
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A self-crosslinking monomer, α-pinene methacrylate: understanding and exploiting hydrogen abstraction†
Olivia R.
Monaghan‡
a,
Stephen T.
Skowron‡
a,
Jonathan C.
Moore‡
a,
María
Pin-Nó
a,
Kristoffer
Kortsen
 a,
Rachel L.
Atkinson
a,
Eduards
Krumins
a,
Joachim C.
Lentz
a,
Fabricio
Machado
ad,
Zeynep
Onat
a,
Adam
Brookfield
b,
David
Collison
b,
Andrei N.
Khlobystov
a,
Davide
De Focatiis
c,
Derek J.
Irvine
c,
Vincenzo
Taresco
*a,
Robert A.
Stockman
*a and
Steven M.
Howdle
*a
a,
Rachel L.
Atkinson
a,
Eduards
Krumins
a,
Joachim C.
Lentz
a,
Fabricio
Machado
ad,
Zeynep
Onat
a,
Adam
Brookfield
b,
David
Collison
b,
Andrei N.
Khlobystov
a,
Davide
De Focatiis
c,
Derek J.
Irvine
c,
Vincenzo
Taresco
*a,
Robert A.
Stockman
*a and
Steven M.
Howdle
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, Nottingham, UK. E-mail: vincenzo.taresco@nottingham.ac.uk; robert.stockman@nottingham.ac.uk; steve.howdle@nottingham.ac.uk
bDepartment of Chemistry, The University of Manchester, Manchester M13 9PL, UK
cFaculty of Engineering, University Park, Nottingham NG7 2RD, Nottingham, UK
dInstitute of Chemistry, University of Brasília, Campus Universitário Darcy Ribeiro, 70910-900, Brasília, DF, Brazil
First published on 1st September 2022
Abstract
Crosslinking is a valuable route to creating new polymeric materials and normally involves introduction of a cross linker or some form of secondary processing. Here we report the discovery and analysis of a self-crosslinking sustainable terpene derived monomer, α-pinene methacrylate (αPMA). This monomer undergoes crosslinking during free radical homopolymerisation and with comonomers e.g. methyl methacrylate (MMA). αPMA does not appear to contain any obvious functionality that would induce crosslinking such as vinyl bonds, but we postulate that it may undergo a fortuitous abstraction of a hydrogen atom from the pendant group. A combined computational (DFT) and experimental approach has been applied to investigate this. Further, we used DFT analysis to predict the behaviour of a related monomer, beta-pinene methacrylate (βPMA). To the best of our knowledge this is the first-time that self-crosslinking has been observed in free radical polymerisation of methacrylates via chain transfer through hydrogen abstraction from a pendant group. We have exploited this crosslinking to generate new, renewable poly high internal phase emulsions (polyHIPEs) that could rival those derived from fossil-based styrene- polyHIPEs and we have done this in a process which does not require any additional cross-linking agent.
Introduction
The drive towards producing polymers from renewable sources has become much more prevalent in recent years. Currently, petroleum based polymers account for around 99% of the global polymer market.1 The anticipated future depletion of fossil fuels and their associated environmental impact requires this figure to be significantly reduced. In addition, there is now a very strong drive to develop and utilise biosourced and renewable materials to provide greener and more consumer acceptable plastics. Terpenes are naturally abundant renewable hydrocarbons sourced from existing waste streams such as the production of citrus juice and as by products from the paper industries.2 The direct polymerisation of terpenes via free radical and cationic polymerisation has been widely studied.3–7However, most terpenes do not readily undergo free radical polymerisation even in the presence of comonomers. Extreme conditions are needed and generally result only in low molecular weight polymers.8–11 Recently, there has been a flurry of papers reporting other routes to polymerisation of terpenes.12–23 We reported the development of new (meth)acrylate based monomers directly derived from terpenes24 with these monomers readily polymerising via free radical polymerisation, in the presence of a thiol chain transfer agent (CTA),25 to produce polymers with a wide range of physical properties. Most significantly poly(α-pinene methacrylate) (Pα-PMA) exhibited a Tg of ∼180 °C, much higher than those observed in the well-known commodity petroleum based polymers such as PS (100 °C),26 PMMA (105 °C)26 and a value that even approaches that of poly(isobornyl methacrylate), IBMA, (199 °C).27
Surprisingly, under free radical conditions in the absence of any chain transfer agent, our polymerisations of αPMA yielded insoluble crosslinked polymers in bulk and in some of the monomer/solvent dilutions we adopted (Fig. 2 and S1†). On the other hand, when the same synthetic conditions were applied for the free radical polymerisation of the model monomers, MMA and IBMA, no crosslinking was observed.
Covalent crosslinking or branching of methacrylate polymers is most commonly induced by incorporation of a multi-functional monomer such as allyl methacrylate,28,29 or by utilising a crosslinking agent such as divinyl benzene or ethylene glycol dimethacrylate.30 Branching or crosslinking can also occur by inter and/or intramolecular chain transfer reactions and these have been extensively studied in the thermal polymerisations of alkyl (meth)acrylates.31,32
Moreover, crosslinking/branching can occur when a propagating radical abstracts a hydrogen from a tertiary carbon atom in the polymer backbone, forming a mid-chain radical (MCR). This can happen either by backbiting, where the propagating radical abstracts the hydrogen from its own backbone (intramolecular chain transfer) or via chain transfer to a neighbouring chain (intermolecular chain transfer).33 It has been suggested that intramolecular hydrogen transfer most likely prefers a 6-membered transition state, and therefore is known as 1,5 backbiting (Fig. 1).34,35
 | ||
| Fig. 1 Formation of mid-chain radicals via intermolecular and intramolecular hydrogen transfer in acrylate monomers. | ||
Intermolecular and intramolecular chain transfer reactions leading to a mid-chain radical are more likely to occur for acrylates than methacrylates because of the less stable secondary propagating radical (SPR).33,36 Through abstraction of a hydrogen from a tertiary carbon, the SPR is transferred to become a more stable tertiary radical (Fig. 1). On the other hand, for methacrylates, these chain transfer reactions are far less likely because of the more stable tertiary propagating radical (TPR).35 TPRs on the methacrylates are unlikely to cause backbiting (intramolecular chain transfer) because they are more sterically hindered by the methyl group on the backbone, preventing formation of the preferred 6-membered transition state.31,33
The formation of these mid chain radicals has been proven in the past for acrylates by monitoring polymerisations with electron paramagnetic resonance (EPR)37–40 to determine the location of the radical centres on the propagating polymer chain. In the polymerisation of alkyl acrylate monomers, MCRs and SPRs will be present simultaneously, which brings in significant band overlap in the EPR spectra and makes deciphering the relevant signals difficult. A number of studies have utilised pulsed laser polymerisation-electron paramagnetic resonance (PLP-EPR) to resolve this issue, as the rate of termination of SPRs is much higher than for MCRs.41
To investigate this, we explore the reactivity of αPMA at different reaction times and monomer dilutions and compare to the well-known behaviours of MMA and IBMA and their bulk polymerisation kinetics. We also investigate use of αPMA as a co-monomer with MMA/and IBMA. To better understand the unusual reactivity αPMA, a range of experimental and computational strategies were adopted to establish a possible mechanistic pathway.
To further broaden the process, we adopted the same computational approach to predict the possible self-crosslinking tendency of βPMA. Finally, we exploit the self-crosslinking of αPMA to yield a renewable based methacrylate PolyHIPE (poly high internal phase emulsion) without any additional crosslinking agent.
Experimental
Materials
α-Pinene methacrylate (α-PMA) was used as provided by Cornelius Specialities. 1-Dodecanethiol (98%) (DDM) was used as provided from Alfa Aesar. 2,2′-Azobis(2-methylpropionitrile) (98%) (AIBN) and Deuterated chloroform (CDCl3) (99%), were used as received from Sigma-Aldrich. Methanol (Reagent Grade), Tetrahydrofuran (Reagent Grade) and Tetrahydrofuran (HPLC Grade) were used as received from Fisher Scientific. Cyclohexanone was used as received from Acros Organics. 2,2′-Azobis (4-methoxy 2,4 dimethylvaleronitrile) (V-70) was used as received from Wako.Size exclusion chromatography
Molecular weight analysis was performed using an Agilent 1260 infinity multidetector GPC/SEC system equipped with a Wyatt Optilab light scattering detector. A PLGEL 5 μm guard column (7.5 mm × 50 mm) and 2× Agilent PLGEL 5 μm Mixed D columns (7.5 mm × 300 mm) were used in succession. HPLC grade THF was used as the eluent at a flow rate of 1 mL min−1 with a dn/dc of 0.106 mL g−1 for poly(α-pinene methacrylate).Nuclear magnetic resonance
1H NMR and 2H NMR spectra were recorded on a Bruker AV400 or AV3400hd spectrometer (400 MHz for 1H NMR and 61.4 MHz for 2H NMR). Chemical shifts (δ) are expressed in ppm relative to residual solvent signals (CHCl3, 1H NMR 7.26), (CDCl3, 2H NMR 7.26) as the internal standard. MestReNova 6.0.2 copyright 2009 (Mestrelab Research S. L.) was used for analysing the spectra.Electron paramagnetic resonance
In situ X band (9.4 GHz) continuous wave EPR spectra were recorded on a Bruker EMX Micro EPR spectrometer equipped with a Bruker ER4122-SHQ resonator. Elevated temperatures of 65 °C were maintained using liquid nitrogen boil-off controlled via a Bruker 4131VT temperature controller.Synthesis of poly(α-pinene methacrylate) and poly(α-pinene methacrylate)-d1 via free radical polymerisation with AIBN
α-PMA (1.00 g, 4.5 mmol) was placed in a 10 mL Schlenk tube with a magnetic stirrer. The AIBN initiator (0.5 wt%, 5.00 mg, 0.0305 mmol) was dissolved in cyclohexanone (1.5 mL) and then added to the Schlenk tube. The Schlenk tube was sealed and the mixture was degassed via 3 free-pump-thaw cycles. The reaction was then purged with argon and stirred at 65 °C for 20–120 minutes or until crosslinking occurred. The reaction was stopped by exposure to air. All non-crosslinked polymers were precipitated out into methanol at 0 °C, these polymers were then characterised using 1H-NMR and GPC.Synthesis of poly(α-pinene methacrylate) polyHIPEs
PolyHIPEs of αPMA were formed by preparation of a water in oil emulsion (HIPE) where the oil phase is the αPMA monomer with AIBN (initiator) and SPAN 80 was used as the emulsifier. αPMA, AIBN (initiator) and the emulsifier SPAN 80 were stirred in a conical flask. The internal phase (water) at varying volume, was added dropwise over 20 minutes. The resultant HIPE was cured in the oven at 65 °C to produce a crosslinked PolyHIPE of PαPMA. The volume of the internal phase was modified from 80 to 90% to understand the effect of the internal phase (water) on the resultant polyHIPEs, and in particular their overall stability. The electrolyte concentration was kept consistent throughout. A series of polyHIPEs were prepared where the volume of the internal phase (water) was increased from 80–90% and the emulsifier (SPAN 80) was varied from 5–20 wt%.Computational methods
DFT calculations were performed using the Q-Chem 5.0 quantum chemistry software package,42 using the dispersion-corrected range-separated hybrid ωB97X-D exchange–correlation functional43 and a 6-311G** basis set. Spin-unrestricted calculations were performed for all radical species. Following geometry optimisation of all species at this level of theory, H-abstraction energies Eabs for the reaction MH + R. → M. + RH were calculated as Eabs = (EM + ERH) − (EMH + ER), where M is the monomer unit (αPMA, βPMA, or IBMA) and R is the radical initiator species (the AIBN-derived isobutyronitrile radical). For calculations of transition-state structures and energy barriers, reactants and products were first optimised at the same level of theory, and then approximate reaction paths and transition states were found with the freezing string method,44 using a quasi-Newton line search method with approximate Hessians updated by the Broyden–Fletcher–Goldfarb–Shanno method (FSM-BFGS).45 The transition state was then optimized with the partitioned rational-function optimization (P-RFO) algorithm using the approximate Hessian and confirmed with a finite difference Davidson method.46Results and discussion
Solvent free polymerisations of MMA, IBMA αPMA and their mixture
PαPMA is a promising material derived from bioresources24,25,47 with excellent thermal and mechanical properties and it can be polymerised successfully with different CTAs leading to homo- and block co-polymerisations in solution.25,47As might be expected, simple solvent free bulk polymerisation led only to solid reaction mixtures for αPMA, MMA and IBMA. All the polymerisations were carried out using AIBN at 65 °C and in all the cases solidification of the reaction mixture was observed. This behaviour was expected considering the high glass transition temperatures of the three growing polymer chains, 105–120 °C for PMMA, 110–200 °C for PIBMA48 and 85–168 °C for PαPMA24,25 (depending upon reaction conditions and thermal analysis approach). The main difference observed was the time at which each sample became completely solid. In fact, MMA needed around 3 h to produce a glassy solid block (Fig. S1A†), IBMA (Fig. S1B†) after circa 30 min while αPMA (Fig. S1C†) solidified in only 10 min.
When we attempted to analyse the molecular weights of the product polymers, we found that whilst PMMA and PIBMA samples were easily soluble in THF (as the GPC eluent), the PαPMA was found to be completely insoluble, and after a few hours started to swell; this same swelling was also observed in a range of solvents (Fig. S1D†). The process was repeated several times with different batches but on each occasion free radical polymerisation led to an apparently crosslinked product. This was surprising given the absence of alkene functionalities in the pendant group, which would ordinarily be the cause of crosslinking in methacrylate monomers.28,29
In order to explore further, a series of mixtures of the monomers were also polymerised with AIBN at 65 °C. The MMA/IBMA mixture solidified during the polymerisation after ca. 40 min, but the polymer was found to be readily soluble in THF. By contrast, all the solid products produced with αPMA were insoluble and showed a tendency to swell in THF (Fig. S1E-F,† PIBAM-PαPMA 70/30% mol mol−1 and PMMA-PαPMA 70/30% mol mol−1 respectively).
αPMA as a PMMA crosslinker
To further probe this surprising crosslinking ability of αPMA, a series of mixtures with variable αPMA/MMA feed ratios (namely, 2.5/97.5; 6/94; 12.5/87.5 and 30/70) were prepared and allowed to polymerise. The kinetics of the reaction for each mixture were followed until solidification and each was then repeated three times (Fig. 2). The data show that the solid/gel point (measured as the time at which the magnetic stirrer failed due to the high viscosity of the medium and stopped stirring) was reached at longer reaction times as the amount of αPMA decreased (Fig. 2). For formulations with αPMA higher than 12.5/87.5 mol mol−1, complete insolubility of the product in THF was observed, while for αPMA/MMA at 2.5/97.5 and 6/94 we observed different degrees of swelling and partial solubility. | ||
| Fig. 2 Solid/gel point observed time and visible solubility in THF for MMA/αPMA reaction mixtures at different %mol/mol of the two monomers. Specifically, 2.5/97.5 and 6/94 mixtures (inside purple triangle) showed partial solubility in THF, while 12.5/87.5 and 30/70 (red rectangle) were completely insoluble in THF. | ||
These exciting preliminary results hint at the possibility that αPMA might be utilised as a green alternative to traditional vinyl cross-linkers in the manufacturing of fully or partially crosslinked polymers. It is well-known that crosslinked PMMA shows improved mechanical and thermal performance when compared to the linear counterpart.49,50
Effect of dilution on αPMA polymerisations
When a higher monomer concentration is employed, the likelihood of intermolecular chain transfer is increased, leading to long chain branching. At lower monomer concentrations intramolecular chain transfer becomes more likely, leading to linear or short chain branching.51,52 Applying this general principle, we investigated the effect of dilution on the αPMA crosslinking in an attempt to establish whether the process passes through an initial branching phase of the growing chains. Cyclohexanone (CHE) was selected as solvent for the free radical process because of its high boiling point and good miscibility with αPMA. When high dilution conditions were adopted, namely αPMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CHE equal to 1:4 and 1:6, no gelling/solidification of the reaction solutions was observed, and high monomer conversion was noted after 24 h (>95%, Fig. S2†). The dispersity of the two polymers, PαPMA 1:4 and PαPMA 1:6 remained largely similar (Đ ∼ 3.00–3.20) while the molecular weight dropped from 52 kDa (PαPMA 1:4) to 40 kDa (PαPMA 1:6). When the αPMA:CHE ratio was reduced to 1:1.5 the gel point was reached within the first 30 min of the reaction. However, for a closer comparison, kinetic information was collected up to 24 h. PαPMA 1:1.5 as prepared showed slow but complete solubility in organic solvents allowing further analysis. From 1H-NMR the conversion of monomer into polymer was calculated to be quantitative (Fig. S2†) with Mn ∼ 177 kDa and Đ ∼ 4.70 detected by GPC (Table 1). Interestingly, the dispersities of all the samples were generally higher than 3.00 and increased as the monomer concentration changed from PαPMA 1:4 to PαPMA 1:1.5. These broad Đ values may be indicative of the onset of branching occurring, in agreement with previous literature examples.53,54 Further reduction of the amount of solvent to 1:1 αPMA:CHE ratio led to a gel/solidification point within the first 20 min of the reaction and the final material was only partially soluble in organic solvents and tended to swell in the same way as the solid produced under bulk conditions. A final dilution condition of 1:0.5 αPMA:CHE volume ratio led to a completely insoluble gel like system.
:solvent dilution ratios
:CHE equal to 1:4 and 1:6, no gelling/solidification of the reaction solutions was observed, and high monomer conversion was noted after 24 h (>95%, Fig. S2†). The dispersity of the two polymers, PαPMA 1:4 and PαPMA 1:6 remained largely similar (Đ ∼ 3.00–3.20) while the molecular weight dropped from 52 kDa (PαPMA 1:4) to 40 kDa (PαPMA 1:6). When the αPMA:CHE ratio was reduced to 1:1.5 the gel point was reached within the first 30 min of the reaction. However, for a closer comparison, kinetic information was collected up to 24 h. PαPMA 1:1.5 as prepared showed slow but complete solubility in organic solvents allowing further analysis. From 1H-NMR the conversion of monomer into polymer was calculated to be quantitative (Fig. S2†) with Mn ∼ 177 kDa and Đ ∼ 4.70 detected by GPC (Table 1). Interestingly, the dispersities of all the samples were generally higher than 3.00 and increased as the monomer concentration changed from PαPMA 1:4 to PαPMA 1:1.5. These broad Đ values may be indicative of the onset of branching occurring, in agreement with previous literature examples.53,54 Further reduction of the amount of solvent to 1:1 αPMA:CHE ratio led to a gel/solidification point within the first 20 min of the reaction and the final material was only partially soluble in organic solvents and tended to swell in the same way as the solid produced under bulk conditions. A final dilution condition of 1:0.5 αPMA:CHE volume ratio led to a completely insoluble gel like system.
:solvent dilution ratios
Proposed hydrogen abstraction and rearrangement
We speculate that the source of crosslinking in the FRP of αPMA is via abstraction of a hydrogen from the pendant group, creating a tertiary radical α to the CH3 group upon the 6-membered ring (Fig. 3 and 4). Indeed, one might expect this C–H bond to have a low bond dissociation enthalpy (BDE) because of the stability of the resultant tertiary radical. There are three other tertiary hydrogens upon the α-pinene methacrylate pendant group (Fig. 3 and 4), but similar C–H are also present in the structure of isobornyl methacrylate (IBMA), a commercial monomer that can also be derived from renewable terpene sources. Crosslinking of isobornyl methacrylate under free radical conditions has never been reported in the literature and, in our hands, when IBMA was polymerised under the same conditions as αPMA crosslinking was not detected.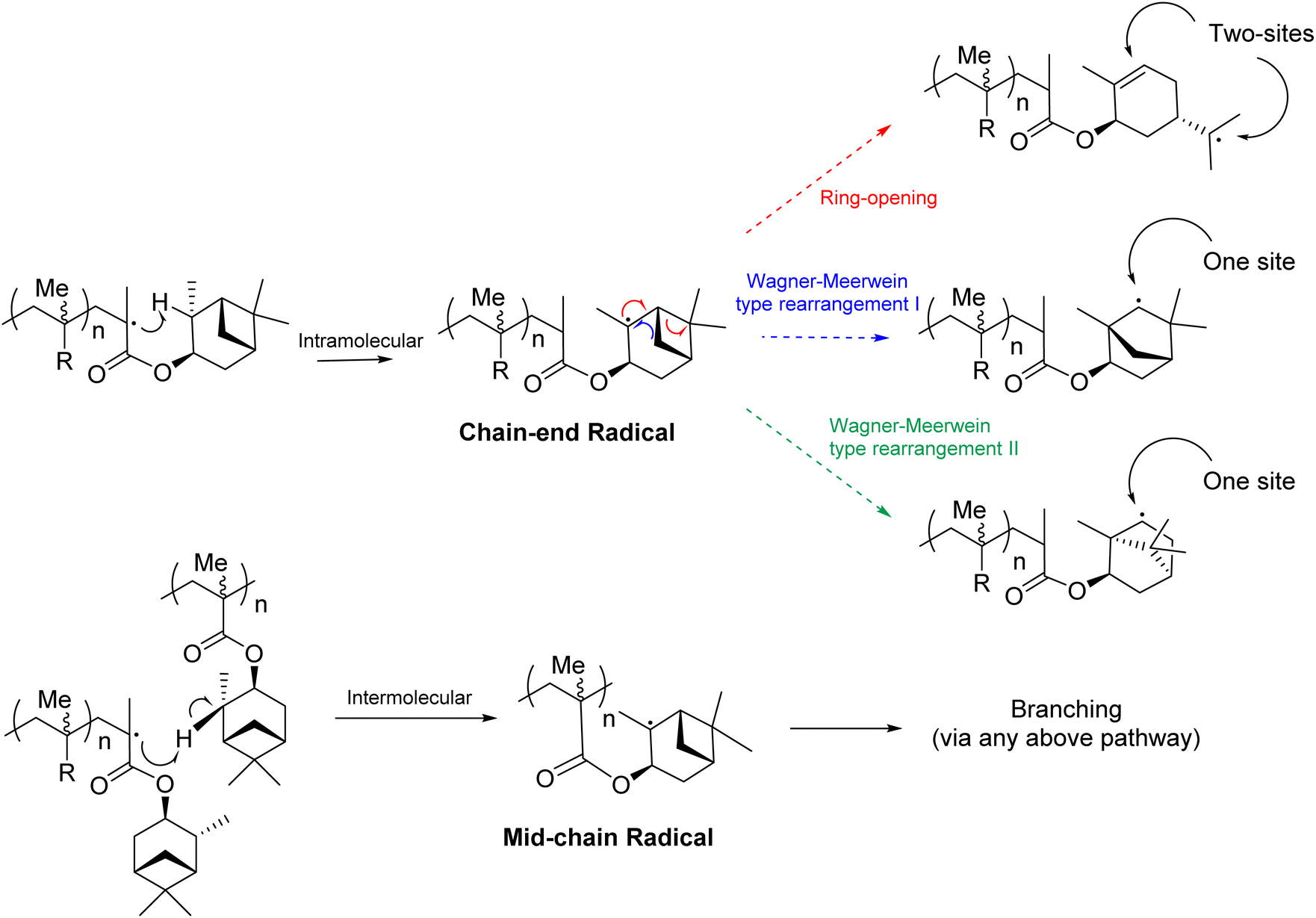 | ||
| Fig. 3 Plausible radical rearrangement pathways following intramolecular or intermolecular hydrogen abstraction from the pendant α-pinene moiety in αPMA. | ||
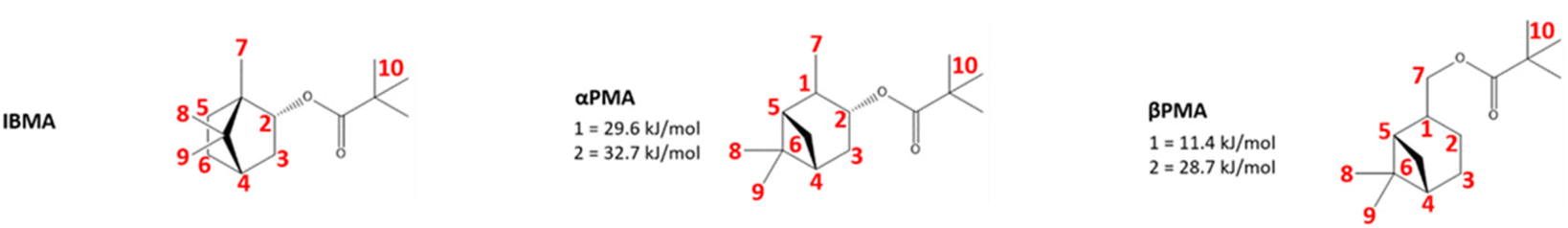 | ||
| Fig. 4 The structures used computationally to represent the IBMA, αPMA, and βPMA moieties. Hydrogen atoms for which abstraction energies were calculated are labelled in red, with relative energies given in kJ mol−1 for the lowest energy abstractions (those substantially below 40 kJ mol−1). All other hydrogen abstraction energies are reported in Table S1,† and the conformers used are attached as supplementary coordinate files. | ||
The abstraction of this hydrogen might be expected to lead to rearrangement of the pendant α-pinene derived moiety on the monomer. For α-pinene itself, such rearrangements are well known in the presence of HCl.55,56 One established pathway is the Wagner–Meerwein rearrangement, which proceeds via formation of a tertiary carbocation when the α-pinene double bond is protonated. There is then a rearrangement into a more favoured secondary carbocation to relieve the strain caused by the 4 membered ring, ultimately forming bornyl chloride (Fig. S3†).
The abstraction of this hydrogen in the pendant α-pinene moiety of the monomer would lead to formation of a tertiary radical and, as with the above published α-pinene rearrangement, this could ultimately lead to an isobornyl structure to relieve ring strain (Fig. 3). Indeed, radical variants of the Wagner–Meerwein rearrangement have been reported on similar terpene derived structures.57 Specifically, rearrangement could occur via migration of the methylene bridge (blue pathway) or the gem-dimethyl bridge (green pathway). Alternatively, a radical ring opening of the cyclobutane would lead to an exocyclic tertiary radical (red pathway). We considered that the initial hydrogen atom abstraction could occur in an intramolecular, or intermolecular fashion. The intramolecular pathway would result in a chain-end radical, from which only the ring opening (red pathway) would be expected to lead to cross-linking, as two sites would be generated for further polymerisation. By contrast, an intermolecular hydrogen abstraction would generate a mid-chain radical, and hence any of the aforementioned rearrangement pathways could result in cross-linking.
DFT analysis to confirm crosslinking speculations
In order to investigate the feasibility of the proposed hydrogen abstraction and radical rearrangement processes, we performed density functional theory (DFT) calculations to explore each step. To identify the most likely site of abstraction, all hydrogen atoms in αPMA and IBMA were considered, with a model system of the monomer units (Fig. 4) with the backbone capped by methyl groups. Relative hydrogen abstraction energies were calculated as detailed in the methods section and are reported in (Fig. 4 and Table S1†).For αPMA, the lowest energy H-abstraction is indeed from the tertiary carbon (1) leading to a radical that is α to the methyl group on the 6-membered ring (with an energy of 30 kJ mol−1). Formation of the radical α to the oxygen atom (2) was very close to, but slightly higher in energy at 33 kJ mol−1, while all other H-abstractions required much more energy in the range 53–81 kJ mol−1. For IBMA the story is very different, and all the hydrogen abstractions are at significantly higher energies. For example, formation of the radical α to the oxygen atom (2) is the lowest in energy at 44 kJ mol−1, and all other abstraction energies lie in the range 47–82 kJ mol−1.
Having confirmed the radical species that is most likely to form, the thermodynamics and kinetics of the three proposed rearrangements can be compared with a transition state analysis to establish the most likely pathway. The energetics of each reaction, calculated as described in the Methods section, can be shown (Fig. 5). The Wagner–Meerwein type rearrangement I has a very large barrier of 208 kJ mol−1 and will be inaccessible at 65 °C, despite having a strong overall thermodynamic driving force of −52 kJ mol−1. The ring-opening and Wagner–Meerwein type II rearrangements have much lower barriers of 69 and 63 kJ mol−1, respectively, both of which would be expected to be accessible at the reaction temperature 65 °C. However, while the ring-opening reaction results in an energy gain of −31 kJ mol−1, the Wagner–Meerwein type rearrangement II has a stronger thermodynamic driving force of −52 kJ mol−1 (comparable to the Wagner–Meerwein type rearrangement I). This results in a barrier for the reverse reaction being 100 kJ mol−1 for the ring-opening reaction, which may be accessible at 65 °C; and 115 kJ mol−1 for the Wagner–Meerwein type rearrangement II, which is hence very likely to be irreversible under our conditions. Based on these calculations, the observed cross-linking of αPMA can be rationalised by either intermolecular hydrogen atom abstraction followed by radical ring opening or via intramolecular hydrogen atom abstraction followed by Wagner–Meerwein type rearrangement II (Fig. 3).
 | ||
| Fig. 5 (a) The energetic pathways for the three potential subsequent radical rearrangements, showing the energies of the reactive hydrogen-abstracted αPMA unit, the transition state, and the product radical in each case. (b) A table of energies comparing the thermodynamics and kinetics of each of the processes in (a). | ||
In summary, from the DFT transition state analysis we would expect the Wagner–Meerwein type rearrangement I to be inaccessible (both forwards and in reverse); the ring-opening reaction to be plausible (forwards reaction accessible, but possibly reversible); and the Wagner–Meerwein type rearrangement II to be the most likely (forwards reaction the most accessible of the three considered, and irreversible). Thus, the overall reaction combining the most likely hydrogen abstraction and subsequent structural rearrangement is calculated to be thermodynamically favourable by −23 kJ mol−1.
We also attempted to look for the radical structures using EPR spectroscopy, but these were not conclusive (see ESI, EPR section and Fig. S4†).
Prediction and experimental βPMA hydrogen abstraction
Hydrogen abstraction energy calculations were repeated for all hydrogen atoms of the beta pinene methacrylate (βPMA) monomer unit (Fig. 4). As with αPMA, there are two particularly low energy hydrogen abstractions available: formation of radicals (1) and (2). These two hydrogen abstractions are the lowest energy of any available to the three molecules IBMA, αPMA, and βPMA, with relative energies of 11 and 29 kJ mol−1, respectively. The fact that an extremely low energy might be required for hydrogen abstraction from the tertiary carbon would suggest that similar cross-linking of βPMA is plausible (in contrast to IBMA which does not crosslink in our hands) although this is also likely to be dependent on the reactivity of the resulting radical species and the energetics of any radical rearrangements. To corroborate these DFT calculations on possible hydrogen abstraction and crosslinking during the free radical polymerisation of βPMA, a series of experiments were performed. As with αPMA, the βPMA polymerisation product showed different solubility behaviour according to the dilution of the reaction. When βPMA was polymerised in the absence of solvent, an insoluble glass-like solid was produced within the first 60 min of reaction. When the dilution was raised to 1:1.5 (monomer:solvent) the final solid product was soluble in THF with Mn ∼ 111000 g mol−1 and Đ ∼ 2.60. These preliminary observations appear to confirm the outputs of the DFT calculation and hint at a possible hydrogen abstraction phenomenon also present in the polymerisation of βPMA.
Renewable polyHipes
PolyHIPEs are porous emulsion-templated polymeric structures, synthesised within a high internal phase emulsion (HIPE).58 They are formed from water in oil (W/O) high internal phase emulsions, where the major internal phase (water) is greater than 74% of the total volume, and is dispersed within the continuous external phase. The aqueous phase will generally contain an electrolyte such as potassium sulfate, the electrolyte reduces the propensity for Ostwald ripening, leading to a more stable emulsion and smaller droplet sizes. An emulsifier is used to stabilise the major phase within the minor phase, most commonly SPAN 80 is used.58–60 PolyHIPEs are formed when monomer is incorporated into the external phase. Typically, the addition of a cross linker is required to lightly crosslink the external phase to increase the stability of the resultant polyHIPE, such crosslinkers include DVB and EGDMA. Here, we show that αPMA offers a promising renewable alternative approach to that of conventional polyHIPEs and could provide an excellent alternative to the commonly used and fossil derived styrene/divinyl benzene system.61It should also be pointed out that a similar monomer, isobornyl acrylate (IBA), has also been utilised as an additive in polyHIPES;62 in order to exploit its high hydrophobicity to ensure formation of a stable HIPE which is a requirement to form a homogeneous polyHIPE. Since, αPMA has a similar pendant group to IBA and similar hydrophobicity, we anticipate that αPMA monomer could thus play a dual role in both cross-linking and hydrophobic stabilisation of the HIPE.
For 5% SPAN-80, all three internal phase volumes produced an unstable emulsion and a PolyHIPE was not formed (Fig. S5†), most likely because large separate water droplets are being formed at this low surfactant concentration. At 20% SPAN-80, a stable emulsion was formed for all three volumes, but well-defined windows and voids were not observed under SEM (Fig. S6†). However, at 10% SPAN-80, a stable HIPE was formed for all three volumes of water (80, 85 and 90%) and well-defined windows and voids were observed under SEM (Fig. 6). The average void size was measured to be 3.7, 4.7 and 3.5 μm respectively for 80, 85 and 90% internal phase (water and electrolyte) and the average window size was found to be 0.45, 0.30 and 0.63 μm respectively.
 | ||
| Fig. 6 SEM images of PαPMA polyHIPE(3.7, 4.7 and 3.5 μm respectively) at 80, 85 and 90 v% water (internal phase) polymerised using 10 wt% SPAN-80 emulsifier. | ||
The optimised conditions to form a stable polyHIPE of PαPMA were found to be 10% SPAN-80 (surfactant), with up to 90% internal phase volume. Analysis showed that the resultant polyHIPEs were crosslinked, as indicated by the insolubility of these PαPMA polyHIPEs in THF indicating that we have exploited the unique crosslinking property of αPMA without the need to add an additional and separate crosslinking agent. Future experimental efforts will be directed towards improving the stability of the polyHIPEs, which could be achieved by altering the crosslinking density. In fact, by exploiting the convenient self-crosslinking of αPMA, the crosslinking density could be tuned by simply altering reaction time.
Further study should look into finer control of the window and void diameters and the introduction of new terpene based renewable (meth)acrylates with added functionality and perhaps even the use of renewably sourced surfactants.63,64
Conclusion
We have demonstrated that through judicious choice of conditions, polymerisation and self cross-linking of αPMA can be achieved. Both experimental and DFT calculations suggest that the origin of the cross-linking can be rationalised by either intermolecular hydrogen atom abstraction followed by radical ring opening or via intramolecular hydrogen atom abstraction followed by Wagner–Meerwein type rearrangement II. DFT analysis was used to corroborate our understanding of the experimental observations for IBMA and αPMA and also to infer the possible hydrogen abstraction and crosslinking of βPMA which was subsequently shown to occur under free radical polymerisation. The crosslinking of αPMA was then exploited to produce a completely renewable polyHIPE with the αPMA taking the roles of monomer, crosslinker and also likely as the hydrophobe stabilising the HIPE in water. We conclude that αPMA is a very promising versatile, renewable and sustainably sourced monomer.Author contributions
Olivia R. Monaghan, Zeynep Onat and Rachel L. Atkinson, Kristoffer Kortsen, Eduards Krumins, Fabricio Machado and Joachim C. Lentz designed, synthesised and characterised all the polymers. María Pin-Nó synthesised αPMA monomer. Olivia R. Monaghan, Adam Brookfield and David Collison designed, performed and analysed the EPR data. Jonathan Moore and Robert A. Stockman rationalised the radical rearrangement pathways and assisted in the design, analysis and characterisation of the polymers. Derek Irvine, Vincenzo Taresco and Steven Howdle conceptualised and supervised the research project. Stephen T. Skowron performed the DFT calculation and analysis. Andrei N Khlobystov and Davide De Focatiis supported on computational and experimental data analysis. Vincenzo Taresco, Robert A Stockman and Steve Howdle rationalised the manuscript layout. All authors contributed to the writing process. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council [grant numbers EP/N019784/1] and the School of Chemistry, University of Nottingham. S. T. S. is grateful to the High Performance Computing (HPC) Facility at the University of Nottingham for providing computational time. We also thank the EPSRC National Research Facility for EPR Spectroscopy [NS/A000055/1] for EPR analysis. FM thanks the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Process no 202176/2020-7) for providing a scholarship to pursue a Post-Doctoral Training at University of Nottingham. Steve Howdle is delighted to have published a paper with David Collison. David was my undergraduate “personal tutor” from 1983-1986 and is a person who I deeply respect who had a profound influence on me becoming an academic.References
- M. R. Thomsett, T. E. Storr, O. R. Monaghan, R. A. Stockman and S. M. Howdle, Progress in the synthesis of sustainable polymers from terpenes and terpenoids, Green Mater., 2016, 4(3), 115–134 CrossRef.
- F. Della Monica and A. W. Kleij, From terpenes to sustainable and functional polymers, Polym. Chem., 2020, 11(32), 5109–5127 RSC.
- M. I. Hulnik, I. V. Vasilenko, A. V. Radchenko, F. Peruch, F. Ganachaud and S. V. Kostjuk, Aqueous cationic homo- and co-polymerizations of β-myrcene and styrene: a green route toward terpene-based rubbery polymers, Polym. Chem., 2018, 9(48), 5690–5700 RSC.
- P. Sahu and A. K. Bhowmick, Redox Emulsion Polymerization of Terpenes: Mapping the Effect of the System, Structure, and Reactivity, Ind. Eng. Chem. Res., 2019, 58(46), 20946–20960 CrossRef CAS.
- N. Bauer, J. Brunke and G. Kali, Controlled Radical Polymerization of Myrcene in Bulk: Mapping the Effect of Conditions on the System, ACS Sustainable Chem. Eng., 2017, 5(11), 10084–10092 CrossRef CAS.
- T. Nishida, K. Satoh, S. Nagano, T. Seki, M. Tamura, Y. Li, K. Tomishige and M. Kamigaito, Biobased Cycloolefin Polymers: Carvone-Derived Cyclic Conjugated Diene with Reactive exo-Methylene Group for Regioselective and Stereospecific Living Cationic Polymerization, ACS Macro Lett., 2020, 9(8), 1178–1183 CrossRef CAS PubMed.
- M. Palenzuela, D. Sánchez-Roa, J. Damián, V. Sessini and M. E. G. Mosquera, Chapter Two - Polymerization of terpenes and terpenoids using metal catalysts, in Advances in Organometallic Chemistry, ed. P. J. Pérez, Academic Press, 2021, vol. 75, pp. 55–93 Search PubMed.
- W. J. Roberts and A. R. Day, A Study of the Polymerization of α- and β-Pinene with Friedel—Crafts Type Catalysts, J. Am. Chem. Soc., 1950, 72(3), 1226–1230 CrossRef CAS.
- J. Lu, M. Kamigaito, M. Sawamoto, T. Higashimura and Y.-X. Deng, Cationic polymerization of β-pinene with the AlCl3/SbCl3 binary catalyst: Comparison with α-pinene polymerization, J. Appl. Polym. Sci., 1996, 61(6), 1011–1016 CrossRef CAS.
- J. Lu, M. Kamigaito, M. Sawamoto, T. Higashimura and Y.-X. Deng, Living Cationic Isomerization Polymerization of β-Pinene. 2. Synthesis of Block and Random Copolymers with Styrene or p-Methylstyrene, Macromolecules, 1997, 30(1), 27–31 CrossRef CAS.
- T. Higashimura, J. Lu, M. Kamigaito, M. Sawamoto and Y.-X. Deng, Cationic polymerization of α-pinene with the binary catalyst AlCl3/SbCl3, Makromol. Chem., 1992, 193(9), 2311–2321 CrossRef CAS.
- M. von Czapiewski, K. Gugau, L. Todorovic and M. A. R. Meier, Synthesis of polyacrylates from limonene by catalytic oxidation and multi-component reaction, Eur. Polym. J., 2016, 83, 359–366 CrossRef CAS.
- N. Bensabeh, A. Moreno, A. Roig, O. R. Monaghan, J. C. Ronda, V. Cádiz, M. Galià, S. M. Howdle, G. Lligadas and V. Percec, Polyacrylates Derived from Biobased Ethyl Lactate Solvent via SET-LRP, Biomacromolecules, 2019, 20(5), 2135–2147 CrossRef CAS PubMed.
- M. A. Droesbeke and F. E. Du Prez, Sustainable Synthesis of Renewable Terpenoid-Based (Meth)acrylates Using the CHEM21 Green Metrics Toolkit, ACS Sustainable Chem. Eng., 2019, 7(13), 11633–11639 CrossRef CAS.
- S. Noppalit, A. Simula, N. Ballard, X. Callies, J. M. Asua and L. Billon, Renewable Terpene Derivative as a Biosourced Elastomeric Building Block in the Design of Functional Acrylic Copolymers, Biomacromolecules, 2019, 20(6), 2241–2251 CrossRef CAS PubMed.
- A. Stamm, A. Biundo, B. Schmidt, J. Brücher, S. Lundmark, P. Olsén, L. Fogelström, E. Malmström, U. T. Bornscheuer and P.-O. Syrén, A Retro-biosynthesis-Based Route to Generate Pinene-Derived Polyesters, ChemBioChem, 2019, 20(13), 1664–1671 CrossRef CAS PubMed.
- A. Stamm, M. Tengdelius, B. Schmidt, J. Engström, P. O. Syrén, L. Fogelström and E. Malmström, Chemo-enzymatic pathways toward pinene-based renewable materials, Green Chem., 2019, 21(10), 2720–2731 RSC.
- T. Castagnet, G. Aguirre, J. M. Asua and L. Billon, Bioinspired Enzymatic Synthesis of Terpenoid-Based (Meth)acrylic Monomers: A Solvent-, Metal-, Amino-, and Halogen-Free Approach, ACS Sustainable Chem. Eng., 2020, 8(19), 7503–7512 CrossRef CAS.
- D. MO'Brien, R. L. Atkinson, R. Cavanagh, A. A. C. Pacheco, R. Larder, K. Kortsen, E. Krumins, A. J. Haddleton, C. Alexander, R. A. Stockman, S. M. Howdle and V. Taresco, A ‘greener’ one-pot synthesis of monoterpene-functionalised lactide oligomers, Eur. Polym. J., 2020, 109516 CrossRef.
- M. A. Droesbeke, A. Simula, J. M. Asua and F. E. Du Prez, Biosourced terpenoids for the development of sustainable acrylic pressure-sensitive adhesives via emulsion polymerisation, Green Chem., 2020, 22(14), 4561–4569 RSC.
- S. Noppalit, A. Simula, L. Billon and J. M. Asua, On the nitroxide mediated polymerization of methacrylates derived from bio-sourced terpenes in miniemulsion, a step towards sustainable products, Polym. Chem., 2020, 11(6), 1151–1160 RSC.
- U. Montanari, D. Cocchi, T. M. Brugo, A. Pollicino, V. Taresco, M. Romero Fernandez, J. C. Moore, D. Sagnelli, F. Paradisi, A. Zucchelli, S. M. Howdle and C. Gualandi, Functionalisable Epoxy-rich Electrospun Fibres Based on Renewable Terpene for Multi-Purpose Applications, Polymers, 2021, 13(11), 1804 CrossRef CAS PubMed.
- M. R. Yarolimek, H. R. Bookbinder, B. M. Coia and J. G. Kennemur, Ring-Opening Metathesis Polymerization of δ-Pinene: Well-Defined Polyolefins from Pine Sap, ACS Macro Lett., 2021, 10(6), 760–766 CrossRef CAS PubMed.
- M. F. Sainz, J. A. Souto, D. Regentova, M. K. G. Johansson, S. T. Timhagen, D. J. Irvine, P. Buijsen, C. E. Koning, R. A. Stockman and S. M. Howdle, A facile and green route to terpene derived acrylate and methacrylate monomers and simple free radical polymerisation to yield new renewable polymers and coatings, Polym. Chem., 2016, 7(16), 2882–2887 RSC.
- R. L. Atkinson, O. R. Monaghan, M. T. Elsmore, P. D. Topham, D. T. W. Toolan, M. J. Derry, V. Taresco, R. A. Stockman, D. S. A. De Focatiis, D. J. Irvine and S. M. Howdle, RAFT polymerisation of renewable terpene (meth)acrylates and the convergent synthesis of methacrylate–acrylate–methacrylate triblock copolymers, Polym. Chem., 2021, 12(21), 3177–3189 RSC.
- Polymerdatabase.com, Accesssed on 2nd October 2018.
- J. M. Yu, P. Dubois and R. Jérôme, Poly[alkyl methacrylate-b-butadiene-b-alkyl methacrylate] Triblock Copolymers: Synthesis, Morphology, and Mechanical Properties at High Temperatures, Macromolecules, 1996, 29(26), 8362–8370 CrossRef CAS.
- A. Matsumoto, K. Kodama, Y. Mori and H. Aota, Emulsion Crosslinking Polymerization of Allyl Methacrylate. Journal of Macromolecular Science, Part A, 1998, 35(9), 1459–1472 Search PubMed.
- A. Matsumoto, T. Shimatani and H. Aota, Emulsion Crosslinking Polymerization of Vinyl Methacrylate as Compared with Allyl Methacrylate, Polym. J., 2000, 32, 871 CrossRef CAS.
- Y. Yoo, G.-H. Hong, S.-R. Hur, Y. S. Kim, S.-G. Lee, H.-J. Kim and J. H. Lee, Preparation of acrylic copolymers and crosslinking agents and properties as a film, J. Appl. Polym. Sci., 2009, 112(3), 1587–1594 CrossRef CAS.
- T. Junkers and C. Barner-Kowollik, The role of mid-chain radicals in acrylate free radical polymerization: Branching and scission, J. Polym. Sci., Part A: Polym. Chem., 2008, 46(23), 7585–7605 CrossRef CAS.
- A. M. van Herk, Historic Account of the Development in the Understanding of the Propagation Kinetics of Acrylate Radical Polymerizations, Macromol. Rapid Commun., 2009, 30(23), 1964–1968 CrossRef CAS PubMed.
- S. Liu, S. Srinivasan, M. C. Grady, M. Soroush and A. M. Rappe, Backbiting and β-scission reactions in free-radical polymerization of methyl acrylate, Int. J. Quantum Chem., 2014, 114(5), 345–360 CrossRef CAS.
- A. N. Nikitin and R. A. Hutchinson, The Effect of Intramolecular Transfer to Polymer on Stationary Free Radical Polymerization of Alkyl Acrylates, Macromolecules, 2005, 38(5), 1581–1590 CrossRef CAS.
- C. Barner-Kowollik, S. Beuermann, M. Buback, P. Castignolles, B. Charleux, M. L. Coote, R. A. Hutchinson, T. Junkers, I. Lacík, G. T. Russell, M. Stach and A. M. van Herk, Critically evaluated rate coefficients in radical polymerization – 7. Secondary-radical propagation rate coefficients for methyl acrylate in the bulk, Polym. Chem., 2014, 5(1), 204–212 RSC.
- J. S. Town, G. R. Jones and D. M. Haddleton, MALDI-LID-ToF/ToF analysis of statistical and diblock polyacrylate copolymers, Polym. Chem., 2018, 9(37), 4631–4641 RSC.
- J. Barth, M. Buback, P. Hesse and T. Sergeeva, Termination and Transfer Kinetics of Butyl Acrylate Radical Polymerization Studied via SP-PLP-EPR, Macromolecules, 2010, 43(9), 4023–4031 CrossRef CAS.
- B. C. Gilbert, J. R. L. Smith, E. C. Milne, A. C. Whitwood and P. Taylor, Kinetic and structural EPR studies of radical polymerization. Monomer, dimer, trimer and mid-chain radicals formed via the initiation of polymerization of acrylic acid and related compounds with electrophilic radicals (˙OH, SO4−˙ and Cl2−˙), J. Chem. Soc., Perkin Trans. 2, 1994,(8), 1759–1769 RSC.
- B. Yamada and K. Sakamoto, ESR study of radical polymerization of styrene 7. Hyperfine structures of ESR spectra of propagating radicals from substituted styrenes, Polymer, 2000, 41(15), 5619–5623 CrossRef CAS.
- M. Buback, P. Hesse, T. Junkers, T. Sergeeva and T. Theis, PLP Labeling in ESR Spectroscopic Analysis of Secondary and Tertiary Acrylate Propagating Radicals, Macromolecules, 2008, 41(2), 288–291 CrossRef CAS.
- J. Barth, M. Buback, C. Barner-Kowollik, T. Junkers and G. T. Russell, Single-pulse pulsed laser polymerization–electron paramagnetic resonance investigations into the termination kinetics of n-butyl acrylate macromonomers, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(22), 4740–4748 CrossRef CAS.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z.-Q. You, Y. Zhu, B. Alam, B. J. Albrecht, A. Aldossary, E. Alguire, J. H. Andersen, V. Athavale, D. Barton, K. Begam, A. Behn, N. Bellonzi, Y. A. Bernard, E. J. Berquist, H. G. A. Burton, A. Carreras, K. Carter-Fenk, R. Chakraborty, A. D. Chien, K. D. Closser, V. Cofer-Shabica, S. Dasgupta, M. D. Wergifosse, J. Deng, M. Diedenhofen, H. Do, S. Ehlert, P.-T. Fang, S. Fatehi, Q. Feng, T. Friedhoff, J. Gayvert, Q. Ge, G. Gidofalvi, M. Goldey, J. Gomes, C. E. González-Espinoza, S. Gulania, A. O. Gunina, M. W. D. Hanson-Heine, P. H. P. Harbach, A. Hauser, M. F. Herbst, M. H. Vera, M. Hodecker, Z. C. Holden, S. Houck, X. Huang, K. Hui, B. C. Huynh, M. Ivanov, Á. Jász, H. Ji, H. Jiang, B. Kaduk, S. Kähler, K. Khistyaev, J. Kim, G. Kis, P. Klunzinger, Z. Koczor-Benda, J. H. Koh, D. Kosenkov, L. Koulias, T. Kowalczyk, C. M. Krauter, K. Kue, A. Kunitsa, T. Kus, I. Ladjánszki, A. Landau, K. V. Lawler, D. Lefrancois, S. Lehtola, R. R. Li, Y.-P. Li, J. Liang, M. Liebenthal, H.-H. Lin, Y.-S. Lin, F. Liu, K.-Y. Liu, M. Loipersberger, A. Luenser, A. Manjanath, P. Manohar, E. Mansoor, S. F. Manzer, S.-P. Mao, A. V. Marenich, T. Markovich, S. Mason, S. A. Maurer, P. F. McLaughlin, M. F. S. J. Menger, J.-M. Mewes, S. A. Mewes, P. Morgante, J. W. Mullinax, K. J. Oosterbaan, G. Paran, A. C. Paul, S. K. Paul, F. Pavošević, Z. Pei, S. Prager, E. I. Proynov, Á. Rák, E. Ramos-Cordoba, B. Rana, A. E. Rask, A. Rettig, R. M. Richard, F. Rob, E. Rossomme, T. Scheele, M. Scheurer, M. Schneider, N. Sergueev, S. M. Sharada, W. Skomorowski, D. W. Small, C. J. Stein, Y.-C. Su, E. J. Sundstrom, Z. Tao, J. Thirman, G. J. Tornai, T. Tsuchimochi, N. M. Tubman, S. P. Veccham, O. Vydrov, J. Wenzel, J. Witte, A. Yamada, K. Yao, S. Yeganeh, S. R. Yost, A. Zech, I. Y. Zhang, X. Zhang, Y. Zhang, D. Zuev, A. Aspuru-Guzik, A. T. Bell, N. A. Besley, K. B. Bravaya, B. R. Brooks, D. Casanova, J.-D. Chai, S. Coriani, C. J. Cramer, G. Cserey, A. E. DePrinceIII, R. A. DiStasioJr., A. Dreuw, B. D. Dunietz, T. R. Furlani, W. A. GoddardIII, S. Hammes-Schiffer, T. Head-Gordon, W. J. Hehre, C.-P. Hsu, T.-C. Jagau, Y. Jung, A. Klamt, J. Kong, D. S. Lambrecht, W. Liang, N. J. Mayhall, C. W. McCurdy, J. B. Neaton, C. Ochsenfeld, J. A. Parkhill, R. Peverati, V. A. Rassolov, Y. Shao, L. V. Slipchenko, T. Stauch, R. P. Steele, J. E. Subotnik, A. J. W. Thom, A. Tkatchenko, D. G. Truhlar, T. V. Voorhis, T. A. Wesolowski, K. B. Whaley, H. L. WoodcockIII, P. M. Zimmerman, S. Faraji, P. M. W. Gill, M. Head-Gordon, J. M. Herbert and A. I. Krylov, Software for the frontiers of quantum chemistry: An overview of developments in the Q-Chem 5 package, J. Chem. Phys., 2021, 155(8), 084801 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10(44), 6615–6620 RSC.
- A. Behn, P. M. Zimmerman, A. T. Bell and M. Head-Gordon, Efficient exploration of reaction paths via a freezing string method, J. Chem. Phys., 2011, 135(22), 224108 CrossRef PubMed.
- S. Mallikarjun Sharada, P. M. Zimmerman, A. T. Bell and M. Head-Gordon, Automated Transition State Searches without Evaluating the Hessian, J. Chem. Theory Comput., 2012, 8(12), 5166–5174 CrossRef CAS PubMed.
- S. M. Sharada, A. T. Bell and M. Head-Gordon, A finite difference Davidson procedure to sidestep full ab initio hessian calculation: Application to characterization of stationary points and transition state searches, J. Chem. Phys., 2014, 140(16), 164115 CrossRef PubMed.
- U. Montanari, V. Taresco, A. Liguori, C. Gualandi and S. M. Howdle, Synthesis of novel carvone (meth)acrylate monomers for the production of hydrophilic polymers with high terpene content, Polym. Int., 2021, 70(5), 499–505 CrossRef CAS.
- O. Kokkorogianni, P. Kontoes-Georgoudakis, M. Athanasopoulou, N. Polizos and M. Pitsikalis, Statistical Copolymers of N-Vinylpyrrolidone and Isobornyl Methacrylate via Free Radical and RAFT Polymerization: Monomer Reactivity Ratios, Thermal Properties, and Kinetics of Thermal Decomposition, Polymers, 2021, 13(5), 778 CrossRef CAS PubMed.
- Y. Suzuki, D. S. Cousins, Y. Shinagawa, R. T. Bell, A. Matsumoto and A. P. Stebner, Phase separation during bulk polymerization of methyl methacrylate, Polym. J., 2019, 51(4), 423–431 CrossRef CAS.
- K. Min, M. Silberstein and N. R. Aluru, Crosslinking PMMA: Molecular dynamics investigation of the shear response, J. Polym. Sci., Part B: Polym. Phys., 2014, 52(6), 444–449 CrossRef CAS.
- E. Sato, T. Emoto, P. B. Zetterlund and B. Yamada, Influence of Mid-Chain Radicals on Acrylate Free Radical Polymerization: Effect of Ester Alkyl Group, Macromol. Chem. Phys., 2004, 205(14), 1829–1839 CrossRef CAS.
- N. M. Ahmad, F. Heatley and P. A. Lovell, Chain Transfer to Polymer in Free-Radical Solution Polymerization of n-Butyl Acrylate Studied by NMR Spectroscopy, Macromolecules, 1998, 31(9), 2822–2827 CrossRef CAS.
- N. T. Nguyen, K. J. Thurecht, S. M. Howdle and D. J. Irvine, Facile one-spot synthesis of highly branched polycaprolactone, Polym. Chem., 2014, 5(8), 2997–3008 RSC.
- F. Y. Hern, A. Hill, A. Owen and S. P. Rannard, Co-initiated hyperbranched-polydendron building blocks for the direct nanoprecipitation of dendron-directed patchy particles with heterogeneous surface functionality, Polym. Chem., 2018, 9(14), 1767–1771 RSC.
- A. Trukhin, F. Kruchkov, L. K. Hansen, R. Kallenborn, A. Kiprianova and V. Nikiforov, Toxaphene chemistry: Separation and characterisation of selected enantiomers of the Polychloropinene mixtures, Chemosphere, 2007, 67(9), 1695–1700 CrossRef CAS PubMed.
- A. Y. Sidorenko, A. Aho, J. Ganbaatar, D. Batsuren, D. B. Utenkova, G. M. Sen'kov, J. Wärnå, D. Y. Murzin and V. E. Agabekov, Catalytic isomerization of α-pinene and 3-carene in the presence of modified layered aluminosilicates, Mol. Catal., 2017, 443, 193–202 CrossRef CAS.
- S. Jana, C. Guin and S. C. Roy, Radical Promoted Wagner−Meerwein-Type Rearrangement of Epoxides in Camphoric Systems Using a Ti(III) Radical Source, J. Org. Chem., 2005, 70(20), 8252–8254 CrossRef CAS PubMed.
- A. Barbetta and N. R. Cameron, Morphology and Surface Area of Emulsion-Derived (PolyHIPE) Solid Foams Prepared with Oil-Phase Soluble Porogenic Solvents: Span 80 as Surfactant, Macromolecules, 2004, 37(9), 3188–3201 CrossRef CAS.
- A. J. Wright, M. J. Main, N. J. Cooper, B. A. Blight and S. J. Holder, Poly High Internal Phase Emulsion for the Immobilization of Chemical Warfare Agents, ACS Appl. Mater. Interfaces, 2017, 9(37), 31335–31339 CrossRef CAS PubMed.
- H. Karimian and M. R. Moghbeli, Conducting poly(styrene-co-divinylbenzene)/polypyrrole PolyHIPE composite foam prepared by chemical oxidative polymerization, J. Appl. Polym. Sci., 2013, 127(1), 804–811 CrossRef CAS.
- M. S. Silverstein, PolyHIPEs: Recent advances in emulsion-templated porous polymers, Prog. Polym. Sci., 2014, 39(1), 199–234 CrossRef CAS.
- R. Owen, C. Sherborne, T. Paterson, N. H. Green, G. C. Reilly and F. Claeyssens, Emulsion templated scaffolds with tunable mechanical properties for bone tissue engineering, J. Mech. Behav. Biomed. Mater., 2016, 54, 159–172 CrossRef CAS PubMed.
- M. d'Almeida Gameiro, A. Goddard, V. Taresco and S. M. Howdle, Enzymatic one-pot synthesis of renewable and biodegradable surfactants in supercritical carbon dioxide (scCO2), Green Chem., 2020, 22(4), 1308–1318 RSC.
- A. R. Goddard, E. A. Apebende, J. C. Lentz, K. Carmichael, V. Taresco, D. J. Irvine and S. M. Howdle, Synthesis of water-soluble surfactants using catalysed condensation polymerisation in green reaction media, Polym. Chem., 2021, 12(20), 2992–3003 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00878e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |