
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Linear ABC amphiphilic triblock copolymers for complexation and protection of dsRNA†
Charlotte E.
Pugsley
 *a,
R. Elwyn
Isaac
b,
Nicholas J.
Warren
a and
Olivier J.
Cayre
*a
*a,
R. Elwyn
Isaac
b,
Nicholas J.
Warren
a and
Olivier J.
Cayre
*a
aSchool of Chemical and Process Engineering, University of Leeds, Leeds, LS2 9JT, UK. E-mail: ce.pugsley@gmail.com; o.j.cayre@leeds.ac.uk
bSchool of Biology, Faculty of Biological Sciences, University of Leeds, Leeds, LS2 9JT, UK
First published on 15th September 2022
Abstract
We herein report the synthesis and characterisation of linear ABC triblock copolymers, investigation of their self-assembly in aqueous solution, and complexation and protection with double stranded-RNA (dsRNA). The amphiphilic triblock copolymers were synthesised via reversible addition–fragmentation chain transfer (RAFT) polymerisation. The precisely controlled polymerisation allowed for modification of the degree of polymerisation of quaternised 2-(dimethylamino)ethyl methacrylate (QDMAEMA, Q), tert-butyl acrylamide (tBAA, B) and N,N-dimethyl acrylamide (DMA, D) blocks, tailoring hydrophobicity. The Q homopolymer was synthesised as a macromolecular chain-transfer agent. The cationic functionality provides the ability for electrostatic interaction of the triblock copolymers with anionic biomolecules, such as dsRNA, for therapeutic or agrochemical delivery applications. The B second block was designed to provide strong anchoring of the assembled structures for enhanced stability. As illustrated by 1H NMR spectroscopy, Q-b-B-b-D linear ABC triblock copolymers were prepared with molecular weights 30, 37 and 44 kDa. The self-assembly of these amphiphilic triblock copolymers in aqueous solution was confirmed by dynamic light scattering (DLS) and transmission electron microscopy (TEM). Furthermore, the potential of these tailored block copolymers as vehicles for dsRNA delivery was demonstrated through complexation and protection of the anionic biomolecule dsRNA against destabilisation at high salt concentration and enzymatic degradation by RNase A, confirmed by ethidium bromide exclusion and agarose gel electrophoresis assays.
Introduction
Amphiphilic block copolymers are suited as vehicles for cargo delivery in therapeutic or agrochemical applications. These polymers consist of, at least, one hydrophilic block and one hydrophobic block, which typically self-assemble in aqueous environments to form aggregated objects such as micelles or vesicles.1,2The advent of controlled ‘living’ polymerisation has brought about a number of reversible deactivation radical polymerisation (RDRP) techniques, which allow the precise design of block copolymers,3–5 where functionalities can be tailored through the choice of monomers to suit the intended application. Additional moieties can be incorporated through further chemical reaction post-polymerisation, for example, conjugation of a folic acid moiety to enhance cellular targeting.6,7 Our group utilises a commonly practiced RDRP technique, reversible addition–fragmentation chain transfer (RAFT) polymerisation, to synthesise precisely controlled block copolymers.
Block copolymers with cationic moieties have been widely explored for their potential for nucleic acid delivery.8–13 Nucleic acids, such as DNA, double stranded-RNA (dsRNA), short interfering-RNA (siRNA) and short hairpin-RNA (shRNA) etc., require delivery in therapeutic applications such as gene therapy14–16 or CRISPR/Cas9 genome editing,17 and agrochemical applications such as in species-specific bioinsecticides.9,18 Nucleic acids are anionic, water-soluble molecules with negatively charged phosphate groups present along the backbone of the nucleotide chain. The hydrophilicity and anionic character of nucleic acids make them suitable as cargo for polymeric micelles. Inclusion of a cationic polymer block as a component of the block copolymer induces electrostatic interaction with the nucleic acid.19 The DNA or RNA can therefore be carried within the hydrophilic corona of the micelle, or within the hydrophilic core of the vesicle. In particular, RNA is a fragile biomolecule that can be easily degraded by RNase nucleases present in the environment and within in vivo subjects.20 Encapsulation of the RNA biomolecule within a polymeric delivery vehicle can provide protection against ex vivo or in vivo degradation by RNase nucleases.21
As far as we are aware, triblock copolymers have not yet been synthesised to complex dsRNA. Instead, research thus far has focused on the complexation, protection and delivery of either short interfering-RNA (siRNA) or plasmid-DNA (pDNA). As demonstrated by Brissault et al., the efficient delivery of pDNA and siRNA requires different polymer architectures depending on the specific cargo. In their work using ABA triblock copolymers (where B refers to the hydrophobic polymer block), the fully water soluble polymer was more effective for pDNA transfection, whereas a self-assembled polymer (due to a longer hydrophobic block) was instead more effective at delivering siRNA for gene knock-down.22
Recent studies on the use of amphiphilic triblock copolymers have investigated the impact of the hydrophilic, neutral block (typically poly(ethylene glycol) (PEG)) on the self-assembled objects themselves or when complexed to pDNA, siRNA or messenger-RNA (mRNA).8,11,23–25 It was found that as hydrophilic block length is increased, the size of the self-assembled objects decreases (with or without nucleic acid).11,24,25 A more compact complex (polyplex or micelleplex) is linked to improved complexation with nucleic acids,26 and the incorporation of a hydrophilic, neutral block has been shown to enhance colloidal stability, and shield the charge of the formed complex.11,24 In some cases, such as work by Cheng et al. and Gary et al., a longer hydrophilic, neutral block protected complexes against destabilisation, and enhanced gene silencing.8,24 However, in work by Sharma et al., the introduction of a PEG block led to weakened binding to pDNA and less efficient transfection.23
There is a clear need for further investigation into the impact of different polymer block lengths on the complexation, stability and protection of nucleic acids when using amphiphilic triblock copolymers, particularly for the delivery of longer dsRNA, as the impact of the nucleic acid subject to delivery has been shown to be significant. It is also worth exploring alternative polymer blocks, such as N,N-dimethyl acrylamide.
In this work, we describe the synthesis and characterisation of a series of novel linear ABC amphiphilic triblock copolymers via RAFT polymerisation. A cationic and hydrophilic quaternised poly(2-(dimethylamino) ethyl methacrylate) (PQDMAEMA, Q) block was first synthesised as a macromolecular chain-transfer agent (macro-CTA). The macro-CTA was successively chain-extended with a hydrophobic poly(tert-butyl acrylamide) (PtBAA, B) block and a hydrophilic poly(N,N-dimethyl acrylamide) (PDMA, D) block thereafter. The degree of polymerisation (DP), and thus the length, of the B and D blocks were varied in order to modify the extent of hydrophobicity of each amphiphilic triblock copolymer in the series, as well as to explore the influence of each polymer block. The amphiphilic triblock copolymers were characterised by 1H NMR spectroscopy (400 MHz) in order to ascertain degree of polymerisation by characteristic peak analysis. Subsequently, dynamic light scattering (DLS) and transmission electron microscopy (TEM) were employed in aqueous environments to determine the self-assembling properties of the triblock copolymers and to perform size analysis.
Further to the synthesis and characterisation of the ABC triblock copolymer series, we investigated the complexation of dsRNA by the polymeric delivery vehicles, in salt-free and high salt concentration environments, and the resulting protection against degradation by a RNA-specific nuclease (RNase A). Therefore, we illustrate the promising potential of these linear ABC amphiphilic triblock copolymers as delivery vehicles for cargo such as nucleic acids for therapeutic or agrochemical applications. The triblock copolymers showed enhanced stability in high salt concentration environments, with respect to previous work on diblock copolymer/dsRNA polyplexes,27 and thus may prove more suitable for commercial formulations.
Materials and methods
Materials
[2-(Methacryloyloxy)ethyl] trimethylammonium chloride solution (QDMAEMA, 75 wt% in Milli-Q water), tert-butyl acrylamide (tBAA, 97%) N,N-dimethylacrylamide (DMA, 99%), sodium chloride (NaCl, 99.5%), D2O (99.9%), methanol-d4 (MeOD, 99.8%) and hydrochloric acid (HCl, 12 M) were purchased from Sigma Aldrich. 4-((((2-Carboxyethyl)thio)carbonothioyl)thio)-4-cyano-pentanoic acid (CCCP, 95%) was purchased from Boron Molecular. 4,4′-Azobis(4-cyanovaleric acid) (ACVA, 97%) was purchased from Acros Organics. V-ATPase 222 bp dsRNA was synthesised by Genolution AgroRNA (4.68 μg μL−1), sequence specific to the pest insect, Drosophila suzukii. Ethidium bromide (EB, 10 mg mL−1) and regenerated cellulose dialysis membrane (molecular weight cut off (MWCO) < 3500 g mol−1) were purchased from Fisher Scientific. 100 bp DNA ladder (500 μg ml−1) and RNase A (20 mg mL−1) were purchased from New England Biolabs. Blue/orange loading dye (6×) was purchased from Promega. Ultrapure Milli-Q water (resistivity of minimum 18.2 MΩ cm) was used for solution preparation and dialysis, and nuclease-free water was used for biological assays.Synthesis of quaternised poly(2-dimethylamino ethyl methacrylate) macro-chain transfer agent
The homopolymer Q macro-CTA was synthesised by RAFT polymerisation, as shown in the scheme in Fig. 1. | ||
| Fig. 1 Reaction scheme for the aqueous RAFT polymerisation of quaternised poly(2-dimethylamino ethyl methacrylate). | ||
DQMAEMA (6.6 g, 75 wt% in Milli-Q water, 32 mmol), CCCP (78 mg, 0.25 mmol) and ACVA (7.1 mg, 0.025 mmol) were dissolved in Milli-Q water, at a ratio of [DQMAEMA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CCCP]:[ACVA] = 126:1:0.1 and 50 wt% in solution. The solution was degassed with N2 and then stirred at 70 °C for 1.5 h. The reaction was quenched by exposure to air. Product was purified by dialysis against Milli-Q water (MWCO < 3500 g mol−1) and subsequent lyophilisation.
:[CCCP]:[ACVA] = 126:1:0.1 and 50 wt% in solution. The solution was degassed with N2 and then stirred at 70 °C for 1.5 h. The reaction was quenched by exposure to air. Product was purified by dialysis against Milli-Q water (MWCO < 3500 g mol−1) and subsequent lyophilisation.
Chain extension of quaternised poly(2-dimethylamino ethyl methacrylate) with tert-butyl acrylamide
Amphiphilic diblock copolymers were prepared via chain extension of the Q100 macro-CTA with tert-butyl acrylamide (see scheme in Fig. 2).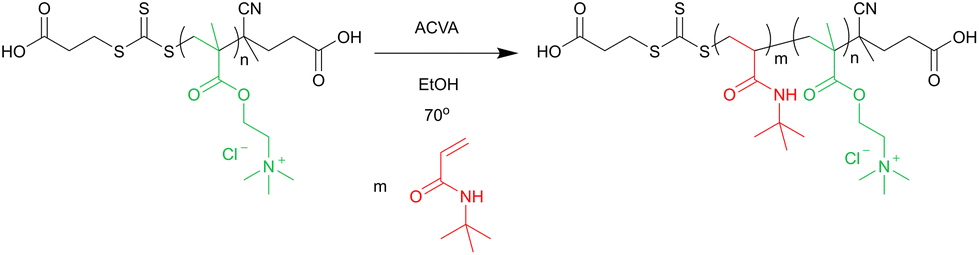 | ||
| Fig. 2 Reaction scheme for the chain extension of Q macro-CTA with tert-butyl acrylamide by RAFT polymerisation. | ||
An example of the reaction procedure is as follows: Q100 macro-CTA (0.40 g, 0.017 mmol), ACVA (1.0 mg, 0.0036 mmol) and tBAA (0.11 g, 0.87 mmol) were dissolved in 100% ethanol to 30 wt%, giving a ratio of [tBAA]:[macro-CTA]:[ACVA] = 50:1:0.2. The solution was degassed with N2 for 15 min and then stirred at 70 °C for 24 h before being quenched by exposure to air. The product was purified by dialysis against 100% ethanol (MWCO < 3500 g mol−1), and remaining solvent was removed by rotary evaporation.
Chain extension of amphiphilic diblock copolymers with N,N-dimethyl acrylamide
Linear triblock copolymers were prepared via chain extension of the Q-b-B macro-CTA with N,N-dimethyl acrylamide (see scheme in Fig. 3).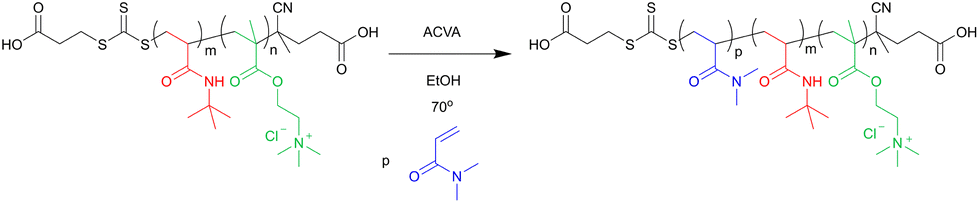 | ||
| Fig. 3 Reaction scheme for the chain extension of Q-b-B macro-CTA with N,N-dimethyl acrylamide by RAFT polymerisation. | ||
An example of the reaction procedure is as follows: Q-b-B macro-CTA (0.22 g, 8.0 μmol), ACVA (1.0 mg, 3.0 μmol) and DMA (0.10 g, 10 mmol) were dissolved in 100% ethanol to 30 wt%, giving a ratio of [DMA]:[macro-CTA]:[ACVA] = 250:1:0.4. The solution was degassed with N2 for 15 min and then stirred at 70 °C for 1.5 h before being quenched by exposure to air. The reaction solution became cloudy after ∼1 h. The product was purified by dialysis against Milli-Q water (MWCO < 3500 g mol−1). This solvent switch induced particle formation, and the resulting particles were lyophilised.
1H NMR spectroscopy
1H NMR spectroscopy was conducted post-purification at 400 MHz. Q homopolymer was diluted in D2O, and Q-b-B and Q-b-B-b-D were diluted in MeOD, with concentrations of 5 mg mL−1. The DP was confirmed in each case by comparison of characteristic peaks. For Q macro-CTA, a peak on the pendant amine group (b) was compared to a peak from the RAFT-end group (d). Q-b-B: a peak from the B block (g) was compared with a peak on the pendant amine group (b). Q-b-B-b-D: a peak from the D block (e) was compared to a peak on the pendant amine group (b) (see Fig. 4 for example spectra, and Fig. S1 and S2 in ESI† for remaining spectra). | ||
| Fig. 4 1H NMR (400 MHz) spectra of the homopolymer Q100, the diblock copolymer Q100-b-B44 and the triblock copolymer Q100-b-B44-b-D99, respectively. | ||
Dynamic light scattering
To prevent dust contamination upon sample preparation, all glass vials, lids and stirrer bars were washed 3× with filtered ultrapure Milli-Q water (filtered through two 0.2 μm pore-size nylon membrane non-sterile Fisherbrand® filters mounted in series) and filtered isopropanol (IPA) (filtered through two 0.2 μm pore-size polytetrafluoroethylene (PTFE) membrane non-sterile Fisherbrand® filters mounted in series) before drying at ∼50 °C in a dust-free environment. Samples were measured in the pre-washed glass light scattering (LS) tubes (rimless Pyrex® culture tubes 75 × 10 mm).Experiments were performed with a 3D LS spectrometer (LS instruments, Switzerland) using the ‘2D mode’. The spectrometer is fitted with a diode-pumped solid state (DPSS) laser operating at 660 nm with a maximum power of 105 mW (Cobolt FlamencoTM, Cobalt). Laser attenuation was automated, and two avalanche photodiode detectors were used, the light was vertically polarised. All experiments were performed at a temperature of 25 ± 0.5 °C controlled by using a water bath. A pseudo-cross correlation mode was used. The angle of measurement was altered from 30–130°.
A dynamic light scattering instrument (Zetasizer Nano-ZS, Malvern) was used for measurement of the hydrodynamic radii of complexes under the influence of varying NaCl concentration. This instrument uses back scatter (173°) detection angles with measurements in triplicate.
The associated scattering vector was calculated using eqn (1).
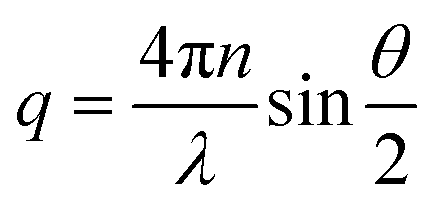 | (1) |
The intensity auto-correlation (IAC) data were fitted using the Levenberg–Marquardt algorithm to eqn (2).28
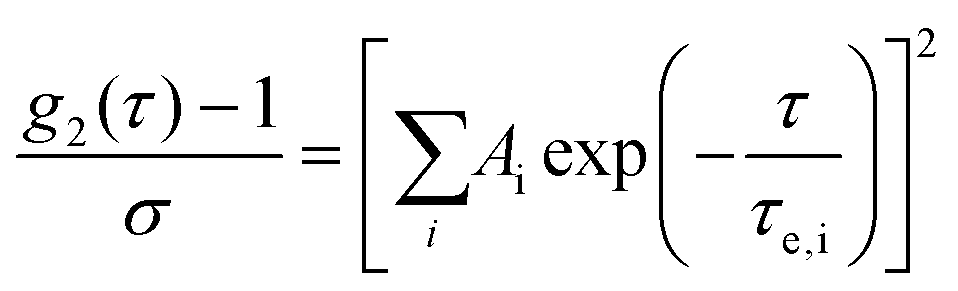 | (2) |
Diffusion coefficients, D, and hydrodynamic radii, RH, were subsequently calculated from the decay rate, Γ, and Stokes–Einstein equations (eqn (3) and (4)), in which η is solvent viscosity.
 | (3) |
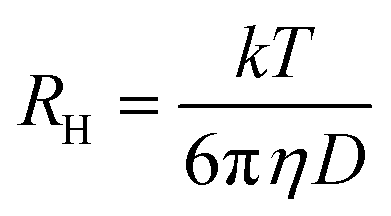 | (4) |
Electrophoretic mobility
Electrophoretic mobility was measured at 25 ± 0.5 °C using the phase analysis light scattering technique. Measurements were carried out using a standard folded capillary cell (DTS1070, Malvern) with a Zetasizer Nano-ZS instrument (Malvern). Data were collected in triplicate with the average taken over three runs. ζ potential, when used, was calculated by the instrument as determined by the Henry equation using the Smoluchowski approximation. Aqueous suspensions were prepared at concentrations of 0.1 (when complexed with dsRNA) or 1 g L−1, 24 h before measurement.Transmission electron microscopy
Triblock copolymer solutions were formulated by directly mixing Q-b-B-b-D in Milli-Q water to a concentration of 10 g L−1. Solutions were vortexed to ensure thorough mixing, and were prepared immediately prior to deposition onto 400-mesh carbon-coated copper grids. Deposition was conducted by addition of 5 μL of triblock copolymer solution onto the grid, prior to washing with Milli-Q water and staining with 1% uranyl acetate. Images were captured using an FEI Tecnai G2-Spirit microscope, with a Gatan Ultrascan 4000 CCD camera, operated at 120 keV with a tungsten filament.Ethidium bromide exclusion
Ethidium bromide (EB) was used as a nucleic acid-intercalating fluorophore. EB solution was stored in an opaque container at 4 °C prior to use. Fluorescence intensity was determined using an Omega FLUOstar® (BMG LABTECH GmbH) multi-mode micro-plate reader, with λex set at 320 nm and λem set at 594 nm. Samples were measured in a Corning® Costar 96-well opaque microplate. Gain was set at 1600–1900.Endpoint measurements were taken with 10 flashes per well. The volume of each well was made up to 200 μL with nuclease-free water. For all samples, 8 μL (0.468 g L−1) dsRNA were added to each well alongside 2.9 μL of EB (0.4 mg mL−1) that provided sufficient fluorescence intensity with the Omega FLUOstar® (BMG LABTECH GmbH) at the ratio [EB]:[P] = 0.12 (molar concentration of EB in relation to molar concentration of dsRNA phosphate groups, approximately one molecule of intercalated EB per four pairs of dsRNA bases). The dsRNA-EB solutions were left to incubate for at least 10 min prior to analysis for full intercalation of EB. An equilibration time was incorporated after each polymer addition prior to endpoint measurement.
Fluorescence intensity (FI) was normalised using eqn (5) with respect to the fluorescence intensity of dsRNA-EB alone (F0), subtracting the weak fluorescence intensity of EB in water (FEB).
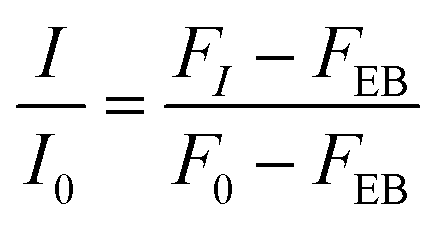 | (5) |
Agarose gel electrophoresis retardation assay
Aliquots of Q-b-B-b-D were added to 1 μg dsRNA, in quantities to vary the N/P ratio, with solutions left to incubate at RT for 1.5 h to allow for complexation. 2 μL of 6× blue/orange loading dye were added to each sample, and solutions were loaded onto a 2% (w/w) agarose gel containing 3.5 μL of EB, prepared with 1× TAE (Tris base, acetic acid and EDTA) buffer. Assays were run for 25 min at 90 V. A 1 μL aliquot of a DNA ladder, alongside 1 μL 6× purple non-SDS dye and 4 μL nuclease-free water, was run for comparison. The gel was imaged under a UV transilluminator at 365 nm. When RNase A (0.5 μL, 5 g L−1) was added to the polyplex solutions, the samples were incubated at 37 °C for 30 min prior to analysis.Results and discussion
Polymer synthesis
Linear, amphiphilic ABC triblock copolymers were synthesised by reversible addition–fragmentation chain transfer (RAFT) polymerisation, allowing control over chain length to produce triblock copolymers with multiple blocks of differing functionalities. The synthesis of amphiphilic block copolymers can be challenging as the combination of both hydrophobic and hydrophilic blocks demands careful consideration of solvent choices for their synthesis and subsequent purification method. The solvent must be carefully selected to ensure solubility of starting reagents and final products, and, typically, care must be taken to purify the desired product at each stage of the synthesis.29 The solubility of monomers, homopolymers, diblock and triblock copolymers were tested in a variety of solvents to determine the optimal conditions for each stage of the polymerisation, the results of which can be found in Table S1 in the ESI.†In the synthesis, aqueous RAFT polymerisation was first used to prepare the macro-CTA, Q. The product was purified by dialysis in Milli-Q water and lyophilised. The second polymerisation, of the hydrophobic B block, was conducted in 100% ethanol, with purification by dialysis against 100% ethanol. The amphiphilic diblock copolymer (Q-b-B) was isolated by evaporation in vacuo. The final polymerisation of the third and hydrophilic polymer block (D) was conducted in 100% ethanol. The final purification was performed by dialysis in Milli-Q water, which is further discussed in the next section.
Due to the charged nature and aqueous self-assembly of the triblock copolymers, gel permeation chromatography (GPC) analysis was inaccessible. The reaction efficiency was instead assessed as percentage of target DP achieved via1H NMR spectroscopy (400 MHz) as described in the Materials and Methods section. The 1H NMR spectra for each stage of the synthesis of the Q100-b-B44-b-D99 triblock copolymer are shown in Fig. 4 (spectra for the remaining triblock copolymers in the series can be found in Fig. S1 and S2 of the ESI†).
Peak (b) represents the Q block of the triblock copolymer, with peaks (g) (in the red spectrum) and (e) (in the blue spectrum) representing the B and D blocks, respectively. Molecular weight (Mn) was calculated via block composition analysis. A summary of the percentage of target DP achieved and composition analysis of each triblock copolymer is included in Table 1.
| Triblock copolymer code | M n/kDa | Q-block % of target DP | B-block % of target DP | D-block % of target DP | Wt% Q | Wt% B | Wt% D |
|---|---|---|---|---|---|---|---|
| Q100-b-B17-b-D212 | 44.1 | 80 | 64 | 85 | 47 | 5 | 48 |
| Q100-b-B25-b-D55 | 29.7 | 80 | 50 | 22 | 71 | 11 | 19 |
| Q100-b-B44-b-D99 | 36.5 | 80 | 44 | 40 | 57 | 15 | 27 |
Fig. 5 provides a schematic representation of the relative degrees of polymerisation of the polymer blocks that make up each ABC triblock copolymer, to aid visualisation.
 | ||
| Fig. 5 Schematic illustration of the relative proportions of polymer blocks within each amphiphilic triblock copolymer, according to the degree of polymerisation as confirmed via characteristic peak analysis of 1H NMR spectra. Green = Q block, red = B block, blue = D block. This illustration is included to give perspective on the proportions of each polymer block, and is not representative of the true size/length of the triblock copolymers. | ||
Characterisation of triblock copolymer self-assembly in aqueous environment
Following RAFT polymerisation of the final D block of the ABC triblock copolymers, purification was accomplished via dialysis. This stage of the synthesis was carried out in 100% ethanol and dialysis was subsequently conducted in Milli-Q water using a MWCO < 3500 g mol−1 regenerated cellulose membrane. Throughout the dialysis procedure, self-assembly was observed with the appearance of some precipitation due to the solvent switch taking place as the 100% ethanol was slowly replaced with Milli-Q water. Lyophilisation of the purified polymer resulted in dried white-coloured triblock copolymer particles, which could be re-dispersed in pure water with no subsequent precipitation observed. Direct mixing of the lyophilised particles into aqueous solution at desired concentrations was used to assess the characteristics of the self-assembled triblock copolymers.Particle size analysis was performed via DLS measurement of 1 g L−1 aqueous solutions (see Table 2).
| Triblock copolymer code | Hydrodynamic radius of objects in aqueous solution (c = 0.1 mg mL−1)/nm | Electrophoretic mobility/μm cm V s−1 |
|---|---|---|
| Q100-b-B17-b-D212 | 119 ± 4 | 2.3 ± 0.2 |
| Q100-b-B25-b-D55 | 214 ± 10 | 3.5 ± 0.1 |
| Q100-b-B44-b-D99 | 154 ± 20 | 3.2 ± 0.1 |
These measurements demonstrate that the triblock copolymers form self-assembled objects that can also be identified in the TEM images, which suggest a large polydispersity of the self-assembled particles, Fig. 6.
 | ||
| Fig. 6 Representative Transmission Electron Micrographs obtained for the 3 triblock copolymers (A) Q100-b-B17-b-D212, (B) Q100-b-B25-b-D55 and (C) Q100-b-B44-b-D99. Solutions were formulated at 10 g L−1, with 5 μL deposited onto 400-mesh carbon-coated copper grids. Grids were then washed with Milli-Q water and stained with 1% uranyl acetate. | ||
Q100-b-B25-b-D55, the triblock copolymer with the greatest proportion of charged Q block, resulted in the formation of the largest polymeric objects with an average apparent radius of >200 nm as measured by DLS. However, the particles appeared to be the most polydisperse in size when investigated by TEM, with some large objects present with radii ∼500 nm, Fig. 6B.
Q100-b-B17-b-D212, the triblock copolymer with the largest proportion of hydrophilic, uncharged D block, appeared to form the smallest objects by DLS and visually by TEM, Fig. 6A. These objects were less polydisperse than in the case of the Q100-b-B25-b-D55 polymer. Similar results have been reported elsewhere, with longer hydrophilic, neutral blocks inducing smaller self-assembled object formation.25
The triblock copolymer with the highest proportion of hydrophobic B block, Q100-b-B44-b-D99 (Fig. 6C), formed particles that were more polydisperse than Q100-b-B17-b-D212 and slightly larger in size on average, but were smaller in apparent size (on average) than the Q100-b-B25-b-D55 particles.
Measuring the electrophoretic mobility of the self-assembled objects revealed a positive surface charge in all cases, however, Q100-b-B17-b-D212 particles had a lower electrophoretic mobility from a dampened positive surface charge. This is in agreement with literature, in which the hydrophilic, neutral block is found to shield charge on the surface of self-assembled objects.25
Complexation of triblock copolymers with dsRNA
Amphiphilic triblock copolymers can be used as delivery vehicles for genetic material such as double stranded-RNA (dsRNA). The Q-b-B-b-D triblock copolymers synthesised in this work contain a hydrophilic and positively charged polymer block, Q, which can electrostatically interact with the anionic phosphodiester backbone of the dsRNA. The interaction is entropically favourable due to the release of counterions, and leads to the formation of complexes that we term here ‘micelleplexes’.30,31A common tool used to interpret the strength of dsRNA binding by polycations is the quenching of ethidium bromide (EB) fluorescence. EB intercalates between the base pairs of double-stranded genetic material, inducing a strong fluorescence in comparison to its weak fluorescence in water,32 and upon addition of a competitive cationic macromolecule, EB is displaced from the genetic material resulting in the observation of fluorescence quenching.32–38 The relative amount of quenching can be used as a measure of binding strength of the polycation to the genetic material.
In this work, dsRNA was combined with EB and incubated for full intercalation. Triblock copolymer solutions were then incorporated to complex with dsRNA. As the N/P ratio (ratio of ammonium groups of the polymer with respect to the phosphate groups of the dsRNA) increases from 0 (with no polymer added to the dsRNA solution) to 1 (equal proportion of number of ammonium to number of phosphate groups) in Fig. 7, the fluorescence intensity (normalised, I/I0) decreases to 50–65%.
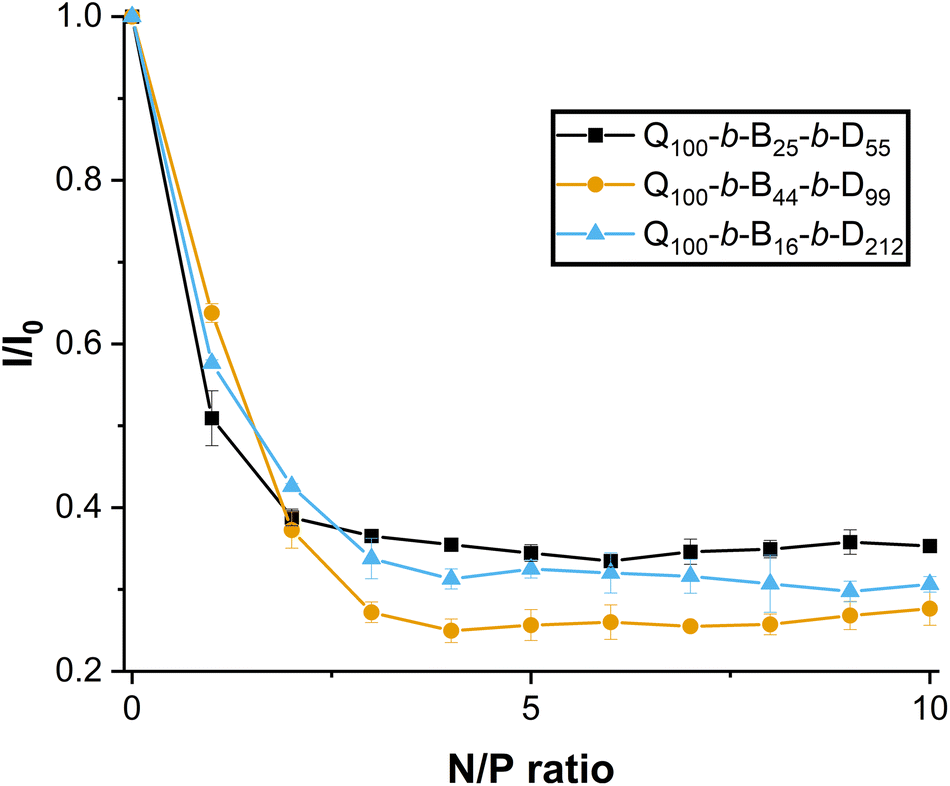 | ||
| Fig. 7 Normalised fluorescence intensity (I/I0, where I0 is the fluorescence intensity of dsRNA-EB complexes prior to triblock copolymer addition) of micelleplexes formed between different amphiphilic triblock copolymers (Q100-b-B25-b-D55, Q100-b-B44-b-D99 and Q100-b-B17-b-D212) and 222 bp V-ATPase dsRNA at varying N/P ratios 0–10. Standard deviation is shown, n = 3. | ||
As N/P ratio is further increased, the normalised fluorescence intensity decreases to a plateau (25–35%) reached at N/P ratio = 3. Interestingly, the Q100-b-B44-b-D99-based micelleplex appears to quench EB fluorescence more significantly over the N/P ratio = 3–8, whereas Q100-b-B25-b-D55 and Q100-b-B17-b-D212 triblock copolymers are not significantly different from one another (except at N/P ratios 4, 9 and 10). These results suggest that a proportionally higher hydrophobicity of triblock copolymers may strengthen binding to dsRNA. It has been previously found that a hydrophilic, neutral block may weaken binding to pDNA.23 However, in this present study, it is clear that all amphiphilic triblock copolymers indeed complex with dsRNA at N/P ratio ≥ 2. In addition, the results of the EB assay suggest that the binding of triblock copolymers with dsRNA appears to be stronger than the binding of homopolymer or diblock copolymer, containing the same hydrophilic blocks, with dsRNA (with no hydrophobic block present), as reported in our previous work.27
The hydrodynamic radii of micelleplexes formulated at N/P ratios = 1, 5 and 10 were measured by DLS (see Table 3).
| Triblock copolymer code | N/P ratio | ||
|---|---|---|---|
| 1 | 5 | 10 | |
| a Indicates that aggregates were visibly forming in solution and thus LS is inaccessible. | |||
| Q100-b-B17-b-D212 | 91 ± 22 | 100 ± 10 | 122 ± 11 |
| Q100-b-B25-b-D55 | 126 ± 3 | 160 ± 5 | |
| Q100-b-B44-b-D99 | 92 ± 9 | 120 ± 12 | 134 ± 9 |
The Q100-b-B25-b-D55-based micelleplex, at N/P ratio = 1, formed aggregates that were visible to the naked eye. We previously found that a similar phenomenon occurs with cationic homopolymers when electrostatically interacting with an anionic macromolecule at a charge ratio close to 1, which is likely due to electro-neutralisation and thus precipitation of the formed complexes.27 In the case of Q100-b-B25-b-D55, the neutral D block may not be of significant length to counteract the hydrophobicity introduced by the B block and electro-neutralisation of the Q block. Despite the longer hydrophobic B block in the Q100-b-B44-b-D99 polymer, the length of the D block is likely sufficient to stabilise the complexes formed with dsRNA at N/P ratio = 1. These results indicate that the length (or DP) of the neutral, hydrophilic block is critical for the stabilisation of RNA-based complexes near the isoelectric point, and must make up more than 19 wt% in amphiphilic ABC linear triblock copolymers.
As N/P ratio is increased from 1 to 10, the average hydrodynamic radii of the micelleplexes increases. In comparison to the triblock copolymers self-assembled in aqueous solution without complexation of dsRNA, the micelleplexes are measured by DLS to be smaller. This suggests that significant chain rearrangement of the initially self-assembled triblock copolymer objects occurs during the complexation process with dsRNA. This chain rearrangement is further confirmed through TEM (see Fig. S3 and S4 in ESI†) which identifies significantly altered structures as compared to the micellar aggregates of the triblock copolymers alone in aqueous solution. Further studies of the detailed morphologies of complexes formed between triblock copolymers and dsRNA is required, and future work will focus on additional transmission electron microscopy and small-angle X-ray/neutron scattering to elucidate their structure further.
With increasing N/P ratio, all micelleplexes undergo a negative to positive charge inversion, Fig. 8. At N/P ratio = 1, Q100-b-B44-b-D99 and Q100-b-B17-b-D212 micelleplexes have electrophoretic mobilities that are close to 0 or slightly negative. As N/P ratio is increased to 5 and 10, Q100-b-B44-b-D99 and Q100-b-B17-b-D212 micelleplexes have positive electrophoretic mobility that is not significantly different over this N/P ratio range. Whereas, Q100-b-B25-b-D55 micelleplexes show a slight increase in electrophoretic mobility between N/P ratio 5 and 10. In literature, the N/P ratio at which an electrophoretic mobility plateau occurs corresponds to ‘full’ complexation, meaning the minimum concentration of polymer able to complex all the dsRNA present in the solution. These results suggest that Q100-b-B25-b-D55 micelleplexes may require higher N/P ratios for full complexation, however this does not agree with the observed EB exclusion around N/P ratio ≥ 2 for these micelleplexes. Once again, Q100-b-B17-b-D212-based micelleplexes have a dampened surface charge in comparison to Q100-b-B25-b-D55 and Q100-b-B44-b-D99 micelleplexes, likely due to shielding of charge by the D polymer block.11,24,25
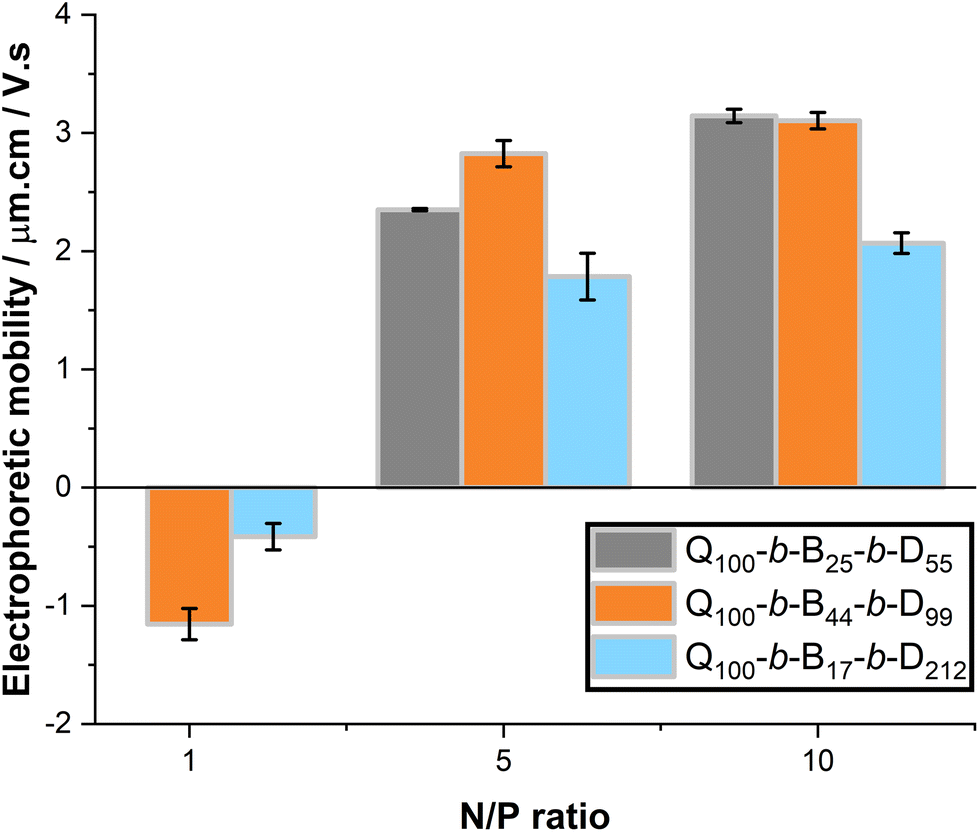 | ||
| Fig. 8 Electrophoretic mobility of micelleplexes, at N/P ratio 1, 5 and 10. Triblock copolymer solutions were kept at constant concentration (0.1 mg mL−1), with dsRNA concentration varied to alter the N/P ratio. Formulations were prepared at least 24 h prior to measurement, which were taken at 25 ± 0.5 °C. Standard deviation is shown, n = 3. | ||
Protection of dsRNA by triblock copolymers
Following successful complexation of triblock copolymers with dsRNA, the protection afforded to dsRNA by the amphiphilic triblock copolymers was tested using the synthetic enzyme, RNase A. As a biomolecule, RNA is inherently unstable and susceptible to degradation in the environment or upon application in therapeutic or agrochemical settings.39,40 Thus to ensure successful delivery of dsRNA, and to minimise loss of costly dsRNA during the delivery process, the dsRNA must be suitably protected to prevent enzymatic degradation.Agarose gel electrophoresis retardation assays are used to confirm the complexation of the triblock copolymers with dsRNA, and to assess the degradation. Free dsRNA is able to migrate freely in the agarose gel toward the anode, arriving at a specific location that is relative to the length (number of base pairs) of the dsRNA. The dsRNA used in the present work is 222 bp, and runs slightly above the 200 bp band of the DNA ladder. Thus, if complexation is unsuccessful, the free dsRNA will migrate in the gel lane. If complexation is only partial, the complexed dsRNA will be retained in the loading well, and the free un-complexed dsRNA will migrate towards the anode, resulting in a ‘smear’ effect. If complexation of the triblock copolymer with dsRNA is complete, dsRNA migration will be entirely prevented, and the dsRNA will be retarded in the loading well. It is important to note that, as described above, EB fluorescence is quenched upon strong binding of a polycation with dsRNA. Therefore, as N/P ratio is increased from 0 to 5, fluorescence is reduced due to this binding effect. Degradation of dsRNA is assessed in comparison to the control sample, comparing the fluorescence intensity of the dsRNA when RNase A is added.
In the case of all three of the triblock copolymer micelleplexes, an N/P ratio ≥ 2 is required for full complexation to be achieved. This is confirmed by the fluorescence retardation in the well of the gel at this N/P ratio and above, shown in Fig. S5, S6 and S7 in the ESI,† lanes 13, 12 and 13, respectively. Prior to this N/P ratio, smearing is observed along the gel lane, indicating only partial complexation.
The fluorescence intensity (normalised with respect to dsRNA alone) of free dsRNA that has migrated through the gel is shown in Fig. 9. The intensity is measured via ImageJ analysis of the gel images provided in the ESI.† The sharp decrease in free dsRNA at N/P ratio = 2 in Fig. 9 demonstrates that N/P ratio ≥ 2 is required for full complexation. There is a decrease in free dsRNA at N/P ratios = 0.75 and 1, particularly with Q100-b-B44-b-D99 micelleplexes. This complements EB exclusion data (Fig. 7) in suggesting the stronger binding of Q100-b-B44-b-D99 to dsRNA.
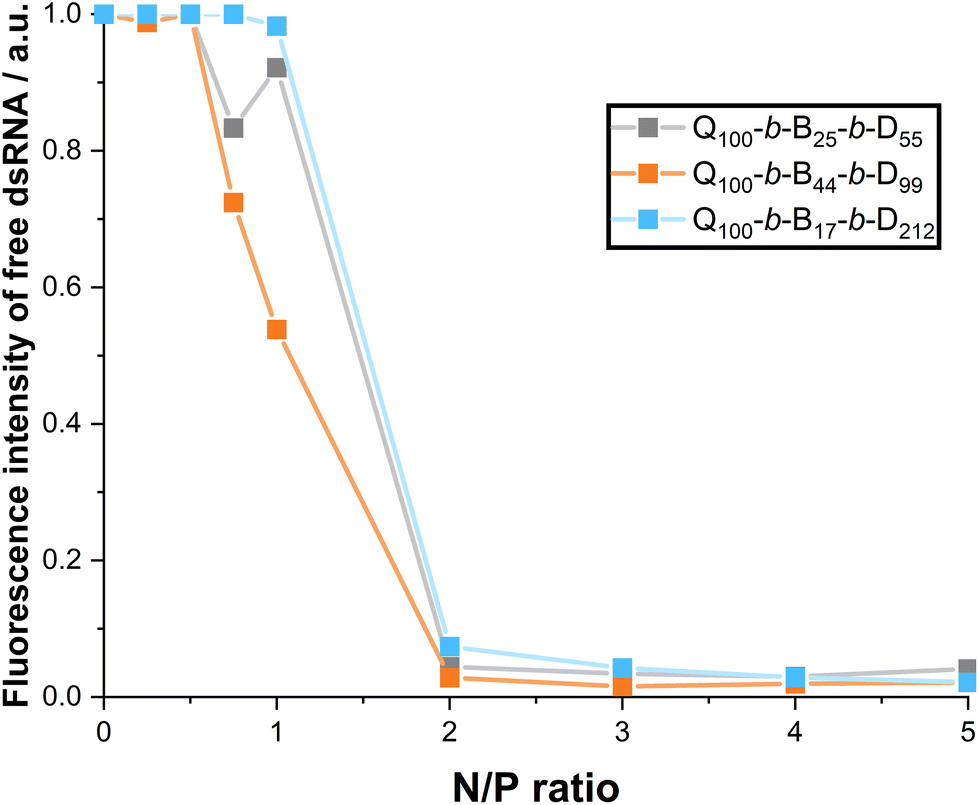 | ||
| Fig. 9 Proportion of free dsRNA when complexed with the three triblock copolymers as determined via gel electrophoresis fluorescence intensity. Data are normalised against naked dsRNA fluorescence and calculations are run using ImageJ. | ||
Fig. 10 shows an example of the protection of dsRNA upon full complexation with triblock copolymer. The free dsRNA is degraded by the presence of RNase A (Fig. 10A). When partial complexation occurs the free dsRNA is degraded by RNase A (Fig. 10B), however the complexed dsRNA remains intact. Upon full complexation (e.g. at N/P ratio = 3 in Fig. 10C), the complexed dsRNA remains intact, with a similar fluorescence level in the well of the gel lane as that shown for the control micelleplex.
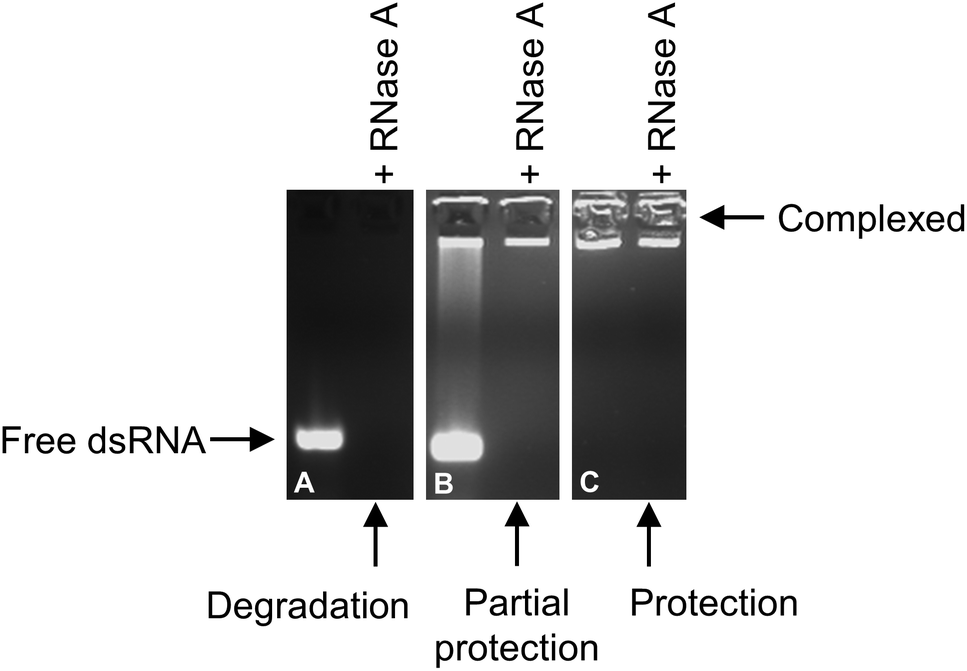 | ||
| Fig. 10 Degradation of free dsRNA (A) when in the presence of RNase A, in comparison to the partial protection of dsRNA when complexed with triblock copolymer at N/P ratio = 1 (B) and the protection of dsRNA when complexed with triblock copolymer at N/P ratio = 3 (C). This example shows the complexation of Q100-b-B17-b-D212 with dsRNA, at final concentrations 273 and 67 ng μL−1, respectively. | ||
Impact of salt concentration
It is also important to consider the impact of bulk salt concentration on the stability of the micelleplexes, as formulations for both therapeutic and agrochemical applications typically contain various electrolytes as part of a buffer solution, or as adjuvants to aid application. Here, we use NaCl as a simple model salt to increase ionic strength of the aqueous solution, measuring the resulting changes in hydrodynamic radii via DLS.Despite the stronger binding between Q100-b-B44-b-D99 and dsRNA in salt-free, aqueous environments, the resulting micelleplexes appear to be the least stable as NaCl concentration (CNaCl) increases, see Fig. 11. Above 150 mM, the apparent average hydrodynamic radius of the micelleplexes steadily increases, suggesting that the charge screening from the increased ionic strength of the solution is causing aggregation between the micelleplexes.
 | ||
| Fig. 11 Apparent hydrodynamic radii of micelleplexes formed between dsRNA and each triblock copolymer, measured with DLS, as NaCl concentration is increased from 0 to 700 mM. | ||
Q100-b-B25-b-D55 and Q100-b-B17-b-D212-based micelleplexes, however, remain stable until CNaCl = 650–700 mM. In comparison to our previous work on the salt stability of hydrophilic homopolymer and diblock copolymer polyplexes, the two triblock copolymers with the highest proportion of hydrophilic polymer blocks (Q or D, respectively) provide stability over a greater salt concentration range.27 Sharma et al. similarly found that amphiphilic triblock copolymer-based micelleplexes stabilised DNA more efficiently than diblock copolymer or homopolymer polyplexes, and protected more effectively against DNase enzymes.23
Sharma et al. demonstrate the resistance of their ABC triblock copolymer complexes with pDNA to aggregation in salt over 1 h. However, a salt concentration of 150 mM only is examined.23 Whilst this may be suitable for some therapeutic applications (for example, mammalian Na+ and Cl− extracellular concentrations reach 145 mM and 116 mM, respectively41,42), demonstrating stability at higher salt concentration, as is shown here, is valuable for assessing formulations of agrochemicals that may require significantly higher salt concentrations.
In our previous work, using diblock copolymers (AC, cationic–hydrophilic) to complex with the same dsRNA, a similar complexation pattern to the ABC triblock copolymers was observed.27 An N/P ratio = 1 was not sufficient to provide full complexation in either case, however full complexation was achieved at N/P ratio ≥ 2. The hydrodynamic radii of polyplexes formed between the AC diblock copolymers and dsRNA ranged from 57–125 nm, and showed no significant change in size across an N/P ratio range of 1–10. In comparison, whilst the triblock copolymer/dsRNA complexes showed similar hydrodynamic radii, they did show variation in size with changing N/P ratio, seeing an increase in radii with an increasing N/P ratio. The AC diblock copolymers did not exhibit any self-assembly prior to the addition of dsRNA, whereas the ABC triblock copolymers do show self-assembly in aqueous solution. However, the similarity in size of diblock copolymer/dsRNA complexes and triblock copolymer/dsRNA complexes suggests that the triblock copolymers undergo substantial chain rearrangement in order to complex with dsRNA. This is corroborated by TEM observations indicating the breakdown of the self-assembled micellar structures observed for the triblock copolymers in aqueous solution, when complexed with dsRNA. The benefit of ABC triblock copolymers for complexation and protection of dsRNA can only be seen when considering the stability of these complexes against increased ion concentration. The more hydrophilic triblock copolymers showed increased stability, in comparison to AC diblock copolymers, in the presence of salt concentrations up to 700 mM.
Conclusions
In this research we demonstrate the RAFT polymerisation synthesis of novel linear ABC triblock copolymers, and their formation of self-assembled objects in aqueous solution. We examine the potential of these triblock copolymers as polymeric delivery vehicles for dsRNA cargo, and investigate the impact of the proportion of each polymer block: hydrophilic, cationic PQDMAEMA (Q) block, hydrophobic PtBAA (B) block and hydrophilic, neutral PDMA (D) block. The D block was found to be the most important for formation of more compact micelleplexes and influenced electrophoretic mobility by shielding the charge. This proved beneficial in producing micelleplexes with greater stability at high salt concentrations, up to 700 mM. In comparison, the hydrophobic B block was found to induce the strongest binding to dsRNA, however the micelleplexes were only stable until CNaCl = 150 mM. The triblock copolymer with the highest proportion of charged Q block formed the most polydisperse self-assembled objects, and also the largest objects when complexed with dsRNA. It was discovered that, in the case of the triblock copolymers tested in this work, >19 wt% hydrophilic, neutral block is required to stabilise micelleplexes at N/P ratio = 1. All triblock copolymers fully complexed with dsRNA at N/P ratios ≥ 2, and successfully protected dsRNA against enzymatic degradation. We envision that ABC triblock copolymers could be useful delivery vehicles for dsRNA, with optimised architecture to prevent destabilisation at high salt concentrations, for commercially viable therapeutic or agrochemical applications.Author contributions
CEP was responsible for project administration, investigation, formal analysis, visualisation and writing (original draft). REI, NJW and OJC were responsible for the conceptualisation, funding acquisition and supervision of the project, and writing (review & editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support of Dr Rosa Domínguez-Espinosa and colleagues at Syngenta. The authors would like to acknowledge the support of Adan Yusuf, for assistance with TEM imaging, and Eleanor Hilton, Dr Yasmeen Jhons and Samuel Turvey for assistance with 1H NMR spectroscopy. The authors would like to acknowledge Dr Johan Mattsson and Dr Daniel Baker for support and use of their photon correlation spectrometer facility. The authors would like to acknowledge the support of the EPSRC (through grant EP/S023631/1) and Syngenta for funding this research.References
- D. Lombardo, M. A. Kiselev, S. Magazù and P. Calandra, Amphiphiles Self-Assembly: Basic Concepts and Future Perspectives of Supramolecular Approaches, Adv. Condens. Matter Phys., 2015, 2015, e151683 CrossRef.
- Y. Xiao, K. Shi, Y. Qu, B. Chu and Z. Qian, Engineering Nanoparticles for Targeted Delivery of Nucleic Acid Therapeutics in Tumor, Mol. Ther.–Methods Clin. Dev., 2018, 12, 1–18 CrossRef PubMed.
- S. Perrier, 50th Anniversary Perspective: RAFT Polymerization—A User Guide, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, RAFT Agent Design and Synthesis, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- D. J. Keddie, A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- A. C. Holley, K. H. Parsons, W. Wan, D. F. Lyons, G. R. Bishop, J. J. Correia, F. Huang and C. L. McCormick, Block ionomer complexes consisting of siRNA and aRAFT-synthesized hydrophilic-block-cationic copolymers: the influence of cationic block length on gene suppression, Polym. Chem., 2014, 5, 6967–6976 RSC.
- H. Willcock and R. K. O'Reilly, End group removal and modification of RAFT polymers, Polym. Chem., 2010, 1, 149–157 RSC.
- D. J. Gary, H. Lee, R. Sharma, J.-S. Lee, Y. Kim, Z. Y. Cui, D. Jia, V. D. Bowman, P. R. Chipman, L. Wan, Y. Zou, G. Mao, K. Park, B.-S. Herbert, S. F. Konieczny and Y.-Y. Won, Influence of Nano-Carrier Architecture on in Vitro siRNA Delivery Performance and in Vivo Biodistribution: Polyplexes vs Micelleplexes, ACS Nano, 2011, 5, 3493–3505 CrossRef CAS PubMed.
- C. E. Pugsley, R. E. Isaac, N. J. Warren and O. J. Cayre, Recent Advances in Engineered Nanoparticles for RNAi-Mediated Crop Protection Against Insect Pests, Front. Agron., 2021, 3 DOI:10.3389/fagro.2021.652981.
- Z. Tan, Y. Jiang, W. Zhang, L. Karls, T. P. Lodge and T. M. Reineke, Polycation Architecture and Assembly Direct Successful Gene Delivery: Micelleplexes Outperform Polyplexes via Optimal DNA Packaging, J. Am. Chem. Soc., 2019, 141, 15804–15817 CrossRef CAS PubMed.
- Y. Jiang, T. P. Lodge and T. M. Reineke, Packaging pDNA by Polymeric ABC Micelles Simultaneously Achieves Colloidal Stability and Structural Control, J. Am. Chem. Soc., 2018, 140, 11101–11111 CrossRef CAS PubMed.
- A. J. Convertine, C. Diab, M. Prieve, A. Paschal, A. S. Hoffman, P. H. Johnson and P. S. Stayton, pH-Responsive Polymeric Micelle Carriers for siRNA Drugs, Biomacromolecules, 2010, 11, 2904–2911 CrossRef CAS PubMed.
- A. W. York, S. E. Kirkland and C. L. McCormick, Advances in the synthesis of amphiphilic block copolymers via RAFT polymerization: Stimuli-responsive drug and gene delivery, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef CAS PubMed.
- G. A. R. Gonçalves and R. de M. A. Paiva, Gene therapy: advances, challenges and perspectives, Einstein, 2017, 15, 369–375 CrossRef PubMed.
- T. Friedmann, A brief history of gene therapy, Nat. Genet., 1992, 2, 93–98 CrossRef CAS.
- T. Friedmann and R. Roblin, Gene Therapy for Human Genetic Disease?, Science, 1972, 175, 949–955 CrossRef CAS PubMed.
- J. A. Doudna and E. Charpentier, Genome editing. The new frontier of genome engineering with CRISPR-Cas9, Science, 2014, 346, 1258096 CrossRef PubMed.
- S. Whyard, A. D. Singh and S. Wong, Ingested double-stranded RNAs can act as species-specific insecticides, Insect Biochem. Mol. Biol., 2009, 39, 824–832 CrossRef CAS PubMed.
- M. Thomas and A. M. Klibanov, Non-viral gene therapy: polycation-mediated DNA delivery, Appl. Microbiol. Biotechnol., 2003, 62, 27–34 CrossRef CAS.
- K. Wang, Y. Peng, J. Pu, W. Fu, J. Wang and Z. Han, Variation in RNAi efficacy among insect species is attributable to dsRNA degradation in vivo, Insect Biochem. Mol. Biol., 2016, 77, 1–9 CrossRef CAS.
- C. W. Scales, F. Huang, N. Li, Y. A. Vasilieva, J. Ray, A. J. Convertine and C. L. McCormick, Corona-Stabilized Interpolyelectrolyte Complexes of SiRNA with Nonimmunogenic, Hydrophilic/Cationic Block Copolymers Prepared by Aqueous RAFT Polymerization †, Macromolecules, 2006, 39, 6871–6881 CrossRef CAS.
- B. Brissault, C. Leborgne, D. Scherman, C. Guis and A. Kichler, Synthesis of Poly(propylene glycol)-block-Polyethylenimine Triblock Copolymers for the Delivery of Nucleic Acids, Macromol. Biosci., 2011, 11, 652–661 CrossRef CAS PubMed.
- R. Sharma, J.-S. Lee, R. C. Bettencourt, C. Xiao, S. F. Konieczny and Y.-Y. Won, Effects of the incorporation of a hydrophobic middle block into a PEG-polycation diblock copolymer on the physicochemical and cell interaction properties of the polymer-DNA complexes, Biomacromolecules, 2008, 9, 3294–3307 CrossRef CAS PubMed.
- C. Cheng, A. J. Convertine, P. S. Stayton and J. D. Bryers, Multifunctional triblock copolymers for intracellular messenger RNA delivery, Biomaterials, 2012, 33, 6868–6876 CrossRef CAS PubMed.
- T. K. Endres, M. Beck-Broichsitter, O. Samsonova, T. Renette and T. H. Kissel, Self-assembled biodegradable amphiphilic PEG–PCL–lPEI triblock copolymers at the borderline between micelles and nanoparticles designed for drug and gene delivery, Biomaterials, 2011, 32, 7721–7731 CrossRef CAS PubMed.
- T. Segura and J. A. Hubbell, Synthesis and in Vitro Characterization of an ABC Triblock Copolymer for siRNA Delivery, Bioconjugate Chem., 2007, 18, 736–745 CrossRef CAS PubMed.
- C. E. Pugsley, R. E. Isaac, N. J. Warren, J. S. Behra, K. Cappelle, R. Dominguez-Espinosa and O. J. Cayre, Protection of Double-Stranded RNA via Complexation with Double Hydrophilic Block Copolymers: Influence of Neutral Block Length in Biologically Relevant Environments, Biomacromolecules, 2022, 23(6), 2362–2373 CrossRef CAS.
- J. Behra, Structure-property relationships of sodium carboxymethyl cellulose (Na CMC) in pure water and formulated solutions, University of Leeds, 2018 Search PubMed.
- M. H. Stenzel, C. Barner-Kowollik, T. P. Davis and H. M. Dalton, Amphiphilic Block Copolymers Based on Poly(2-acryloyloxyethyl phosphorylcholine) Prepared via RAFT Polymerisation as Biocompatible Nanocontainers, Macromol. Biosci., 2004, 4, 445–453 CrossRef CAS.
- V. Incani, A. Lavasanifar and H. Uludağ, Lipid and hydrophobic modification of cationic carriers on route to superior gene vectors, Soft Matter, 2010, 6, 2124–2138 RSC.
- M. A. Cohen Stuart, N. A. M. Besseling and R. G. Fokkink, Formation of Micelles with Complex Coacervate Cores, Langmuir, 1998, 14, 6846–6849 CrossRef.
- M. J. Waring, Complex formation between ethidium bromide and nucleic acids, J. Mol. Biol., 1965, 13, 269–282 CrossRef CAS.
- W. Chen, N. J. Turro and D. A. Tomalia, Using Ethidium Bromide To Probe the Interactions between DNA and Dendrimers, Langmuir, 2000, 16, 15–19 CrossRef CAS.
- R. J. Douthart, J. P. Burnett, F. W. Beasley and B. H. Frank, Binding of ethidium bromide to double-stranded ribonucleic acid, Biochemistry, 1973, 12, 214–220 CrossRef CAS PubMed.
- R. Galindo-Murillo and T. E. Cheatham III, Ethidium bromide interactions with DNA: an exploration of a classic DNA–ligand complex with unbiased molecular dynamics simulations, Nucleic Acids Res., 2021, 49, 3735–3747 CrossRef CAS PubMed.
- V. A. Izumrudov, M. V. Zhiryakova and A. A. Goulko, Ethidium Bromide as a Promising Probe for Studying DNA Interaction with Cationic Amphiphiles and Stability of the Resulting Complexes, Langmuir, 2002, 18, 10348–10356 CrossRef CAS.
- J.-B. Lepecq and C. Paoletti, A fluorescent complex between ethidium bromide and nucleic acids: Physical—Chemical characterization, J. Mol. Biol., 1967, 27, 87–106 CrossRef CAS PubMed.
- J. Olmsted and D. R. Kearns, Mechanism of ethidium bromide fluorescence enhancement on binding to nucleic acids, Biochemistry, 1977, 16, 3647–3654 CrossRef CAS PubMed.
- R. Whitfield, A. Anastasaki, N. P. Truong, A. B. Cook, M. Omedes-Pujol, V. Loczenski Rose, T. A. H. Nguyen, J. A. Burns, S. Perrier, T. P. Davis and D. M. Haddleton, Efficient Binding, Protection, and Self-Release of dsRNA in Soil by Linear and Star Cationic Polymers, ACS Macro Lett., 2018, 7, 909–915 CrossRef CAS PubMed.
- S. Dubelman, J. Fischer, F. Zapata, K. Huizinga, C. Jiang, J. Uffman, S. Levine and D. Carson, Environmental Fate of Double-Stranded RNA in Agricultural Soils, PLoS One, 2014, 9, e93155 CrossRef PubMed.
- H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore and J. Darnell, in Molecular Cell Biology, W. H. Freeman & Co., New York, 4th edn., 2000, ch. 21, pp. 921–924 Search PubMed.
- B. Liu, B. Poolman and A. J. Boersma, Ionic Strength Sensing in Living Cells, ACS Chem. Biol., 2017, 12, 2510–2514 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00914e |
| This journal is © The Royal Society of Chemistry 2022 |