
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Macroscopic volume phase transitions in supramolecular gels directed by covalent crosslinking†
Santanu
Panja
 ,
Simona
Bianco
,
Bart
Dietrich
and
Dave J.
Adams
*
,
Simona
Bianco
,
Bart
Dietrich
and
Dave J.
Adams
*
School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
First published on 2nd August 2022
Abstract
Reaction-driven modification of the chemical composition of gels is an interesting approach to synthesize useful materials. Of the different reaction-based systems, crosslinking of the components through the formation of covalent bonds is used to provide mechanical stability to the materials. Here, we show that covalent crosslinking in multicomponent gels can be an effective strategy to synthesize new types of functional materials with spatiotemporal dynamic properties.
Introduction
Gels are ubiquitous in many areas including materials and medicine. Supramolecular gels are formed when small molecules undergo self-assembly involving various noncovalent interactions such as π-stacking, hydrogen bonding, ionic and hydrophobic interactions, resulting in a fibrous network that immobilizes the solvent.1,2 To explore these materials for task-specific applications, the responsive behaviour of the gels towards external stimuli such as UV-light, pH, heat, and chemical analytes has been extensively studied.3–6 However, unlike polymeric gels where the molecules are connected by covalent bonds, one major issue with these supramolecular systems is that they often suffer from poor mechanical stability and tend to break at low strain, limiting many practical applications.6–9 For example, exploring gels as drug delivery vehicles demands sufficient stiffness of the materials for a controlled and sustainable drug release.10 In cell and tissue engineering, the mechanical properties of the scaffold should mimic the cells’ environment and thereby determine the outcome of cellular differentiation.11 Similarly, for 3D printable gels, the material should have sufficient strength to be extruded through a needle without much deviation from its native properties.12To overcome this, chemical reaction-driven in situ crosslinking of the gelator molecules through the formation of covalent bonds is receiving attention to improve the final material properties. The design of such gels incorporates a reaction centre on the gelator skeleton where the chemical reaction proceeds post-gelation. Exposure of these gels to a stimulus makes the system reactive and instigates the components to crosslink covalently leading to a permanent change in the chemical composition of the materials. Several methods such as photodimerization and photopolymerization,13,14 disulfide formation,15 amide16 and imine17,18 bond formation have been used to achieve covalent crosslinking in supramolecular gels.
Typically, such gels are formed by the self-assembly of a single component. However, multicomponent supramolecular gels formed by the combination of two or more organic molecules have potential as soft functional materials with potential in organic electronics, drug delivery, actuators, and tissue engineering.3,19,20 In synthesizing multicomponent gels, mixing two organic compounds bearing oppositely pH-responsive functionalities (for example, carboxylic acid and amine group) is promising.19,21 Such systems can form hierarchical structures on perturbation of pH, enabling modification of material properties simply by controlling the surface charge on the fibres through pH change.22–25 While considering these ionic complementary multicomponent systems, chemical crosslinking is possible in many ways (Fig. 1). One possibility is to instigate crosslinking between the two different components by a covalent bond. Another possibility is to carry out selective crosslinking with only one of the components.
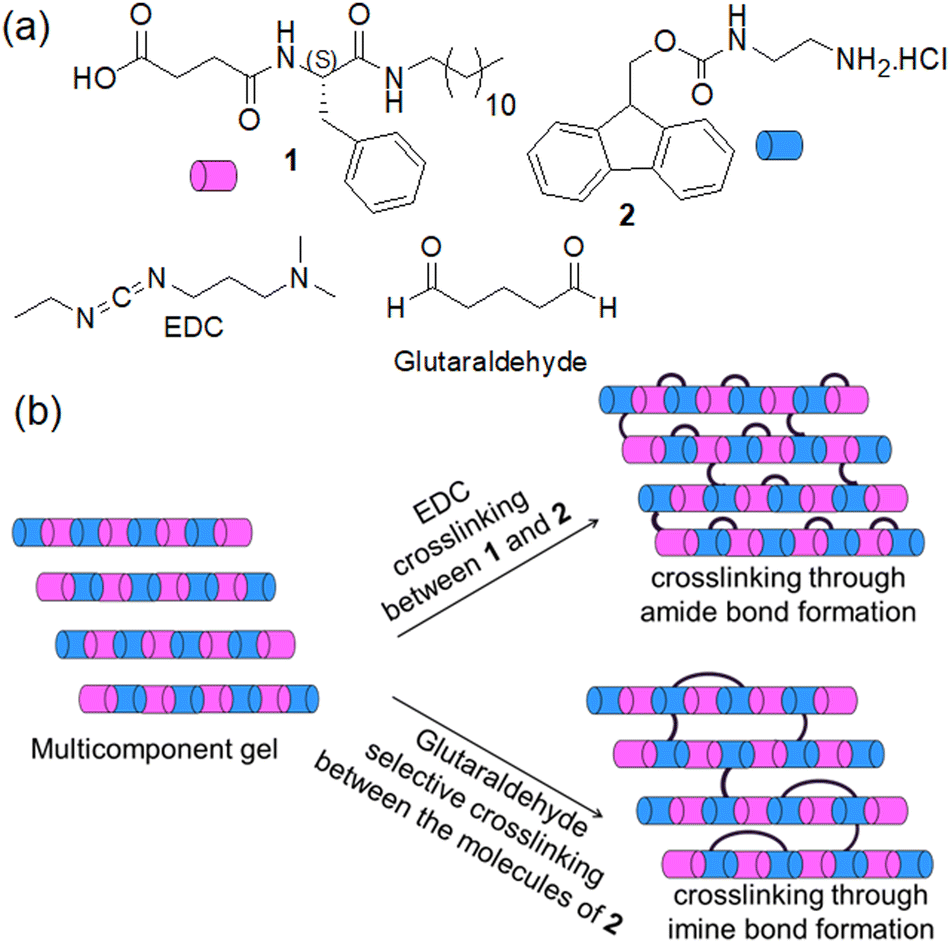 | ||
| Fig. 1 (a) Chemical structures of compounds 1, 2, and the crosslinking agents EDC (used as hydrochloride salt) and glutaraldehyde. (b) Conceptual cartoon schematic showing different possibilities of covalent crosslinking between the components bearing a carboxylic acid and amine functionality in a multicomponent gel system. For ease of visualisation, an alternating structure is shown but random coassembly is likely to occur. | ||
Here, we explored both possibilities in an ionic-complementary multicomponent system comprising gelators with carboxylic acid (1) and an amine group (2). We employed 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) as a coupling reagent to achieve crosslinking through amide bond formation between components 1 and 2 (Fig. 1). In contrast, glutaraldehyde was incorporated into the system to accomplish selective crosslinking between the molecules of 2 involving an imine bond formation reaction (Fig. 1). Depending on the pH of the medium, the multicomponent system (1 + 2) responds differently towards the crosslinking reagents. Different types of crosslinking allow controlling the mechanical properties to different extents with a change in the macroscopic volume of the gels which is uncommon for such supramolecular systems. To the best of our knowledge, this is the first report on shrinking/swelling in multicomponent gels tuned by covalent crosslinking.
Results and discussion
In our previous work on crosslinked gels, phenylalanine-functionalized small molecule gelators were studied.16,17 There was no change in the volume of the gels after crosslinking. Generally, the shrinking or swelling behaviour of supramolecular gels is linked to the change in hydrophobicity of the individual molecules in response to an applied stimulus.26–28 However, this is not always true. There are precedents where rather than the hydrophobicity of individual molecules, it is the hydrophobicity of self-assembled fibres that regulates the macroscopic volume change in gels.25,29–32 There is possibly a rearrangement of the three-dimensional self-assembled structures driven by the change in hydrophobicity of the network causing macroscopic volume phase transitions in gels.Keeping this in mind, here we functionalized the phenylalanine residue with a dodecyl chain to synthesize compound 1. In DMSO/H2O (20/80, v/v), 1 self-assembled into needle-like fibres leading to a gel with a pH of 3.5 (Fig. 2a–c and Fig. S1, ESI†). During self-assembly, while the dodecyl segment exerts hydrophobic interactions, the amide groups are involved in intermolecular hydrogen bonding. The hydrogel of 1 is pH-responsive and formed a free-flowing solution in presence of base due to deprotonation of the terminal carboxylic acid. Compared to 1, the Fmoc-salt 2 exhibits opposite response to pH perturbation. In this case, deprotonation of the ammonium group of 2 generates the corresponding amine.33 Visually, the solution of 2 turned into a gel (pH 10.4) upon addition of equimolar amounts of NaOH (Fig. 2a and Fig. S2, ESI†). The gel consists of spherulitic domains of fibres as observed in confocal and optical microscopy imaging (Fig. S2, ESI†).
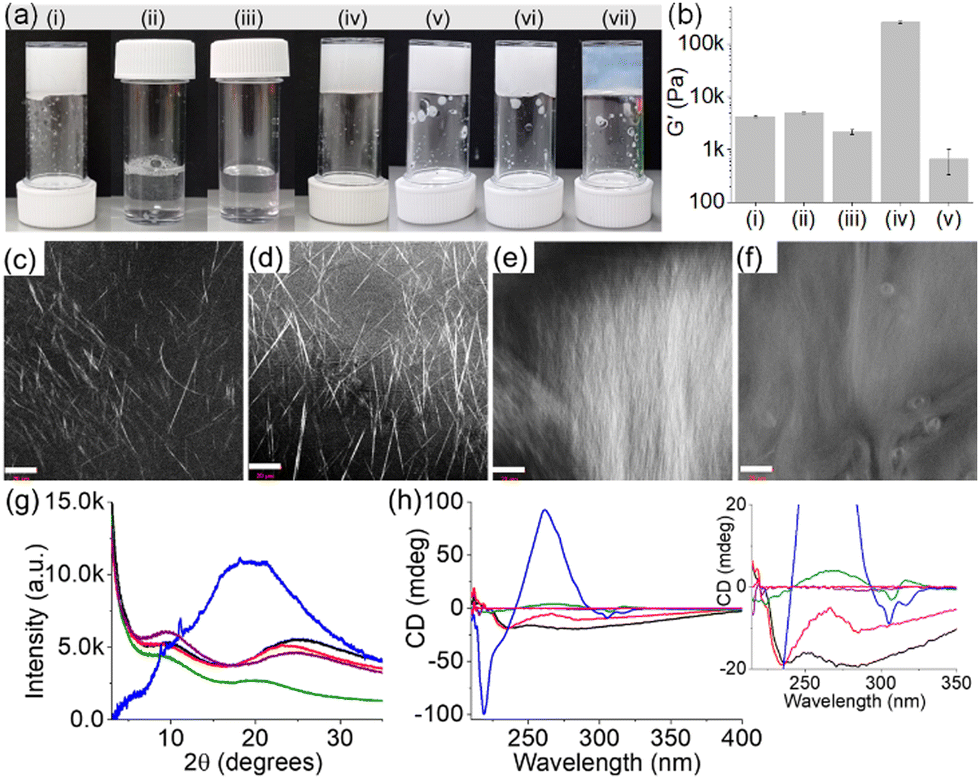 | ||
Fig. 2 (a) Photographs of the phase changes of 1 (i and ii) and 2 (iii and iv) in the absence (i and iii) and presence (ii and iv) of 1 equiv. of NaOH. (v–vii) Photographs of the multicomponent gels of (1 + 2) at pH (v) 3.3 (with no NaOH), (vi) 9.3 (with 1 equiv. of NaOH) and (vii) 10.8 (with 2 equiv. of NaOH). (b) Bar graph showing the changes in stiffness (G′, calculated at 0.2% strain from strain sweeps) of the hydrogel of (i) 1, (ii) 2 with NaOH, and the multicomponent gels of (1 + 2) at pH (iii) 3.3, (iv) 9.3 and (v) 10.8. (c–f) Confocal microscopy images of the hydrogel of 1 (c) and the multicomponent gels of (1 + 2) at pH (d) 3.3, (e) 9.3 and (f) 10.8 (scale bars represent 20 μm). (g) PXRD data and (h) CD spectra for the hydrogels of 1 (black), 2 (purple), and the multicomponent gels of (1 + 2) at pH 3.3 (red), 9.3 (green) and 10.8 (blue). For (h), the magenta data represent the solution of 2. Inset of (h) represents expanded section of the graph. In all cases, concentration of 1 is 10 mg mL−1 and the molar ratio of 1 and 2 is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Solvent is DMSO/H2O (20/80, v/v). :1. Solvent is DMSO/H2O (20/80, v/v). | ||
The multicomponent gel (1 + 2) was prepared by mixing equimolar amounts of 1 and 2 in the absence and presence of different concentrations of NaOH (Fig. 2a and b). Simple dilution of the mixture of 1 and 2 in DMSO with water resulted in a gel (pH 3.3) with a microstructure similar to the hydrogel of 1 alone (Fig. 2d and Fig. S3, ESI†). However, spectroscopic studies confirmed that the fibres are formed by co-assembly. In the presence of 2, the amide carbonyl stretches of 1 moved from 1641 cm−1 and 1697 cm−1 to 1643 cm−1 and 1692 cm−1, respectively (Fig. S4, ESI†). Moreover, the shoulder peak at 1720 cm−1 from the carboxylic carbonyl of 1 shifted to a higher region by 15 cm−1 and appeared at 1735 cm−1 in the multicomponent gel. Noticeably, the carbamate carbonyl stretching of 2 at 1715 cm−1 became too broad to distinguish in the multicomponent gel, signifying the involvement of 2 in self-assembly which was further verified by fluorescence studies. The solution of 2 exhibited a sharp emission at 320 nm with a shoulder band near 365 nm corresponding to the monomer and excimer emissions of the fluorenyl group, respectively (Fig. S5, ESI†).33,34 In the gel (1 + 2), the monomer peak was red-shifted by 3 nm with a significant increase in the relative intensity of the excimer emission band. These results indicate coassembly of 1 and 2.33 To confirm this, powder X-ray diffraction (PXRD) study of the wet gels was conducted (Fig. 2g). In both cases, no sharp diffraction peak was seen, suggesting less ordered molecular packing in the gels. However, the broad diffraction peaks at 2θ = 24.88° and 9.92° for the hydrogel of 1 were shifted to 2θ = 23.46° and 9.43°, respectively in the multicomponent gel. The differences in the peak positions corroborate a coassembly-driven change in hydrogen-bonded structures of 1 in the presence of 2. Again, from circular dichroism (CD) spectroscopy, while compound 2 is CD inactive both in the solution and gel state, a 4 nm blue shift of the intense negative peak for 1 at 234 nm further supports a change in the H-bonded secondary structure of 1 in the multicomponent system (Fig. 2h). Rheologically, the stiffness (storage modulus, G′) of the hydrogel (1 + 2) was reduced by >50% compared to the gel of 1 alone (Fig. 2b and Fig. S6, ESI†).
We next increased the pH of the multicomponent system using NaOH. The large difference between the pKa of 1 and 2 (5.8 and 8.4 respectively) enables control over the degree of deprotonation of the components (Fig. S7, ESI†). In presence of 1 equivalent of NaOH, the formation of the carboxylate of 1 triggered the salt-bridge formation involving charge–charge interactions with the ammonium group of 2 resulting in a gel (pH is 9.3) which shows an unusually high stiffness (G′) of >2.5 × 105 Pa (Fig. 2b and Fig. S6, ESI†).24,25 Under these conditions, the pH of the gel was higher than the apparent pKa of 2 suggesting that the gel at pH 9.3 is dominated by the amine form (>50%) than the ammonium form of 2. Analysis of the equilibrium position using Henderson–Hasselbalch equation35,36 reveals ∼80% deprotonation of 2 at pH 9.3. On further increasing the pH to 10.8 (with 2 equivalents of NaOH), the stiffness (G′) of the material drops dramatically due to the complete loss of charge on the ammonium group (∼100% deprotonation of 2) and thereby destruction of the salt-bridge between 1 and 2 (Fig. 2b and Fig. S6, ESI†).24,25 However, the extent of aromatic stacking in the gels increases with the increase of pH as evident from fluorescence and CD spectroscopy. On increasing the pH of the multicomponent gel from pH 3.3 to 10.8, the monomer emission of 2 at 323 nm gradually disappeared and the intensity of the excimer emission peaks in the region 425–500 nm increased considerably (Fig. S8, ESI†). The negative CD signal at 305 nm derived from the fluorenyl group37 of 2 was also intensified on increasing the pH to 10.8 suggesting aromatic stacking plays a pivotal role in maintaining the assembled structures at high pH (Fig. 2h). Moreover, different CD signals in the region 218–284 nm for the system (1 + 2) at different pH signify a change in the underlying molecular packing of the gels. In UV-vis, a gradual red shift of the peak from 299 nm to 303 nm with increasing pH further supports this proposition (Fig. S8, ESI†). FTIR spectroscopy also shows a gradual decrease of amide carbonyl stretching of 1 from 1643 cm−1 to 1636 cm−1 along with simultaneous broadening of the peak near 1735 cm−1 on changing the pH from 3.3 to 10.8, validating the existence of different hydrogen-bonding interactions between the components at different pH (Fig. S9, ESI†). Both the gels at pH 9.3 and 10.8 showed long fibres as their microstructures in contrast to needle-shaped fibres at pH 3.3 (Fig. 2d–f and Fig. S10, ESI†). In this context, proton NMR of the gels confirms the stability of the components at high pH (Fig. S11, ESI†). Taking together the pH data and the spectroscopic observations, the existence of different structures of 1 and 2 responsible for gelation for the system (1+2) at different pH is illustrated in Scheme S2 (ESI†). Interestingly, PXRD data of the multicomponent gel at pH 10.8 was eclipsed by a broad band centred at 2θ = 19.48° along with a number of sharp peaks (2θ = 8.81°, 9.43°, 11.13°, 18.14°) implying that the gel formed at pH 10.8 is more crystalline (Fig. 2g). These results further suggest that the multicomponent gelation at both pH 9.3 and 10.8 is again driven by co-assembly.
To achieve covalent crosslinking in the gels as shown in Fig. 1, we added aqueous solutions of EDC (1 equivalent as the hydrochloride salt) or glutaraldehyde (0.5 equivalent) separately on top of the gels and allowed the systems to react for ∼24 h. To check the EDC-triggered coupling between 1 and 2, the 1H NMR spectra of the EDC-treated gels were compared with the original gel of (1 + 2) (Fig. S12, ESI†). At pH 3.3, the gels obtained before and after treatment with EDC exhibited identical NMR spectra, suggesting no chemical reaction occurred between 1 and 2 at pH 3.3. However, in the EDC-treated gels at pH 9.3 and 10.8 (proton NMR was recorded after acidifying the samples), disappearance of the peak at 8.02 ppm corresponding to the ammonium protons of 2 indicates the crosslinking between 1 and 2via amide bond formation in both cases. Moreover, HRMS analysis of the samples showed the expected mass for the amide product 3 and confirms the crosslinking at both pH 9.3 and 10.8 (Fig. S13, ESI†). Interestingly, instead of post assembly functionalization, a direct addition of EDC to the mixture of 1 and 2 in presence of base brought about an identical proton NMR, further confirming that cross-linking between 1 and 2 occurred through amide bond formation (Fig. S14 and S15, ESI†). Unlike EDC, glutaraldehyde triggered the crosslinking between the molecules of 2 through imine bond formation at pH 3.3. This was confirmed by 1H NMR of the glutaraldehyde-treated gel that shows disappearance of the ammonium protons at 8.02 ppm along with generation of a broad peak near 7.61 ppm (Fig. S16, ESI†).17 Moreover, by FTIR, a new band appearing at 1714 cm−1 after the chemical reaction also supports the crosslinking (Fig. S17, ESI†). Along the same line, emergence of the stretching band near 1714 cm−1 for the gels at pH 9.3 and 10.8 (Fig. S18, ESI†) further endorses that crosslinking is indeed taking place via imine bond formation in both cases.
Different types of crosslinking at different pH perturb the systems to different extents. Rheological experiments were carried out to monitor the effects of covalent crosslinking in the gels. Time sweep rheology at pH 3.3 indicated that diffusion of glutaraldehyde into the gel resulted in an increase of both storage (G′) and loss modulus (G′′) over time (Fig. S19, ESI†). We propose that glutaraldehyde interacts with the molecules of 2 leading to alteration of the primary assembled structures in the network yielding a gel with improved mechanical properties (Fig. S19, ESI†). A considerable decrease in monomer emission accompanied by the simultaneous increase in relative intensity of excimer emission of the fluorenyl group was recorded in the crosslinked gel (Fig. S20, ESI†), indicating that reaction with glutaraldehyde brought about a change in the intermolecular interactions between the molecules which was also evident from the disappearance of the negative CD signal at 284 nm in the crosslinked gel (Fig. S21, ESI†). Microscopically, the crosslinked gel exhibited needle-like fibres similar to the original gel (1 + 2), however, the density of the fibres increased considerably after crosslinking (Fig. S22, ESI†). Importantly, the emergence of sharp peaks at 2θ = 19.78° to 24.97° in PXRD experiments implies a change in the distribution of the fibres at longer length scale, yielding a gel with higher crystallinity upon crosslinking (Fig. S21, ESI†). Visually, the colour of the gel turned slight yellow with apparently no change in volume (Fig. S19, ESI†).
As mentioned earlier, at pH 9.3, electrostatic interactions between the carboxylate of 1 and the ammonium group of 2 play a crucial role in maintaining the gel structure. Crosslinking using either EDC or glutaraldehyde (through amide and imine bond formation, respectively) destroys the salt bridge between the gelators. Despite this, there were no significant changes in the mechanical properties of the gels after crosslinking in both cases (Fig. 3a, b and Fig. S23, S24, ESI†). This is highly suggestive of inter-fibre crosslinking that enables the systems to retain their mechanical properties after crosslinking. The negative CD signal at 305 nm as well as the broad positive peak centred at 266 nm in the hydrogel remain unaltered after the crosslinking, further agreeing with the inter-fibre coupling of molecules (Fig. 3e). Microscopic studies revealed a dramatic change in the microstructures of the gels upon crosslinking. The resulting crosslinked gels exhibited fibres which are more intertwined than the pristine gel, validating the lateral association of the fibres in presence of both EDC and glutaraldehyde (Fig. 3k and l). Surprisingly, the resulting crosslinked gels exhibited slight increase in gel volume (Fig. 3c and d). The volume of the gel increased by ∼4% after treatment with EDC. When glutaraldehyde was used as a crosslinking agent, the resulting gel showed ∼6% swelling compared to the pristine gel. Here the swelling can be ascribed to the crosslinking-driven change in hydrophobicity of the co-assembled fibres that enables a rearrangement of the self-assembled structures and causes water molecules to enter into the hydrogel network resulting in swelling.29–32 By PXRD, the broad peak at 2θ = 8.81° in the original gel became prominent and shifted to around 2θ = 9.69° upon crosslinking (Fig. 3g). Moreover, shifting of the broad peak centred at 2θ = 19.69° to 2θ = 24.21° and 2θ = 20.85° after crosslinking using EDC and glutaraldehyde, respectively, suggests a change in the three-dimensional network structure in the resulting crosslinked gels. Interestingly, unlike in the EDC-coupling, a reddish-brown colouration appeared in the glutaraldehyde treated gel which was supported from the shoulder band near 350 nm in the UV-vis (Fig. 3a and Fig. S25, ESI†). However, both the crosslinked gels exhibited similar emission profiles (Fig. S25, ESI†). A comparison of normalized emission spectra of the gels revealed a decrease in relative intensity of monomer emission of 2 at 323 nm after crosslinking.
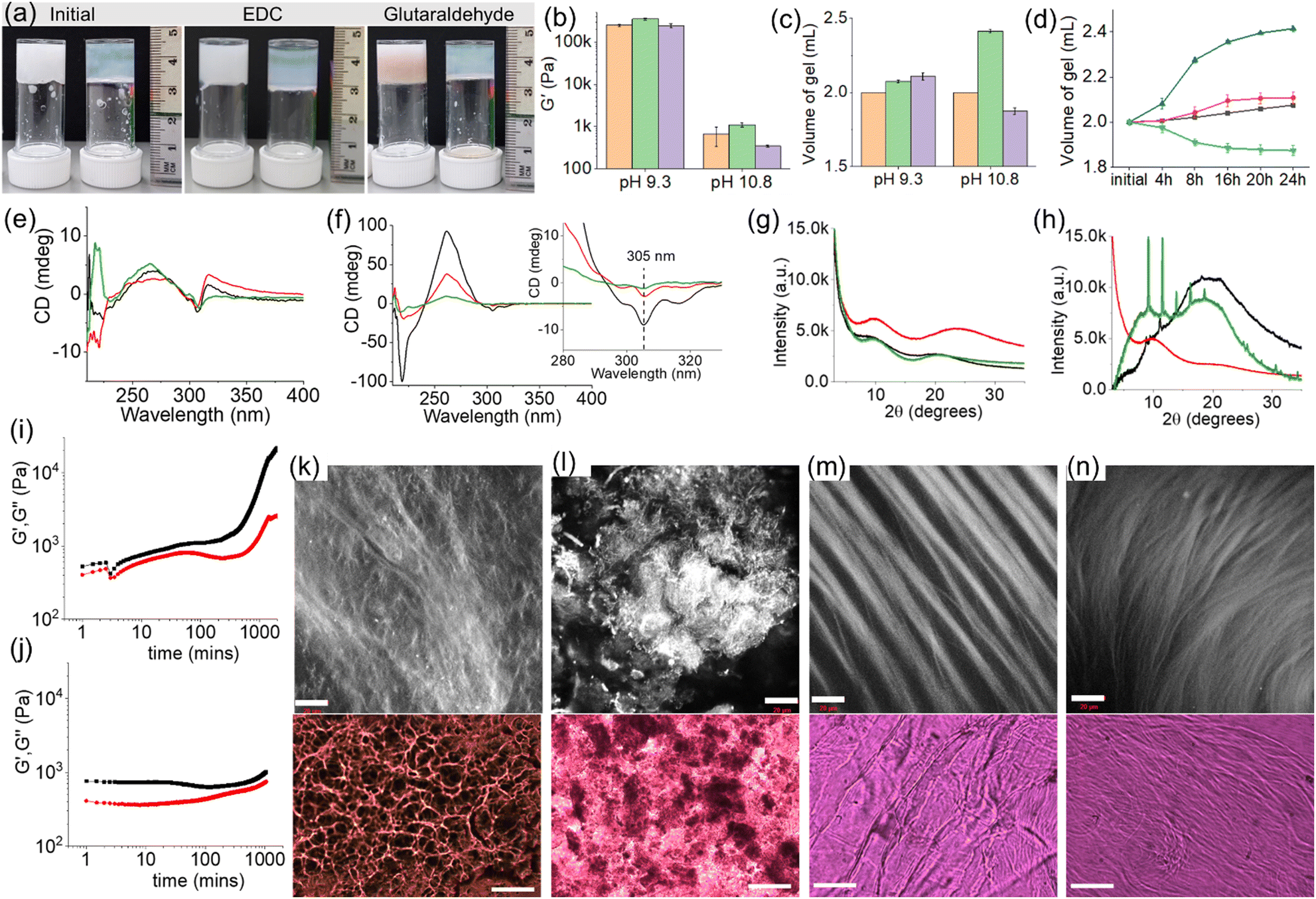 | ||
| Fig. 3 (a) Photographs showing the changes in gel volume of the multicomponent gels of (1 + 2) after treatment with EDC and glutaraldehyde at pH 9.3 (left vial) and 10.8 (right vial). (b–c) Bar graph showing the changes in (b) stiffness and (c) final volume of the multicomponent gel (1 + 2) (orange) after treatment with EDC (green) and glutaraldehyde (purple) at pH 9.3 and 10.8. (d) Time variable change in the volume of the multicomponent gels of (1 + 2) in presence of EDC (black, blue) and glutaraldehyde (red, green) at pH 9.3 (black, red) and 10.8 (blue, green). (e and f) CD spectra and (g and h) PXRD data for the multicomponent gels of (1 + 2) (black), and the hydrogels obtained after treatment with EDC (red) and glutaraldehyde (green) at pH 9.3 (e and g) and 10.8 (f and h). Inset of (f) represents expanded section of the graph. (i and j) Variation of G′ (black) and G′′ (red) with time for the hydrogels of (1 + 2) at pH 10.8 upon addition of EDC (i) and glutaraldehyde (j). (k–n) Confocal (top, scale bars are 20 μm) and optical (bottom, scale bars are 100 μm) microscopy images of the crosslinked gels obtained by treatment with EDC (k and m) and glutaraldehyde (l and n) at pH 9.3 (k and l) and 10.8 (m and n). In all cases, initial concentration of 1 is 10 mg mL−1 and the molar ratios of 1, 2, EDC and glutaraldehyde are 1:1:1:0.5. Solvent is DMSO/H2O (20/80, v/v). | ||
On further increasing the pH to 10.8, the multicomponent gel of (1 + 2) responds differently to the crosslinking agents. While reaction with EDC resulted in a gradual increase of both the rheological moduli (G′ and G′′) of the gel over time accompanied by a ∼21% expansion in gel volume, crosslinking by glutaraldehyde caused a substantial reduction in stiffness of the material along with a ∼7% contraction in volume (Fig. 3a–j and Fig. S26, ESI†). The crosslinked gels are more reproducible as noted from small error bars in the rheological data (Fig. 3b). Both the EDC and glutaraldehyde-treated gels exhibit long fibres as their microstructures (Fig. 3m and n). The CD spectra of the resulting crosslinked gels showed a decrease in peak intensity at 218 nm and 261 nm signifying a change in the hydrogen-bonded secondary structure in the gel network driven by the chemical reactions (Fig. 3f). Moreover, a decrease in intensity of the negative peak at 305 nm suggests a modulation of aromatic stacking involving the fluorenyl groups after crosslinking.37 By fluorescence, a decrease in intensity of the fluorenyl excimer peaks in the region of 425–500 nm further agrees with the CD data (Fig. S27, ESI†). From PXRD, the broad peak centred at 2θ = 19.48° (before crosslinking) almost disappeared after EDC coupling and a broad band near 2θ = 9.93° became predominant (Fig. 3h). In contrast, the glutaraldehyde treated gel exhibited sharp peaks at 2θ = 9.18° and 11.51°. An illustration of the PXRD patterns of the crosslinked gels implies that the multicomponent system evolved differently in the presence of EDC and glutaraldehyde leading to different packing of the supramolecular assemblies. Such a change in the underlying network structures is responsible for the opposite phase behaviour (shrinking/swelling) of the gels.29–32
Conclusions
In conclusion, we have explored the possibilities of different types of covalent crosslinking in a multicomponent system consisting of components bearing a carboxylic acid and amine group where we are able to crosslink either both components or selectively one of the components to target materials with different final properties. Depending on the pH of the medium, the system reacts to the crosslinking reagents to different extents enabling us not only to control the mechanical properties but also the macroscopic volume of the resulting gels. Such control over these systems is unprecedented and results in materials with unusual properties, for instance rare examples of such gels that can swell. Considering the growing interests in adaptive functional materials,6–9,27 we envisage that the insights provided here would be effective in designing gel-based actuators, conducting materials, and drug delivery systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SP and SB thank the University of Glasgow for funding. BD and DA thank the EPSRC for funding (EP/S019472/1).Notes and references
- P. Terech and R. G. Weiss, Low Molecular Mass Gelators of Organic Liquids and the Properties of Their Gels, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Water Gelation by Small Organic Molecules, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- C.-W. Chu and C. A. Schalley, Recent Advances on Supramolecular Gels: From Stimuli-Responsive Gels to Co-Assembled and Self-Sorted Systems, Org. Mater., 2021, 03, 025–040 CrossRef CAS.
- S. Panja and D. J. Adams, Stimuli responsive dynamic transformations in supramolecular gels, Chem. Soc. Rev., 2021, 50, 5165–5200 RSC.
- J. Hoque, N. Sangaj and S. Varghese, Stimuli-Responsive Supramolecular Hydrogels and Their Applications in Regenerative Medicine, Macromol. Biosci., 2019, 19, 1800259 CrossRef PubMed.
- Z. Deng, R. Yu and B. Guo, Stimuli-responsive conductive hydrogels: design, properties, and applications, Mater. Chem. Front., 2021, 5, 2092–2123 RSC.
- A. Döring, W. Birnbaum and D. Kuckling, Responsive hydrogels – structurally and dimensionally optimized smart frameworks for applications in catalysis, micro-system technology and material science, Chem. Soc. Rev., 2013, 42, 7391–7420 RSC.
- A. Roy, K. Manna and S. Pal, Recent advances in various stimuli-responsive hydrogels: from synthetic designs to emerging healthcare applications, Mater. Chem. Front., 2022 10.1039/D2QM00469K.
- J. Shang, X. Le, J. Zhang, T. Chen and P. Theato, Trends in polymeric shape memory hydrogels and hydrogel actuators, Polym. Chem., 2019, 10, 1036–1055 RSC.
- K. Basu, A. Baral, S. Basak, A. Dehsorkhi, J. Nanda, D. Bhunia, S. Ghosh, V. Castelletto, I. W. Hamley and A. Banerjee, Peptide based hydrogels for cancer drug release: modulation of stiffness, drug release and proteolytic stability of hydrogels by incorporating d-amino acid residue(s), Chem. Commun., 2016, 52, 5045–5048 RSC.
- L. Saunders and P. X. Ma, Self-Healing Supramolecular Hydrogels for Tissue Engineering Applications, Macromol. Biosci., 2019, 19, 1800313 CrossRef PubMed.
- H. Jian, M. Wang, Q. Dong, J. Li, A. Wang, X. Li, P. Ren and S. Bai, Dipeptide Self-Assembled Hydrogels with Tunable Mechanical Properties and Degradability for 3D Bioprinting, ACS Appl. Mater. Interfaces, 2019, 11, 46419–46426 CrossRef CAS PubMed.
- M. N. Tahir, A. Nyayachavadi, J.-F. Morin and S. Rondeau-Gagné, Recent progress in the stabilization of supramolecular assemblies with functional polydiacetylenes, Polym. Chem., 2018, 9, 3019–3028 RSC.
- Y. Ding, Y. Li, M. Qin, Y. Cao and W. Wang, Photo-Cross-Linking Approach to Engineering Small Tyrosine-Containing Peptide Hydrogels with Enhanced Mechanical Stability, Langmuir, 2013, 29, 13299–13306 CrossRef CAS PubMed.
- L. Aulisa, H. Dong and J. D. Hartgerink, Self-Assembly of Multidomain Peptides: Sequence Variation Allows Control over Cross-Linking and Viscoelasticity, Biomacromolecules, 2009, 10, 2694–2698 CrossRef CAS PubMed.
- S. Panja and D. J. Adams, Chemical crosslinking in ‘reactive’ multicomponent gels, Chem. Commun., 2022, 58, 5622–5625 RSC.
- L. J. Marshall, O. Matsarskaia, R. Schweins and D. J. Adams, Enhancement of the mechanical properties of lysine-containing peptide-based supramolecular hydrogels by chemical cross-linking, Soft Matter, 2021, 17, 8459–8464 RSC.
- M. A. Khalily, M. Goktas and M. O. Guler, Tuning viscoelastic properties of supramolecular peptide gels via dynamic covalent crosslinking, Org. Biomol. Chem., 2015, 13, 1983–1987 RSC.
- L. Li, R. Sun and R. Zheng, Tunable morphology and functionality of multicomponent self-assembly: A review, Mater. Des., 2021, 197, 109209 CrossRef CAS.
- B. O. Okesola and A. Mata, Multicomponent self-assembly as a tool to harness new properties from peptides and proteins in material design, Chem. Soc. Rev., 2018, 47, 3721–3736 RSC.
- L. E. Buerkle and S. J. Rowan, Supramolecular gels formed from multi-component low molecular weight species, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- W. Edwards and D. K. Smith, Enantioselective Component Selection in Multicomponent Supramolecular Gels, J. Am. Chem. Soc., 2014, 136, 1116–1124 CrossRef CAS PubMed.
- S. Toksoz, R. Mammadov, A. B. Tekinay and M. O. Guler, Electrostatic effects on nanofiber formation of self-assembling peptide amphiphiles, J. Colloid Interface Sci., 2011, 356, 131–137 CrossRef CAS PubMed.
- M. Tena-Solsona, S. Alonso-de Castro, J. F. Miravet and B. Escuder, Co-assembly of tetrapeptides into complex pH-responsive molecular hydrogel networks, J. Mater. Chem. B, 2014, 2, 6192–6197 RSC.
- S. Panja, A. Seddon and D. J. Adams, Controlling hydrogel properties by tuning non-covalent interactions in a charge complementary multicomponent system, Chem. Sci., 2021, 12, 11197–11203 RSC.
- N. E. Shi, H. Dong, G. Yin, Z. Xu and S. H. Li, A Smart Supramolecular Hydrogel Exhibiting pH-Modulated Viscoelastic Properties, Adv. Funct. Mater., 2007, 17, 1837–1843 CrossRef CAS.
- B. Xue, M. Qin, T. Wang, J. Wu, D. Luo, Q. Jiang, Y. Li, Y. Cao and W. Wang, Electrically Controllable Actuators Based on Supramolecular Peptide Hydrogels, Adv. Funct. Mater., 2016, 26, 9053–9062 CrossRef CAS.
- S.-L. Zhou, S. Matsumoto, H.-D. Tian, H. Yamane, A. Ojida, S. Kiyonaka and I. Hamachi, pH-Responsive Shrinkage/Swelling of a Supramolecular Hydrogel Composed of Two Small Amphiphilic Molecules, Chem. – Eur. J., 2005, 11, 1130–1136 CrossRef CAS PubMed.
- F. Xie, L. Qin and M. Liu, A dual thermal and photo-switchable shrinking–swelling supramolecular peptide dendron gel, Chem. Commun., 2016, 52, 930–933 RSC.
- A. M. Castilla, M. Wallace, L. L. E. Mears, E. R. Draper, J. Doutch, S. Rogers and D. J. Adams, On the syneresis of an OPV functionalised dipeptide hydrogel, Soft Matter, 2016, 12, 7848–7854 RSC.
- J. Chen, T. Wang and M. Liu, A hydro-metallogel of an amphiphilic l-histidine with ferric ions: shear-triggered self-healing and shrinkage, Inorg. Chem. Front., 2016, 3, 1559–1565 RSC.
- B. K. Das, B. Pramanik, S. Chowdhuri, O. A. Scherman and D. Das, Light-triggered syneresis of a water insoluble peptide-hydrogel effectively removes small molecule waste contaminants, Chem. Commun., 2020, 56, 3393–3396 RSC.
- S. Panja, B. Dietrich, A. J. Smith, A. Seddon and D. J. Adams, Controlling Self-Sorting versus Co-assembly in Supramolecular Gels, ChemSystemsChem, 2022, 4, e202200008 CrossRef CAS.
- J. W. Sadownik, J. Leckie and R. V. Ulijn, Micelle to fibre biocatalytic supramolecular transformation of an aromatic peptide amphiphile, Chem. Commun., 2011, 47, 728–730 RSC.
- W. Smith, M. Pouvreau, K. Rosso and A. E. Clark, pH dependent reactivity of boehmite surfaces from first principles molecular dynamics, Phys. Chem. Chem. Phys., 2022, 24, 14177–14186 RSC.
- D. Yi, H. Yang, M. Zhao, L. Huang, G. Camino, A. Frache and R. Yang, A novel, low surface charge density, anionically modified montmorillonite for polymer nanocomposites, RSC Adv., 2017, 7, 5980–5988 RSC.
- C. Gila-Vilchez, M. C. Mañas-Torres, J. A. González-Vera, F. Franco-Montalban, J. A. Tamayo, F. Conejero-Lara, J. M. Cuerva, M. T. Lopez-Lopez, A. Orte and L. Álvarez de Cienfuegos, Insights into the co-assemblies formed by different aromatic short-peptide amphiphiles, Polym. Chem., 2021, 12, 6832–6845 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qm00599a |
| This journal is © the Partner Organisations 2022 |