uncorrected Mw's up to 140000 again in combination with high dispersities (>3). A major shortcoming of these useful REP catalysts is their toxicity, which is unknown, but predictably higher than that of the commercial standard catalyst SnOct2.
To overcome or reduce this problem, the Kricheldorf group explores synthesis and application of cyclic Sn(II) bisphenoxides. At first catechols, 2,2′-dihydroybiphenyl or 1,1-bisnachtol were added to SnOct2-catalyzed polymerizations of LLA in bulk at 160 °C. Indeed, cyclic PLLAs were obtained in contrast to analogous experiments with monophenols as cocatalysts.176 This observation prompted the synthesis of the cyclic Sn(II) bisphenoxides listed in Scheme 35. Numerous REPs conducted in bulk at 160 of 180 °C were performed in such a way, that either the isolated catalysts or the combination of SnOct2 and catechol or 4-tert-butylcatecol were used under identical conditions.177 Surprisingly, higher molar masses (Mw's up to 170000) and perfect cyclization were found for the combination of SnOct2 and catechols. Analogous results were obtained with salicylic acid as cocatalyst.179 However, a lower risk of discoloration was observed for salicylic acid. Catechol and salicylic acid were selected as cocatalyst, because they are natural products with low and well-known toxicity.
SnBiph and SnNaph proved to be the most active cyclic tin catalysts studied so far even slightly more reactive than SnOct2.180 When REPs were performed in bulk at 160 or 180 °C with SnBiph conversions were complete within 15 min at LLA/Cat ratio of 1000/1 and within 1 h at a LLA/Cat ratio of 10000/1. Cyclic PLLAs free of racemization and with Mw values up to 305000 were obtained. Finally, polymerization experiments with the spirocyclic Sn(IV) compounds SnNaph2 should be commented. In a first paper (also including experiments with analogous Ge and Zr compounds) the authors reported that SnNaph2 is a rather sluggish catalyst yielding low molar mass cycles with low dispersity and a conspicuous predominance of even-numbered species.181 However, this interpretation turned out to be a mistake, because masses of cyclic PLLAs and linear chains having a dihydoxybisnaphthol end group (Scheme 38) are nearly identical, so that mass spectrometry alone does not suffice for trustworthy distinction. This analytical problem does not exist for other monomers, and in a recent publication including glycolide and ε-caprolactone the authors have revised their original misinterpretation of MALDI TOF mass spectra and clarified that all monomers yield linear chains having the structures formulated in Scheme 38.182 Nonetheless, the strong predominance of even-numbered chains which reaches levels >90% at low polymerization temperatures, was an interesting phenomenon and a mechanistic explanation has been discussed.
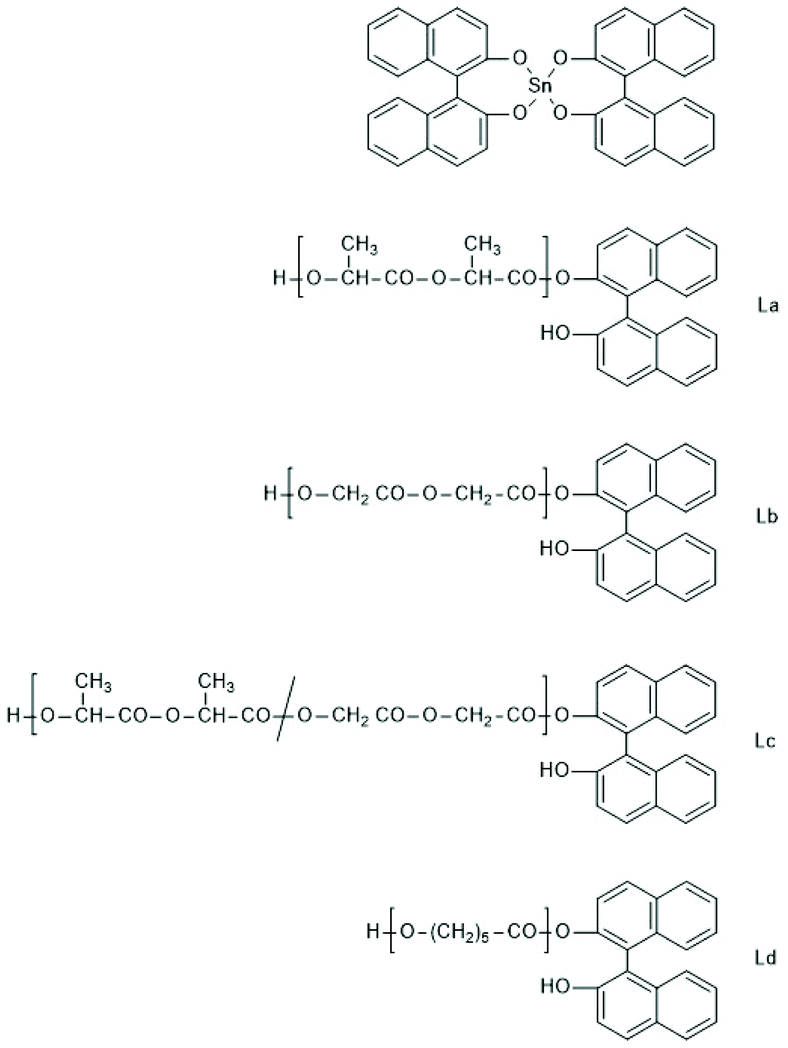 | ||
| Scheme 38 Structures of the SnNaph2 catalyst and of the linear chains obtained from it via REP followed by hydrolytic cleavage of the Sn–O bonds (adapted from ref. 182 with permission from Wiley). | ||
6. Ring-opening polymerization with simultaneous polycondensation (ROPPOC)
6.1. Tin mercaptides and phenoxides as catalysts
A normal ROP is characterized by an initiation reaction which yield a stable end group, usually an amide or ester group in the case of PLLAs, whereas chain growth continues at the nucleophilic chain end, typically a Sn–O–CH(CH)3 group. Condensation steps, and thus, a ROPPOC mechanism come into play, when the CO-X chain end is electrophilic/reactive enough to react with the nucleophilic end group. Depending on chain length and concentration more intermolecular condensation steps, resulting in chain growth, or more intramolecular condensation steps, yielding cyclic PLLAs are the consequence. Two review articles, the second one with a more detailed definition and theoretical analysis, have been published.183,184Since the Sn–S bond is more stable than the Sn–O bond, condensation reactions were expected when the initiator is a Sn mercaptide or thiophenolate yielding a Sn–SR end group. Using three Sn–SR compounds displayed in Scheme 39, the first ROPPOC syntheses of more or less cyclic PLLAs were achieved via the mechanism outlined in Scheme 40.185 Under identical conditions the extend of cyclization was higher for the catalysts derived from acidic SH-compounds (BuSnMBT and BuSnSPF) than that from benzyl mercaptane, as expected for an ete-cyclization. At temperatures above 130 °C BuSnMBT has the shortcoming to cause slight racemization of LLA. Moreover, the basicity of the N-heterocycle is low. Therefore, the BuSnSPF catalyst proved to be the most useful Sn–S type ROPPOC catalyst and yielded cyclic PLLAs with Mw's up to 200000 without racemization.
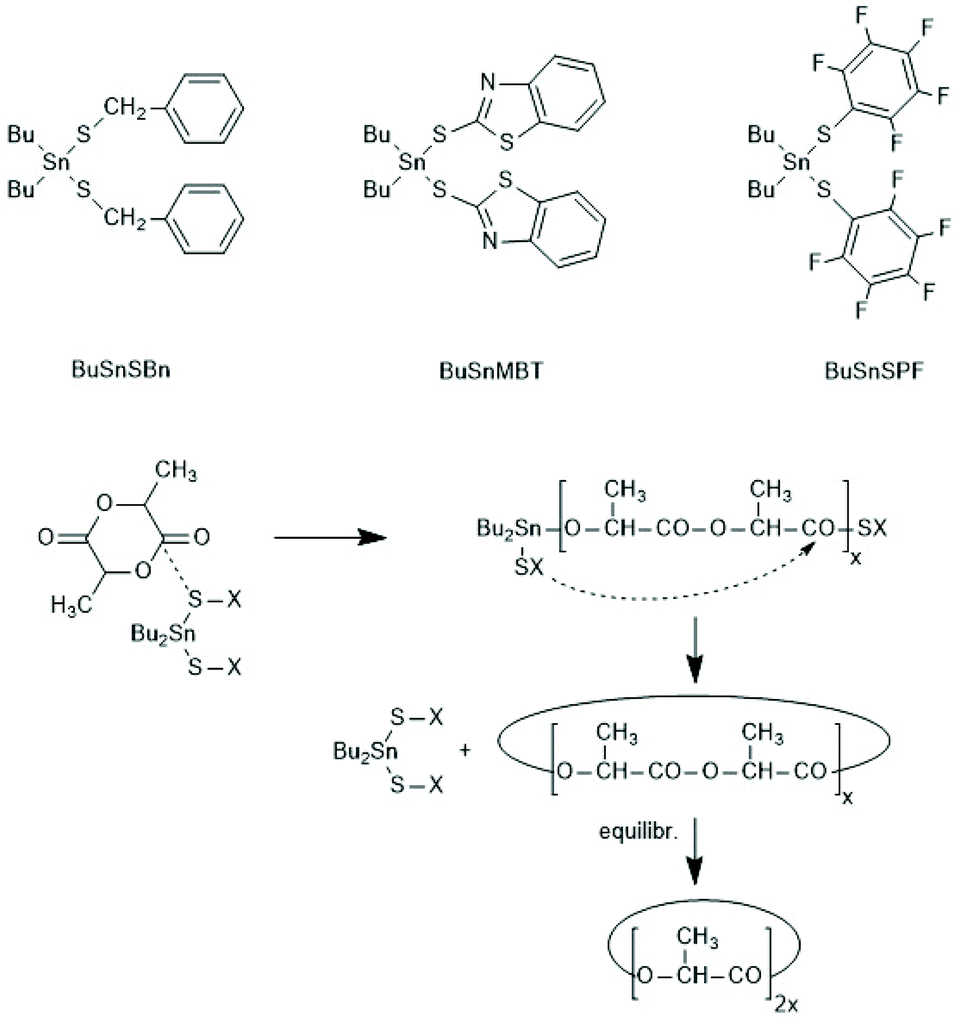 | ||
| Scheme 39 Dibutyltin bis-thiolates prepared and used by Kricheldorf et al. for ROPPOCs of LLA and the ROPPOC mechanism catalyzed by dibutyltin bis-thiolates.185 | ||
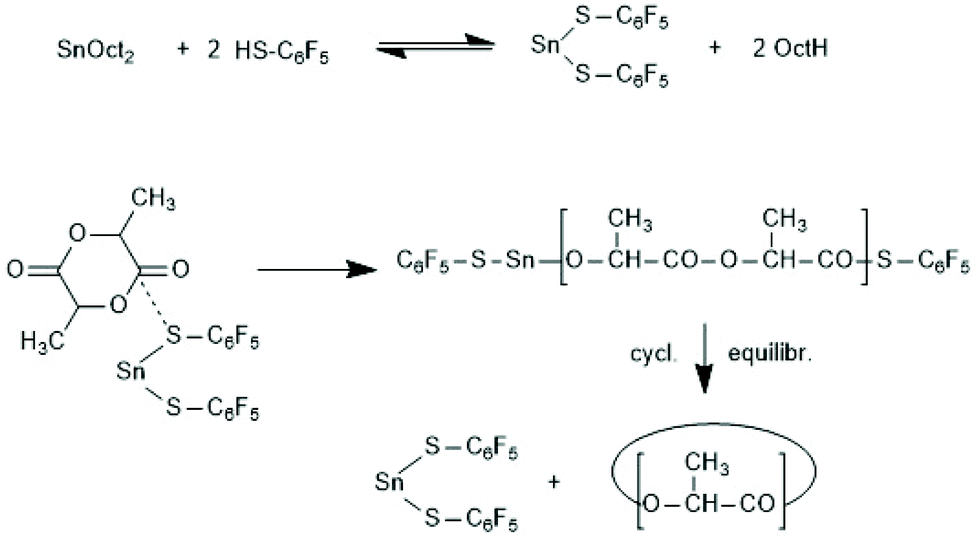 | ||
| Scheme 40 ROPPOC mechanism resulting from the in situ combination of SnOct2 and pentafluoro thiophenol.185 | ||
Experiments with Sn(II) mercaptides or thiophenolates have not been reported yet, but the following results indicate that also Sn(SR)2 compounds may react as ROPPOC catalysts.186 When pentafluorothiophenol was added to a SnOct2 catalyzed polymerization of LLA at 160 °C in bulk, formation of linear chains having SPF end groups as the first reaction products was detected by MALDI mass spectrometry. At longer reaction times (>4 h) complete transformation into cyclic PLLA was observed. These observations suggested the mechanism outlined in Scheme 40 (and steps analogous to those formulated in Scheme 39). The assumption that the thiophenol almost completely substitutes the octanoate groups is based on the fact that the Sn–S bond is in general thermodynamically more stable than analogous Sn–O bonds. Yet, compelling evidence requires, of course, the isolation of Sn(SPF)2.
In a subsequent paper the authors studied reactivity and usefulness of dibutyltin bisphenoxides ((b)–(d) in Scheme 41) in comparison with dibutyltin bisbenzyloxide ((a) Scheme 41) and in comparison with BuSnSPF (Scheme 39).186 Polymerizations performed in bulk at 160 °C revealed that the efficiency of cyclization strongly increased with the acidity of the R–OH used for the preparation of the catalyst. This means, in turn, the electrophilicity of the CO-chain end is decisive for the success in agreement with the ROPPOC mechanism and ete-cyclization. Since all PLLA chains have identical Bu2Sn–O–CH end groups, formation of cycles should be independent of the CO-end group when the formation of cycles exclusively occurs via “back-biting” from the nucleophilic chain end. As evidenced by MALDI TOF mass spectrometry and measurements of intrinsic viscosities the most reactive catalyst, BuSnOPF, yielded almost completely cyclized PLLAs with Mw's up to 200000. Furthermore, it was observed, that BuSnOPF is considerably more reactive than BuSnSPF and allowed polymerizations in solution at 120 °C, whereupon the resulting molar masses can roughly be controlled via the initial monomer concentration. However, an unexplained phenomenon is the tendency towards increasing side reactions at lower catalyst concentration, so that relatively large amounts of catalyst are needed for successful syntheses of cyclic PLLAs (LLA/Cat = 100/1–400/1).
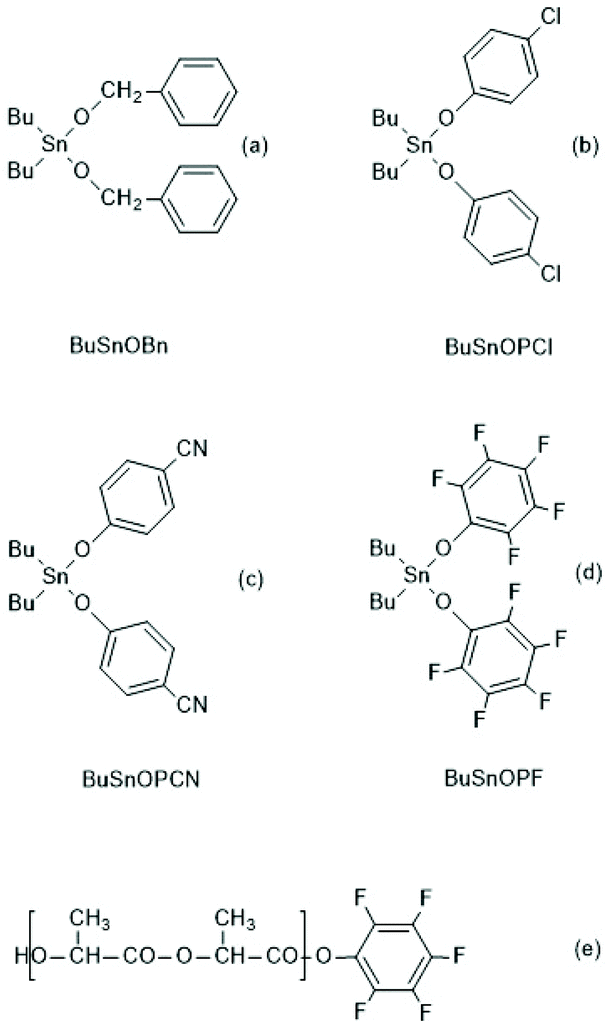 | ||
| Scheme 41 Dibutyltin bisbenzyloxide and bisphenoxides prepared and used by Kricheldorf et al. for ROPPOCs of LLA, and a linear intermediate identified at low temperature. | ||
6.2. Tin carboxylates as catalysts
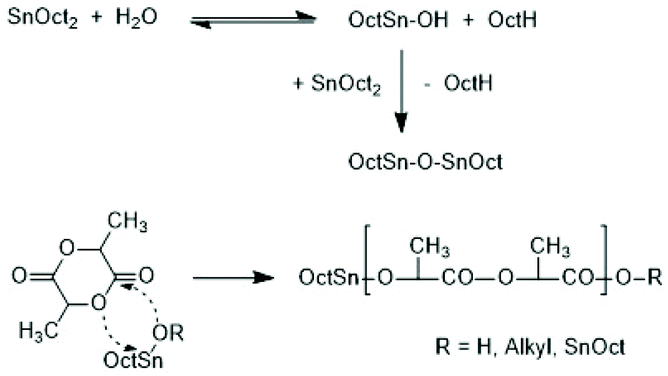
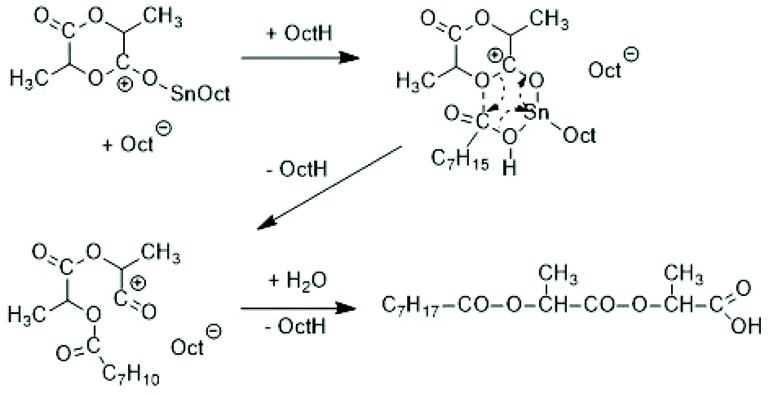
Activation of the CO-chain end in the form of a mixed anhydride via initiation with tin carboxylates is another variant of the ROPPOC approach. However, a mixed anhydride consists of two more or less electrophilic CO-groups and thus, the polymerization may, in principle, proceed in two different directions as illustrated for SnOct2 as catalyst in Scheme 42. When the CO-group of the lactyl unit is more electrophilic, cycles will be the main reaction product (course (a) in Scheme 42), but when the CO group of the initiator is more electrophilic, linear chains having COOH end groups will be the sole or predominant polymerization product (b). In other words, the ratio of the rate constants ka/kb should be high to yield cyclic PLLAs. In the case of SnOct2 the CO group of hexanoic acid is for electronic and steric reasons far less reactive than that of lactyl units. The high sensitivity of LLA or PLLAs to hydrolysis in contrast to alkyl alkanoates is indicative for this scenario. Hence, the authors expected that SnOct2 might be a useful ROPPOC catalyst.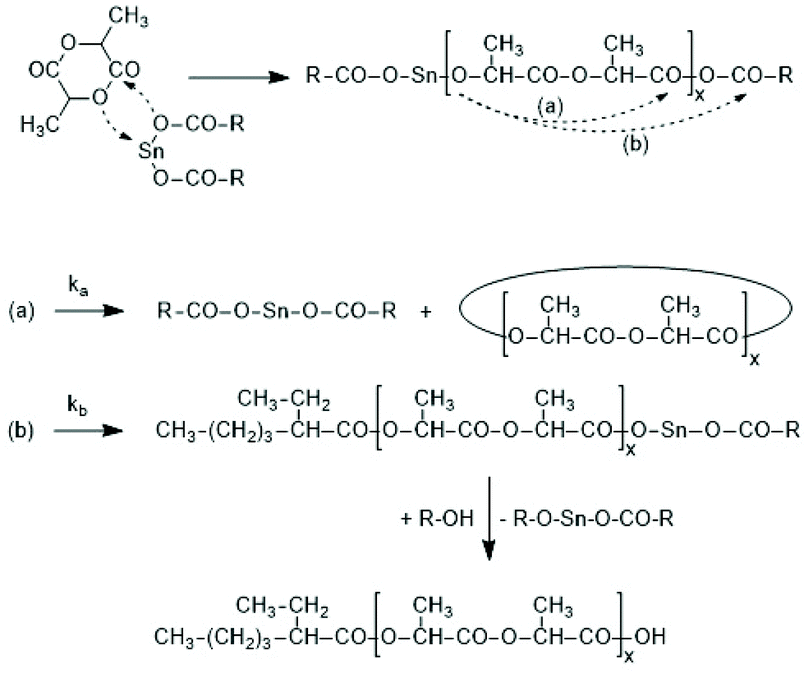 | ||
| Scheme 42 ROPPOC mechanism catalyzed by SnOct2 as proposed by Kricheldorf et al.73 | ||
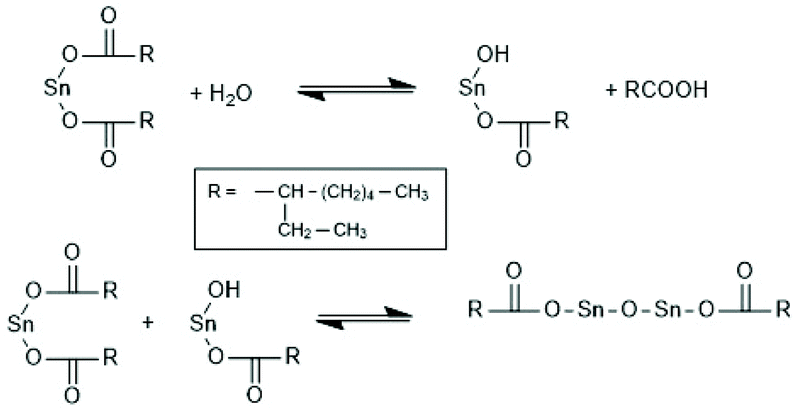
Polymerizations of LLA and racLA catalyzed by neat SnOct2 were performed by numerous research groups61,66,69,70,115,187–205 beginning in 1972 with a report of Sinclair and Gynn.66 High molar mass PLLA (Mw > 500000) were prepared by Pennings et al. and used for the characterization of physical and mechanical properties and of potential applications in medicine.61,187–192 That group performed, for example, a detailed study how the thermal history influences Tm and ΔHm, and extrapolated the equilibrium Tm0 of an ideal crystal.188,191 Much work was also dedicated to fibre spinning.189,190,193 The influence of triphenyl phosphine on the molar mass and thermal properties was studied by Degée et al.204 and Kaur et al.205 Several research groups proposed initiation mechanisms as illustrated in Schemes 43–46. Witzke et al.202 and Shinno et al.203 contributed detailed kinetic studies concerning polymerizations conducted in bulk. Most authors who commented on the polymerization mechanism assumed that traces of water or other R–OH impurities play the role of cocatalysts/initiators (Schemes 43 and 44). Nijenhuis et al. proposed the cationic mechanism outlined in Scheme 45. Kricheldorf et al. assumed an initial formation of a mixed anhydride group (Scheme 46). However, a complete polymerization mechanism was never formulated, and the formation of linear PLLAs was never put into question.
 | ||
| Scheme 43 Initiation mechanism of SnOct2-catalyzed ROP of LLA proposed by Pennings et al.192 (with permission from Wiley). | ||
 | ||
| Scheme 44 Initiation mechanism of SnOct2-catalyzed ROPs of LLA proposed by Dahlmann and Rafler198–200 (with permission from Wiley). | ||
 | ||
| Scheme 45 Cationic polymerization mechanism proposed by Nijenhuis et al., (with permission from ref. 194, Copyright 1992 American Chemical Society). | ||
 | ||
| Scheme 46 Initiation mechanism proposed by Kricheldorf et al. for ROPs of LLA catalyzed by neat SnOct269 (with permission from Elsevier). | ||
Quite recently, polymerizations performed by the authors with water-free SnOct2 revealed that the structure of the polymerization products can only be explained by the ROPPOC mechanism formulated in Scheme 42.73 At extremely low LLA/Cat ratios (50/1 and lower) linear chains terminated by one octanoate and one COOH group were the main reactions products. With increasing LLA/Cat ratio the fraction of cycles increased at the expense of linear chains and at LLA/Cat ratios >400/1 PLLAs almost exclusively consisting of cycles were obtained. The cyclic topology was proven by MALDI mass spectrometry, NMR spectroscopy and by comparison of intrinsic viscosity with the corresponding properties of a commercial linear PLLA. Furthermore, analogous polymerizations were performed with SnAc2, and nearly identical results were found. Mw's in the range of 250000–333000 were achieved.73,77 These findings have the important advantage that a technical production of high molar mass cyclic PLLA is easily feasible, because a change of the catalyst is not necessary. The advantage of low toxicity, high efficiency, absence of discoloration and identical catalyst poison (needed at the end of the polymerization process) all remain. These results also mean, in turn, that all those research groups, that used water-free SnOct2 in fact synthesised cyclic PLLAs without knowing it. Since the water content of the SnOct2 was not determined and the end groups of the resulting PLLAs were not characterized, it is not possible to retrace which group studied cyclic PLLAs or mainly linear species.
In this context, a recent paper of Louisy et al. should be mentioned.206 These authors reported on the production of glass fibre reinforced composites by polymerization of LLA with neat SnOct2 in bulk. That work is a typical example of studies of physical and mechanical properties of PLLA without knowing, whether the sample under investigation has a linear or cyclic topology. This issue is aggravated by the problem that the initially formed cyclic topology may change to a linear one, due to chain scission by thermomechanical processing, by reaction with moisture or additives such as catalyst poison, stabilizers, and pigments. Only a characterization after completion of the processing can reveal whether the final product had cyclic or linear topology.
Finally, it is worth noting that Bu2SnAc2 and Oct2SnAc2 also yield cyclic PLLA when used in bulk at 130–160 °C.77 Yet in contrast to SnOct2 and SnAc2, cyclic PLLAs are best achieved at low LLA/Cat ratios (e.g. 50/1). Furthermore, Mw's in the range of 20000–80000 were obtained, complementary to the high Mw's typical for the Sn(II) carboxylates. Unfortunately, the bisalkyl tin acetates are certainly more toxic than the Sn2+ based catalyst and thus, their preparative usefulness is restricted to academic research.
6.3. Tin chlorides as catalysts
SnCl4-catalyzed polymerizations belong to the oldest examples of tin based ROPs of LLA.11,207 Since SnCl4 is a strong Lewis acid a cationic polymerization mechanism was assumed, but without any experimental evidence. Mass spectrometry was not available and thus, no information about the topology of SnCl4-catalyzed PLLA was published and the polymerization mechanism is still unknown (see below) In the case of SnCl2 a recent study has elucidated that ROPs conducted in bulk >120 °C yield mixtures of cycles and linear chains having unidentified end groups.156 A recent examination of various commercial alky- or aryl tin chlorides gave the following results.208 Dimethyl tin dichloride and dibutyltin dichloride yielded reaction products mainly consisting of cycles, but the ratio of cyclic/linear chains was difficult to reproduce. BuSnCl3 gave mixtures of cyclic and several linear species. However, when Bu3SnCl was used as catalyst at 160 °C in bulk, cyclic PLLA was reproducibly prepared at LLA/Cat ratios in the range of 100/1–600/1 (Scheme 47), whereas linear by-products became detectable at higher LLA/Cat ratios.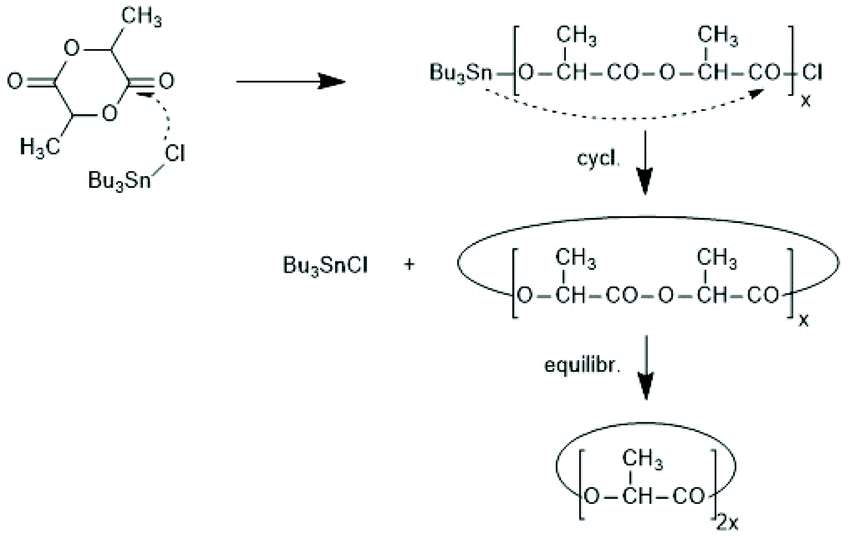 | ||
| Scheme 47 ROPPOC mechanism catalyzed by tributyltin chloride proposed by Kricheldorf et al.208 (with permission from Wiley). | ||
Bu3SnCl is a sluggish catalyst and reaction times around 20 h were required to reach maximum conversion and Mw's up to 80000. Far more reactive proved Ph2SnCl2 which allowed maximum conversion within 1 h at 160 °C. Here again, low LLA/Cat ratios are beneficial, because at LLA/Cat ratios >200/1 more and more linear by-products are formed. The reasons for this unusual correlation (also observed for BuSnOPF) were not explored. Although, such tin chlorides have the advantage to be inexpensive commercial products, further studies of their properties are of minor interest because of their high toxicity.
6.4 Various tin compounds as catalysts
Cyclic tin(II) complexes such as the bisphenoxide complexes 1a–g or the salicylaldiminato complexes 2a–c in Scheme 48 were synthesized by the Phomphrai group and characterized by various methods including X-ray structure analyses.209–211 In most polymerization experiments with LLA or εCL cyclic polyesters were obtained and their formation was ascribed to a zwitterionic mechanism, which is another version of ROPPOC mechanism as explained and discussed before. Those authors assumed that the lone electron pair of the tin atom stabilizes the acylium ion of the electrophilic chain end in analogy to a carbene. However, convincing evidence for such a mechanism is lacking. These authors ignored the more likely alternative, which is a REP mechanism via a Sn–O bond in analogy to the cyclic tin bisphenoxides compiled in Scheme 35 and in analogy to the REP mechanism formulated for cyclic tin sulphides in Scheme 36. A zwitterionic mechanism causes racemization of LLA even below 50 °C like anionic ROPs. Yet, the cyclic tin(II) bisphenoxides outlined in Scheme 35 yielded optically pure PLLAs up to 180 °C clearly indicating that such tin(II) catalysts do not initiate a zwitterionic ROP.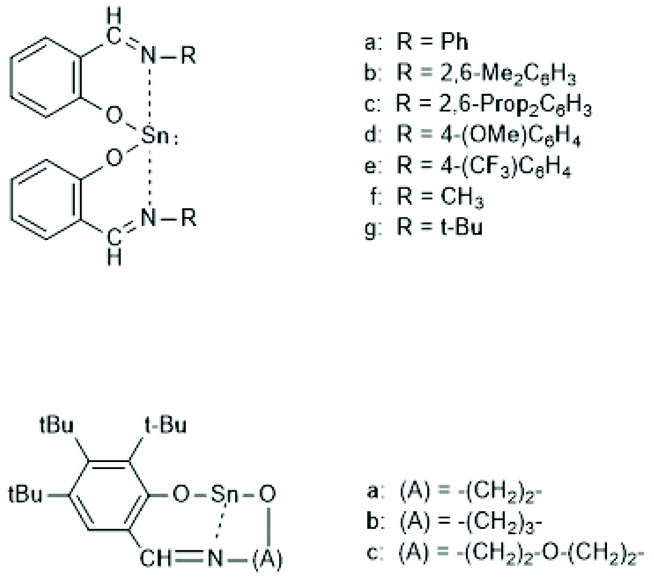 | ||
| Scheme 48 Cyclic tin(II) catalysts used by the Phomphrai group for the preparation of cyclic polylactides210 (with permission from Wiley). | ||
7. Various catalysts and unknown polymerization mechanisms
7.1 SnCl4 and SnCl2 as catalysts
This part summarizes reports on catalysts, of which the polymerization mechanism has not clearly been identified. In this context, the first academic paper, published in 1959 by Kleine and Kleine should be mentioned, because those authors used SnCl4 in addition to catalysts based on other metals.11 The polymerizations were conducted in bulk at 140 °C and yielded low molar mass poly(L-lactides) as estimated from the viscosity measurements (SEC was not available at that time). No information on end groups or hypothetical reaction mechanism was provided. Twelve years later Dittrich and Schulz reported on SnCl4-catalyzed ROPs of L-lactide in solution at temperatures between 35 and 110 °C.207 More than 20 years later Feijen and co-workers tried to detect the formation of lactide or glycolide complexes with SnCl4 by means of IR spectroscopy, but failed.212 Since SnCl4 is a liquid, which is aggressive, hygroscopic and difficult to handle, it has not found much interest in later years. Those researchers, who used it as catalyst for ROPs of lactones classified it as cationic catalyst, because it is a strong Lewis acid.207,212,213 However, several decades later it was demonstrated by Lillie and Schulz60 and Kricheldorf et al.161,162 that Lewis acids or carbenium salts do not catalyse ROPs of lactide (in contrast to ROPs of εCL). Only protic super acids such as fluorosulfonic acid and trifluoromethane sulfonic acid were able to catalyse a true cationic polymerization of lactide. The mechanism was elucidated about twenty years later by Bourissou and co-workers.34 Therefore, SnCl4-catalyzed polymerizations do certainly not proceed via a cationic mechanism.In the case of SnCl2 the majority of papers deal with its application as polycondensation catalyst, an aspect which will be discussed in part 8. Only a handful ROPs of LLA catalyzed with neat SnCl2 were studied by Zavrazhnov et al.214 Those experiments were conducted in bulk at 120–160 °C with a high concentration of catalyst. Only low Mw's < 16000 were obtained whereas SnOct2 gave higher Mw's under identical conditions. In a more detailed study of the authors the same trend was found, but higher LLA/Cat ratios were used. Mw values up to 190000 were achieved.156 Yet, regardless if neat or in combination with alcohols (see part 4) SnCl2 proved to be inferior to SnOct2 under all polymerization conditions with regard to molar masses and homogeneity of the topology of the resulting PLLAs. However, when SnCl2 was compared with other acidic metal chlorides, such as MgCl2, ZnCl2, FeCl3, BiCl3 and HfCl4- in ROPs conducted in bulk at 160 °C, SnCl2 proved to be the best catalyst.
7.2 SnPh4 as catalyst
SnPh4 is another catalyst which was used by a couple of research groups in the early years of polylactide research.196,212,215–217 Kulkarni et al. conducted polymerizations of racLA in bulk only for preparative purposes, because those authors were only interested in properties and medical applications of polylactides.215,216 However, Feijen and co-workers were interested in optimization of the reaction conditions and elucidation of the polymerization mechanism.196,217 Their attempts to identify complex formation of SnPh4 and lactide via IR spectroscopy failed and 1H NMR spectroscopy was not used, so that end groups were not identified. However, numerous polymerizations of racLA were performed in bulk under various conditions and the resulting polylactides were characterized by SEC measurements. Using the universal calibration with polystyrene standards a Mark–Houwink–Sakurada equation was elaborated. High molar masses (Mw up to 200000) were obtained with broad dispersities. A systematic dependence of the molar masses on the LA/Cat ratio was not found and the authors concluded that no living chain ends survived the polymerization process. Due to the poor reproducibility and control of the SnPh4-catalyzed ROPs of lactide, the Feijen group and other researchers abandoned to work with this catalyst and turned their interest to SnOct2-catalyzed polymerizations with or without addition of alcohols. Further studies of SnPh4-catalyzed ROPs of racLA were contributed by Dahlmann and Rafler in 1993.200 Later, the first author studied SnBu4-catalyzed ROPs of ε-caprolactone and found that the activity of this tin compound depended on its exposure to oxygen, apparently due an intermediate formation of a Sn–OBu group.218 Possibly, the catalytic activity of SnPh4 obeys the same reaction pathway.
7.3 Various tin compounds
A third Sn(IV) compound which was used for a few ROP experiments by the Pennings group was the Sn acetylacetonate complex.194 High molar mass PLLA was obtained, but since the reactivity of this catalyst proved to be lower than that of SnOct2, it has not found interest in later years. A group of cyclic tin(IV) complexes containing a Sn–NR2 group as active site was synthesized by the Gibson group (Scheme 49).41 The ROP of racLA yield low molar mass mixtures of several species with dimethyl amide terminated chains as the main polymerization product. Similar complexes containing Sn–alkoxide groups were discussed in part 3.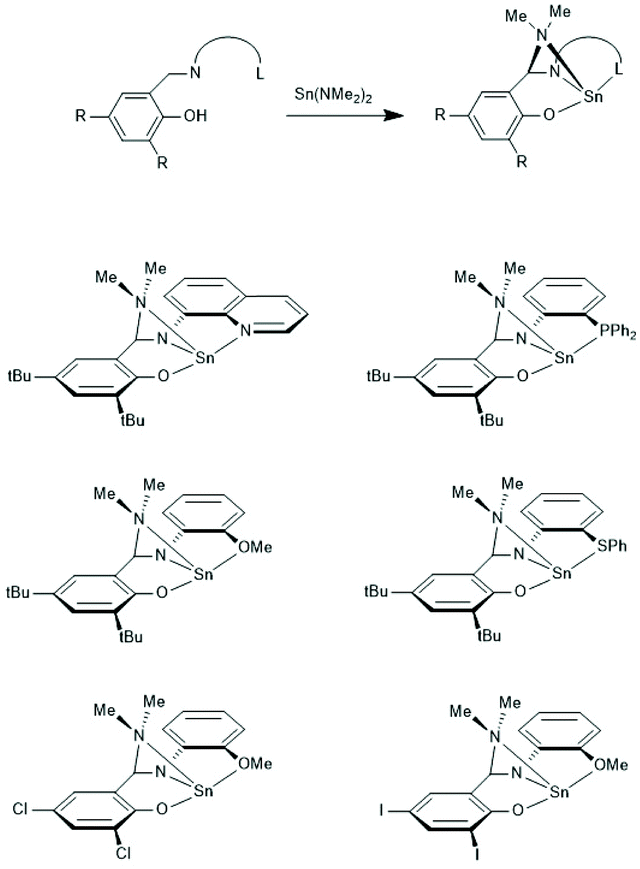 | ||
| Scheme 49 Syntheses of Sn(IV)amine complexes as reported by the Gibson group (reproduced with permission from ref. 41, Copyright 2008 American Chemical Society). | ||
8. Polycondensations
The first synthesis of a polylactide was published in 1833 by the French chemists Gay-Lussac and Pelouze.219 They tried to isolate pure lactic acid by concentration of an aqueous solution at 140 °C in vacuo. Although those authors did not understand the consequences of their experiment (even the formula of lactic acid was unknown at that time) the description of the experiment and the properties of the reaction product clearly indicate that low molar mass poly(D,L-lactic acid) was obtained.A systematic study of the polycondensation of L-lactic acid was launched more than 150 years later by chemists of Mitsui Chemicals Co.220–222 Their method was based on polycondensation in inert solvents with azeotropic removal of water and recycling of the liberated L-lactide. The rate of polycondensation increased with higher boiling points of the solvents and diphenyl ether proved to be particularly useful. Different catalysts were compared, and the best results (Mw = 230000) were achieved with SnO and SnCl2. These polycondensation experiments were extended to various ratios of D- and L-lactic acid. The synthetic method of the Mitsui group has certain disadvantages, such as the need of a relatively expensive solvent, which is difficult to remove completely from the polylactides, and long reaction times compared to the ROP of LLA.
In order to get rid of the solvent and to shorten the reaction time, Kimura and co-workers have studied the polycondensation in bulk.203,223–226 Screening of different metal catalysts showed that SnCl2–2H2O gave the best results and an examination of several acidic cocatalysts revealed that a 1:1 combination with 4-toluene sulfonic acid (TSA) was the most efficient combination.223 However, even with this combination reaction times around 5 h were required for polycondensations at 180 °C to achieve Mw's up to 85000, whereupon partial racemization occurred, whereas discoloration was avoidable. In a later study SnCl2–2H2O was also combined with various metal oxides or alkoxides and Ge(OEt)4 was found to be the best partner of the tin chloride, but the results were not better than those obtained with TSA as cocatalyst.226 A breakthrough was achieved with a three-step procedure based on the SnCl2–2H2O/TSA catalyst.224,225 The first step consisted in a polycondensation in the melt at 180 °C for a short time and yielded a low molar mass PLLA (Mw up to 13000). In a second step crystallization followed for 2 h at 105 °C and, finally, solid state polycondensation was conducted at 150 °C. Mw values up to 600000 were achieved by this technique without significant racemization. However, annealing for 20 h was required. The most likely mechanism of SnCl2-catalyzed esterification of carboxylic groups is outlined in Scheme 50, and a formulation of SnCl2 + TSA-catlyzed reactions in the amorphous phase are presented in Scheme 51. This Scheme might be difficult to understand, but represents the original version published by Moon et al.225 A few years later Shyamroy et al. reported on polycondensations of lactic acid catalyzed by SnPh4 or tetramethyl dichlorodistannoxane.227 No solid state polycondensation (SSP) was applied and all Mw's were below 43000.
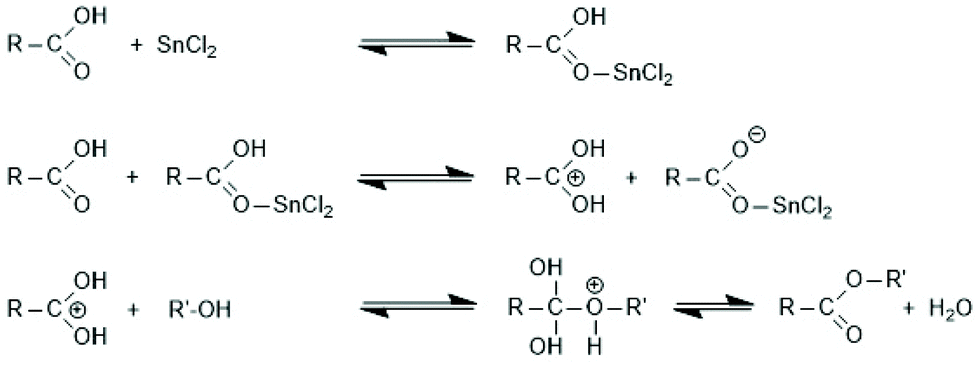 | ||
| Scheme 50 Mechanism of SnCl2-catalyzed esterification of carboxylic acids and alcohols. | ||
 | ||
| Scheme 51 Hypothetical illustration of transesterification and esterification reactions proceeding in the amorphous phase of PLLAs prepared by polycondensation of LLA according to Moon et al.225 (with permission from Elsevier). | ||
When these results are compared with ROPs of LLA, the following aspects need to be considered.
1. The polycondensation process avoids the costs resulting from the preparation of LLA.
2. The “Moon-Kimura process” requires three steps at different temperatures with total reaction times >15 h, whereas the technical ROP of LLA requires only 0.5–1.5 h.
3. The ROP of LLA allows for control of the molar mass via the LLA/In ratio as discussed in part 4.1. Although this control is not precise at high LLA/In ratios, it is easy to perform, but difficult to achieve in the polycondensation process. However, it is worth noting that the role of chain terminators in the polycondensation process has not been studied yet.
4. The ROP of LLA allows for the preparation of various copolymers, for instance by mixing LLA with glycolide, ε-caprolactone or trimethylene carbonate. ROP in the melt is best suited for the preparation of (nearly) random copolymers, whereas ROP in solution with sequential addition of the comonomer can be used to synthesize block copolymers. The polycondensation process cannot compete in this regard.
In addition to homopolylactide various copolyesters of lactide were prepared by SnCl2-catalyzed polycondensations. Matos et al. reported on syntheses of biodegradable and completely biosourced copolyesters by transesterification and condensation of bishydroxyethyl 2,5-difuranate with oligopolylactides (Scheme 52).228
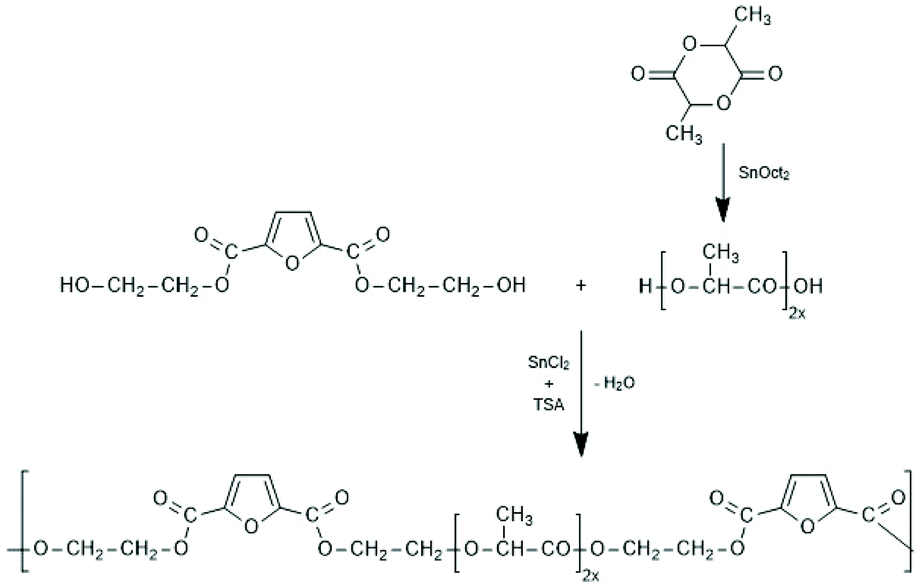 | ||
| Scheme 52 SnCl2-Catalyzed synthesis of copolyesters of LLA; 2,5-tetrahydrofuran dicarboxyclic acid and ethylene glycol (reproduced from ref. 228 with permission from Wiley). | ||
Sb2O3 and SnCl2 + TAS gave similar results, but the Mw's remained below 10000 despite reaction temperatures up to 210 °C. However, these temperatures were perhaps too high and caused degradation. Further polycondensation methods were reported which involve lactide as comonomer. Copolyesters of L-lactide, isosorbide, and isophthalic, 4-tert-butyl isophthalic and terephthalic acid were prepared in a two-step/one-pot process.229,230 At first, isosorbide was used as initiator of a SnOct2 or SnCl2-catalyzed oligomerization of L-lactide, and in the second step the resulting telechelic oligomers were condensed with the corresponding acid chlorides (Scheme 53). Mw's above 100000 were achieved in several experiments, but this approach had the shortcoming of considerable discoloration and the use of chlorobenzene as reaction medium. The purpose of that study was the preparation of biodegradable and largely biosourced materials with high Tg's (>90 °C), because most engineering plastics used in everyday life (PMMA, amorphous polystyrene, polycarbonate etc.) possess these properties. Depending on the composition, Tg's in the range off 90–170 °C were indeed achieved. Mechanical measurements proved that the mechanical properties of these copolylactides were better than those of commercial PMMA, but a convenient, inexpensive synthesis suited for technical production is still lacking.231
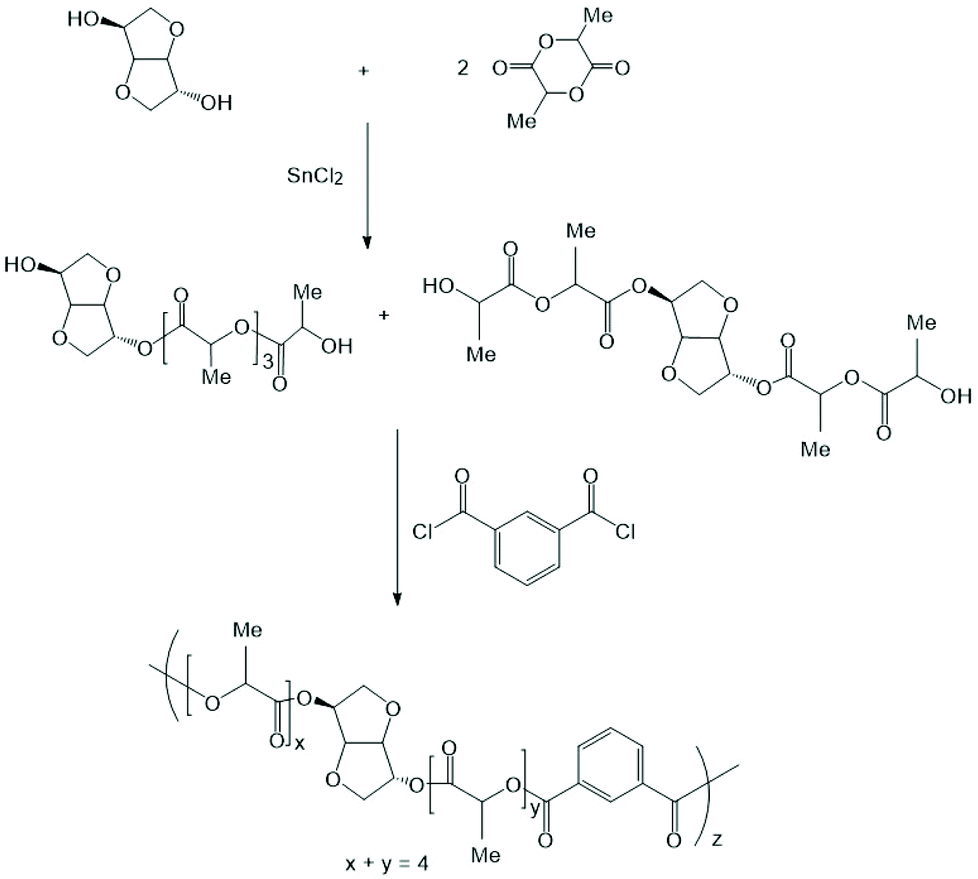 | ||
| Scheme 53 SnCl2-Catalyzed synthesis of copolyesters from LLA, isosorbide and isophthaloylchloride (reproduced fromref. 229 and 230 with permission from Wiley). | ||
The approach outlined in Scheme 53 is flexible and allows for the preparation of unsaturated copolyesters in two ways.232 Either the isosorbide-initiated oligolactides reacted with fumaroyl chloride ((a) Scheme 54) or unsaturated diols were used as initiators ((b) and (c), Scheme 54) The double bond can be used for radical grafting of styrene or other vinyl monomers, and for addition of thiols or bromine. In the latter case, the resulting copolyester ((d) Scheme 54) has flame-retardant properties. Furthermore, addition of the oligolactides onto aromatic diisocyanates is feasible and yield biodegradable polyurethanes having high Tg values above 100 °C (Scheme 55).233 SnOct2 and SnCl2 catalyse both oligomerization of lactide and polyaddition of the resulting oligomers. This approach avoids expensive acid chlorides and liberation of HCl. But the thermostability of the resulting polyurethanes is approximately 100 °C lower than those of the afore mentioned copolyesters, because the urethane bond begins to dissociate above 180 °C. However, application as glues or lacquers (in suitable solvents) are imaginable.
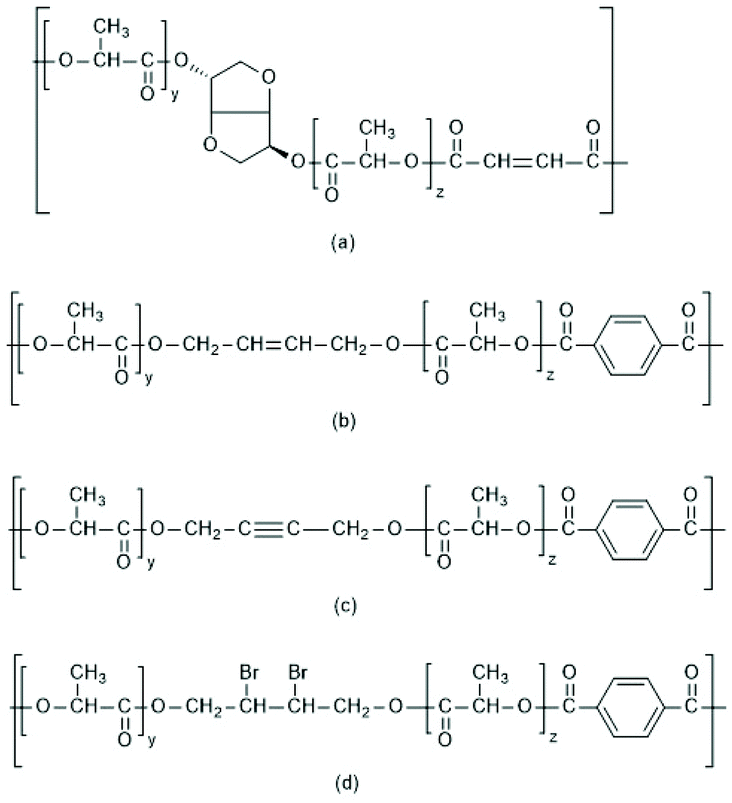 | ||
| Scheme 54 Unsaturated copolyesters of LLA prepared via SnCl2-catalyzed ROP followed by polycondensation of the resulting oligolactides with dicarboxylic acid dichlorides.232 | ||
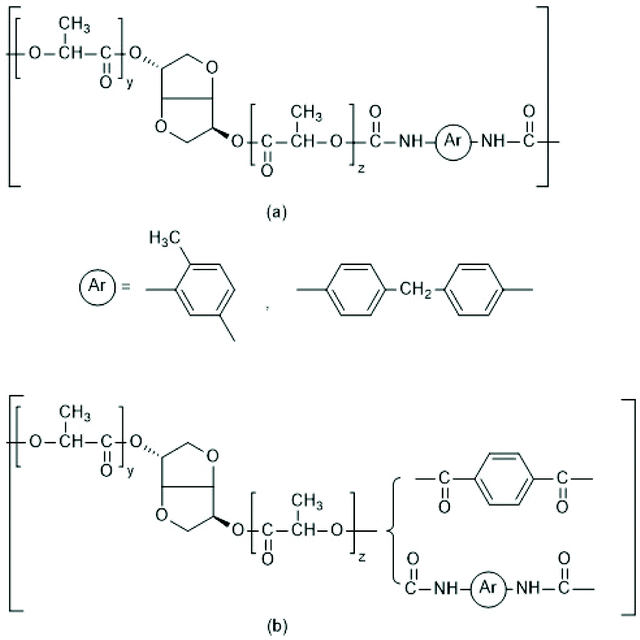 | ||
| Scheme 55 Poly(ester-urethan)es prepared from isosorbide, LLA and aromatic diisocyanates233 (with permission from Wiley). | ||
Furthermore, a paper of Katiyar and Nanavati should be mentioned which describes synthesis of OH-terminated oligolactides followed by SSP, whereupon uncorrected Mw's up to 228000 were achieved.235 The mechanism presented in Scheme 56 was postulated.
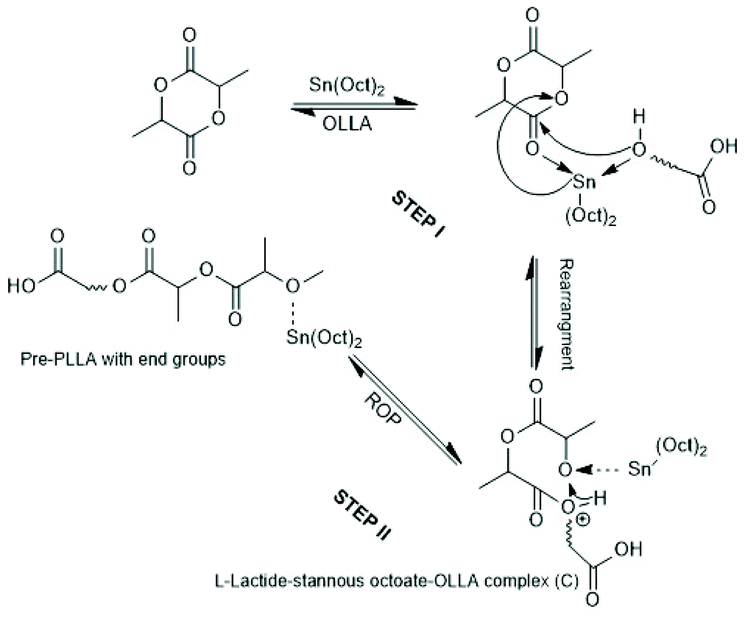 | ||
| Scheme 56 Reaction mechanism postulated by Katiyar and Nanavati shown in ref. 235 (with permission from Wiley). | ||
Finally, polycondensations involving alkyl ester end groups need to be mentioned. The alkyl ester end groups resulting from alcohol initiated ROPs discussed in part 4 are considered to be stable. This classification is correct, when short reaction times are considered (<2 h). But recent experiments with ethyl L-lactate have shown that this liquid, which is stable for years < 30 °C, undergoes slow polycondensation when heated with SnOct2 and even faster with BuSnOPF.234 This means that heating of alkyl ester terminated oligo and polylactides at temperatures of 160 or 180 °C may cause chain growth by polycondensation with the consequence of higher dispersities. However, a detailed study in this direction has not yet been published.
9. Chemical reactions in solid PLLA
One reason for the numerous applications of polylactides in various areas is the broad variation of physical properties such as glass transition (Tg), melting temperature (Tm) and the degree of crystallinity paralleling the melting enthalpy (ΔHm). Above all, three parameters are responsible for the variation of these fundamental parameters.1. Since lactic acid is a chiral monomer, the optical purity, or in other words, the ratio of D and L units is of great influence on all physical properties. PolyLLA with a D-content >15% turns amorphous and a lower content has a strong influence on Tm and other physical properties.53,54,56,236,237
2. Quite analogous to other semi crystalline polymers the thermal history (heating and cooling rates, annealing time and temperature) plays an important role for the physical properties.72,92,93,188,191,237–245
3. Due to the polyester backbone, polylactides can undergo transesterification reactions not only in the melt and in solution, but also in the solid state. At temperatures below 200 °C nature and efficiency of transesterification reactions depend on the presence of catalysts. Hence, catalyst activity and concentration are important for rate and extent of transesterification.
Whereas the first two parameters have intensively been studied over the past thirty years by numerous research groups, research activities concerning the third parameter are relatively new.77,246–248
Most research groups reported Tm values in the range of 174–180 °C for annealed optically pure polyLLAs.53,54,56,72,92,93,188,191,236–245 In agreement with those reports, the authors found for three commercial PLLA samples (Purapol L105, NW 3251D, NW 3001D) Tm's in the range of 173.5–180.3 °C after annealing at 160 °C for 10 h or at 170 °C for 24 h.248 Those commercial polyLLAs were prepared by SnOct2, but as usual for the technical production, a catalyst poison was added to prevent severe degradation upon thermal processing.
The morphology responsible for Tm's around or below 180 °C has now been labelled as low-Tm (LTm) morphology to allow for a clear and rapid distinction from the high Tm (HTm) morphology which shows Tm's above 190 °C. PLLA having a Tm > 190 °C were first reported by Kalb and Pennings, but without any characterization of the morphology and without any explanation.188,191 The authors have found that HTm morphology is formed when catalysts yielding cyclic PLLA either via the REP or via the ROPPOC mechanism are used, and when the time is extended to >20 h, even when the conversion was complete after 1–3 h. Non-cyclic catalysts such as SnOct2 also yield the HTm morphology, but the Tm's are slightly lower under identical conditions.248 A poorly understood phenomenon is the observation that the highest Tm's were achieved by direct isothermal crystallization from the polymerization process. Annealing did never enhance the Tm but yielded higher crystallinities (up to 90%).247,248 Another unexpected and unexplained phenomenon is the finding that alcohol initiated ROPs are less favourable for the formation of the HTm morphology. Particularly unfavourable is the standard catalyst system, namely the combination of alcohol and SnOct2.77,246 When alcohols are used as initiators, only cyclic Sn(II) catalysts, (above all SnBiph) yield the HTm morphology.248 Furthermore, it was found that the most active cyclic catalysts allow for a transformation of LTm PLLA into HTm PLLA by annealing at 170 °C for 8–24 h.77,247
SAXS measurements revealed that the formation of the HTm morphology involves growth of the crystal thickness by a factor 2–3. Furthermore, smoothing of the crystal surface, which reduces the surface free energy, takes place. An additional effect and consequence of these processes is a higher 3-dimensional order of the lamellar crystals in the spherulites. Three transesterification reactions which explain these phenomena are outlined in Schemes 57–59. The consequences for the shapes of the crystallites and their 3-dimensional order are schematically illustrated in Scheme 60.
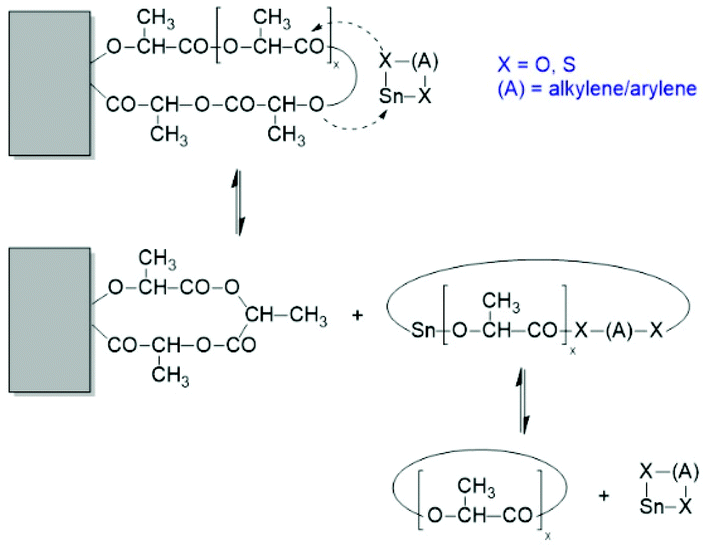 | ||
| Scheme 57 Transesterification resulting in shortening or growing of loops on a crystal surface.247 | ||
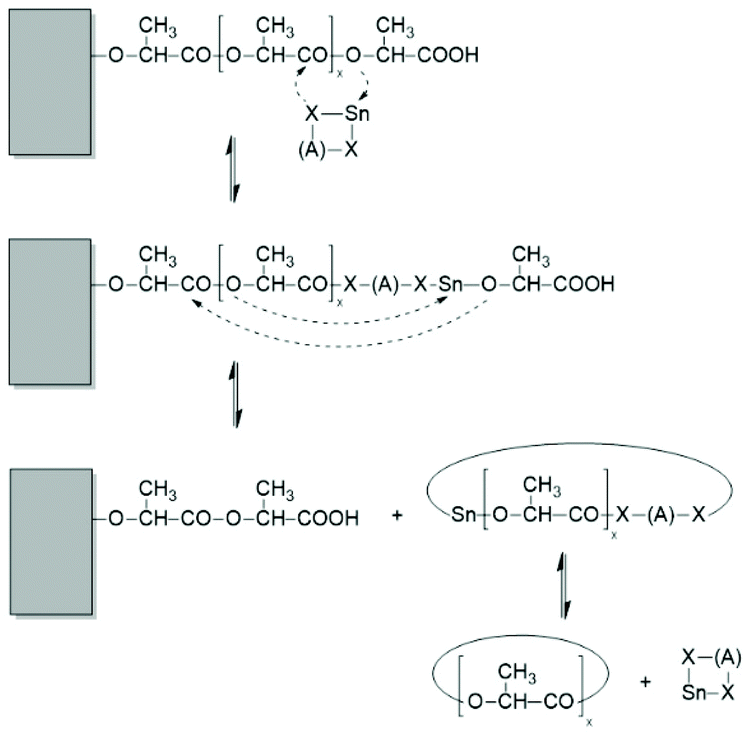 | ||
| Scheme 58 Transesterification resulting in shortening or growing of linear chains sticking out from the surface of a crystallite.247 | ||
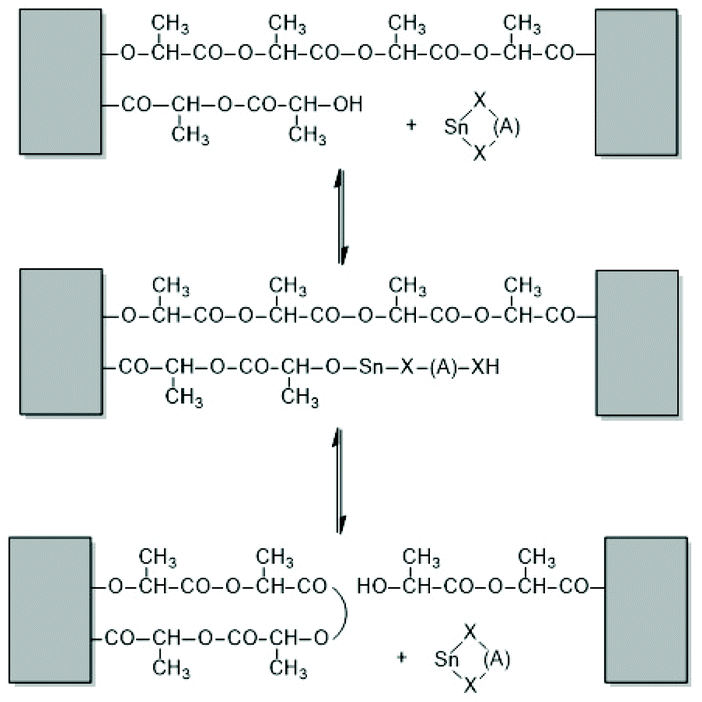 | ||
| Scheme 59 Transesterification resulting in formation of a loop and cleavage of a tie molecule.247 | ||
 | ||
| Scheme 60 Simplified model of “smoothing” of lamellar surfaces combined with thickness growth.247 | ||
Finally, it should be noted that a discussion of chemical reactions in solid PLLA was first published by the Kimura group in connection with the polycondensation of lactic acid.225 As already mentioned in part 8, those authors found a considerable chain growth by annealing in the solid state and postulated the existence of condensation reactions (SSP). Scheme 51 was presented to illustrate the progress of condensation reactions in the amorphous phase. An exact reaction mechanism was not formulated. Since it is well known that carboxylic acids can protonate themselves at higher temperatures, so that esterification is self-catalyzed, the mechanism outlined in Scheme 50 is the most likely reaction pathway. The complexation of COOH groups with SnCl2 enhances their acidity and thus, accelerates the reaction with alcoholic OH groups. This mechanism also explains the catalytic effect of TSA. A detailed study of SSP was later reported by Katiyar and Nanavati, who polymerized LLA by means of SnOct2 and low oligolactides as initiators.235 Those authors observed a dramatic growth of the molar masses upon SSP at 150 °C, whereupon Tm's up to 189.5 °C were reached. The two-step reaction mechanism of Scheme 56 combining ROP with a C–I mechanism and a condensation reaction was formulated. The existence of condensation reactions of course does not exclude, that in the presence of tin catalysts transesterification reactions as outlined in Schemes 57–59 also occur.
Author contributions
HRK – conceptualization, supervision, investigation, writing – original draft, writing – review & editing, SMW – investigation, methodology, visualization, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Bundesanstalt für Materialforschung und-prüfung (BAM) for technical support.Notes and references
- G. Kharas, F. Sanchez-Riera and D. Severson, in Plastics from Microbes, ed. D. P. Mobley, Hanser Gardner Publications, Inc. p, 1994 Search PubMed.
- D. Garlotta, J. Polym. Environ., 2001, 9, 63–84 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- L.-T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS.
- A. Gupta and V. Kumar, Eur. Polym. J., 2007, 43, 4053–4074 CrossRef CAS.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- R. A. Auras, L.-T. Lim, S. E. Selke and H. Tsuji, Poly (lactic acid): synthesis, structures, properties, processing, and applications, John Wiley & Sons, 2011 Search PubMed.
- S. P. Dubey, H. A. Abhyankar, V. Marchante, J. L. Brighton and K. Blackburn, Int. Res. J. Pure Appl. Chem, 2016, 1–20 CAS.
- J. A. Byers, A. B. Biernesser, K. R. Delle Chiaie, A. Kaur and J. A. Kehl, in Advances in Polymer Science, ed. M. L. Di Lorenzo and R. Androsch, Springer, Cham, 2017, vol. 279, pp. 67–118 Search PubMed.
- K. Masutani and Y. Kimura, in Poly(lactic acid) Science and Technology: Processing, Properties, Additives and Applications, ed. A. Jiménez, M. Peltzer and R. Ruseckaite, Royal Society of Chemistry, 2014, pp. 1–36 Search PubMed.
- V. J. Kleine and H. H. Kleine, Makromol. Chem., 1959, 30, 23–38 CrossRef.
- H. R. Kricheldorf and A. Serra, Polym. Bull., 1985, 14, 497–502 CrossRef CAS.
- Tin poisoning, https://e.wikipedia.org/wiki/Tin-poisoning.
- S. Blunden and T. Wallace, Food Chem. Toxicol., 2003, 41, 1651–1662 CrossRef CAS PubMed.
- P. J. Boogaard, M. Boisset, S. Blunden, S. Davies, T. J. Ong and J.-P. Taverne, Food Chem. Toxicol., 2003, 41, 1663–1670 CrossRef CAS PubMed.
- F. Nederberg, E. F. Connor, M. Möller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712–2715 CrossRef CAS PubMed.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- R. C. Pratt, B. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- B. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade and R. M. Waymouth, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- H. Li and L. Gu, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 968–976 CrossRef CAS.
- C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687–1699 RSC.
- Y.-L. Chung, J. V. Olsson, R. J. Li, C. W. Frank, R. M. Waymouth, S. L. Billington and E. S. Sattely, ACS Sustainable Chem. Eng., 2013, 1, 1231–1238 CrossRef CAS.
- L. Mezzasalma, A. P. Dove and O. Coulembier, Eur. Polym. J., 2017, 95, 628–634 CrossRef CAS.
- A. Nachtergael, O. Coulembier, P. Dubois, M. Helvenstein, P. Duez, B. Blankert and L. Mespouille, Biomacromolecules, 2015, 16, 507–514 CrossRef CAS PubMed.
- S. Moins, P. Loyer, J. Odent and O. Coulembier, RSC Adv., 2019, 9, 40013–40016 RSC.
- M. L. Di Lorenzo and R. Androsch, in Advances in Polymer Science, 282, ed. M. L. Di Lorenzo and R. Androsch, Springer International Publishing, Cham, 2018 Search PubMed.
- K. C. Frisch and S. L. Reegen, Ring-opening polymerization, M. Dekker, 1969 Search PubMed.
- T. Ouhadi, J.-P. Bioul, C. Stevens, R. Warin, L. Hocks and P. Teyssié, Inorg. Chim. Acta, 1976, 19, 203–208 CrossRef CAS.
- T. Ouhadi, C. Stevens and P. Teyssié, Makromol. Chem., Suppl., 1975, 1, 191–201 CrossRef.
- P. Dubois, C. Jacobs, R. Jérôme and P. Teyssie, Macromolecules, 1991, 24, 2266–2270 CrossRef CAS.
- H. R. Kricheldorf, J. M. Jonté and M. Berl, Makromol. Chem., Suppl., 1985, 12, 25–38 CrossRef CAS.
- H. R. Kricheldorf, M. Berl and N. Scharnagl, Macromolecules, 1988, 21, 286–293 CrossRef CAS.
- H. R. Kricheldorf, J. M. Jonte and R. Dunsing, Makromol. Chem., 1986, 187, 771–785 CrossRef CAS.
- D. Bourissou, B. Martin-Vaca, A. Dumitrescu, M. Graullier and F. Lacombe, Macromolecules, 2005, 38, 9993–9998 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 7359–7370 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 689–695 CrossRef CAS.
- S. Penczek, A. Duda, A. Kowalski, J. Libiszowski, K. Majerska and T. Biela, Macromol. Symp., 2000, 157, 61–70 CrossRef CAS.
- A. P. Dove, V. C. Gibson, E. L. Marshall, A. J. White and D. J. Williams, Chem. Commun., 2001, 283–284 RSC.
- A. P. Dove, V. C. Gibson, E. L. Marshall, H. S. Rzepa, A. J. White and D. J. Williams, J. Am. Chem. Soc., 2006, 128, 9834–9843 CrossRef CAS PubMed.
- N. Nimitsiriwat, V. C. Gibson, E. L. Marshall, A. J. White, S. H. Dale and M. R. Elsegood, Dalton Trans., 2007, 4464–4471 RSC.
- N. Nimitsiriwat, V. C. Gibson, E. L. Marshall and M. R. Elsegood, Inorg. Chem., 2008, 47, 5417–5424 CrossRef CAS PubMed.
- N. Nimitsiriwat, V. C. Gibson, E. L. Marshall and M. R. Elsegood, Dalton Trans., 2009, 3710–3715 RSC.
- K. B. Aubrecht, M. A. Hillmyer and W. B. Tolman, Macromolecules, 2002, 35, 644–650 CrossRef CAS.
- V. Poirier, T. Roisnel, S. Sinbandhit, M. Bochmann, J. F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2012, 18, 2998–3013 CrossRef CAS PubMed.
- L. Wang, C. E. Kefalidis, S. Sinbandhit, V. Dorcet, J. F. Carpentier, L. Maron and Y. Sarazin, Chem. – Eur. J, 2013, 19, 13463–13478 CrossRef CAS PubMed.
- L. Wang, S.-C. Roşca, V. Poirier, S. Sinbandhit, V. Dorcet, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2014, 43, 4268–4286 RSC.
- L. F. Wang, M. Bochmann, R. D. Cannon, J. F. Carpentier, T. Roisnel and Y. Sarazin, Eur. J. Inorg. Chem., 2013, 5896–5905 CrossRef CAS.
- L. Wang, V. Poirier, F. Ghiotto, M. Bochmann, R. D. Cannon, J.-F. o. Carpentier and Y. Sarazin, Macromolecules, 2014, 47, 2574–2584 CrossRef CAS.
- W. Limwanich, P. Meepowpan, N. Kungwan and W. Punyodom, React. Kinet., Mech. Catal., 2016, 119, 381–392 CrossRef CAS.
- M. Sriyai, T. Chaiwon, R. Molloy, P. Meepowpan and W. Punyodom, RSC Adv., 2020, 10, 43566–43578 RSC.
- T. Mekpothi, P. Meepowpan, M. Sriyai, R. Molloy and W. Punyodom, Polymers, 2021, 13, 3374 CrossRef CAS PubMed.
- M. Sriyai, J. Tasati, R. Molloy, P. Meepowpan, R. Somsunan, P. Worajittiphon, D. Daranarong, J. Meerak and W. Punyodom, ACS Omega, 2021, 6, 28788–28803 CrossRef CAS PubMed.
- E. Fischer, H. J. Sterzel and G. Wegner, Kolloid Z u. Z. Polymere, 1973, 251, 980–990 CrossRef CAS.
- D. M. Bigg, presented at the Annual Technical Conference of the Society of Plastic Engineers (ANTEC), 1996, 2028–2038.
- J. J. Kolstad, J. Appl. Polym. Sci., 1996, 62, 1079–1091 CrossRef CAS.
- L.-I. Palade, H. J. Lehermeier and J. R. Dorgan, Macromolecules, 2001, 34, 1384–1390 CrossRef CAS.
- J. R. Dorgan, J. Janzen, D. M. Knauss, S. B. Hait, B. R. Limoges and M. H. Hutchinson, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3100–3111 CrossRef CAS.
- N. Yui, P. J. Dijkstra and J. Feijen, Makromol. Chem., 1990, 191, 481–488 CrossRef CAS.
- N. Sugai, T. Yamamoto and Y. Tezuka, ACS Macro Lett., 2012, 1, 902–906 CrossRef CAS.
- E. Lillie and R. C. Schulz, Makromol. Chem., 1975, 176, 1901–1906 CrossRef CAS.
- A. Schindler and D. Harper, J. Polym. Sci., Polym. Lett. Ed., 1976, 14, 729–734 CrossRef CAS.
- A. Schindler and K. Gaetano, J. Polym. Sci., Polym. Lett. Ed., 1988, 26, 47–48 CrossRef CAS.
- H. R. Kricheldorf, C. Boettcher and K. U. Tonnes, Polymer, 1992, 33, 2817–2824 CrossRef CAS.
- W. Limwanich, P. Meepowpan, N. Kungwan and W. Punyodom, Chiang Mai J. Sci., 2018, 45, 2069–2078 CAS.
- A. K. Schneider, US Pat., 2703316, 1973 Search PubMed.
- R. G. Sinclair and G. M. Gynn, Preparation and evaluation of glycolic and lactic acid-based polymers for implant devices used in management of maxillofacial trauma, Battelle Columbus Div OH, 1972 Search PubMed.
- A. Schindler, Y. Hibionada and C. Pitt, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 319–326 CrossRef CAS.
- X. C. Zhang, D. A. Macdonald, M. F. A. Goosen and K. B. McAuley, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2965–2970 CrossRef CAS.
- H. R. Kricheldorf, I. Kreiser-Saunders and C. Boettcher, Polymer, 1995, 36, 1253–1259 CrossRef CAS.
- H. R. Kricheldorf, I. Kreiser-Saunders and A. Stricker, Macromolecules, 2000, 33, 702–709 CrossRef CAS.
- M. Jalabert, C. Fraschini and R. E. Prud'Homme, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1944–1955 CrossRef CAS.
- K. Jamshidi, S.-H. Hyon and Y. Ikada, Polymer, 1988, 29, 2229–2234 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, Polym. Chem., 2020, 11, 5249–5260 RSC.
- S. M. Weidner and H. R. Kricheldorf, Macromol. Chem. Phys., 2017, 218, 1600331 CrossRef.
- S. M. Weidner and H. R. Kricheldorf, Macromol. Chem. Phys., 2018, 219, 1800445 CrossRef.
- S. Sosnowski and P. Lewinski, Polym. Chem., 2015, 6, 6292–6296 RSC.
- S. M. Weidner, A. Meyer, J. Falkenhagen and H. R. Kricheldorf, Eur. Polym. J., 2021, 153, 110508 CrossRef CAS.
- D. Pholharn, Y. Srithep and J. Morris, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 213, 012022 Search PubMed.
- H. R. Kricheldorf and S. M. Weidner, J. Polym. Sci., 2021, 59, 439–450 CrossRef CAS.
- H. Kricheldorf, S. Weidner and F. Scheliga, Macromol. Chem. Phys., 2022, 2100464 CrossRef , published online.
- A. Kowalski, J. Libiszowski, T. Biela, M. Cypryk, A. Duda and S. Penczek, Macromolecules, 2005, 38, 8170–8176 CrossRef CAS.
- R. Itzinger, C. Schwarzinger and C. Paulik, J. Polym. Res., 2020, 27, 1–13 CrossRef.
- Y. Yu, G. Storti and M. Morbidelli, Macromolecules, 2009, 42, 8187–8197 CrossRef CAS.
- Y. Yu, G. Storti and M. Morbidelli, Ind. Eng. Chem. Res., 2011, 50, 7927–7940 CrossRef CAS.
- Y. C. Yu, E. J. Fischer, G. Storti and M. Morbidelli, Ind. Eng. Chem. Res., 2014, 53, 7333–7342 CrossRef CAS.
- K. Karidi, T. Mantourlias, A. Seretis, P. Pladis and C. Kiparissides, Eur. Polym. J., 2015, 72, 114–128 CrossRef CAS.
- Y. Ikada, K. Jamshidi, H. Tsuji and S. H. Hyon, Macromolecules, 1987, 20, 904–906 CrossRef CAS.
- H. Tsuji, S. H. Hyon and Y. Ikada, Macromolecules, 1991, 24, 5651–5656 CrossRef CAS.
- H. Tsuji, F. Horii, S. H. Hyon and Y. Ikada, Macromolecules, 1991, 24, 2719–2724 CrossRef CAS.
- H. Tsuji and Y. Ikada, Macromolecules, 1992, 25, 5719–5723 CrossRef CAS.
- H. Tsuji, S. H. Hyon and Y. Ikada, Macromolecules, 1992, 25, 2940–2946 CrossRef CAS.
- H. Tsuji, F. Horii, M. Nakagawa, Y. Ikada, H. Odani and R. Kitamaru, Macromolecules, 1992, 25, 4114–4118 CrossRef CAS.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS PubMed.
- K. Fukushima and Y. Kimura, Polym. Int., 2006, 55, 626–642 CrossRef CAS.
- H. Bai, S. Deng, D. Bai, Q. Zhang and Q. Fu, Macromol. Rapid Commun., 2017, 38, 1700454 CrossRef PubMed.
- D. Gilding and A. Reed, Polymer, 1979, 20, 1459–1464 CrossRef CAS.
- M. Yılmaz, S. Eğri, N. Yıldız, A. Calımlı and E. Pişkin, Polym. J., 2011, 43, 785–791 CrossRef.
- R. Langer and M. Chasin, in Drugs and the Pharmaceutical Sciences, ed. M. Chasin and R. Langer, Marcel Dekker Inc., NY, 1990, vol. 45 Search PubMed.
- D. Bendix, Polym. Degrad. Stab., 1998, 59, 129–135 CrossRef CAS.
- C. D. A. C. Erbetta, R. J. Alves, J. Magalh, R. F. de Souza Freitas and R. G. de Sousa, J. Biomater. Nanobiotechnol., 2012, 3, 208–225 CrossRef CAS.
- W. Hadasha and D. Bezuidenhout, in Industrial Applications of Poly (lactic acid), Springer, 2017, pp. 51–77 Search PubMed.
- J. Li, J. Ding, T. J. Liu, L. Yan and X. Chen, in Industrial Applications of Poly(lactic acid), ed. M. L. DiLoreno and R. Androsch, 2018 Search PubMed.
- P. J. In't Veld, E. M. Velner, P. Van De Witte, J. Hamhuis, P. J. Dijkstra and J. Feijen, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 219–226 CrossRef.
- S. Pensec, M. Leroy, H. Akkouche and N. Spassky, Polym. Bull., 2000, 45, 373–380 CrossRef CAS.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS PubMed.
- T. Fujiwara, M. Miyamoto and Y. Kimura, Macromolecules, 2000, 33, 2782–2785 CrossRef CAS.
- T. Fujiwara, M. Miyamoto, Y. Kimura, T. Iwata and Y. Doi, Macromolecules, 2001, 34, 4043–4050 CrossRef CAS.
- Y. Min, S. Lee, J.-K. Park, K. Y. Cho and S.-J. Sung, Macromol. Res., 2008, 16, 231–237 CrossRef CAS.
- G. Maglio, A. Migliozzi, R. Palumbo, B. Immirzi and M. G. Volpe, Macromol. Rapid Commun., 1999, 20, 236–238 CrossRef CAS.
- H. R. Kricheldorf, S. Rost, C. Wutz and A. Domb, Macromolecules, 2005, 38, 7018–7025 CrossRef CAS.
- K. Zhu, L. Xiangzhou and Y. Shilin, J. Appl. Polym. Sci., 1990, 39, 1–9 CrossRef CAS.
- K. Zhu, L. Xiangzhou and Y. Shilin, J. Polym. Sci., Part C: Polym. Lett., 1986, 24, 331–337 CrossRef CAS.
- K. Zhu, S. Bihai and Y. Shilin, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2151–2159 CrossRef CAS.
- X. Deng, C. Xiong, L. Cheng and R. Xu, J. Polym. Sci., Part C: Polym. Lett., 1990, 28, 411–416 CrossRef CAS.
- H. R. Kricheldorf and J. Meier-Haack, Makromol. Chem., 1993, 194, 715–725 CrossRef CAS.
- A. Bishara, H. R. Kricheldorf and A. J. Domb, Macromol. Symp., 2005, 225, 17–30 CrossRef CAS.
- W. M. Stevels, M. J. Ankone, P. J. Dijkstra and J. Feijen, Macromol. Chem. Phys., 1995, 196, 3687–3694 CrossRef CAS.
- D. W. Lim and T. G. Park, J. Appl. Polym. Sci., 2000, 75, 1615–1623 CrossRef CAS.
- T. Mukose, T. Fujiwara, J. Nakano, I. Taniguchi, M. Miyamoto, Y. Kimura, I. Teraoka and C. Woo Lee, Macromol. Biosci., 2004, 4, 361–367 CrossRef CAS PubMed.
- C. W. Lee and Y. Kimura, Bull. Chem. Soc. Jpn., 1996, 69, 1787–1795 CrossRef CAS.
- D. Kubies, F. Rypáček, J. Kovářová and F. Lednický, Biomaterials, 2000, 21, 529–536 CrossRef CAS PubMed.
- H. Tsuji, M. Ogawa and Y. Arakawa, J. Phys. Chem. B, 2017, 121, 2695–2702 CrossRef CAS PubMed.
- S. Castillejos, J. Cerna, F. Meléndez, M. E. Castro, R. Aguilar, C. Márquez-Beltrán and M. González, Polymers, 2018, 10, 1184–1193 CrossRef PubMed.
- P. Bruin, G. Veenstra, A. Nijenhuis and A. Pennings, Makromol. Chem., 1988, 9, 589–594 CrossRef CAS.
- T. Kissel, Z. Brich, S. Bantle, I. Lancranjan, F. Nimmerfall and P. Vit, J. Controlled Release, 1991, 16, 27–41 CrossRef CAS.
- S. H. Kim, Y. K. Han, Y. H. Kim and S. I. Hong, Makromol. Chem., 1992, 193, 1623–1631 CrossRef CAS.
- S. H. Kim, Y. K. Han, K. D. Ahn, Y. H. Kim and T. Chang, Makromol. Chem., 1993, 194, 3229–3236 CrossRef CAS.
- S. H. Kim and Y. H. Kim, Polymer, 1996, 20, 528–534 CAS.
- S. H. Lee, S. Hyun Kim, Y. K. Han and Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 973–985 CrossRef CAS.
- H. Korhonen, A. Helminen and J. V. Seppälä, Polymer, 2001, 42, 7541–7549 CrossRef CAS.
- N. Kang and J.-C. Leroux, Polymer, 2004, 45, 8967–8980 CrossRef CAS.
- T. Biela, A. Duda, K. Rode and H. Pasch, Polymer, 2003, 44, 1851–1860 CrossRef CAS.
- M. Danko, J. Libiszowski, T. Biela, M. Wolszczak and A. Duda, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4586–4599 CrossRef CAS.
- T. Biela, A. Duda, H. Pasch and K. Rode, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6116–6133 CrossRef CAS.
- W. Radke, K. Rode, A. V. Gorshkov and T. Biela, Polymer, 2005, 46, 5456–5465 CrossRef CAS.
- Q. Hao, F. Li, Q. Li, Y. Li, L. Jia, J. Yang, Q. Fang and A. Cao, Biomacromolecules, 2005, 6, 2236–2247 CrossRef CAS PubMed.
- L. Wang and C. M. Dong, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2226–2236 CrossRef CAS.
- M. P. Shaver and D. J. Cameron, Biomacromolecules, 2010, 11, 3673–3679 CrossRef CAS PubMed.
- M. R. Perry and M. P. Shaver, Can. J. Chem., 2011, 89, 499–505 CrossRef CAS.
- H. Tsuji, N. Matsumura and Y. Arakawa, Polym. J., 2016, 48, 1087–1093 CrossRef CAS.
- H. Tsuji, R. Ozawa and Y. Arakawa, J. Phys. Chem. B, 2017, 121, 9936–9946 CrossRef CAS PubMed.
- C. Choochottiros, E. Park and I.-J. Chin, J. Ind. Eng. Chem., 2012, 18, 993–1000 CrossRef CAS.
- C. Choochottiros, Macromol. Res., 2016, 24, 838–846 CrossRef CAS.
- Y. Li, J. Nothnagel and T. Kissel, Polymer, 1997, 38, 6197–6206 CrossRef CAS.
- A. Breitenbach and T. Kissel, Polymer, 1998, 39, 3261–3271 CrossRef CAS.
- C. Nouvel, P. Dubois, E. Dellacherie and J. L. Six, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2577–2588 CrossRef CAS.
- T. Ouchi, T. Kontani, T. Saito and Y. Ohya, J. Biomater. Sci., Polym. Ed., 2005, 16, 1035–1045 CrossRef CAS PubMed.
- B. Luo, J. Yang, J. Zhao, C. Hsu, J. Li and C. Zhou, J. Appl. Polym. Sci., 2012, 125, E125–E131 CrossRef CAS.
- C. Zhao, D. Wu, N. Huang and H. Zhao, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 589–598 CrossRef CAS.
- D. W. Lim, S. H. Choi and T. G. Park, Macromol. Rapid Commun., 2000, 21, 464–471 CrossRef CAS.
- T. Okuda, K. Ishimoto, H. Ohara and S. Kobayashi, Macromolecules, 2012, 45, 4166–4174 CrossRef CAS.
- L. M. Pitet, S. B. Hait, T. J. Lanyk and D. M. Knauss, Macromolecules, 2007, 40, 2327–2334 CrossRef CAS.
- F. K. Wolf and H. Frey, Macromolecules, 2009, 42, 9443–9456 CrossRef CAS.
- A. M. Fischer, F. K. Wolf and H. Frey, Macromol. Chem. Phys., 2012, 213, 1349–1358 CrossRef CAS.
- J. L. E. Ivirico, M. Salmerón-Sánchez, J. L. G. Ribelles and M. M. Pradas, Colloid Polym. Sci., 2009, 287, 671–681 CrossRef.
- H. R. Kricheldorf and S. M. Weidner, J. Polym. Environ., 2021, 1–13 Search PubMed.
- H. R. Kricheldorf, S. Weidner and F. Scheliga, Polym. Chem., 2017, 8, 1589–1596 RSC.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Macromol. Chem. Phys., 2017, 218, 1700274 CrossRef.
- H. R. Kricheldorf and S. M. Weidner, Eur. Polym. J., 2018, 105, 158–166 CrossRef CAS.
- M. Novák, J. Turek, Y. Milasheuskaya, Z. Růžičková, Š. Podzimek and R. Jambor, Dalton Trans., 2021, 50, 16039–16052 RSC.
- H. R. Kricheldorf and R. Dunsing, Makromol. Chem., 1986, 187, 1611–1625 CrossRef CAS.
- H. R. Kricheldorf and I. Kreiser, Makromol. Chem., 1987, 188, 1861–1873 CrossRef CAS.
- H. R. Kricheldorf and S. R. Lee, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1994, 35, 502–503 CAS.
- H. R. Kricheldorf and S. R. Lee, Macromolecules, 1995, 28, 6718–6725 CrossRef CAS.
- H. R. Kricheldorf and D. Langanke, Macromol. Chem. Phys., 1999, 200, 1174–1182 CrossRef CAS.
- H. R. Kricheldorf, S. R. Lee and N. Schittenhelm, Macromol. Chem. Phys., 1998, 199, 273–282 CAS.
- H. Li, A. Debuigne, R. Jérome and P. Lecomte, Angew. Chem., Int. Ed., 2006, 45, 2264–2267 CrossRef CAS PubMed.
- H. Li, R. Riva, H. R. Kricheldorf, R. Jérôme and P. Lecomte, Chem. – Eur. J., 2008, 14, 358–368 CrossRef CAS PubMed.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4723–4742 CrossRef CAS.
- H. R. Kricheldorf and D. Langanke, Macromol. Biosci., 2001, 1, 364–369 CrossRef CAS.
- H. R. Kricheldorf and A. Stricker, Macromol. Chem. Phys., 1999, 200, 1726–1733 CrossRef CAS.
- D. Pospiech, H. Komber, D. Jehnichen, L. Haussler, K. Eckstein, H. Scheibner, A. Janke, H. R. Kricheldorf and O. Petermann, Biomacromolecules, 2005, 6, 439–446 CrossRef CAS PubMed.
- H. R. Kricheldorf and B. Fechner, Macromolecules, 2001, 34, 3517–3521 CrossRef CAS.
- H. R. Kricheldorf, S. R. Lee and S. Bush, Macromolecules, 1996, 29, 1375–1381 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 749–759 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1915–1925 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, J. Polym. Environ., 2019, 27, 2697–2706 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and A. Meyer, Mater. Adv., 2022, 3, 1007–1016 RSC.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2056–2063 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Eur. Polym. J., 2019, 116, 256–264 CrossRef CAS.
- S. M. Weidner and H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2730–2738 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Macromol. Chem. Phys., 2021, 2100308 CrossRef CAS.
- H. R. Kricheldorf, Macromol. Rapid Commun., 2009, 30, 1371–1381 CrossRef CAS PubMed.
- H. R. Kricheldorf and S. M. Weidner, Macromol. Rapid Commun., 2020, 41, e2000152 CrossRef PubMed.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3767–3775 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, Eur. Polym. J., 2018, 109, 360–366 CrossRef CAS.
- A. Schindler and D. Harper, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 2593–2599 CrossRef CAS.
- B. Kalb and A. J. Pennings, Polymer, 1980, 21, 607–612 CrossRef CAS.
- B. Eling, S. Gogolewski and A. Pennings, Polymer, 1982, 23, 1587–1593 CrossRef CAS.
- S. Gogolewski and A. Pennings, J. Appl. Polym. Sci., 1983, 28, 1045–1061 CrossRef CAS.
- K. Kishore, R. Vasanthakumari and A. J. Pennings, J. Polym. Sci., Part B: Polym. Phys., 1984, 22, 537–542 CAS.
- J. W. Leenslag and A. J. Pennings, Makromol. Chem., 1987, 188, 1809–1814 CrossRef CAS.
- W. Hoogsteen, A. Postema, A. Pennings, G. Ten Brinke and P. Zugenmaier, Macromolecules, 1990, 23, 634–642 CrossRef CAS.
- A. Nijenhuis, D. Grijpma and A. Pennings, Macromolecules, 1992, 25, 6419–6424 CrossRef CAS.
- F. E. Kohn, J. G. Vanommen and J. Feijen, Eur. Polym. J., 1983, 19, 1081–1088 CrossRef CAS.
- J. Van Dijk, J. Smit, F. Kohn and J. Feijen, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 197–208 CrossRef CAS.
- Y. J. Du, P. J. Lemstra, A. J. Nijenhuis, H. A. Van Aert and C. Bastiaansen, Macromolecules, 1995, 28, 2124–2132 CrossRef CAS.
- J. Dahlmann, G. Rafler, K. Fechner and B. Mehlis, Br. Polym. J., 1990, 23, 235–240 CAS.
- G. Rafler and J. Dahlmann, Acta Polym., 1990, 41, 611–617 CrossRef CAS.
- J. Dahlmann and G. Rafler, Acta Polym., 1993, 44, 103–107 CrossRef CAS.
- G. Schwach, J. Coudane, R. Engel and M. Vert, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3431–3440 CrossRef CAS.
- D. R. Witzke, R. Narayan and J. J. Kolstad, Macromolecules, 1997, 30, 7075–7085 CrossRef CAS.
- K. Shinno, M. Miyamoto, Y. Kimura, Y. Hirai and H. Yoshitome, Macromolecules, 1997, 30, 6438–6444 CrossRef CAS.
- P. Degée, P. Dubois, S. Jacobsen, H. G. Fritz and R. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2413–2420 CrossRef.
- P. Kaur, R. Mehta, D. Berek and S. N. Upadhyay, Macromol. Symp., 2012, 315, 106–111 CrossRef CAS.
- E. Louisy, F. Samyn, S. Bourbigot, G. Fontaine and F. Bonnet, Polymers, 2019, 11, 339 CrossRef PubMed.
- V. W. Dittrich and R. C. Schulz, Angew. Makromol. Chem., 1971, 15, 109–126 CrossRef.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 952–960 CrossRef CAS.
- P. Piromjitpong, P. Ratanapanee, W. Thumrongpatanaraks, P. Kongsaeree and K. Phomphrai, Dalton Trans., 2012, 41, 12704–12710 RSC.
- P. Wongmahasirikun, P. Prom-on, P. Sangtrirutnugul, P. Kongsaeree and K. Phomphrai, Dalton Trans., 2015, 44, 12357–12364 RSC.
- S. Praban, S. Yimthachote, J. Kiriratnikom, S. Chotchatchawankul, J. Tantirungrotechai and K. Phomphrai, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2104–2112 CrossRef CAS.
- F. Kohn, J. Van Ommen and J. Feijen, Eur. Polym. J., 1983, 19, 1081–1088 CrossRef CAS.
- R. Lundberg and E. Cox, in Ring-Opening Polymerization, ed. K. C. Frisch and S. L. Reegen, London, 1969, p. 247 Search PubMed.
- S. Zavrazhnov, V. Fomin, L. Beloded and T. Lobaeva, Russ. J. Appl. Chem., 2012, 85, 1264–1268 CrossRef CAS.
- R. Kulkarni, K. Pani, C. Neuman and F. Leonard, Arch. Surg., 1966, 93, 839–843 CrossRef CAS PubMed.
- R. K. Kulkarni, E. Moore, A. Hegyeli and F. Leonard, J. Biomed. Mater. Res., 1971, 5, 169–181 CrossRef CAS PubMed.
- F. Kohn, J. Van Den Berg, G. Van De Ridder and J. Feijen, J. Appl. Polym. Sci., 1984, 29, 4265–4277 CrossRef CAS.
- H. Kricheldorf and I. Kreiser-Saunders, Polymer, 2000, 41, 3957–3963 CrossRef CAS.
- J. Gay-Lussac and J. Pelouze, Ann. Chim. Phys., 1833, 52, 410–424 Search PubMed.
- M. Ajioka, K. Enomoto, K. Suzuki and A. Yamaguchi, Bull. Chem. Soc. Jpn., 1995, 68, 2125–2131 CrossRef CAS.
- K. Koyanagi, T. Fukushima, Y. Sumihiro, T. Sakai and N. Hashimoto, Polym. Prepr. Jpn-Engl. Ed., 1996, 45, E 904–E 904 Search PubMed.
- G. Shin, J. H. Kim, S. H. Kim and Y. H. Kim, Korea Polym. J., 1997, 5, 19–25 CAS.
- S. I. Moon, C. W. Lee, M. Miyamoto and Y. Kimura, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1673–1679 CrossRef CAS.
- S.-I. Moon, I. Taniguchi, M. Miyamoto, Y. Kimura and C.-W. Lee, High Perform. Polym., 2001, 13, S189–S196 CrossRef CAS.
- S.-I. Moon, C.-W. Lee, I. Taniguchi, M. Miyamoto and Y. Kimura, Polymer, 2001, 42, 5059–5062 CrossRef CAS.
- S. I. Moon and Y. Kimura, Polym. Int., 2003, 52, 299–303 CrossRef CAS.
- S. Shyamroy, B. Garnaik and S. Sivaram, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2164–2177 CrossRef CAS.
- M. Matos, A. F. Sousa, A. C. Fonseca, C. S. Freire, J. F. Coelho and A. J. Silvestre, Macromol. Chem. Phys., 2014, 215, 2175–2184 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, Eur. Polym. J., 2013, 49, 2293–2302 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, Macromol. Chem. Phys., 2013, 214, 726–733 CrossRef CAS.
- Z. Zhang, H. R. Kricheldorf and C. Friedrich, Macromol. Rapid Commun., 2015, 36, 262–268 CrossRef CAS PubMed.
- M. Lahcini, S. M. Weidner, J. Oumayama, F. Scheliga and H. R. Kricheldorf, RSC Adv., 2016, 6, 93496–93504 RSC.
- H. R. Kricheldorf, R. Mix and S. M. Weidner, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 867–875 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and J. Falkenhagen, Polym. Chem., 2021, 12, 5003–5016 RSC.
- V. Katiyar and H. Nanavati, Polym. Eng. Sci., 2011, 51, 2078–2084 CrossRef CAS.
- J. J. Kolstad, J. Appl. Polym. Sci., 1996, 62, 1079–1091 CrossRef CAS.
- J. R. Dorgan, J. Janzen, M. P. Clayton, S. B. Hait and D. M. Knauss, J. Rheol., 2005, 49, 607–619 CrossRef CAS.
- H. Tsuji and Y. Ikada, Polymer, 1995, 36, 2709–2716 CrossRef CAS.
- H. Tsuji, K. Ikarashi and N. Fukuda, Polym. Degrad. Stab., 2004, 84, 515–523 CrossRef CAS.
- Y. Ohtani, K. Okumura and A. Kawaguchi, J. Macromol. Sci., Part B: Phys., 2003, 42, 875–888 CrossRef.
- M. Pyda, R. Bopp and B. Wunderlich, J. Chem. Thermodyn., 2004, 36, 731–742 CrossRef CAS.
- J.-R. Sarasua, N. L. Rodríguez, A. L. Arraiza and E. Meaurio, Macromolecules, 2005, 38, 8362–8371 CrossRef CAS.
- M. L. Di Lorenzo, J. Appl. Polym. Sci., 2006, 100, 3145–3151 CrossRef CAS.
- M. Pyda and A. Czerniecka-Kubicka, Synthesis, Structure and Properties of Poly (lactic acid), 2017, pp. 153–193 Search PubMed.
- B. Lotz, Adv. Polym. Sci., 2018, 279, 273–302 CrossRef.
- H. R. Kricheldorf, S. M. Weidner and A. Meyer, Polym. Chem., 2020, 11, 2182–2193 RSC.
- S. M. Weidner, A. Meyer, S. Chatti and H. R. Kricheldorf, RSC Adv., 2021, 11, 2872–2883 RSC.
- H. R. Kricheldorf, S. M. Weidner and A. Meyer, RSC Adv., 2021, 11, 14093–14102 RSC.
| This journal is © The Royal Society of Chemistry 2022 |