
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of nanosized mordenite and beta zeolites with improved catalytic performance: non-surfactant diquaternary ammonium compounds as structure-directing agents†
Shaojie
Li
,
Rim C. J.
van de Poll
,
Nikolay
Kosinov
 and
Emiel J. M.
Hensen
*
and
Emiel J. M.
Hensen
*
Laboratory of Inorganic Materials and Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.j.m.hensen@tue.nl
First published on 9th May 2022
Abstract
Non-surfactant diquaternary ammonium compounds have already been used for obtaining various zeolites in nanocrystalline form. However, facile synthesis of nanocrystals of mordenite (MOR) and beta (BEA) in this way remains challenging. Here, we present the direct synthesis of nanosized mordernite (MOR) and beta (BEA) zeolites with hexane- and p-xylene-bridged bis-methylpyrrolidinium, -methylpiperidinium and -DABCO diquats, which can be synthesized in a single step from common chemicals. Optimized recipes are presented for nanosized MOR (20–50 nm) and BEA (15–30 nm) zeolites. By investigating the solid products obtained during hydrothermal synthesis, the formation of nanocrystals can be linked to the strong interaction between diquat templates and aluminosilicate species during the induction stage, which limits the amorphous precursor particles to a size below 50 nm. Based on the textural and acidic properties, catalytic performance data are discussed evidencing the clear benefits of these nanosized zeolites over bulk reference samples in Friedel–Crafts reactions and n-alkane hydroconversion.
1. Introduction
Zeolites are a family of crystalline porous materials containing molecule-sized cavities and channels, which are widely used in industrial processes enabling separation, adsorption and enhanced reaction rates (catalysis).1,2 They are useful catalysts because of their tunable acidity, (hydro)thermal stability and shape selectivity.3 The performance of zeolite catalysts is often hampered by the long residence times of reactants and products in the micropore network.4 This can negatively affect the catalytic activity and lead to rapid deactivation due to formation of bulky products that cannot leave and therefore block the micropores. Over the last decades, many approaches have been explored to improve the rate of diffusion in zeolites.4,5 A common aspect of nearly all of these approaches is that the crystallite domain size is reduced to limit the diffusional pathways in the micropore space.6 Besides introducing additional (meso)pores in zeolite crystals,7 it is also effective to decrease the size of zeolite crystals below 100 nm (nanocrystals).8 Another potential benefit of these nanocrystals compared to conventional, often micron-sized zeolites is the much higher external surface area that not only increases the rate of product desorption,9 but also leads to higher conversion rates of reactions that take place on or close to the external surface.10In the last two decades, considerable efforts have been made to prepare nanocrystalline zeolites. Top-down and bottom-up approaches are distinguished based on whether small crystals are obtained after or during zeolite crystallization, respectively.8 Ball-milling is a physical approach of the first category, which is hampered by the need to remove amorphous debris left after subsequent recrystallization of the milled samples.11 Chemical treatment is a more versatile top-down approach. For instance, Corma and co-workers prepared nanosheet zeolites, in which zeolite MWW was swelled with hexadecyltrimethylammonium bromide followed by delamination. A limitation of this approach is that it only can be used for particular zeolite topologies like MWW and FER.12,13
Compared to top-down approaches, bottom-up methods provide more flexibility in the preparation of nanocrystalline zeolites.14 Without being exhaustive, approaches may include a change in the order of mixing the reagents,15,16 replacing traditional heating by microwave irradiation,17 the use of ultra-dense gels crystallized by steam treatment18 and decoupling nucleation from crystal growth via a temperature-staged approach.19,20 Confined space synthesis was also employed to prepare nanosized zeolites, involving crystallization of the zeolite inside a mesoporous matrix.21 Although nanocrystalline zeolites can be obtained by the above methods, major drawbacks remain, usually involving the complexity of the synthesis in the number of steps and the facilities needed. Therefore, it remains highly desirable to prepare nanocrystalline zeolites via facile one-step methods.
As a more facile and effective bottom-up approach, soft-templating methods have been used to prepare nanosized zeolites. Soft templates such as surfactants or polymers can limit both crystal grain growth and Ostwald ripening by adsorbing on the growing crystal surfaces, thereby reducing crystal sizes.22,23 For instance, ferrierite nanosheets were synthesized by a dual-templating method, in which piperidine and cetyltrimethylammonium bromide acted as structure-directing agent (SDA) and morphology modifier, respectively.24 Ryoo and co-workers reported the synthesis of nanocrystalline zeolites such as ZSM-5, BEA and ZSM-12 by amphiphilic surfactants that have two functions, viz. directing zeolite growth by their ammonium groups and limiting crystal grain growth by the long hydrophobic alkyl tails.25,26 A drawback is that synthesis of these surfactants requires multiple steps. From an industrial perspective, it is preferred to prepare nanosized zeolites with relatively cheap organic templates, such as small-sized non-surfactant molecules obtained by one-step synthesis procedure.27 Such agents will not self-assemble into specific micelles, needed for obtaining (ordered) hierarchical structures or nanosized crystals. To prepare nanosized zeolites with non-surfactant molecules, a low amount of alkali together with mild crystallization temperature is often adopted to limit the aggregation of negatively charged sub-colloidal particles.8,28 This can explain the preference for organic templates in their OH form and the use of low synthesis temperatures, which usually increases the time to require fully crystalline products.29–31 For example, Corma and co-workers reported the direct synthesis of nanocrystalline ZSM-5 and BEA zeolites by using non-surfactant alkyl-substituted mono-cationic ammonium cations as templates, which took 14 days at a high template/Si molar ratio of 0.4.32
It has been shown that diquaternary ammonium compounds are better at directing nanocrystalline zeolite formation than conventional monoquaternary ammonium compounds.33–35 These compounds can be tailored in terms of size, functional groups and rigidity to control the crystal morphology and, in some cases, even the zeolite topology.33,36–40 The literature reveals the potential of this approach for preparing nanosized zeolites using relatively cheap non-surfactant organic templates. The synthesis of nanosized forms of commercially important MOR and BEA zeolites using non-surfactant diquaternary ammonium molecules as the sole organic template has only been scarcely explored.41–46 There is little understanding about the underlying crystallization mechanism, which is essential to pave the way to optimization of synthesis of nanosized forms of these zeolites and its generality for synthesis of nanosized zeolites.
In this work, we report the facile and economical synthesis of nanosized MOR and BEA zeolites by using the diquaternary ammonium compound as the sole organic template under conventional hydrothermal synthesis conditions. A total of 6 organic compounds (SDA1–6) were used varying in terms of heterocycle size and geometry of the end groups and the structural rigidity of the linkages (1,6-hexylidene and benzyl) between the quaternary ammonium centers (Fig. 1). These organic templates were synthesized from commercially available chemicals (Table S1†) via a one-step procedure. Synthesis conditions for obtaining nanosized MOR and BEA zeolites with these templates were explored. For selected samples, the crystallization behavior was monitored by studying intermediate solid products. The physicochemical properties such as chemical composition, morphology, texture and acidity were extensively characterized. The benefit of nanosizing these zeolites was evaluated by comparing their catalytic performance to conventional MOR and BEA reference zeolites for several model reactions.
 | ||
| Fig. 1 Organic structure-directing agents (SDAs) used in this work. | ||
2. Experimental section
2.1 Synthesis of organic templates
The purity of the above organic templates was verified by 1H NMR spectroscopy (Fig. S1†).
2.2 Synthesis of zeolites
All zeolite synthesis were performed in 45 ml Teflon-lined stainless-steel autoclave (Parr Instruments) under conventional hydrothermal conditions. In a typical synthesis, 0.56 g of sodium hydroxide (Sigma Aldrich, ≥98%) was dissolved in 15.53 g of deionized water, followed by the addition of the required amount of organic template. After stirring for 5 min, first 0.503 g of AlCl3·6H2O (Alfa Aesar, 99%) was added to the mixture, followed by 3.75 g of Ludox AS-40 (Sigma Aldrich, 40 wt%) under stirring. The resulting synthesis gel had a molar composition of 12SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5Al2O3:3.6Na2O:1.2SDA:480H2O. After vigorous stirring for 3 h at room temperature, the gel was transferred into an autoclave. The autoclave was placed in an oven for hydrothermal treatment at 160 °C under rotation at 50 rpm. The resulting solid products were recovered by centrifugation, thoroughly washed with demi-water until pH < 8 followed by drying at 30 °C overnight in a vacuum oven. The zeolites were calcined at 550 °C (heating rate 1 °C min−1) for 8 h under flowing air to remove the organic species. The calcined zeolites were ion-exchanged three times with 1.0 M NH4NO3 solutions, dried and calcined at 550 °C (heating rate 1 °C min−1) for 4 h in O2:N2 (1:4 vol. ratio) flow. Sample notation was zeolite topology–template name–Si/Al gel ratio. For comparison, bulk reference samples were obtained using appropriate templates. The samples were indicated by the suffix-Con to the zeolite topology. A conventional BEA zeolite was synthesized with tetraethylammonium hydroxide (Sigma Aldrich, 35 wt%) by following a reported procedure.47 This zeolite is denoted by BEA-12-Con.
:0.5Al2O3:3.6Na2O:1.2SDA:480H2O. After vigorous stirring for 3 h at room temperature, the gel was transferred into an autoclave. The autoclave was placed in an oven for hydrothermal treatment at 160 °C under rotation at 50 rpm. The resulting solid products were recovered by centrifugation, thoroughly washed with demi-water until pH < 8 followed by drying at 30 °C overnight in a vacuum oven. The zeolites were calcined at 550 °C (heating rate 1 °C min−1) for 8 h under flowing air to remove the organic species. The calcined zeolites were ion-exchanged three times with 1.0 M NH4NO3 solutions, dried and calcined at 550 °C (heating rate 1 °C min−1) for 4 h in O2:N2 (1:4 vol. ratio) flow. Sample notation was zeolite topology–template name–Si/Al gel ratio. For comparison, bulk reference samples were obtained using appropriate templates. The samples were indicated by the suffix-Con to the zeolite topology. A conventional BEA zeolite was synthesized with tetraethylammonium hydroxide (Sigma Aldrich, 35 wt%) by following a reported procedure.47 This zeolite is denoted by BEA-12-Con.
2.3 Preparation of Pt-containing zeolites
The proton form of the zeolites were loaded with 0.5 wt% Pt using wet impregnation with an aqueous Pt(NH3)4(NO3)2 solution. The resulting samples were dried in air followed by calcination at 450 °C (heating rate 0.5 °C min−1) for 2 h in an O2:N2 (1:4 vol. ratio) flow.
2.4 Characterization
X-ray diffraction (XRD) patterns were recorded on a Bruker D2 Endeavor diffraction system using Cu Kα radiation. Patterns were collected in the 2θ range of 5–40°.The elemental composition of samples was determined by inductively couple plasma optical emission spectrometry (ICP-OES). Prior to the measurement, a 1:1:1 (by weight) mixture of HF (40 wt% in H2O), HNO3 (60 wt% in H2O) and H2O was used to dissolve the samples.
Textural properties were obtained from the Ar physisorption at −186 °C with a Micromeritics ASAP 2020 instrument. Prior to the measurements, samples were outgassed at 400 °C for 6 h. The total pore volume was determined at relative pressure (p/p0) of 0.97. The BET surface area was calculated in the p/p0 range between 0.05–0.25. The micropore volume and external surface area were calculated by t-plot method. The mesopore volume was calculated by Barrett–Joyner–Halenda (BJH) method.
Thermogravimetric analysis (TGA) was performed with a TGA/DSC 1 instrument (Mettler Toledo). The temperature was increased from 40 °C to 800 °C (heating rate 5 °C min−1) in 20 ml min−1 O2 and 40 ml min−1 He flow.
Scanning electron microscope (SEM) images were obtained on a FEI Quanta 200F scanning electron microscope with an accelerating voltage of 3 or 5 kV. Transmission electron microscopy (TEM) images were acquired on a FEI Tecnai 20 at 200 kV. Annular dark field scanning transmission electron microscopy (ADF-STEM) was performed on the TU/e CryoTitan (FEI, now Thermo Fischer Scientific) at 300 kV and room temperature.
IR spectra were obtained with a FTIR spectrometer (Bruker Vertex 70v). The spectra were acquired in the range of 4000–1000 cm−1, with a resolution of 2 cm−1 and an average of 64 scans. The samples were pressed into thin wafers (∼10 mg) and placed into a controlled-environment transmission cell. The samples were first pretreated at 550 °C for 1 h in artificial air. After pretreatment, the samples were cooled down to 150 °C and a spectrum was collected as background. Pyridine was introduced into the cell until the sample was fully saturated. Finally, spectra were collected at 150 °C after outgassing for 1 h at 150 °C, 300 °C and 500 °C, respectively.
Solid-state nuclear magnetic Resonance (NMR) experiments were performed on a 11.7 Tesla Bruker DMX500 NMR spectrometer, operating at 132 MHz for 27Al, 99 MHz for 29Si, 125 MHz for 13C and 500 MHz for 1H. 27Al magic angle spinning (MAS) NMR measurements were performed with a Bruker 2.5 mm MAS probe head and a 2.5 mm zirconia rotor, operated at a spinning speed of 25 kHz. All other measurements were carried out with a Bruker Triple Channel 4 mm MAS probe head and a 4 mm zirconia rotor, under a spinning speed of 10 kHz. A single excitation pulse of 1 μs and a recycle delay of 1 s were used for 27Al NMR measurements. 27Al chemical shift was referred to Al(NO3)3. Quantitative 29Si MAS NMR spectra were collected using a high-power proton decoupling direct excitation (DE) pulse sequence with a 54° pulse duration of 3 μs and a recycle delay of 120 s. 29Si chemical shift was referred to tetramethylsilane (TMS). 1H NMR spectra were collected using a Hahn-echo pulse sequence of p1–τ1–p2–τ2–aq with a 90° pulse p1 = 5 μs, a 180° p2 = 10 μs and τ1 = τ2 = 0.5 μs. A recycle delay of 120 s was applied to obtain quantitative spectra. TMS was used as reference for 1H NMR chemical shift. Prior to 1H NMR measurements, the samples were dehydrated at 350 °C for 6 h under vacuum and then transferred into 4 mm rotors in a glovebox. 1H–13C cross-polarization (CP) MAS NMR spectra were collected with a ramped contact pulse of 3 ms and a recycle delay of 3 s. Solid adamantane was used as reference for 13C chemical shift. Two-dimensional (2D) 1H–29Si and 1H–13C heteronuclear correlation (HETCOR) spectra were collected with a rectangular contact pulse of 4 ms and 3 ms, respectively.
Liquid-state NMR measurements were performed on a Bruker 400 MHz spectrometer. The organic template was first dissolved in deuterated water, and the resulting solution was transferred into a 5 mm NMR tube. 1H NMR spectra were collected with a total of 32 scans and a relaxation delay of 1 s. 13C NMR spectra were collected with a total of 1024 scans and a relaxation delay of 2 s.
2.5 Catalytic activity measurements
3. Results and discussion
3.1 Zeolite synthesis
Fig. 2 shows XRD patterns of the samples obtained at a Si/Al gel ratio of 12 using various templates. Phase-pure MOR zeolite was obtained from a highly alkaline (NaOH/Si = 0.6) inorganic gel without organic templates.48 Highly crystalline MOR zeolites were also obtained with SDA2 and SDA6, whereas the use of SDA3, SDA4 and SDA5 resulted in BEA zeolites. No crystalline product was obtained with SDA1, even when the synthesis was prolonged to 6 days. The XRD patterns of the as-synthesized zeolites prepared with these SDAs display less intense and broader diffraction peaks compared to the reference MOR and BEA zeolites. This is typically attributed to reduction of the size of the crystalline zeolite domains.49 Moreover, when the Si/Al ratio of the gel was raised to 30, BEA zeolites were still obtained with SDA3, SDA4 and SDA5, whereas the use of SDA2 and SDA6 resulted, respectively, in an amorphous sample and a product for which the crystal phase could not be identified (Fig. S2–S4†). From the inorganic gel with a Si/Al ratio of 30, MOR zeolite was obtained in a low yield, pointing to poor incorporation of silicon into the zeolite as apparent from the low final Si/Al ratio and consistent with earlier studies (Table S3†).50,51 | ||
| Fig. 2 XRD patterns of samples synthesized using the templates shown in Fig. 1. | ||
Fig. 3 and 4 show SEM and TEM images of the as-prepared samples, respectively. As shown in Fig. 3b–f, the use of organic templates led to MOR and BEA zeolites in the form of aggregates of nanosized crystals. The nanocrystalline morphology is also apparent from the TEM images. Fig. 4 shows that the MOR-SDA2–12 and MOR-SDA6–12 samples consist of nanoparticles in the 20–50 nm range. Even smaller crystals (15–30 nm) were obtained for BEA-SDA3–12, BEA-SDA4–12 and BEA-SDA5–12. In contrast, MOR-12-Con obtained from a gel without organic template presents large (>100 nm) crystallites. BEA-12-Con prepared using TEAOH as the SDA resulted in large spherical particles (0.5–2 μm in diameter) with a rough surface in line with literature.47,52
 | ||
| Fig. 3 SEM images of calcined zeolites. | ||
 | ||
| Fig. 4 Representative TEM images of calcined zeolites. | ||
The textural properties of the calcined samples were characterized by Ar physisorption. All isotherms show a steep uptake below P/P0 = 0.02, indicating the existence of micropores (Fig. 5).53 MOR-12-Con and BEA-12-Con display type-I isotherms, which are typical for microporous materials.54 Samples prepared with SDA2–6 have a type-IV isotherm with a hysteresis loop, corresponding to the interparticle capillary condensation.45 The resulting textural properties are shown in Table 1. MOR-SDA2–12 and MOR-SDA6–12 present significantly higher external surfaces of 99 and 93 m2 g−1, respectively, as compared to MOR-12-Con (50 m2 g−1). The external surface areas of nanosized BEA prepared with SDA3, SDA4 and SDA5 are all much higher (>240 m2 g−1) than external surface area of the BEA-12-Con reference (65 m2 g−1). Among the BEA zeolites, BEA-SDA3–12 has the highest external surface area of 308 m2 g−1. These data agree with the difference seen in the SEM and TEM images. Thus, the diquaternary ammonium compounds are effective SDAs for synthesizing nanosized MOR and BEA zeolites.
 | ||
| Fig. 5 Ar physisorption isotherms of calcined zeolites. | ||
| Zeolite | S BET (m2 g−1) | V tot (cm3 g−1) | V meso (cm3 g−1) (BJH) | V micro (cm3 g−1) (t-plot) | S ext (m2 g−1) (t-plot) |
|---|---|---|---|---|---|
| MOR-12-Con | 315 | 0.16 | 0.04 | 0.11 | 50 |
| MOR-SDA2–12 | 408 | 0.29 | 0.13 | 0.13 | 99 |
| MOR-SDA6–12 | 398 | 0.25 | 0.10 | 0.12 | 93 |
| BEA-SDA3–12 | 566 | 0.89 | 0.72 | 0.12 | 308 |
| BEA-SDA4–12 | 539 | 0.89 | 0.72 | 0.12 | 257 |
| BEA-SDA5–12 | 528 | 0.92 | 0.75 | 0.12 | 245 |
| BEA-12-Con | 520 | 0.24 | 0.04 | 0.17 | 65 |
13C NMR spectra of the 6 organic templates and the corresponding samples in their dried state are shown in Fig. S5.† These NMR spectra of the as-prepared samples match well with those of the pure SDAs, showing that they are stable during the crystallization process.
The TGA profiles determined in artificial air of the as-prepared samples are presented in Fig. S6.† For MOR-12-Con, the total weight loss below 250 °C of ∼9 wt% can be related to the removal of physisorbed water (<150 °C) and water bonded to the zeolite framework (150–250 °C).55 The corresponding weight loss for the other zeolites was much lower (<4 wt%), which can be attributed to the presence of organic template in the zeolite micropores. The weight loss above 250 °C occurs in two steps due to the combustion of the organic template. The first weight-loss feature between 250–500 °C is likely due to the elimination of organic molecules on and close to external surface. The second weight loss in the range of 500–650 °C relates to the decomposition of organic species within zeolite pores. This aspect will be further investigated below. The TGA curve for amor-SDA1–12 contains only a single feature, consistent with the absence of micropores of this amorphous material. Combined with the 13C NMR data, these results demonstrate that SDA2–6 can direct the formation of nanosized MOR and BEA zeolites. Among the set of MOR and BEA zeolites, MOR-SDA2–12 and BEA-SDA3–12 were selected for further investigation of the crystallization process because of their favorable textural properties, viz. the largest external surface.
3.2 Crystallization process
The above results show that nanosized MOR and BEA zeolite crystals can be obtained by adding different organic templates to inorganic gels that otherwise give rise to bulk MOR. This implies a crucial role of the organic template for obtaining zeolite nanoparticles. It is therefore interesting to understand how the presence of organic template affects the precursors giving rise to zeolites with such distinctive topology and texture. For this purpose, the solid products obtained at different crystallization times were investigated.The crystallization curves of the three zeolite samples determined by XRD are shown in Fig. 6. The intensities of the 4 main diffraction peaks (2θ = 9.8°, 22°, 25.6° and 26.2°) were used to calculate the relative crystallinity of the MOR zeolites. The intensities of the diffraction peaks at 2θ = 7.7° and 22.2° were chosen to calculate the relative crystallinity of the BEA samples. MOR-12-Con-24 and BEA-SDA3–72 were used as fully crystallized references for the MOR and BEA zeolites, respectively. Crystalline MOR zeolite can be obtained in 24 h from an inorganic gel. On the other hand, it took more than 70 h to fully crystallize MOR-SDA2–12 and BEA-SDA3–12. Notably, diffraction peaks are not observed after 48 h crystallization, implying a long induction period (Fig. S7†). The crystal growth times for MOR-12-Con and MOR-SDA2–12 are comparable (∼6 h), while the crystal growth stage of BEA-SDA3–12 was slightly longer.
 | ||
| Fig. 6 Relative crystallinities of obtained solid samples at different crystallization time based on XRD analysis. | ||
The morphology of the intermediate products was determined by SEM and TEM (Fig. 7, 8, S8 and S9†). For MOR-12-Con, worm-like particles with a size of 0.1–2 μm were obtained after 12 h of crystallization (Fig. 7a). This morphology has been observed before for amorphous aluminosilicates.56 Prolonging the crystallization time to 18 h led to an additional phase mainly consisting of plates (Fig. 7b). After 20 h, the fraction of this plate-like material was slightly higher (Fig. 7c). The final MOR-12-Con material consisted of particles with a size in the 100–600 nm range with an inhomogeneous morphology (Fig. 7e). MOR-SDA2–12 consisted of nanoparticles with a primary diameter of 20–50 nm aggregated into secondary particles with a typical size of 200 nm after 12 h hydrothermal treatment (Fig. 8a). The significantly smaller size of the zeolite nanoparticles compared to MOR-12-Con is due to interruption of zeolite growth by the organic template. Prolonging crystallization from 12 h to 64 h did not substantially change this morphology (Fig. 8a–e and S9a†). Slightly smaller nanoparticles (15–40 nm) were observed for the sample obtained after 66 h (Fig. 8f). In the period between 66 h and 72 h, rapid crystal growth took place (Fig. 6), resulting in the final zeolite nanoparticles with sizes between 20 and 50 nm after 72 h hydrothermal treatment (Fig. 8h). For BEA-SDA3–12, aggregated particles with a primary size smaller than 50 nm were formed throughout the whole synthesis process (Fig. S8 and S9b†). These findings show that the size of solid intermediates is significantly reduced from larger than 100 nm to less than 50 nm by the presence of the SDAs, while comparable Si/Al ratios and yields of solid products were observed throughout the synthesis for each synthetic system regardless of the presence of the SDAs (Tables S5–S7†).
 | ||
| Fig. 7 SEM images of solid samples obtained at different crystallization times of MOR-12-Con. | ||
 | ||
| Fig. 8 SEM images of solid samples obtained at different crystallization times of MOR-SDA2–12. | ||
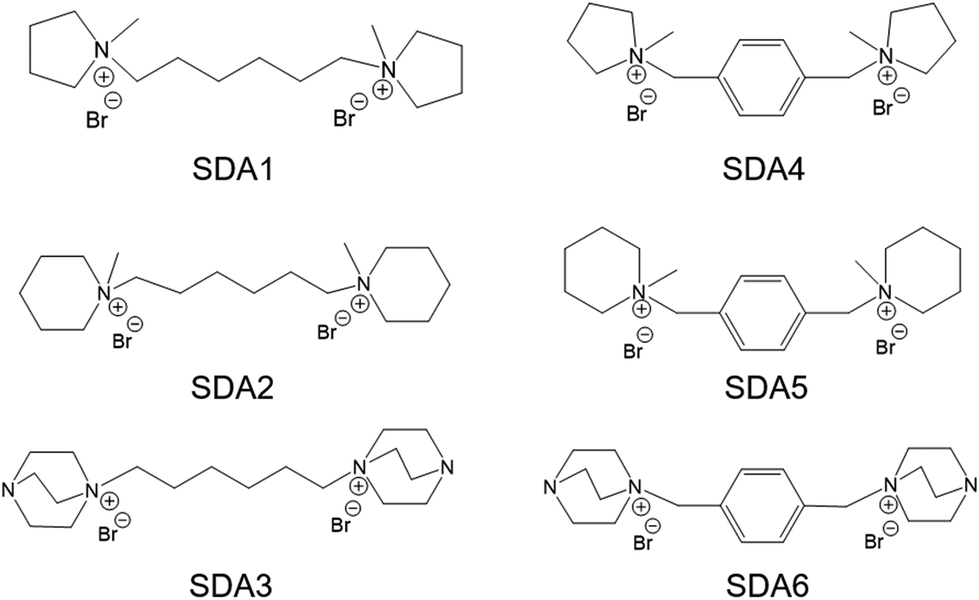
The Si coordination in the solid products was investigated by 29Si MAS NMR spectroscopy. As shown in Fig. 9, the NMR spectra of samples obtained after hydrothermal treatment for 12 h contain a broad feature in the range between −80 and −120 ppm. Main features at 91 ppm, 102 ppm, 106 ppm and 112 ppm can be observed, corresponding to Q2(0Al), Q3(0Al)/Q4(2Al), Q4(1Al) and Q4(0Al) sites, respectively.57,58 Notably, the Si coordination does not substantially change during the induction period. During crystal growth, the relative intensity of the Q4(0Al) peak gradually increased towards the final intensity observed for the fully crystallized zeolite. Together with the XRD data, these findings confirm that zeolite crystallization comprised induction and crystal growth stages with significant condensation mainly taking place during the latter stage. Similar trends in Si speciation have been observed in previous work in which the synthesis of MOR and BEA zeolites by dry gel conversion was studied in detail.59,60
 | ||
| Fig. 9 29Si MAS NMR spectra of samples obtained at different crystallization times: (a) MOR-12-Con, (b) MOR-SDA2–12 and (c) BEA-SDA3–12. | ||
A common view about zeolite synthesis is that precursor units formed during the induction stage assemble into zeolite crystals.58,61,62 The presence of specific features in the solid products was investigated by IR spectroscopy (Fig. 10). A band at 560 cm−1 present during the whole synthesis of MOR-12-Con has been linked to five-membered rings (5MR) silicate species.63 This does not necessarily mean that these species are involved in crystal growth. During the crystal growth stage (20–24 h), two other bands appear at 580 cm−1 and 640 cm−1 assigned to 5MR and four-membered rings (4MR), respectively.63,64 A similar trend in the IR spectra is observed for the MOR sample synthesized with SDA2, showing that the 5MR-containing motives are already present in the induction period, while formation of 4MR goes together with crystal growth. Thus, the synthesis of MOR-12-Con and MOR-SDA2–12 appears to follow a classical mechanism involving induction, nucleation and crystal growth. The longer induction period for MOR-SDA2–12 is likely due to the interactions between the organic template and the aluminosilicate precursors.61 For the sample synthesized with SDA3, the band at 560 cm−1 corresponding to 5MR is also observed during the induction period.58 After prolonging the hydrothermal treatment time from 60 h to 72 h, two characteristic bands of BEA zeolite at 460 cm−1 and 518 cm−1 are visible,65 which corresponds well with the evolution in crystallinity determined by XRD during BEA-SDA-3–12 synthesis.
 | ||
| Fig. 10 IR spectra of solid samples obtained at different crystallization times: (a) MOR-12-Con, (b) MOR-SDA2–12 and (c) BEA-SDA3–12. | ||
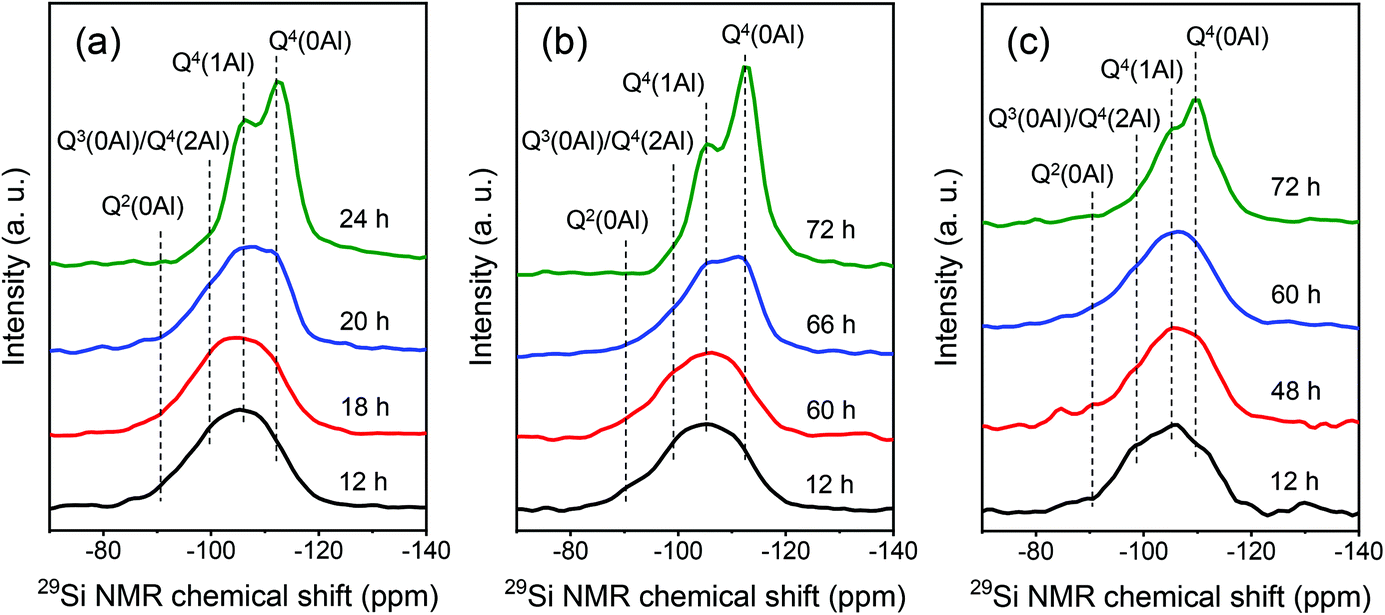
TG-DTG measurements of MOR-SDA2–12 and BEA-SDA3–12 were performed to investigate the interactions between the organic template and the aluminosilicate intermediates. The TG-DTG curves in Fig. 11 show three weight-loss steps. The first weight-loss feature below 250 °C can be attributed to the desorption of water.55 The other two weight-loss features in the 250–660 °C range are mainly due to the oxidation of organics at or close to the zeolite external surface and inside the zeolite micropores. Notably, already during the induction stage both MOR and BEA precursors contain a substantial amount of SDA (>8.6 wt%, Tables S6 and S7†), indicative of the strong interactions between the SDA and the aluminosilicate precursor species. Moreover, the third weight-loss feature becomes stronger and shifts to higher temperature during the crystallization stage. This trend is consistent with the assignment of this weight-loss feature to organics occluded in the micropores formed during crystallization. These results further underpin that SDA2 and SDA3 were included in the micropores during SDA-directed synthesis of MOR and BEA zeolite.
 | ||
| Fig. 11 TG (left) and DTG (right) curves of samples obtained at different crystallization times: (a and b) MOR-SDA2–12 and (c and b) BEA-SDA3–12. | ||
2D 1H–29Si HETCOR NMR spectroscopy measurements were carried out to investigate the zeolite-SDA interaction in more detail. This allows resolving short-range (<1 nm) interactions between 29Si and 1H spins.66–68 The 29Si projection of the spectrum in Fig. 12a contains two main peaks around −106 and −113 ppm, which are due to Q4(1Al) and Q4(0Al) zeolite framework species,57 respectively. The 1H projection contains three peaks, which can all be correlated to SDA2. The 1H peaks in Fig. 12a were identified by recording separate 1H NMR spectrum (Fig. S1b†), 1H–13C CPMAS NMR spectrum (Fig. S5b†) and 1H–13C HETCOR NMR spectrum (Fig. S10a†). The 1H signals at 3.7 ppm and 5.1 ppm are strongly correlated with the signals due to Q4(1Al) and Q4(0Al) framework species, resulting in 4 distinct peaks in the 2D spectrum. Besides, a weak cross-peak between a feature at 2.2 ppm in 1H dimension and a feature at −114 ppm in the 29Si dimension is present in the 2D spectrum. Similarly, strong correlation signals between features assigned to the various protons of SDA3 and Si atoms of the BEA zeolite framework can be distinguished in the 2D NMR spectrum in Fig. 12b. These NMR data further underpin that SDA2 and SDA3 are predominantly present in the micropores of respectively MOR and BEA zeolite.
 | ||
| Fig. 12 1H–29Si HETCOR NMR spectra of as-prepared zeolites: (a) MOR-SDA2–12 and (b) BEA-SDA3–12 (assignment to different H atoms in SDA in top panels). | ||
3.3 Acidity
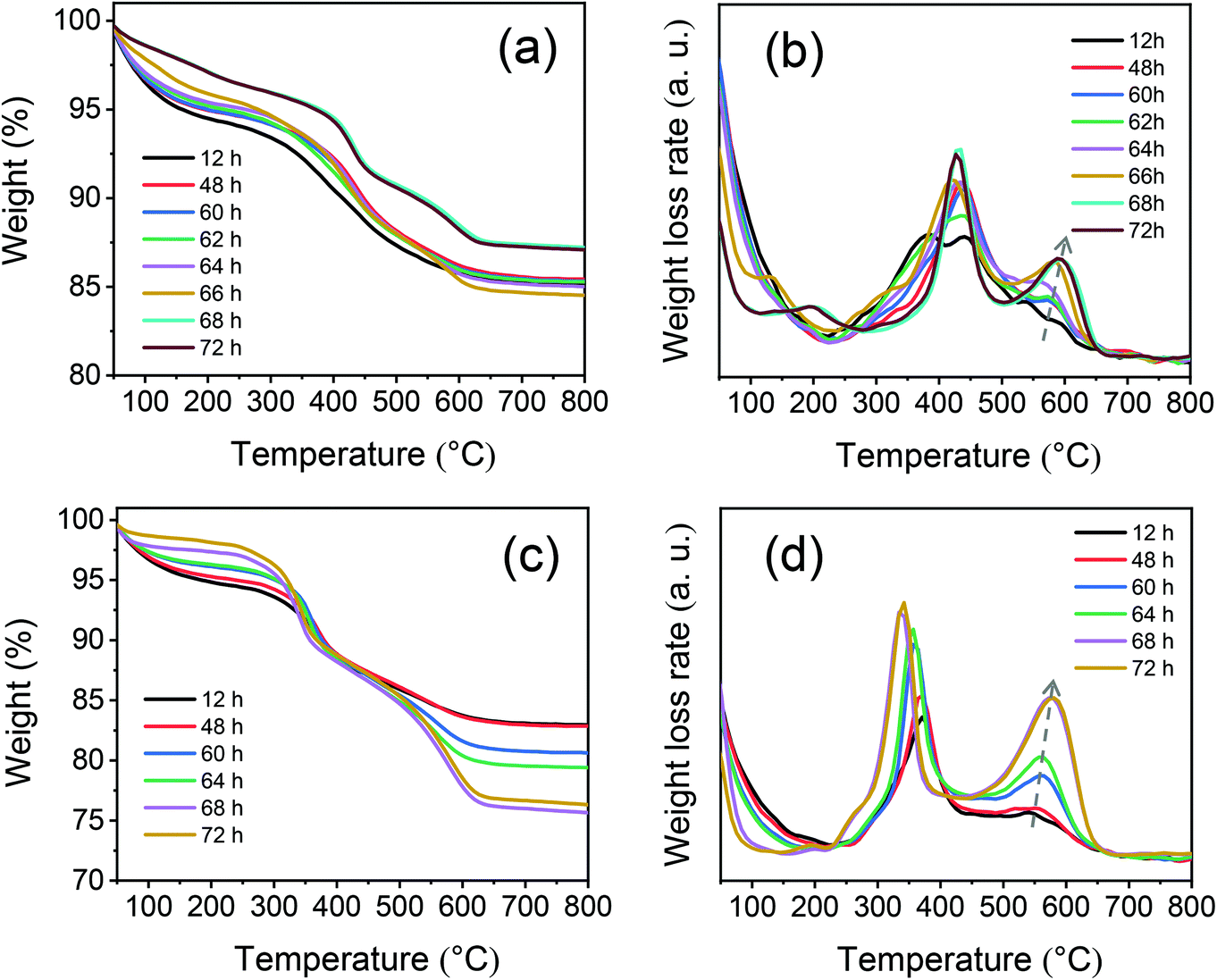
Next, the acidic properties of the calcined zeolites in their proton form were characterized. The Si/Al ratios of MOR-12-Con and MOR-SDA2–12 of respectively 8.4 and 9.2 (Table 2) are both lower than the Si/Al ratio in the initial gels. The Si/Al ratios of BEA-12-Con and BEA-SDA3–12 of 11.3 and 11.7, on the other hand, are close to the initial gel ratio. The Al coordination was investigated by 27Al MAS NMR spectroscopy. The 27Al NMR spectra in Fig. 13a exhibit a strong signal at ∼54 ppm assigned to tetrahedrally coordinated Al species in the zeolite framework (AlF), while the presence of a comparatively weak signal at ∼0 ppm is attributed to extraframework Al species in octahedral coordination.21,67 Deconvoluting these spectra shows that most Al atoms are incorporated into the zeolite framework (Table S8†). The Si coordination environment was studied by 29Si MAS NMR spectroscopy. The relevant spectra in Fig. 13b consist of 4 signals due to asymmetric Q4(0Al), symmetric Q4(0Al), Q4(1Al) and Q4(2Al)/Q3(0Al) sites.57 The fractional contributions obtained by deconvolution were used to calculate the Si/Al ratio of the zeolite framework. As shown in Table S8,† MOR-12-Con has a lower framework Si/Al ratio (6.8) than MOR-SDA2–12 (8.5), while similar framework Si/Al ratios of 10.5 and 10.3 were determined for BEA-12-Con and BEA-SDA3–12, respectively. Notably, the Si/AlF ratios determined by 29Si NMR are lower than the bulk Si/Al ratios. The reason for this discrepancy is that the Q3(0Al) and Q4(2Al) signals strongly overlap. For deconvolution of these spectra, we assumed that there are no Q3(0Al) sites, which can explain the lower Si/AlF ratios. Such a systematic difference when using this assumption has been discussed by others.57,69 The presence of Q3(0Al) in these samples is supported by the relatively higher intensity of the Q4(2Al)/Q3(0Al) sites in comparison to Q4(0Al) sites in the 1H–29Si CPMAS NMR spectra when contrasted to 29Si MAS NMR spectra (Fig. S11†).69,70 | ||
| Fig. 13 (a) 27Al MAS NMR and (b) 29Si MAS NMR spectra of the calcined zeolites. | ||
| Zeolite | Si/Al (ICP) | [BAS]a (μmol g−1) | [BAS]b (μmol g−1) | [LAS]c (μmol g−1) |
|---|---|---|---|---|
| a Concentration of BAS determined by 1H MAS NMR spectra. b Concentration of BAS determined by IR spectra of adsorbed pyridine after evacuation for 1 h at 150 °C. c Concentration of LAS determined by IR spectra of adsorbed pyridine after evacuation for 1 h at 150 °C. | ||||
| MOR-12-Con | 8.4 | 1291 | 1015 | 41 |
| MOR-SDA2–12 | 9.2 | 841 | 753 | 120 |
| BEA-12-Con | 11.3 | — | 961 | 378 |
| BEA-SDA3–12 | 11.7 | — | 704 | 484 |
The acidic properties of zeolites were characterized by IR spectroscopy after adsorption of pyridine and subsequent evacuation at 150 °C, 300 °C and 500 °C. The relevant spectra given in Fig. S12† contain two bands at 1455 cm−1 and 1545 cm−1 due to pyridine adsorbed on Lewis acid sites (LAS) and Brønsted acid sites (BAS),45,71 respectively. The amount of BAS and LAS based on spectra obtained after evacuation at 150 °C are given in Table 2. For BEA zeolites, the corresponding numbers represent the total acidic concentration, because all the pores are accessible for pyridine. As shown in Table 2, BEA-SDA3–12 displays a lower BAS concentration but a higher LAS concentration as compared to BEA-12-Con. For the MOR zeolite, pyridine is too large to probe all of the acid sites. According, we first determined the total acidity for the MOR zeolites using 1H MAS NMR spectroscopy of well-dehydrated samples (Fig. S13† and Table 2). Compared with MOR-12-Con, MOR-SDA2–12 presents a lower total BAS concentration.
It has been demonstrated that there is a distribution of BAS in MOR zeolites related to their location in 12MR straight channels, 8MR side pockets and at the interface of these two locations (8/12MR).50,72,73 The different confinement in these pores can profoundly impact the catalytic performance. For instance, BAS in side-pockets are very active for the carbonylation of dimethyl ether to methyl acetate due to effective stabilization of acetyl intermediates in 8MR channels, whereas BAS in 12MR channels mainly lead to the formation of coke.74,75 Reactants involving bulky reactants mainly occur in 12MR channels and at the external surface.76 Therefore, the BAS distribution in MOR-12-Con and MOR-SDA2–12 were further investigated. The distribution of BAS was evaluated by deconvolution of the hydroxyl stretching region of IR spectra of the dehydrated zeolites.77 As shown in Fig. 14 right and Table S9,† MOR-SDA2–12 contains a higher fraction (64%) of side-pocket BAS as compared to MOR-12-Con (49%). The accessibility of BAS was investigated by tracing the evolution of IR bands assigned to BAS sites upon pyridine adsorption and subsequent desorption at different evacuation temperatures. Fig. 14 left shows that the relative decrease of the band due to BAS is larger for MOR-SDA2–12 as compared to MOR-12-Con. This indicates that more side-pocket BAS of MOR-SDA2–12 are accessible for pyridine. For both samples, the hydroxyl bands shift to higher wavenumbers upon pyridine desorption at 500 °C, suggesting that the recovery of BAS is mainly from 12MR channels and the 8/12MR interfaces.
 | ||
| Fig. 14 (Left) IR spectra of the calcined MOR zeolites: (a) after pyridine adsorption and evacuation at 150 °C, (b) evacuation at 300 °C, (c) evacuation at 500 °C and (d) hydroxyl region of IR spectra of MOR zeolites before pyridine adsorption; (right) deconvolution into contributions of hydroxyl groups due to 12MR, 8MR and 8/12MR interfaces as well as hydroxyl groups connected to EFAl species. | ||
3.4 Catalytic activity
It has been firmly established that pore hierarchization in microporous zeolites can substantially improve the catalytic performance.4,14 Reducing the crystal size of zeolites below 100 nm is one such approach that benefits reactions with bulky reactants and reactions in which competing consecutive reactions of products can lead to lower selectivity to desired products or catalyst deactivation.8 For instance, nanocrystalline zeolites perform better in Friedel–Crafts alkylation and acylation reactions than bulk zeolites.78 As nanosized MOR and BEA zeolites are promising for the alkylation of benzene with benzyl alcohol and the acylation of anisole with acetic anhydride, respectively,76,79 we evaluated the optimized nanosized MOR and BEA zeolites prepared in the present study using these two model reactions compared to the bulk reference zeolites. Pt/BEA zeolite was also evaluated for the hydroconversion of n-C16. Paraffins hydroisomerization and hydrocracking are industrially important reactions for hydrocarbon processing.80,81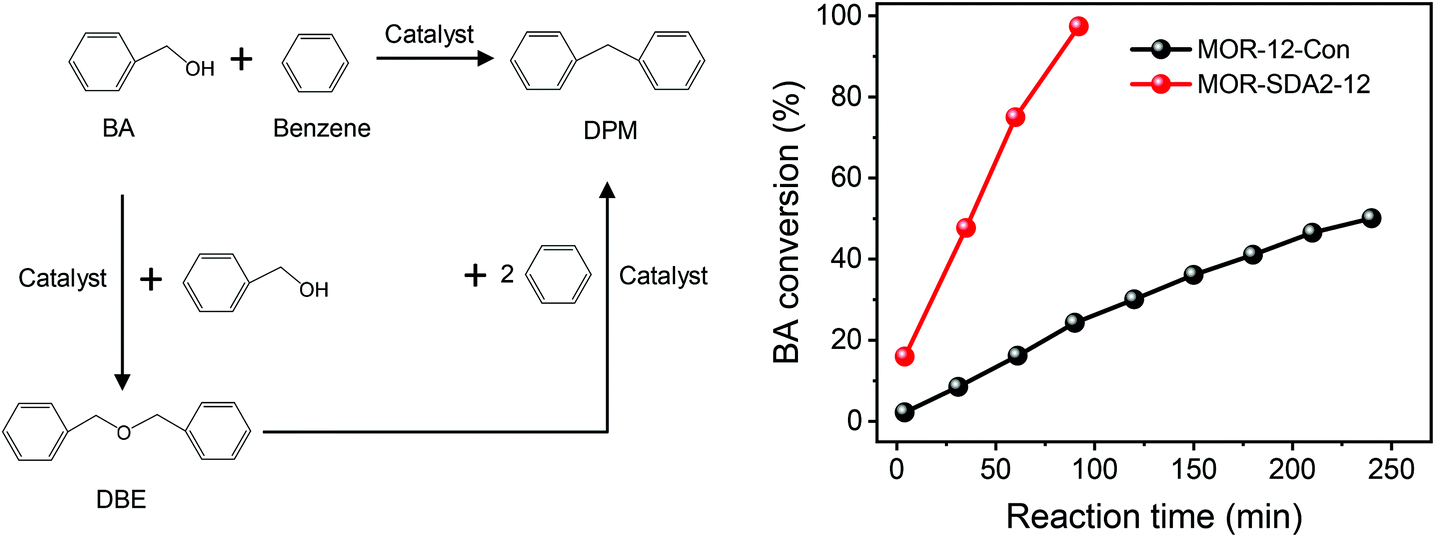 | ||
| Fig. 15 (Left) Reaction pathways for benzylation of benzene with benzyl alcohol (BA) and (right) catalytic conversion of BA over MOR zeolite samples at 80 °C. | ||
 | ||
| Fig. 16 (Left) The mechanism of anisole acylation and (right) catalytic conversion of acetic anhydride over BEA zeolite samples at 70 °C. | ||
Fig. 17a shows the conversion of n-C16 as a function of reaction temperature. Pt/BEA-SDA3–12 displays a lower reaction activity than Pt/BEA-12-Con. This can be attributed to the lower acidity of the nanosized sample in line with the activity in n-alkane hydroisomerization reactions being proportional to the intrinsic acidity.88 It is also common that pore hierarchization of BEA zeolite impedes the amount of BAS and, thus, the activity for hydroconversion of n-alkanes.89 Astafan et al. reported that the activity of n-C16 hydroconversion over Pt/BEA was proportional to the concentration of BAS probed by pyridine, regardless of the zeolite crystal size.90 This can most likely be explained by the fact that the n-C16 hydroconversion reaction is not limited by mass transport limitations under the given reaction conditions. Fig. 17b shows the differences in the product distribution between the two catalysts. For both catalysts, the yield of isomers gradually increases with reactant conversion until a maximum is reached, after which cracking becomes the dominant route. The origin of this dependence of the product distribution is well understood in terms of skeletal isomerization of linear alkanes, leading to multibranched isomers whose olefinic counterparts are easier to crack.81 Notably, the maximum isomer yield of 29.3% for Pt/BEA-SDA3–12 is much higher than the corresponding yield of 14.6% for Pt/BEA-12-Con. The work of Astafan et al. showed that the maximum yield of isomer can be correlated to the BEA zeolite crystal size.90 Reducing the zeolite crystal size reduces the residence time of olefinic intermediates within the zeolite domains and, therefore, limits consecutive reactions.9 As the activity comparison excludes diffusion limitations, it is more likely that the selectivity is improved by the larger external surface, which benefits the desorption of intermediate olefins from the zeolite domains.10,91 Thus, the higher yield of isomers over Pt/BEA-SDA2–12 can be explained by the smaller crystal size. The beneficial effect of nanosizing BEA zeolite is also clear from the more symmetric cracked product distribution for Pt/BEA-SDA2–12 (Fig. S15†).
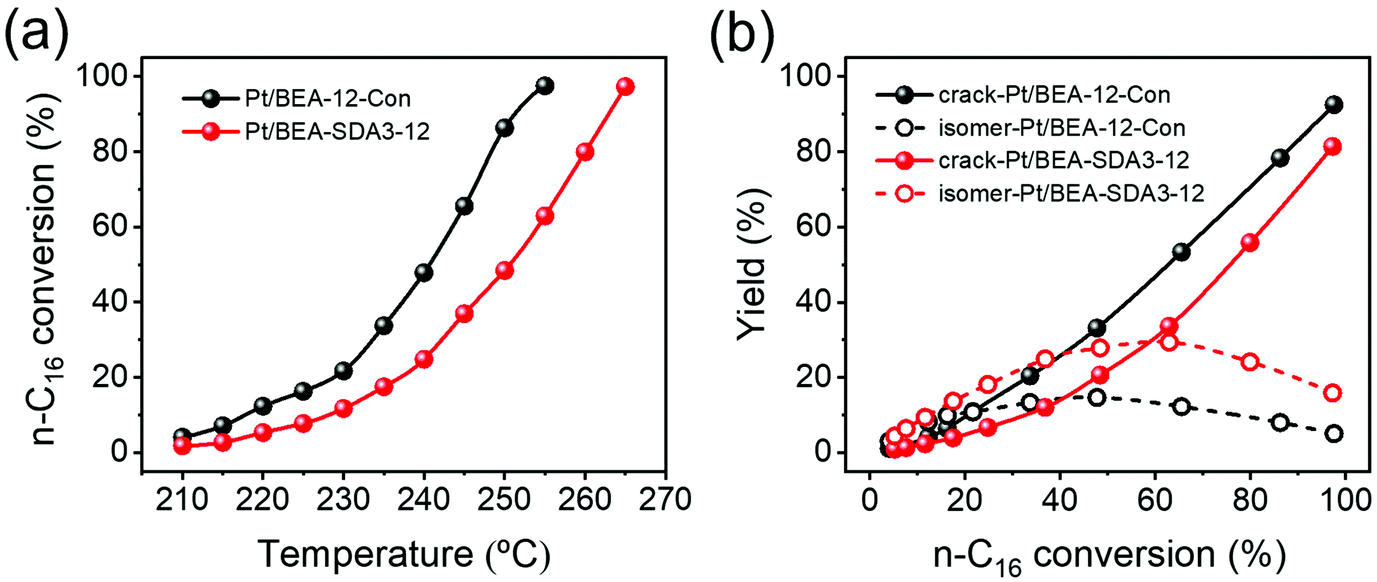 | ||
| Fig. 17 (a) Conversion of n-C16 as a function of the reaction temperature and (b) yield of isomers and cracked hydrocarbon products as a function of the n-C16 conversion. | ||
4. Conclusions
This work describes the facile and economical synthesis of nanosized MOR and BEA zeolites by using simple bromide-form diquaternary ammonium compound as the sole organic template. The specific formation of MOR or BEA zeolites strongly depends on the size, geometry, and structural rigidity of organic templates. SDA2 and SDA6 give MOR zeolites, while SDA3–5 give BEA zeolites. The optimized nanosized zeolites are MOR-SDA2–12 (Si/Al = 9.2, 20–50 nm) and BEA-SDA3–12 (Si/Al = 11.7, 15–30 nm). While bulk MOR-12-Con (>100 nm) can be obtained within 24 h from a completely inorganic gel, the strong interaction between organic templates and aluminosilicate precursors results in an extended induction period (∼48 h) for SDA2 and SDA3, effectively decreasing the aluminosilicate precursor size below 50 nm. MOR-12-Con and MOR-SDA2–12 display a similar crystal growth behavior, viz. a quick crystal growth (∼6 h) via reorganization in the solid state. Specific strong framework stabilization by the pore-filling template molecules is decisive for the final zeolite topology. Nanocrystalline MOR-SDA2–12 and BEA-SDA3–12 are more active than their corresponding bulk counterparts in the alkylation of benzene with benzyl alcohol and the acylation of anisole with acetic anhydride, respectively, due to the enhanced mass transport ability and better accessibility of the Brønsted acid sites. In hydroconversion of n-C16, Pt/BEA-SDA3–12 shows much higher isomer yield than Pt/BEA-12-Con, which is explained by shorter residence times of olefinic intermediates in the smaller zeolite crystals. Overall, this work not only shows a facile route to synthesize nanosized MOR and BEA zeolites with excellent catalytic performance by using simple organic molecules, but also provides better insight into the crystallization behavior.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Shaojie Li acknowledges financial support from the China Scholarship Council. The authors thank A. M. Elemans-Mehring for ICP-OES analysis and Brahim Mezari for valuable discussions.References
- J. Li, A. Corma and J. Yu, Synthesis of new zeolite structures, Chem. Soc. Rev., 2015, 44, 7112–7127 RSC
.
- N. Kosinov, J. Gascon, F. Kapteijn and E. J. M. Hensen, Recent developments in zeolite membranes for gas separation, J. Membr. Sci., 2016, 499, 65–79 CrossRef CAS
- J. Přech, P. Pizarro, D. P. Serrano and J. Čejka, From 3D to 2D zeolite catalytic materials, Chem. Soc. Rev., 2018, 47, 8263–8306 RSC
- D. Kerstens, B. Smeyers, J. Van Waeyenberg, Q. Zhang, J. Yu and B. F. Sels, State of the Art and Perspectives of Hierarchical Zeolites: Practical Overview of Synthesis Methods and Use in Catalysis, Adv. Mater., 2020, 32, 2004690 CrossRef CAS PubMed
- X.-Y. Yang, L.-H. Chen, Y. Li, J. C. Rooke, C. Sanchez and B.-L. Su, Hierarchically porous materials: synthesis strategies and structure design, Chem. Soc. Rev., 2017, 46, 481–558 RSC
- S. Mintova, J.-P. Gilson and V. Valtchev, Advances in nanosized zeolites, Nanoscale, 2013, 5, 6693–6703 RSC
- L.-H. Chen, M.-H. Sun, Z. Wang, W. Yang, Z. Xie and B.-L. Su, Hierarchically Structured Zeolites: From Design to Application, Chem. Rev., 2020, 120, 11194–11294 CrossRef CAS PubMed
- S. Mintova, M. Jaber and V. Valtchev, Nanosized microporous crystals: emerging applications, Chem. Soc. Rev., 2015, 44, 7207–7233 RSC
- L. Meng, G. Vanbutsele, R. Pestman, A. Godin, D. E. Romero, A. J. van Hoof, L. Gao, T. F. Kimpel, J. Chai, J. A. Martens and E. J. M. Hensen, Mechanistic aspects of n-paraffins hydrocracking: Influence of zeolite morphology and acidity of Pd (Pt)/ZSM-5 catalysts, J. Catal., 2020, 389, 544–555 CrossRef CAS
- T. Blasco, A. Chica, A. Corma, W. J. Murphy, J. Agúndez-Rodríguez and J. Pérez-Pariente, Changing the Si distribution in SAPO-11 by synthesis with surfactants improves the hydroisomerization/dewaxing properties, J. Catal., 2006, 242, 153–161 CrossRef CAS
- J. Wang, S. Xu, J. Li, Y. Zhi, M. Zhang, Y. He, Y. Wei, X. Guo and Z. Liu, An approach to prepare nanosized HZSM-22 with enhanced lifetime in the methanol to hydrocarbon (MTH) reaction, RSC Adv., 2015, 5, 88928–88935 RSC
- A. Corma, U. Diaz, M. E. Domine and V. Fornés, AlITQ–6 and TiITQ–6: Synthesis, Characterization, and Catalytic Activity, Angew. Chem., Int. Ed., 2000, 39, 1499–1501 CrossRef CAS
- A. Corma, V. Fornes, S. Pergher, T. L. Maesen and J. J. N. Buglass, Delaminated zeolite precursors as selective acidic catalysts, Nature, 1998, 396, 353–356 CrossRef CAS
- K. Na, M. Choi and R. Ryoo, Recent advances in the synthesis of hierarchically nanoporous zeolites, Microporous Mesoporous Mater., 2013, 166, 3–19 CrossRef CAS
- Z. Chen, Z. Li, Y. Zhang, D. Chevella, G. Li, Y. Chen, X. Guo, J. Liu and J. Yu, A green route for the synthesis of nano-sized hierarchical ZSM-5 zeolite with excellent DTO catalytic performance, Chem. Eng. J., 2020, 388, 124322 CrossRef
- B. O. Hincapie, L. J. Garces, Q. Zhang, A. Sacco and S. L. Suib, Synthesis of mordenite nanocrystals, Microporous Mesoporous Mater., 2004, 67, 19–26 CrossRef CAS
- M. Ansari, A. Aroujalian, A. Raisi, B. Dabir and M. Fathizadeh, Preparation and characterization of nano-NaX zeolite by microwave assisted hydrothermal method, Adv. Powder Technol., 2014, 25, 722–727 CrossRef CAS
- A. Sakthivel, A. Iida, K. Komura, Y. Sugi and K. V. R. Chary, Nanosized β-zeolites with tunable particle sizes: Synthesis by the dry gel conversion (DGC) method in the presence of surfactants, characterization and catalytic properties, Microporous Mesoporous Mater., 2009, 119, 322–330 CrossRef CAS
- G. Zhang, Y. Fan, J. Huang, L. Wang, C. Yang, M. Lyu, H. Liu and Y. Ma, Decoupling nucleation from crystal-growth for the synthesis of nanocrystalline zeolites, Dalton Trans., 2020, 49, 7258–7266 RSC
- C. J. Van Oers, K. Góra-Marek, K. Sadowska, M. Mertens, V. Meynen, J. Datka and P. Cool, In situ IR spectroscopic study to reveal the impact of the synthesis conditions of zeolite β nanoparticles on the acidic properties of the resulting zeolite, Chem. Eng. J., 2014, 237, 372–379 CrossRef CAS
- M. H. Sun, L. H. Chen, S. Yu, Y. Li, X. G. Zhou, Z. Y. Hu, Y. H. Sun, Y. Xu and B. L. Su, Micron–sized zeolite Beta single crystals featuring intracrystal interconnected ordered macro–meso–microporosity displaying superior catalytic performance, Angew. Chem., Int. Ed., 2020, 59, 19582–19591 CrossRef CAS PubMed
- C. Jo, J. Jung, H. S. Shin, J. Kim and R. Ryoo, Capping with multivalent surfactants for zeolite nanocrystal synthesis, Angew. Chem., Int. Ed., 2013, 52, 10014–10017 CrossRef CAS PubMed
- H. Zhu, Z. Liu, D. Kong, Y. Wang and Z. Xie, Synthesis and Catalytic Performances of Mesoporous Zeolites Templated by Polyvinyl Butyral Gel as the Mesopore Directing Agent, J. Phys. Chem. C, 2008, 112, 17257–17264 CrossRef CAS
- T. Xue, S. Li and H. Wu, Surfactant-promoted synthesis of hierarchical zeolite ferrierite nano-sheets, Microporous Mesoporous Mater., 2021, 312, 110748 CrossRef CAS
- W. Kim, J.-C. Kim, J. Kim, Y. Seo and R. Ryoo, External Surface Catalytic Sites of Surfactant-Tailored Nanomorphic Zeolites for Benzene Isopropylation to Cumene, ACS Catal., 2013, 3, 192–195 CrossRef CAS
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. J. N. Ryoo, Stable single-unit-cell nanosheets of zeolite MFI as active and long-lived catalysts, Nature, 2009, 461, 246–249 CrossRef CAS PubMed
- W. Schwieger, A. G. Machoke, T. Weissenberger, A. Inayat, T. Selvam, M. Klumpp and A. Inayat, Hierarchy concepts: classification and preparation strategies for zeolite containing materials with hierarchical porosity, Chem. Soc. Rev., 2016, 45, 3353–3376 RSC
- Y. Xu, X. Shen, C. Peng, Y. Ma, L. Han, P. Wu, H. Peng and S. Che, Synthesis of ultra-small mordenite zeolite nanoparticles, Sci. China Mater., 2018, 61, 1185–1190 CrossRef CAS
- L. Ding and Y. Zheng, Effect of template concentration and gel dilution on crystallization and particle size of zeolite beta in the absence of alkali cations, Microporous Mesoporous Mater., 2007, 103, 94–101 CrossRef CAS
- E. M. Gallego, C. Li, C. Paris, N. Martín, J. Martínez-Triguero, M. Boronat, M. Moliner and A. Corma, Making nanosized CHA zeolites with controlled Al distribution for optimizing methanol-to-olefin performance, Chem. – Eur. J., 2018, 24, 14631–14635 CrossRef CAS PubMed
- X. Zhang, D. Liu, D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi and O. J. S. Terasaki, Synthesis of self-pillared zeolite nanosheets by repetitive branching, Science, 2012, 336, 1684–1687 CrossRef CAS PubMed
- E. M. Gallego, C. Paris, M. R. Díaz-Rey, M. E. Martínez-Armero, J. Martínez-Triguero, C. Martínez, M. Moliner and A. Corma, Simple organic structure directing agents for synthesizing nanocrystalline zeolites, Chem. Sci., 2017, 8, 8138–8149 RSC
- X. Jia, Y. Zhang, Z. Gong, B. Wang, Z. Zhu, J. Jiang, H. Xu, H. Sun, L. Han, P. Wu and S. Che, Bolaform Molecules Directing Intergrown Zeolites, J. Phys. Chem. C, 2018, 122, 9117–9126 CrossRef CAS
- Y. Zhao, Z. Ye, L. Wang, H. Zhang, F. Xue, S. Xie, X.-M. Cao, Y. Zhang and Y. Tang, Engineering Fractal MTW Zeolite Mesocrystal: Particle-Based Dendritic Growth via Twinning-Plane Induced Crystallization, Cryst. Growth Des., 2018, 18, 1101–1108 CrossRef CAS
- A. Molino, K. A. Łukaszuk, D. Rojo-Gama, K. P. Lillerud, U. Olsbye, S. Bordiga, S. Svelle and P. Beato, Conversion of methanol to hydrocarbons over zeolite ZSM-23 (MTT): exceptional effects of particle size on catalyst lifetime, Chem. Commun., 2017, 53, 6816–6819 RSC
- S. H. Keoh, W. Chaikittisilp, K. Muraoka, R. R. Mukti, A. Shimojima, P. Kumar, M. Tsapatsis and T. Okubo, Factors Governing the Formation of Hierarchically and Sequentially Intergrown MFI Zeolites by Using Simple Diquaternary Ammonium Structure-Directing Agents, Chem. Mater., 2016, 28, 8997–9007 CrossRef CAS
- A. Jackowski, S. I. Zones, S.-J. Hwang and A. W. Burton, Diquaternary Ammonium Compounds in Zeolite Synthesis: Cyclic and Polycyclic N-Heterocycles Connected by Methylene Chains, J. Am. Chem. Soc., 2009, 131, 1092–1100 CrossRef CAS PubMed
- X. Hong, W. Chen, G. Zhang, Q. Wu, C. Lei, Q. Zhu, X. Meng, S. Han, A. Zheng, Y. Ma, A.-N. Parvulescu, U. Müller, W. Zhang, T. Yokoi, X. Bao, B. Marler, D. E. De Vos, U. Kolb and F.-S. Xiao, Direct Synthesis of Aluminosilicate IWR Zeolite from a Strong Interaction between Zeolite Framework and Organic Template, J. Am. Chem. Soc., 2019, 141, 18318–18324 CrossRef CAS PubMed
- M. Moliner, F. Rey and A. Corma, Towards the rational design of efficient organic structure–directing agents for zeolite synthesis, Angew. Chem., Int. Ed., 2013, 52, 13880–13889 CrossRef CAS PubMed
- S. B. Hong, Use of flexible diquaternary structure-directing agents in zeolite synthesis: Discovery of zeolites TNU-9 and TNU-10 and their catalytic properties, Catal. Surv. Asia, 2008, 12, 131–144 CrossRef CAS
- H. Xue, X. Huang, E. Ditzel, E. Zhan, M. Ma and W. Shen, Dimethyl ether carbonylation to methyl acetate over nanosized mordenites, Ind. Eng. Chem. Res., 2013, 52, 11510–11515 CrossRef CAS
- S. Inagaki, Y. Watanabe, Y. Nishita and Y. Kubota, Synthesis of Mordenite Nanocrystals by Using a Hydrophobic Structure-directing Agent, Chem. Lett., 2013, 42, 186–188 CrossRef CAS
- K. Na, M. Choi and R. Ryoo, Cyclic diquaternary ammoniums for nanocrystalline BEA, MTW and MFI zeolites with intercrystalline mesoporosity, J. Mater. Chem., 2009, 19, 6713–6719 RSC
- K. Zhang, Z. Liu, X. Yan, X. Hao, M. Wang, C. Li and H. Xi, In Situ Assembly of Nanoparticles into Hierarchical Beta Zeolite with Tailored Simple Organic Molecule, Langmuir, 2017, 33, 14396–14404 CrossRef CAS PubMed
- R. Martínez-Franco, C. Paris, M. E. Martínez-Armero, C. Martínez, M. Moliner and A. Corma, High-silica nanocrystalline Beta zeolites: efficient synthesis and catalytic application, Chem. Sci., 2016, 7, 102–108 RSC
- R. Kore and R. Srivastava, Synthesis of zeolite Beta, MFI, and MTW using imidazole, piperidine, and pyridine based quaternary ammonium salts as structure directing agents, RSC Adv., 2012, 2, 10072–10084 RSC
- J. Zhang, P. Cao, H. Yan, Z. Wu and T. Dou, Synthesis of hierarchical zeolite Beta with low organic template content via the steam-assisted conversion method, Chem. Eng. J., 2016, 291, 82–93 CrossRef CAS
- L. Zhang, A. N. C. v. Laak, P. E. d. Jongh and K. P. d. Jong, Synthesis of large mordenite crystals with different aspect ratios, Microporous Mesoporous Mater., 2009, 126, 115–124 CrossRef CAS
- R. Kore, B. Satpati and R. Srivastava, Synthesis of dicationic ionic liquids and their application in the preparation of hierarchical zeolite Beta, Chem. – Eur. J., 2011, 17, 14360–14365 CrossRef CAS PubMed
- A. Bolshakov, D. E. Romero Hidalgo, A. J. F. van Hoof, N. Kosinov and E. J. M. Hensen, Mordenite nanorods prepared by an inexpensive pyrrolidine–based mesoporogen for alkane hydroisomerization, ChemCatChem, 2019, 11, 2803–2811 CrossRef CAS
- Y. Yuan, L. Wang, H. Liu, P. Tian, M. Yang, S. Xu and Z. Liu, Facile preparation of nanocrystal-assembled hierarchical mordenite zeolites with remarkable catalytic performance, Chin. J. Catal., 2015, 36, 1910–1919 CrossRef CAS
- J. Zhang, L. Wang, G. Wang, F. Chen, J. Zhu, C. Wang, C. Bian, S. Pan and F.-S. Xiao, Hierarchical Sn-Beta Zeolite Catalyst for the Conversion of Sugars to Alkyl Lactates, ACS Sustainable Chem. Eng., 2017, 5, 3123–3131 CrossRef CAS
- S. Storck, H. Bretinger and W. F. Maier, Characterization of micro- and mesoporous solids by physisorption methods and pore-size analysis, Appl. Catal., A, 1998, 174, 137–146 CrossRef CAS
- L. Meng, X. Zhu, W. Wannapakdee, R. Pestman, M. G. Goesten, L. Gao, A. J. F. van Hoof and E. J. M. Hensen, A dual-templating synthesis strategy to hierarchical ZSM-5 zeolites as efficient catalysts for the methanol-to-hydrocarbons reaction, J. Catal., 2018, 361, 135–142 CrossRef CAS
- K. Zhang, S. Luo, Z. Liu, C. Li, Z. Ke, X. Yan, Y. Wu and H. Xi, In Situ Fabrication of Hierarchical MTW Zeolite via Nanoparticle Assembly by a Tailored Simple Organic Molecule, Chem. – Eur. J., 2018, 24, 8133–8140 CrossRef CAS PubMed
- M. Pan, J. Zheng, Y. Ou, Q. Wang, L. Zhang and R. Li, A facile approach for construction of hierarchical zeolites via kinetics, Microporous Mesoporous Mater., 2021, 316, 110983 CrossRef CAS
- T. I. Korányi and J. B. Nagy, Distribution of Aluminum in Different Periodical Building Units of MOR and BEA Zeolites, J. Phys. Chem. B, 2005, 109, 15791–15797 CrossRef PubMed
- T. Ikuno, W. Chaikittisilp, Z. Liu, T. Iida, Y. Yanaba, T. Yoshikawa, S. Kohara, T. Wakihara and T. Okubo, Structure-Directing Behaviors of Tetraethylammonium Cations toward Zeolite Beta Revealed by the Evolution of Aluminosilicate Species Formed during the Crystallization Process, J. Am. Chem. Soc., 2015, 137, 14533–14544 CrossRef CAS PubMed
- W. Gao, C. C. Amoo, G. Zhang, M. Javed, B. Mazonde, C. Lu, R. Yang, C. Xing and N. Tsubaki, Insight into solvent-free synthesis of MOR zeolite and its laboratory scale production, Microporous Mesoporous Mater., 2019, 280, 187–194 CrossRef CAS
- S. Inagaki, K. Nakatsuyama, Y. Saka, E. Kikuchi, S. Kohara and M. Matsukata, Elucidation of Medium-Range Structure in a Dry Gel-Forming *BEA-Type Zeolite, J. Phys. Chem. C, 2007, 111, 10285–10293 CrossRef CAS
- J. Twu, P. K. Dutta and C. T. Kresge, Vibrational spectroscopic examination of the formation of mordenite crystals, J. Phys. Chem., 1991, 95, 5267–5271 CrossRef CAS
- X. Zhu, M. G. Goesten, A. J. J. Koekkoek, B. Mezari, N. Kosinov, G. Filonenko, H. Friedrich, R. Rohling, B. M. Szyja, J. Gascon, F. Kapteijn and E. J. M. Hensen, Establishing hierarchy: the chain of events leading to the formation of silicalite-1 nanosheets, Chem. Sci., 2016, 7, 6506–6513 RSC
- P. Van Geem, K. Scholle and G. Van der Velden, Study of the transformation of small-port into large-port mordenite by magic-angle spinning NMR and infrared spectroscopy, J. Phys. Chem., 1988, 92, 1585–1589 CrossRef CAS
- M. Cui, L. Wang, Y. Zhang, Y. Wang and C. Meng, Changes of medium-range structure in the course of crystallization of mordenite from diatomite, Microporous Mesoporous Mater., 2015, 206, 52–57 CrossRef CAS
- D. Zhao, W. Chu, Y. Wang, X. Zhu, X. Li, S. Xie, J. An, W. Xin, S. Liu and L. Xu, Organic promoter-driven fast synthesis of zeolite beta and its acceleration mechanism, J. Mater. Chem. A, 2018, 6, 24614–24624 RSC
- K. Na, C. Jo, J. Kim, K. Cho, J. Jung, Y. Seo, R. J. Messinger, B. F. Chmelka and R. Ryoo, Directing zeolite structures into hierarchically nanoporous architectures, Science, 2011, 333, 328–332 CrossRef CAS PubMed
- K. Lu, J. Huang, L. Ren, C. Li, Y. Guan, B. Hu, H. Xu, J. Jiang, Y. Ma and P. Wu, High ethylene selectivity in methanol–to–olefin (MTO) reaction over MOR nanosheets, Angew. Chem., Int. Ed., 2020, 59, 6258–6262 CrossRef CAS PubMed
- M. Kumar, Z. J. Berkson, R. J. Clark, Y. Shen, N. A. Prisco, Q. Zheng, Z. Zeng, H. Zheng, L. B. McCusker, J. C. Palmer, B. F. Chmelka and J. D. Rimer, Crystallization of Mordenite Platelets using Cooperative Organic Structure-Directing Agents, J. Am. Chem. Soc., 2019, 141, 20155–20165 CrossRef CAS PubMed
- H.-M. Kao, C.-Y. Yu and M.-C. Yeh, Detection of the inhomogeneity of Brønsted acidity in H-mordenite and H-β zeolites: a comparative NMR study using trimethylphosphine and trimethylphosphine oxide as 31P NMR probes, Microporous Mesoporous Mater., 2002, 53, 1–12 CrossRef CAS
- R. Hajjar, Y. Millot, P. P. Man, M. Che and S. Dzwigaj, Two kinds of framework Al sites studied in BEA zeolite by X-ray diffraction, Fourier transform infrared spectroscopy, NMR techniques, and V probe, J. Phys. Chem. C, 2008, 112, 20167–20175 CrossRef CAS
- X. Wang, R. Li, C. Yu, Y. Liu, L. Liu, C. Xu, H. Zhou and C. Lu, Influence of Acid Site Distribution on Dimethyl Ether Carbonylation over Mordenite, Ind. Eng. Chem. Res., 2019, 58, 18065–18072 CrossRef CAS
- M. Wang, S. Huang, J. Lü, Z. Cheng, Y. Li, S. Wang and X. Ma, Modifying the acidity of H–MOR and its catalytic carbonylation of dimethyl ether, Chin. J. Catal., 2016, 37, 1530–1538 CrossRef CAS
- Y. Li, M. Yu, K. Cai, M. Wang, J. Lv, R. F. Howe, S. Huang and X. Ma, Template-induced Al distribution in MOR and enhanced activity in dimethyl ether carbonylation, Phys. Chem. Chem. Phys., 2020, 22, 11374–11381 RSC
- Z. Cheng, S. Huang, Y. Li, K. Cai, Y. Wang, M.-y. Wang, J. Lv and X. Ma, Role of Brønsted Acid Sites within 8-MR of Mordenite in the Deactivation Roadmap for Dimethyl Ether Carbonylation, ACS Catal., 2021, 11, 5647–5657 CrossRef CAS
- A. Bhan, A. D. Allian, G. J. Sunley, D. J. Law and E. Iglesia, Specificity of sites within eight-membered ring zeolite channels for carbonylation of methyls to acetyls, J. Am. Chem. Soc., 2007, 129, 4919–4924 CrossRef CAS PubMed
- M.-N. Liu, Z.-X. Xie, Q.-X. Luo, J. Zhang, H. Chen, L. Xu, M. Sun, X. Ma and Q.-Q. Hao, Synthesis of Nanosized Mordenite with Enhanced Catalytic Performance in the Alkylation of Benzene with Benzyl Alcohol, Ind. Eng. Chem. Res., 2022, 61, 1078–1088 CrossRef CAS
- N. Cherkasov, T. Vazhnova and D. B. Lukyanov, Quantitative infra-red studies of Brønsted acid sites in zeolites: Case study of the zeolite mordenite, Vib. Spectrosc., 2016, 83, 170–179 CrossRef CAS
- G. Sartori and R. Maggi, Use of solid catalysts in Friedel−Crafts acylation reactions, Chem. Rev., 2006, 106, 1077–1104 CrossRef CAS PubMed
- E. G. Derouane, I. Schmidt, H. Lachas and C. J. H. Christensen, Improved Performance of Nano-Size H-BEA Zeolite Catalysts for the Friedel–Crafts Acetylation of Anisole by Acetic Anhydride, Catal. Lett., 2004, 95, 13–17 CrossRef CAS
- W. Wang, C.-J. Liu and W. Wu, Bifunctional catalysts for the hydroisomerization of n-alkanes: the effects of metal–acid balance and textural structure, Catal. Sci. Technol., 2019, 9, 4162–4187 RSC
- J. Weitkamp, Catalytic Hydrocracking—Mechanisms and Versatility of the Process, ChemCatChem, 2012, 4, 292–306 CrossRef CAS
- K. Leng, Y. Wang, C. Hou, C. Lancelot, C. Lamonier, A. Rives and Y. J. J. o. c. Sun, Enhancement of catalytic performance in the benzylation of benzene with benzyl alcohol over hierarchical mordenite, J. Catal., 2013, 306, 100–108 CrossRef CAS
- Y. Li, C. Sun, W. Fan, Y. Wang, A. Lan, P. Han, X. Li and T. Dou, One-pot synthesis of hierarchical mordenite and its performance in the benzylation of benzene with benzyl alcohol, J. Mater. Sci., 2015, 50, 5059–5067 CrossRef CAS
- M. Guidotti, C. Canaff, J.-M. Coustard, P. Magnoux and M. Guisnet, Acetylation of aromatic compounds over H-BEA zeolite: the influence of the substituents on the reactivity and on the catalyst stability, J. Catal., 2005, 230, 375–383 CrossRef CAS
- U. Freese, F. Heinrich and F. Roessner, Acylation of aromatic compounds on H-Beta zeolites, Catal. Today, 1999, 49, 237–244 CrossRef CAS
- E. G. Derouane, C. J. Dillon, D. Bethell and S. B. Derouane-Abd Hamid, Zeolite Catalysts as Solid Solvents in Fine Chemicals Synthesis: 1. Catalyst Deactivation in the Friedel–Crafts Acetylation of Anisole, J. Catal., 1999, 187, 209–218 CrossRef CAS
- G. Huang, P. Ji, H. Xu, J.-G. Jiang, L. Chen and P. Wu, Fast synthesis of hierarchical Beta zeolites with uniform nanocrystals from layered silicate precursor, Microporous Mesoporous Mater., 2017, 248, 30–39 CrossRef CAS
- E. J. M. Hensen, D. G. Poduval, V. Degirmenci, D. A. J. M. Ligthart, W. Chen, F. Maugé, M. S. Rigutto and J. A. R. van Veen, Acidity Characterization of Amorphous Silica–Alumina, J. Phys. Chem. C, 2012, 116, 21416–21429 CrossRef CAS
- H. Sammoury, J. Toufaily, K. Cherry, Y. Pouilloux, T. Hamieh and L. Pinard, Impact of Chain Length on the Catalytic Performance in Hydroisomerization of n-Alkanes Over Commercial and Alkaline Treated* BEA Zeolites, Catal. Lett., 2018, 148, 3051–3061 CrossRef CAS
- A. Astafan, Y. Pouilloux, J. Patarin, N. Bats, C. Bouchy, T. Jean Daou and L. Pinard, Impact of extreme downsizing of *BEA-type zeolite crystals on n-hexadecane hydroisomerization, New J. Chem., 2016, 40, 4335–4343 RSC
- J. Zecevic, G. Vanbutsele, K. P. De Jong and J. A. Martens, Nanoscale intimacy in bifunctional catalysts for selective conversion of hydrocarbons, Nature, 2015, 528, 245–248 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qi00696k |
| This journal is © the Partner Organisations 2022 |