
A multifunctional material with distinct mechanochromic and piezochromic properties: π-stacking in play†
Zeyang
Ding
ab,
Tong
Lu
b,
Changjiang
Bi
b,
Bao
Li
 b,
Shi-Tong
Zhang
b,
Weiqing
Xu
b and
Shimei
Jiang
*ab
b,
Shi-Tong
Zhang
b,
Weiqing
Xu
b and
Shimei
Jiang
*ab
aEngineering Research Center of Organic/Polymer Optoelectronic Materials, Ministry of Education, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China. E-mail: smjiang@jlu.edu.cn
bState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China
First published on 16th November 2021
Abstract
Controlling different types of forces to achieve materials with different emission responses remains a challenge. In this work, polymorphic organic crystals with blue (1-B) and green (1-G) emission based on N-(4-(1-cyano-2-phenylvinyl)phenyl)benzamide are prepared. In 1-G, the molecules form continuous π-stacking through π–π interactions, while no π–π interactions are found in 1-B. When shearing and UV irradiation are applied to 1-G, the emission blue shifts to a shorter wavelength region. Experimental investigations show that the shearing causes a crystal-to-crystal transition from 1-G to 1-B, while the UV irradiation promotes the occurrence of a photocycloaddition reaction. The external and internal factors contribute to the destruction of molecular π-stacking. In sharp contrast, under isotropic pressing, 1-G exhibits an obviously red-shifted emission owing to the formation of a tighter packing structure where the π–π interactions are reinforced. This work not only regulates molecular packing and controls emission variations in a versatile manner but also enriches the insights into mechanochromic and multifunctional materials.
Introduction
Organic solid-state luminescent materials, by virtue of their mysterious and complex photoelectronic nature in the excited state, have become a research hotspot.1–3 Their intrinsic multiple stimulus-responsive feature endows such materials with wide potential applications in the fields of sensors, optical recording, and security engineering.4–8 Upon application of external stimuli, such as mechanical force, heat, vapor, light, the emission intensity and color of materials exhibit remarkable changes owing to the variation of aggregation structures.9–13 Thus, the establishment of a “structure–function” relationship is the core pursuit of scientists. To date, most reported mechanisms of stimulus-responsive behaviors include molecular conformation or packing transformation, molecular chemical structure variation, etc.14–17 In particular, molecular π–π interactions, which can lower energy level effectively and promote intermolecular coupling, plays a significant role in emission modulation.18,19 Therefore, an effective means of adjusting molecular π-stacking is urgently demanded, which is not only of academic interest but also of the development of multiple stimulus-responsive luminescent materials.Mechanical force provides an efficient strategy to regulate molecular packing. Materials with emission changes in response to mechanical forces are known as mechanochromic or piezochromic materials, which are still in their infancy.20–25 Mechanical forces possess numerous species like shearing, tensioning, smashing and pressing. The difference in the direction and strength of various mechanical forces, for instance, anisotropic shearing and isotropic pressing, provides opportunity for diversity of molecular packing and emission regulation. However, most reported materials show consistent red-shifted emissions under anisotropic shearing and isotropic pressing owing to the conformation planarization, and increased interactions or charge transfer effect26–30 although many efforts are made to achieve the directional switching of the emission wavelength through tailoring of the molecular structure.22,31–34 Distinct emission response behaviors based on the same molecule remain rather scarce and worth investigating. For instance, in 2013, Yamaguchi et al. pioneered the distinct emission responses to anisotropic shearing and isotropic pressing based on 2,3,4,5-tetra(2-thiazolyl)thiophene molecule through regulating its dimeric structure.35 In 2018, Tian et al. reported another material with such a phenomenon by changing both π–π and charge transfer interactions.36 The inspiring results promote the development of mechanochromic materials, but it still lacks an intuitive understanding of which factor determines the emission changes. Another question that such materials meet is a crystal-to-amorphous transition after external stimuli in most cases.37–40 The amorphous powders can only offer an ambiguous explanation for molecular packing and emission changes, which is adverse to “structure–function” relationship exploration.
With the consideration of the above challenges, crystal, which can provide effective information on the critical factors that influence the emission of material, serves as an ideal candidate.41,42 As for the molecular design strategy, one is to guarantee strong emission in the solid state, while the other is to make the structure sensitive to environment.43–46 Herein, we modify the cyanostilbene skeleton with benzamide and prepare N-(4-(1-cyano-2-phenylvinyl)phenyl)benzamide (compound 1). The π-conjugate structure affords the possibility for π-stacking. The cyano group with steric hindrance and N atom with sp3 hybridization facilitate twisted conformation of the molecule, leading to a loose packing mode and benefiting phase transition upon external stimuli.47–49 As expected, compound 1 can form polymorphic crystals with blue and green emission (Scheme 1a). Under anisotropic shearing, the green emissive crystal shows a unique emission blue shift and enhancement (a transition to blue emissive crystal, crystal-to-crystal transition), while an emission red shift with decrease is discovered upon isotropic pressing (Scheme 1b). Furthermore, such green emissive crystals can also undergo similar blue-shifted emission upon UV irradiation and heating. Distinct emission variations are attributed to the destruction and reinforce of molecular π–π interactions, respectively. Such multifunctional materials reflect the significance of molecular π-stacking and enrich the scope of mechanochromic materials.
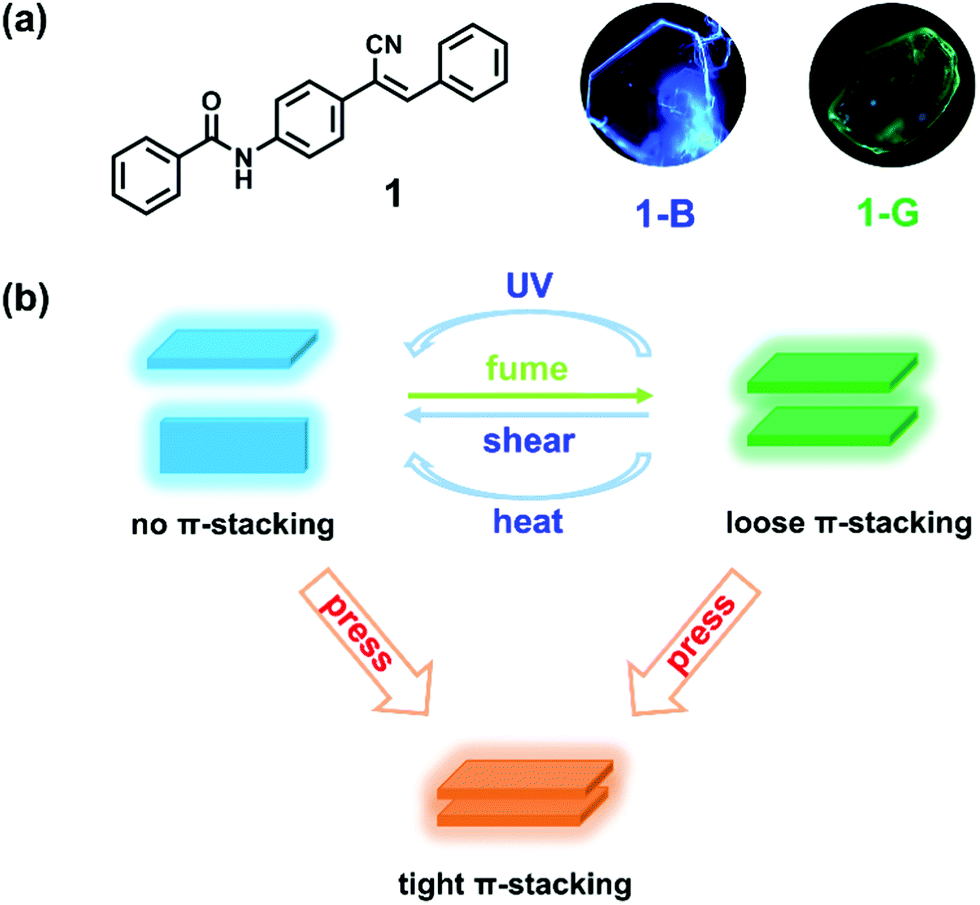 | ||
| Scheme 1 (a) Chemical structure of compound 1 and fluorescence microscopic images of its polymorphic crystals: 1-B and 1-G. (b) Schematic representation of π-stacking regulation and tunable luminescence under multifunctional stimuli. | ||
Results and discussion
N-(4-(1-Cyano-2-phenylvinyl)phenyl)benzamide (compound 1) was synthesized by introducing benzoyl into cyanostilbene skeleton according to our previous work (Fig. S1 and S2, ESI†).50 In view of the rotatable single bond (C–C, C–N), single crystals of compound 1 were cultivated to find the possibility of polymorphs. Fortunately, blue emissive plate crystals (1-B, λem = 441 nm) and green emissive crystals with bulk and sheet shapes (1-G, λem = 486 nm) were obtained through vacuum deposition and solvent evaporation methods (Scheme 1a, Fig. 1a and Fig. S3, ESI†). Detailed processes of crystal growth are presented in the ESI.† Differential scanning calorimetry (DSC) results revealed that 1-B and 1-G both displayed the same strong endothermic peak at about 228 °C, corresponding to the melting points of compound 1 (Fig. S4, ESI†). A small endothermic peak at 213 °C was also noticed in the curve of 1-G, which can be considered as the phase transformation temperature.51 However, such temperature was not found in the curve of 1-B, revealing a more stable thermodynamic state of 1-B. In addition to the different emission colors of the two polymorphs, there was also great distinction in the fluorescence quantum yield of 1-B (φF = 31%) and 1-G (φF = 5%), which may be ascribed to the different molecular packing. Detailed photophysical data of 1-B and 1-G are listed in Table S1 (ESI†). | ||
| Fig. 1 (a) Fluorescence spectra and (b) XRD patterns of 1-B and 1-G. | ||
To understand the molecular packing of 1-B and 1-G as a whole, X-ray diffraction (XRD) experiments were performed by placing the two polymorphs on the substrate surface horizontally. Sharp and high diffraction peaks of both were noticed, indicating well-ordered crystalline structures (Fig. 1b). Also, such patterns of the two polymorphs displayed obvious layer structures.52 According to single-crystal X-ray diffraction results, the upper surfaces of crystals were considered to be (200) face and (002) for 1-B and 1-G, respectively. The distance of the layer structure in 1-B was calculated to be 18.7 Å, while that in 1-G was calculated to be 18.6 Å (Fig. S5, ESI†). Both distances were larger than the length of molecule (17.7 Å, Fig. S6, ESI†), suggesting a loose layer structure in 1-B and 1-G. Such loose structures were conducive to the regulation of molecular packing upon external stimuli.
Shearing force was applied to the two polymorphs by grinding. No change was observed in 1-B, indicating that 1-B was stable enough to resist external shearing force. However, upon grinding 1-G, an obvious blue-shifted emission from green (λem = 486 nm) to blue (λem = 448 nm) was observed (Fig. 2a and b). Meanwhile, the quantum yield increased from 5% to 25% dramatically. More interestingly, both emission wavelength and quantum yield of the resultant ground solids were similar to those of 1-B (440 nm, 31%), implying a possible crystal-to-crystal transition. To confirm the structure transformation, XRD was employed again. After grinding, the diffraction peaks broadened and some new peaks appeared in the pattern of the ground solids (1-G-ground), suggesting the molecular arrangement in 1-G underwent reorganization to a new crystallization state. Such a pattern of 1-G-ground was full matched with the simulated one from 1-B, revealing the same packing structure between 1-G-ground and 1-B, as well as a crystal-to-crystal transition from 1-G to 1-B (Fig. 2c).53,54 After treating the ground solids (1-G-ground) with THF vapor, not only the emission wavelength was restored to 475 nm but also the XRD pattern was transformed. The recovered XRD pattern was almost in accordance with that of the simulated one of 1-G, indicating a reversible transition between the two polymorphs. The solvent vapors probably play a role in dissolution of molecules, and followed by recrystallization.14 It is noticed that heat failed to induce the reverse transition because the ground solids (1-G-ground) were stable after heating. On the contrary, by heating the initial 1-G at 215 °C for 30 minutes and cooling down to room temperature naturally, a cracked crystal with blue-shifted emission to 439 nm was obtained, demonstrating heat can only affect the packing structure of 1-G (Fig. S7, ESI†).
 | ||
| Fig. 2 (a) Fluorescence images and (b) fluorescence spectra of 1-G after grinding and subsequent fuming. (c) XRD patterns of 1-G after grinding (1-G-ground) and subsequent fuming (1-G-ground-fumed), and the simulated results of 1-B and 1-G. (d) Schematic diagram of the anisotropic grinding behavior on 1-G. | ||
To get insights into the structure transformation of the two polymorphs (1-B and 1-G), the molecular conformation and interactions in their crystal structures were investigated. Detailed crystallographic data are listed in Table S2 (ESI†). 1-B held the monoclinic crystal system and belonged to the Cc space group. The molecule conformation was twisted in the vinyl part (dihedral angle, 51.9°) but planar in the amide part (dihedral angle, 16.5°) (Fig. S6, ESI†). Each molecule interacted with neighbouring molecules through multiple C–H⋯π interactions (from 2.63 to 3.17 Å), and one type of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H (3.03 Å) and two types of C
O⋯H (3.03 Å) and two types of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N⋯H (1.65, 2.37 Å) hydrogen bonds (Fig. 3a and b). Two types of interactions, hydrogen bonds and C–H⋯π, extended along almost orthogonal directions to form two kinds of molecular columns (Fig. 3c). It is worth noting that no π–π stacking between molecules was observed in 1-B. As for 1-G, which belonged to the monoclinic crystal system, the P21/c space group, the molecule in 1-G was also highly twisted, which showed a twisted conformation in both the vinyl part (dihedral angle, 43.2°) and the amide part (dihedral angle, 40.6°) (Fig. S6, ESI†). Similar molecular column packing along orthogonal directions was observed in 1-G (Fig. 3d–f). In one molecular column, there also existed the hydrogen bonds, which were composed of one type of CO⋯H (2.57 Å) and two types of CN⋯H (2.29, 2.46 Å). However, in another one, weak π–π interactions (3.52 Å) served as the self-assembly driving forces. The continuous π–π interactions caused a perfect molecular π-stacking with slight slippage. The distance of π–π interactions in 1-G was longer than that of C–H⋯π interactions in 1-B, indicating 1-G was more vulnerable to external disturbance than 1-B.
N⋯H (1.65, 2.37 Å) hydrogen bonds (Fig. 3a and b). Two types of interactions, hydrogen bonds and C–H⋯π, extended along almost orthogonal directions to form two kinds of molecular columns (Fig. 3c). It is worth noting that no π–π stacking between molecules was observed in 1-B. As for 1-G, which belonged to the monoclinic crystal system, the P21/c space group, the molecule in 1-G was also highly twisted, which showed a twisted conformation in both the vinyl part (dihedral angle, 43.2°) and the amide part (dihedral angle, 40.6°) (Fig. S6, ESI†). Similar molecular column packing along orthogonal directions was observed in 1-G (Fig. 3d–f). In one molecular column, there also existed the hydrogen bonds, which were composed of one type of CO⋯H (2.57 Å) and two types of CN⋯H (2.29, 2.46 Å). However, in another one, weak π–π interactions (3.52 Å) served as the self-assembly driving forces. The continuous π–π interactions caused a perfect molecular π-stacking with slight slippage. The distance of π–π interactions in 1-G was longer than that of C–H⋯π interactions in 1-B, indicating 1-G was more vulnerable to external disturbance than 1-B.
 | ||
| Fig. 3 Molecular intermolecular interactions of compound 1 in (a–c) 1-B and (d–f) 1-G. | ||
Based on the crystal structure analysis, a possible transition was proposed (Fig. 2d). The grinding process provided pressing and shearing along different directions towards crystals of 1-G.35 Anisotropic shearing can make the molecular columns formed by the strong hydrogen bonds slide along the opposite directions.36 The weak π–π interactions in another molecular column were destroyed and replaced by the C–H⋯π interactions. We know that π–π interactions can promote intermolecular coupling and make the emission shift to the long wavelength region accordingly. Under external shearing stresses, the efficient π electron overlapped and intermolecular coupling weakened, leading to the unique emission blue shift and enhancement. A structural reorganization from 1-G (loose π-stacking) to 1-B (no π-stacking) was realized.
Unexpectedly, 1-G was also sensitive to UV light. A rapid blue-shifted emission of 1-G was observed in the fluorescence microscope owing to the UV irradiation (365 nm) (Fig. 4a). With the increase of irradiation time, the emission further blue shifted gradually until 449 nm (Fig. 4b). In view of the inherent photosensitivity of cyanostilbene skeleton in compound 1, a possible photochemical reaction would occur in this system. Our previous work proved that a trans–cis isomerization occurred in the solution containing compound 1 upon UV irradiation.50 Hence, 1H NMR spectroscopy measurements were carried out to examine the possible photochemical reaction in the solid state (Fig. 4c). The solid sample (1-G) was irradiated for 30 minutes and dissolved in DMSO-d6 for the 1H NMR test. In the spectrum of the irradiated solid sample (1-G), a series of new proton peaks appeared at high field, which was totally different from that of the irradiated solution, suggesting a different photochemical reaction. Particularly, a new proton peak appeared at 5.4 ppm, corresponding to the cyclobutyl proton, and showed an equal integral proportion with amide proton peak, indicating the formation of cycloadduct.55,56 These results indicated that the [2+2] photocycloaddition reaction occurred in the solid sample upon UV irradiation. The corresponding cycloadduct was further purified by column chromatography method and characterized by 1H NMR and mass spectrometry (Fig. S8 and S9, ESI†). The isolated cycloadduct displayed an emission peak at 467 nm, which was different from the sample after irradiation (1-G-light, 449 nm), implying that the blue-shifted emission did not derive from the cycloadduct, but from other species influenced by the photocycloaddition reaction (Fig. S10, ESI†).
 | ||
| Fig. 4 (a) Fluorescence microscopic images and (b) fluorescence spectra of 1-G under continuous UV irradiation at 365 nm. (c) 1H NMR spectra of the initial state (green line), the solid sample after irradiation (red line), and the solution sample after irradiation (blue line, comparative experiment). Triangle and rhombus represent the proton peaks of the newly generated product. | ||
It is noticed that 1-B failed to obtain any gains upon UV irradiation. According to the aforementioned crystal structure, the vinyl double bonds of neighbouring molecules in 1-G were perfectly parallel and the distance between them was calculated to be 3.95 Å, which was shorter than the maximum distance (4.2 Å) for photocycloaddition, confirming that such a reaction can take place and 1-G was photosensitive (Fig. S11, ESI†).57 While in 1-B, the neighbouring vinyl double bonds were crossed and the distance between them was about 5.38 Å, which was longer than 4.2 Å. Such unsatisfactory packing accounted for the photostability of 1-B. The photochemical reaction which would dissipate the excited energy may provide another piece of evidence for the low quantum yield (5%) of 1-G and higher one (31%) of 1-B.
Now the puzzle was why shearing stress and UV irradiation caused a consistent blue-shifted emission. According to the aforementioned discussion, the shearing stress induced emission variation was attributed to the slippage between neighbouring π-stacking molecules (Fig. 2d). Such external shearing destroyed the π–π interactions and influences the emission. As far as the irradiation experiment was concerned, the blue-shifted emission did not derive from the newly generated cycloadduct, but was similar to that of 1-B. To characterize the packing variation, the XRD pattern after irradiation was measured. There was no obvious change but the intensity weakened, expressing the decrease in the order (Fig. S12, ESI†). Meanwhile, a new diffraction peak at about 23.9° (3.72 Å) was observed, demonstrating the subtle change of molecular packing. Such distance was longer than that of the π–π interactions (3.52 Å) in 1-G, which may imply the breakage of molecular π-stacking. Thus, the UV irradiation can be seen as a kind of “internal shearing”, that is, the generation of the cycloadduct can exert shearing on neighbouring π-stacking molecules probably, leading to a strain in crystalline lattice.58 In such a case, the π-stacking was destroyed and the intermolecular coupling weakened, which would emit emission in the shorter wavelength region. In addition, such a kind of photomechanical idea was supported by the crystal deformation experiments upon UV irradiation (Fig. S13a and Supporting Video 1, ESI†). When the crystal of 1-G with thin sheet shape was irradiated, not only blue-shifted emission but also crystal bending was realized. The property was ascribed to the elongation in the side facing the light, where more cycloadducts enlarged the outmost intermolecular distance (Fig. S13b, ESI†).59–61 Such sheet crystals showed underlying applications in photoactuators.
Based on the above facts, whatever external shearing or light (internal shearing), uniform blue-shifted emissions were realized owing to the destruction of π-stacking. That is, the weak green emission of 1-G was attributed to the intermolecular coupling caused by π–π interactions between neighbouring molecules. This can be proved by the fluorescence lifetime of 1-G and 1-B (Fig. S14, ESI†). 1-G exhibited a longer fluorescence lifetime (4.33 ns) than that of 1-B (2.51 ns), implying the excimer emission in 1-G.35 To rationalize experimentally observed emission phenomena, time-dependent density functional theory (TD-DFT) calculations were carried out to calculate the HOMOs, LUMOs and vertical excitation energies (Fig. 5).62 The monomer and dimer model structures were extracted from 1-B and 1-G crystal, respectively. In the monomer of 1-B, the HOMO and LUMO were located at the same part. However, they were located at different molecules in the dimer of 1-G. The delocalization of the electron cloud density can give rise to the decrease of the band gap (from 3.637 eV to 3.332 eV) and red-shifted calculated excitation wavelength (from 340.9 nm to 372.1 nm, Δλ = 31 nm). Meanwhile, based on the experimental data, both 1-B and 1-G showed excitation peaks at 385 nm, which can be assigned as the monomer excitation (Fig. S15, ESI†). While 1-G showed an extra shoulder excitation peak at 412 nm, which can be assigned as the dimer excitation. The nearly 27 nm red shifts were in agreement with the calculated results, indicating the red-shifted emission of 1-G. The lower oscillator strength (f = 0.0098) in the dimer of 1-G in comparison to that (f = 1.1561) in the monomer of 1-B also manifested a poor emission intensity of 1-G. The results further evidenced that the destruction of π-stacking contributed to the blue-shifted emission accompanied by enhancement.
 | ||
| Fig. 5 Molecular orbitals, vertical excitation energies and calculated excitation wavelength of the monomer in 1-B and the dimer in 1-G. | ||
In comparison to the anisotropic shearing, isotropic hydrostatic pressure possessed more powerful strength and definite directions.28,63 Different mechanical forces may induce different modulation of molecular packing. The high-pressure experiments were performed using a diamond anvil cell, where silicone oil was added as the pressure transmitting medium. Both 1-B and 1-G exhibited significant red shifts in emission under hydrostatic pressure, as shown in Fig. 6 and Fig. S16 (ESI†), respectively. Take 1-B as the example, during the compression process, not only did the emission maximum peak shifted to a longer wavelength region but its intensity also decreased gradually (Fig. 6a). When the pressure reached 10.86 GPa, a nearly quenched emission peak at 583 nm was observed. Meanwhile, the emission of the crystal became dark orange, displaying a high-contrast emission regulation (Fig. 6c). Such a red shift in emission under isotropic hydrostatic pressure was opposite to the emission change caused by anisotropic shearing, implying different molecular packing changes under different forces. In the following decompression process, the emission wavelength of both can recover to the original state (Fig. 6b, Fig. S16 and S17, ESI†). The particular restorability in emission implied that the molecular packing underwent no remarkable changes under the hydrostatic pressure process. However, during the decompression process, more pressure release was needed to reach the original emission wavelength (Fig. S18, ESI†). For example, the emission wavelength reached 509 nm when 1-B was compressed at 8.46 GPa. When 1-B was decompressed from 10.86 GPa to 7.61 GPa, the emission wavelength can only go back to 568 nm. Such spectral residue may be ascribed to the hysteresis effect, that is, from high pressure to ordinary pressure, the molecules suffer from difficulty to their original state.
 | ||
| Fig. 6 Fluorescence spectra of 1-B under reversible (a) increasing and (b) releasing hydrostatic pressures. (c) Fluorescent images of a single crystal of 1-B under hydrostatic pressures. | ||
To provide evidence for packing structure variation under isotropic pressure, high-pressure Raman experiments were recorded (Fig. S19, ESI†). In the initial state, the peak at 1000 cm−1 was assigned as the ring breathing vibration of benzene.64 The peak at 1189 and 1202 cm−1 were attributed to the C–H rocking vibration in-plane.64,65 And the CN stretching vibration peak was observed at 2227 cm−1.66 With the increase of pressure, most peaks expressed obvious blue-shifts, suggesting the decrease of the intermolecular distances as well as the enhanced interactions (π–π and H-bonds). A tighter molecular packing was realized under high pressure.64–66
Putting the shearing and high-pressure experiments together, we can draw a clear picture that 1-G underwent distinct emission variations upon anisotropic shearing and isotropic hydrostatic pressure (Scheme 2). Such novel phenomena depended on the different changes of molecular packing without doubt. Shearing forces with opposite directions resulted in a slippage of molecular columns, which destroyed the π–π interactions and inhibited intermolecular coupling, contributing to the stronger emission in the shorter wavelength region. In sharp contrast, the hydrostatic pressure provided a compressed environment, inducing molecules to form a tighter packing mode, which would reinforce the π-stacking and shorten the distance of π–π interactions. In such cases, stronger intermolecular coupling occurred, leading to the weaker emission in the longer wavelength region.
 | ||
| Scheme 2 Schematic diagram of molecular packing transformation under anisotropic shearing and isotropic pressure. | ||
Conclusions
In summary, we prepared polymorphic crystals with blue (1-B) and green emission (1-G) based on a cyanostilbene derivative successfully. Between them, 1-G exhibited opposite emission responses to anisotropic shearing and isotropic pressure. Crystal analysis results revealed that weak π–π interactions appeared in 1-G, leading to a less stable packing structure with a low fluorescence quantum yield (λem = 486 nm, φF = 5%) while in 1-B, no π–π interactions but C–H···π interactions were observed, leading to a strong emission in the shorter wavelength region (λem = 441 nm, φF = 31%). A crystal-to-crystal transition from 1-G to 1-B was realized under external shearing, which can result in a slippage of neighbouring molecules. In such a case, the π–π interactions were destroyed and stronger C–H⋯π interactions replaced them. The process hence caused the blue-shifted emission accompanied by enhancement. Meanwhile, 1-G underwent a similar blue-shifted emission under UV irradiation owing to the [2+2] photocycloaddition reaction. The generation of a cycloadduct exerted shearing stresses on neighbouring π-stacking molecules, which can also destroy the π–π interactions and result in a blue-shifted emission. In sharp contrast, upon hydrostatic pressure to 1-B and 1-G, a tighter molecular packing was realized, which reinforced the π-stacking and shortened the distance of π–π interactions, contributing to weaker emission in longer wavelength region (λem = 583 nm). Therefore, in our system, by inhibiting and reinforcing the molecular π-stacking, further influencing the intermolecular coupling, different emission variations under various stimuli were realized. This work revealed universal molecular packing regulation rules for controlling emission of materials, which provided a significant guidance on the development of multifunctional materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52173167 and 21935005).Notes and references
- M. K. Bera, P. Pal and S. Malik, Solid-state emissive organic chromophores: design, strategy and building blocks, J. Mater. Chem. C, 2020, 8, 788–802 RSC
.
- Q. Li and Z. Li, Miracles of molecular uniting, Sci. China Mater., 2019, 63, 177–184 CrossRef
- C. Wang and Z. Li, Molecular conformation and packing: their critical roles in the emission performance of mechanochromic fluorescence materials, Mater. Chem. Front., 2017, 1, 2174–2194 RSC
- G. Huang, Q. Xia, W. Huang, J. Tian, Z. He, B. S. Li and B. Z. Tang, Multiple Anti-Counterfeiting Guarantees from a Simple Tetraphenylethylene Derivative - High-Contrasted and Multi-State Mechanochromism and Photochromism, Angew. Chem., Int. Ed., 2019, 58, 17814–17819 CrossRef CAS PubMed
- D. Kim, J. E. Kwon and S. Y. Park, Fully Reversible Multistate Fluorescence Switching: Organogel System Consisting of Luminescent Cyanostilbene and Turn-On Diarylethene, Adv. Funct. Mater., 2018, 28, 1706213 CrossRef
- Y. G. Shi, J. W. Wang, H. Li, G. F. Hu, X. Li, S. K. Mellerup, N. Wang, T. Peng and S. Wang, A simple multi-responsive system based on aldehyde functionalized amino-boranes, Chem. Sci., 2018, 9, 1902–1911 RSC
- S. W. Thomas, G. D. Joly and T. M. Swager, Chemical Sensors Based on Amplifying Fluorescent Conjugated Polymers, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed
- D. Yan and D. G. Evans, Molecular crystalline materials with tunable luminescent properties: from polymorphs to multi-component solids, Mater. Horiz., 2014, 1, 46–57 RSC
- H.-J. Kim, J. Gierschner and S. Y. Park, Tricolor fluorescence switching in a single component mechanochromic molecular material, J. Mater. Chem. C, 2020, 8, 7417–7421 RSC
- H. Liu, Z. Lu, K. Ye, Z. Zhang and H. Zhang, Polymorph-Dependent Luminescence Response to Acid Vapors and Its Application in Safety Protection of File Information, ACS Appl. Mater. Interfaces, 2019, 11, 34526–34531 CrossRef CAS PubMed
- Q. Qi, C. Li, X. Liu, S. Jiang, Z. Xu, R. Lee, M. Zhu, B. Xu and W. Tian, Solid-State Photoinduced Luminescence Switch for Advanced Anticounterfeiting and Super-Resolution Imaging Applications, J. Am. Chem. Soc., 2017, 139, 16036–16039 CrossRef CAS PubMed
- T. Yang, Y. Wang, X. Liu, G. Li, W. Che, D. Zhu, Z. Su and M. R. Bryce, Reversible tricolour luminescence switching based on a piezochromic iridium(iii) complex, Chem. Commun., 2019, 55, 14582–14585 RSC
- W. Zhang, X. Ji, B. J. Peng, S. Che, F. Ge, W. Liu, M. Al-Hashimi, C. Wang and L. Fang, High-Performance Thermoresponsive Dual-Output Dye System for Smart Textile Application, Adv. Funct. Mater., 2020, 30, 1906463 CrossRef CAS
- M. Echeverri, C. Ruiz, S. Gamez-Valenzuela, I. Martin, M. C. Ruiz Delgado, E. Gutierrez-Puebla, M. A. Monge, L. M. Aguirre-Diaz and B. Gomez-Lor, Untangling the Mechanochromic Properties of Benzothiadiazole-Based Luminescent Polymorphs through Supramolecular Organic Framework Topology, J. Am. Chem. Soc., 2020, 142, 17147–17155 CrossRef CAS PubMed
- Y. Gui, X. Yao, I. A. Guzei, M. M. Aristov, J. Yu and L. Yu, A Mechanism for Reversible Solid-State Transitions Involving Nitro Torsion, Chem. Mater., 2020, 32, 7754–7765 CrossRef CAS
- C. Z. Kuo, L. Y. Hsu, Y. S. Chen, K. Goto, S. Maity, Y. H. Liu, S. M. Peng, K. V. Kong, T. Shinmyozu and J. S. Yang, Alkyl
Chain Length- and Polymorph-Dependent Photomechanochromic Fluorescence of Anthracene: Photodimerization in Molecular Crystals: Role of the Lattice Stiffness, Chem. – Eur. J., 2020, 26, 11511–11521 CrossRef CAS PubMed
- Y. Wang, X. Tan, Y. M. Zhang, S. Zhu, I. Zhang, B. Yu, K. Wang, B. Yang, M. Li, B. Zou and S. X. Zhang, Dynamic behavior of molecular switches in crystal under pressure and its reflection on tactile sensing, J. Am. Chem. Soc., 2015, 137, 931–939 CrossRef CAS PubMed
- X. Du, F. Xu, M.-S. Yuan, P. Xue, L. Zhao, D.-E. Wang, W. Wang, Q. Tu, S.-W. Chen and J. Wang, Reversible luminescence color switching in the crystal polymorphs of 2,7-bis(2'-methyl-[1,1'-biphenyl]-4-yl)-fluorenone by thermal and mechanical stimuli, J. Mater. Chem. C, 2016, 4, 8724–8730 RSC
- M.-S. Yuan, D.-E. Wang, P. Xue, W. Wang, J.-C. Wang, Q. Tu, Z. Liu, Y. Liu, Y. Zhang and J. Wang, Fluorenone Organic Crystals: Two-Color Luminescence Switching and Reversible Phase Transformations between π–π Stacking-Directed Packing and Hydrogen Bond-Directed Packing, Chem. Mater., 2014, 26, 2467–2477 CrossRef CAS
- G. Chen and W. Hong, Mechanochromism of Structural-Colored Materials, Adv. Opt. Mater., 2020, 8, 2000984 CrossRef CAS
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Mechanoresponsive Luminescent Molecular Assemblies: An Emerging Class of Materials, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed
- J. Wu, Y. Cheng, J. Lan, D. Wu, S. Qian, L. Yan, Z. He, X. Li, K. Wang, B. Zou and J. You, Molecular Engineering of Mechanochromic Materials by Programmed C-H Arylation: Making a Counterpoint in the Chromism Trend, J. Am. Chem. Soc., 2016, 138, 12803–12812 CrossRef CAS PubMed
- Y. R. Girish, K. Prashantha and K. Byrappa, Recent advances in aggregation-induced emission of mechanochromic luminescent organic materials, Emergent Mater., 2021, 4, 673–724 CrossRef CAS
- S. Ito, Recent Advances in Mechanochromic Luminescence of Organic Crystalline Compounds, Chem. Lett., 2021, 50, 649–660 CrossRef CAS
- L. Wang, L. Liu, B. Xu and W. Tian, Recent Advances in Mechanism of AIE Mechanochromic Materials, Chem. Res. Chin. Univ., 2021, 37, 100–109 CrossRef CAS
- Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Piezochromic luminescence based on the molecular aggregation of 9,10-bis((E)-2-(pyrid-2-yl)vinyl)anthracene, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef CAS PubMed
- Q. Qi, J. Qian, X. Tan, J. Zhang, L. Wang, B. Xu, B. Zou and W. Tian, Remarkable Turn-On and Color-Tuned Piezochromic Luminescence: Mechanically Switching Intramolecular Charge Transfer in Molecular Crystals, Adv. Funct. Mater., 2015, 25, 4005–4010 CrossRef CAS
- X. Wang, C. Qi, Z. Fu, H. Zhang, J. Wang, H.-T. Feng, K. Wang, B. Zou, J. W. Y. Lam and B. Z. Tang, A synergy between the push–pull electronic effect and twisted conformation for high-contrast mechanochromic AIEgens, Mater. Horiz., 2021, 8, 630–638 RSC
- H. Yuan, K. Wang, K. Yang, B. Liu and B. Zou, Luminescence Properties of Compressed Tetraphenylethene: The Role of Intermolecular Interactions, J. Phys. Chem. Lett., 2014, 5, 2968–2973 CrossRef CAS PubMed
- B. Shao, R. Jin, A. Li, Y. Liu, B. Li, S. Xu, W. Xu, B. Xu and W. Tian, Luminescent switching and structural transition through multiple external stimuli based on organic molecular polymorphs, J. Mater. Chem. C, 2019, 7, 3263–3268 RSC
- R. Yoshii, K. Suenaga, K. Tanaka and Y. Chujo, Mechanofluorochromic materials based on aggregation-induced emission-active boron ketoiminates: regulation of the direction of the emission color changes, Chem. – Eur. J., 2015, 21, 7231–7237 CrossRef CAS PubMed
- B. Li, K. Seth, B. Niu, L. Pan, H. Yang and H. Ge, Transient-Ligand-Enabled ortho-Arylation of Five-Membered Heterocycles: Facile Access to Mechanochromic Materials, Angew. Chem., Int. Ed., 2018, 57, 3401–3405 CrossRef CAS PubMed
- T. Ishi-i, H. Tanaka, R. Youfu, N. Aizawa, T. Yasuda, S.-i. Kato and T. Matsumoto, Mechanochromic fluorescence based on a combination of acceptor and bulky donor moieties: tuning emission color and regulating emission change direction, New J. Chem., 2019, 43, 4998–5010 RSC
- S. Nagai, M. Yamashita, T. Tachikawa, T. Ubukata, M. Asami and S. Ito, Efficient and versatile mechanochromic luminescence of phenanthroimidazolylbenzothiadiazoles: tricolor switching and directional control over the chromism, J. Mater. Chem. C, 2019, 7, 4988–4998 RSC
- K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, Distinct responses to mechanical grinding and hydrostatic pressure in luminescent chromism of tetrathiazolylthiophene, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef CAS PubMed
- Y. Liu, Q. Zeng, B. Zou, Y. Liu, B. Xu and W. Tian, Piezochromic Luminescence of Donor-Acceptor Cocrystals: Distinct Responses to Anisotropic Grinding and Isotropic Compression, Angew. Chem., Int. Ed., 2018, 57, 15670–15674 CrossRef CAS PubMed
- X. Liu, A. Li, W. Xu, Z. Ma and X. Jia, An ESIPT-based fluorescent switch with AIEE, solvatochromism, mechanochromism and photochromism, Mater. Chem. Front., 2019, 3, 620–625 RSC
- Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, Material Design for Piezochromic Luminescence: Hydrogen-Bond-Directed Assemblies of a Pyrene Derivative, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS PubMed
- L. Wilbraham, M. Louis, D. Alberga, A. Brosseau, R. Guillot, F. Ito, F. Labat, R. Metivier, C. Allain and I. Ciofini, Revealing the Origins of Mechanically Induced Fluorescence Changes in Organic Molecular Crystals, Adv. Mater., 2018, 30, 1800817 CrossRef PubMed
- P. Xu, Q. Qiu, X. Ye, M. Wei, W. Xi, H. Feng and Z. Qian, Halogenated tetraphenylethene with enhanced aggregation-induced emission: an anomalous anti-heavy-atom effect and self-reversible mechanochromism, Chem. Commun., 2019, 55, 14938–14941 RSC
- A. K. Nangia and G. R. Desiraju, Crystal Engineering: An Outlook for the Future, Angew. Chem., Int. Ed., 2019, 58, 4100–4107 CrossRef CAS PubMed
- O. Sato, Dynamic molecular crystals with switchable physical properties, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Aggregation-Induced Emission: Together We Shine, United We Soar!, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed
- S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Design amphiphilic dipolar pi-systems for stimuli-responsive luminescent materials using metastable states, Nat. Commun., 2014, 5, 4013 CrossRef CAS PubMed
- S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, Multistimuli Two-Color Luminescence Switching via Different Slip-Stacking of Highly Fluorescent Molecular Sheets, J. Am. Chem. Soc., 2010, 49, 13675–13683 CrossRef PubMed
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Aggregation-Induced Emission: New Vistas at the Aggregate Level, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed
- B. Liu, H. Liu, H. Zhang, Q. Di and H. Zhang, Crystal Engineering of a Hydrazone Molecule toward High Elasticity and Bright Luminescence, J. Phys. Chem. Lett., 2020, 11, 9178–9183 CrossRef CAS PubMed
- M. Martinez-Abadia, R. Gimenez and M. B. Ros, Self-Assembled α-Cyanostilbenes for Advanced Functional Materials, Adv. Mater., 2018, 30, 1704161 CrossRef PubMed
- M. Martínez-Abadía, S. Varghese, P. Romero, J. Gierschner, R. Giménez and M. B. Ros, Highly Light-Sensitive Luminescent Cyanostilbene Flexible Dimers, Adv. Opt. Mater., 2017, 5, 1600860 CrossRef
- Z. Ding, Y. Zhang, Y. Gao, B. Yang and S. Jiang, Tunable morphologies and emission of photosensitive supramolecular self-assemblies through positional and trans-cis isomerization, Nanoscale, 2020, 12, 2071–2080 RSC
- J.-Y. Zhu, C.-X. Li, P.-Z. Chen, Z. Ma, B. Zou, L.-Y. Niu, G. Cui and Q.-Z. Yang, A polymorphic fluorescent material with strong solid state emission and multi-stimuli-responsive properties, Mater. Chem. Front., 2020, 4, 176–181 RSC
- Z. Zhang, X. Song, S. Wang, F. Li, H. Zhang, K. Ye and Y. Wang, Two-Dimensional Organic Single Crystals with Scale Regulated, Phase-Switchable, Polymorphism-Dependent, and Amplified Spontaneous Emission Properties, J. Phys. Chem. Lett., 2016, 7, 1697–1702 CrossRef CAS PubMed
- Y. Liu, A. Li, S. Xu, W. Xu, Y. Liu, W. Tian and B. Xu, Reversible Luminescent Switching in an Organic Cocrystal: Multi-Stimuli-Induced Crystal-to-Crystal Phase Transformation, Angew. Chem., Int. Ed., 2020, 59, 15098–15103 CrossRef CAS PubMed
- S. A. Sharber, A. Mann, K. C. Shih, W. J. Mullin, M. P. Nieh and S. W. Thomas, 3rd, Directed Polymorphism and Mechanofluorochromism of Conjugated Materials through Weak Non-Covalent Control, J. Mater. Chem. C, 2019, 7, 8316–8324 RSC
- B. B. Rath, G. K. Kole, S. A. Morris and J. J. Vittal, Rotation of a helical coordination polymer by mechanical grinding, Chem. Commun., 2020, 56, 6289–6292 RSC
- P. Wei, J. X. Zhang, Z. Zhao, Y. Chen, X. He, M. Chen, J. Gong, H. H. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, Multiple yet Controllable Photoswitching in a Single AIEgen System, J. Am. Chem. Soc., 2018, 140, 1966–1975 CrossRef CAS PubMed
- V. Ramamurthy and J. Sivaguru, Supramolecular Photochemistry as a Potential Synthetic Tool: Photocycloaddition, Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS PubMed
- J. W. Chung, Y. You, H. S. Huh, B.-K. An, S.-J. Yoon, S. H. Kim, S. W. Lee and S. Y. Park, Shear- and UV-Induced Fluorescence Switching in Stilbenic π-Dimer Crystals Powered by Reversible [2 + 2] Cycloaddition, J. Am. Chem. Soc., 2009, 131, 8163–8172 CrossRef CAS PubMed
- P. Li, J. Wang, P. Li, L. Lai and M. Yin, Minor alkyl modifications for manipulating the fluorescence and photomechanical properties in molecular crystals, Mater. Chem. Front., 2021, 5, 1355–1363 RSC
- S. Li and D. Yan, Tuning Light-Driven Motion and Bending in Macroscale-Flexible Molecular Crystals
Based on a Cocrystal Approach, ACS Appl. Mater. Interfaces, 2018, 10, 22703–22710 CrossRef CAS PubMed
- H. Wang, J. Liu, K. Ye, Q. Li, J. Zhang, H. Xing, P. Wei, J. Sun, F. Ciucci, J. W. Y. Lam, R. Lu and B. Z. Tang, Positive/Negative Phototropism: Controllable Molecular Actuators with Different Bending Behavior, CCS Chem., 2021, 3, 1491–1500 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09 Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed
- J. Li, L. Gao, T. Lu, Z. Feng, D. Jiang, C. Du, K. Wang, P. Lu and B. Zou, Luminogens Based on Cyano-Substituted Anthracene Isomers: Different Molecular Packing and Distinct Piezochromic Properties, Adv. Opt. Mater., 2021, 9, 2100813 CrossRef CAS
- A. Li, Z. Ma, J. Wu, P. Li, H. Wang, Y. Geng, S. Xu, B. Yang, H. Zhang, H. Cui and W. Xu, Pressure-Induced Wide-Range Reversible Emission Shift of Triphenylamine-Substituted Anthracene via Hybridized Local and Charge Transfer (HLCT) Excited State, Adv. Opt. Mater., 2018, 6, 1700647 CrossRef
- A. Li, J. Wang, Y. Liu, S. Xu, N. Chu, Y. Geng, B. Li, B. Xu, H. Cui and W. Xu, Remarkable pressure-induced emission enhancement based on intermolecular charge transfer in halogen bond-driven dual-component co-crystals, Phys. Chem. Chem. Phys., 2018, 20, 30297–30303 RSC
- J. Wang, A. Li, S. Xu, B. Li, C. Song, Y. Geng, N. Chu, J. He and W. Xu, Tunable luminescence of a novel organic co-crystal based on intermolecular charge transfer under pressure, J. Mater. Chem. C, 2018, 6, 8958–8965 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2097646 and 2097647. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qm01443a |
| This journal is © the Partner Organisations 2022 |