
Enantioselective total syntheses of marine natural products (+)-cylindricines C, D, E and their diastereomers†
Ying-Hong
Huang‡
,
Zhan-Jiang
Liu‡
and
Pei-Qiang
Huang
 *
*
Department of Chemistry, Fujian Provincial Key Laboratory of Chemical Biology, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, P. R. China. E-mail: pqhuang@xmu.edu.cn
First published on 9th November 2021
Abstract
A seven-step total synthesis of (+)-cylindricine D and epi-cylindricine D is described. The efficiency relies on the highly diastereoselective reductive bis-functionalization of N,O-protected (S)-pyroglutaminol by our recently improved protocol to build the N-α-tert-alkylamine motif, and a telescoped one-step four-reaction protocol to build the fused bicyclic ring system. (+)-Cylindricine C and its 2-epimer were synthesized by essentially the same sequence in eight steps. Acetylation of the latter then delivered (+)-cylindricine E and its 2-epimer, respectively.
Efficiency is one of the major themes in organic synthesis.1 In the field of the total synthesis of natural products, the efficiency not only depends on the synthetic strategy employed, but also relies in efficient synthetic methods.2 Amides are a class of readily available and highly stable compounds which have long been employed as privileged starting materials and synthetic intermediates in the total synthesis of alkaloids and N-containing medicinal agents. However, as the least reactive class of carboxylic acid derivatives, the direct, chemoselective transformation of amides is challenging. Hence multistep methods have to be employed which dramatically reduced the synthetic efficiency.3 During the last decade, we have developed a series of methods for the direct transformations of amides,4 which include an unique reaction for the one-pot conversion of tertiary amides (A) into α-tert-alkylamines (B)5,6 (reductive di- and bis-alkylation)4a (Scheme 1a). On the basis of the amide transformation reactions (amide strategy), we have achieved the concise total syntheses of several alkaloids.4b,d,e
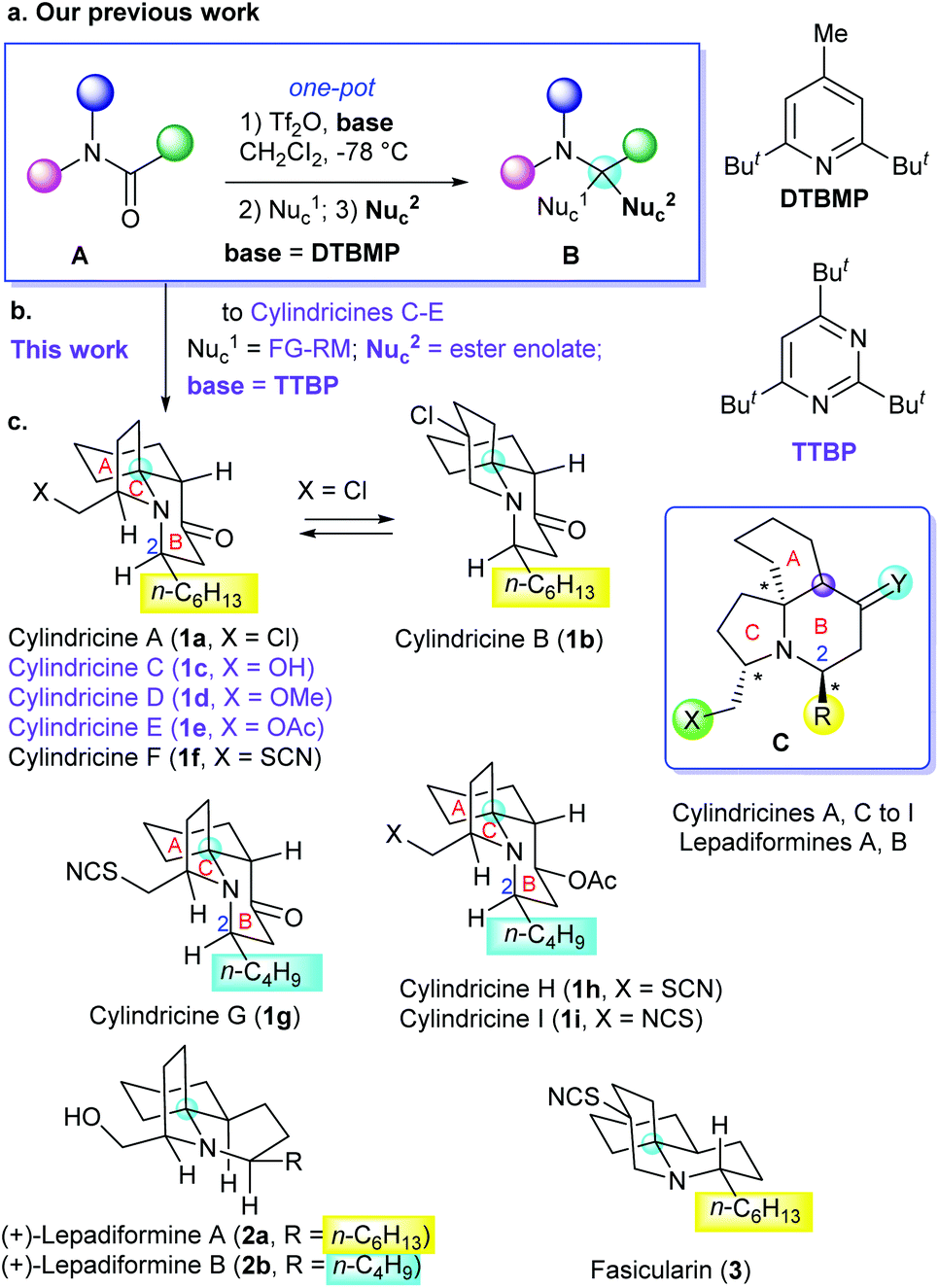 | ||
| Scheme 1 (a) Our previous work; (b) modification of the amide reductive bis-alkylation reaction towards new targets; (c) the alkaloids of the cylindricine/fasicularin/lepadiformine family. | ||
Cylindricines A–K are a group of eleven tricyclic alkaloids (Scheme 1c, only the structures of 1a–1i shown) isolated from the ascidian Clavelina cylindrica collected from the East Coast of Tasmania.7 Cylindricines A, C–I, and K possess a new pyrrolo[2,1-j]quinoline framework, whereas cylindricines B and J share a pyrido[2,1-j]quinoline system. All of them possess a cis-fused 1-azadecalin A/B-ring system. Surprisingly, neither optical rotations nor the absolute configurations of this group of natural products have been described in the literature. Despite of the lack of information on the absolute stereochemistry and of unknown bioactivities (except for toxicity in a brine shrimp assay), the intriguing structures of cylindricines have attracted considerable attention from synthetic community.8–10 More than twenty research groups including those of Snider,9a Weinreb,9b Heathcock,9c Molander,10a Kibayashi,10c,f Trost,10b Ciufolini,10d Hsung,10e,g–i Shibasaki,10j–l Donohoe,9e Renaud,9d Pandey,10m and Chida/Sato6c have been engaged in the total syntheses of cylindricines, which resulted in many elegant approaches.8–10 However, efficient and flexible enantioselective synthetic approaches are still demanding.8c In connection with our longstanding interest in the divergent total synthesis of alkaloids and their diastereomers,4b,d,e,11 we decided to explore a concise and flexible approach to (+)-cylindricines C–E (1c, 1d, and 1e), and the results are reported herein.
Because the absolute configurations of the natural cylindricines are unknown, before starting our enantioselective total synthesis, we have to firstly decide the enantiomers to be synthesized. In this regard, it is worth mentioning that several structurally related (to cylindricines) tricyclic alkaloids such as lepadiformine (2)12 and fasicularin (3)13 have been isolated. Although their absolute configurations have not been determined by the phytochemists,12,13 through the enantioselective total syntheses of lepadiformine (2), Kibayashi14 and Weinreb15 have independently arrived at the conclusion that natural lepadiformine is dextrorotatory having (2R,5S,10S,13S) configuration (with the structure shown by 2). Thus, Weinreb has assumed that all the other alkaloids of the cylindricine/fasicularin/lepadiformine family have the same absolute stereochemistry as lepadiformine.8b On the basis of these considerations, we opted for the putative natural (+)-cylindricines C–E (1c–1e) with the absolute configurations shown in Scheme 1 as our targets.
Our synthetic strategy to (+)-cylindricines C–E (1c–1e) stemmed from the identification of the α-tert-alkylamine motif-bearing prolinol moiety as the key structural feature, and inspired from the diversity of the alkaloids of the cylindricine/fasicularin/lepadiformine family as marked by the generic structure C (Scheme 1). Thus, in the light of the amide reductive bis-alkylation reaction,4aL-pyroglutamic acid derived lactam (+)-9 was selected as our chiral pool. In line with this strategy, the retrosynthetic analysis of (+)-cylindricine D (1d) is outlined in Scheme 2, which implies the last stage formation of A ring from bicyclic precursor 4, and a later stage formation of B ring from (S)-prolinol derivative 5. The employment of Weinreb amide167 would allow for the introduction of different alkyl groups and thus affords the flexibility for the synthesis of different alkaloids bearing either an n-hexyl or an n-butyl side chain and analogues. Ester 8 was envisioned to be accessible in one-pot from chiral lactam (S)-9 by the amide reductive bis-alkylation method that we developed previously.4a
 | ||
| Scheme 2 Retrosynthetic analysis of (+)-cylindricine D. | ||
To start our investigation, the known (S)-N-benzylpyroglutaminol methyl ether (9)17 was prepared from commercially available (S)-N-benzylpyroglutaminol [(S)-11] (NaH, MeI, 0 °C) in 90% yield. Successive treatment of a solution of lactam (S)-9 (1.0 equiv.) and DTBMP (1.2 equiv., 2,6-di-tert-butyl-4-methylpyridine) in CH2Cl2 with Tf2O (1.1 equiv.), Grignard reagent 10 (1.0 equiv.), and sodium enolate generated in situ by deprotonation of ethyl acetate (1.5 equiv.) with NaHMDS (1.8 equiv., sodium bis(trimethylsilyl)amide), afforded the desired functionalized prolinol derivative 8 and its diastereomer in 60% yield (dr = 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Scheme 3). Replacement of the Tf2O/DTBMP combination by Tf2O/TTBP (2,4,6-tri-tert-butylpyrimidine), an amide activation system that has recently been shown to be versatile for the transformations of both secondary and tertiary amides,18 resulted in an improvement of the yield from 60% to 75% (dr = 7:1). The stereochemistry of the major diastereomer 8 was assumed to be trans in analogy with our previous results,4a which was confirmed by its ultimate transformation into (+)-cylindricine D (1d) (vide infra). The stereochemical outcome can be understood in terms of the trans-addition of the enolate from the less hindered β-face of the pyrrolidinium ion intermediate.
:1) (Scheme 3). Replacement of the Tf2O/DTBMP combination by Tf2O/TTBP (2,4,6-tri-tert-butylpyrimidine), an amide activation system that has recently been shown to be versatile for the transformations of both secondary and tertiary amides,18 resulted in an improvement of the yield from 60% to 75% (dr = 7:1). The stereochemistry of the major diastereomer 8 was assumed to be trans in analogy with our previous results,4a which was confirmed by its ultimate transformation into (+)-cylindricine D (1d) (vide infra). The stereochemical outcome can be understood in terms of the trans-addition of the enolate from the less hindered β-face of the pyrrolidinium ion intermediate.
 | ||
| Scheme 3 Total syntheses of (+)-cylindricine D, 2-epi-cylindricine D, and an intermediate of cylindricines C and E. | ||
Next, we proceeded to the chain elongation of ester. Employing Liotta's method,19 the Weinreb amide 7 was obtained in excellent yield. Subjection of N-benzylpyrrolidine derivative 7 to catalytic hydrogenolysis in the presence of Pearlman's catalyst [20% Pd(OH)2/C] and Boc2O resulted in the switch of N-protecting group from benzyl to Boc (6) in 93% yield. The reaction of Weinreb's amide 6 with alkynyllithium, generated in situ from oct-1-yne (3.0 equiv.) and n-butyllithium (3.0 equiv.), proceeded smoothly to give the desired ynone 5 in almost quantitative yield. Next, partial saturation of the alkynyl group was investigated. After several trials, it was found that in the presence of a catalytic amount of Lindlar catalyst (5% Pd/CaCO3 poisoned with Pb, 30 mol%), partial hydrogenation of ynone 5 in toluene/1-hexene proceeded smoothly to give the desired (Z)-α,β-enone 17, which after a simple filtration, was exposed to trifluoroacetic acid in methylene chloride (0 °C, 1 h). After removal of CH2Cl2 and TFA under reduced pressure, the residue was treated with K2CO3 in methanol at 60 °C for 8 hours, which yielded bicyclic alcohol 18 in 83% yield as an inseparable 1:1 diastereomeric mixture (the diastereomeric ratio was determined by integrating the peaks at 3.37 and 3.35 ppm of the 1H NMR spectrum of the mixture). Comparing with the reported excellent diastereoselectivity from the enamine-based SNi reaction,10c the stereochemical outcome of our intramolecular aza-conjugate addition is unexpected. This is attributable to the conformational flexibility of both the pyrrolidine ring and the conjugate addition reaction. Although we were unable to control the formation of the newly formed stereocenter, telescoping four reactions [(1) partial reduction of ynone 5 to the presumed (Z)-enone 17; (2) N-deprotection; (3) O-deprotection; (4) intramolecular aza-conjugate addition] into one step rendered the synthesis highly efficient. Finally, O-mesylation of the diastereomeric mixture 18 and treating the resultant keto mesylate with potassium tert-butoxide in THF produced (+)-cylindricine D (1d) and 2-epi-cylindricine D (2-epi-1d) (dr = 1:1) in a combined yield of 80%. The physical {1d: [α]20D +22.5 (c 0.25, CHCl3); natural sample:7b data not reported; synthetic sample: lit.10e [α]23D +21.8 (c 0.57, CHCl3); 2-epi-1d: [α]25D +7.4 (c 0.1, CHCl3)} and spectroscopic data of our synthetic compounds match those reported in the literature.10e
Having achieved a short total synthesis of (+)-cylindricine D, we proceeded to the syntheses of (+)-cylindricines C and E. For this purpose, the known benzyl ether 1217 was prepared from (S)-N-benzylpyroglutaminol [(S)-11] by exposing to NaH and BnBr at 0 °C, which afforded 12 in 94% yield (Scheme 3). Pleasantly, the Tf2O/TTBP-promoted one-pot reductive bis-functionalization of 12 with Grignard reagent 10 and sodium enolate generated in situ from ethyl acetate and NaHMDS afforded the desired functionalized prolinol derivative 13 in 80% yield and in excellent diastereoselectivity (dr ≥ 20:1) (Scheme 3). The observed higher diastereoselectivity as compares with that obtained from lactam 9 may be due to beneficial steric hindrance of the benzyl group in lactam 12. Amidation of ester 13 [Me(MeO)NH-HCl, iPrMgCl, THF, −20 °C, yield: 96%] followed by 20% Pd(OH)2/C-catalyzed hydrogenolysis of Weinreb amide 14 in the presence of (Boc)2O produced selectively O-t-butyl carbamate 15 in 95% yield. Alkynylation of Weinreb amide 15 yielded ynone 16 in 88% yield (Scheme 3). Telescoped one-step Lindlar hydrogenation of ynone 16, cleavage of the N-Boc with trifluoroacetic acid and treating the resultant mixture with K2CO3 yielded indolizidinone 20, which was O-tosylated to yield tosylate 21 in 77% yield (from 16, Scheme 4) as an inseparable 1.1:1 diastereomeric mixture (the diastereomeric ratio was determined by integrating the peaks at 3.27 and 3.08 ppm of the 1H NMR spectrum of the mixture). KHMDS-mediated cyclization of 21 in THF at −78 °C afforded diastereomeric mixture 22 (dr = 1.1:1) in 94% yield. O-Debenzylation of 22 under acidic conditions (10% Pd/C, H2, MeOH/H2O/AcOH, rt, 24 h) proceeded smoothly to afford (+)-cylindricine C (1c) and 2-epi-cylindricine C (2-epi-1c) both in 41% yield. The physical {1c: [α]25D +56.9 (c 0.25, CHCl3); natural sample:7b data not reported; synthetic sample: lit.10m [α]22D +60.2 (c 0.5, CHCl3); 2-epi-1c: [α]25D +46.2 (c 1.0, CHCl3), lit.10d [α]25D −39 (c 0.5, CH2Cl2)} and spectroscopic data of our synthetic compounds match those reported in the literature.10m
 | ||
| Scheme 4 Completion of the total syntheses of (+)-cylindricines C, E, and C-2 epimers. | ||
Finally, O-acetylation of 1c (Ac2O, Et3N, DMAP, CH2Cl2) produced (+)-cylindricine E (1e) in 97% yield. Similarly, O-acetylation of 2-epi-cylindricine C (2-epi-1c) yielded 2-epi-cylindricine E (2-epi-1e) in excellent yield. The physical {1e: [α]25D +27.5 (c 0.25, CHCl3), lit.10m [α]22D +28.3 (c 0.15, CHCl3); 2-epi-1e: [α]25D +29.1 (c 1.0, CHCl3)} and spectroscopic data of our synthetic compounds matched those reported in the literature.9a
Conclusions
To summarize, on the basis of the amide strategy and telescoped one-step four-reaction transformation, we have developed a concise approach to cylindricines C–E. The total synthesis of cylindricine D (1d) was achieved in 7 steps and 20.9% overall yield from the known compound (S)-9 [8 steps, 18.8% overall yield from commercial compound (S)-11]. Similarly, (+)-cylindricine C (1c) was synthesized in 8 steps, 19.1% overall yield from (S)-12 [9 steps, 17.9% overall yield from (S)-11], and (+)-cylindricine E (1e) was synthesized in 9 steps, 18.5% overall yield from (S)-12 [10 steps, 17.4% overall yield from (S)-11]. Thus, we have accomplished one of the shortest and the most efficient total syntheses of (+)-cylindricines C–E so far reported.8,20 In addition, alongside with cylindricines C–E, their C-2 epimers were obtained concurrently in similar overall yields. Work is on progress towards the total synthesis of other members and analogues of alkaloids of the cylindricine/fasicularin/lepadiformine family.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21931010) and the National Key R&D Program of China (grant No. 2017YFA0207302).Notes and references
- T. R. Hoye and E. Sizova, The Evolution of Chemistry through Synthesis (and of Synthesis in Chemistry), in Chemical Evolution II: From the Origins of Life to Modern Society, ACS Symposium Series, American Chemical Society, Washington, DC, 2010, ch. 10, pp. 181–203 Search PubMed.
- (a) Efficiency in Natural Product Total Synthesis, ed. P.-Q. Huang, Z.-J. Yao and R. P. Hsung, John Wiley & Sons, Inc., Hoboken, 2018 Search PubMed; (b) A. L. Zografos, Angew. Chem., Int. Ed., 2021, 60, 2738 CrossRef CAS.
- For an example, see: C. Kibayashi, S. Aoyagi and H. Abe, Total Synthesis of Bioactive Tricyclic Marine Alkaloids, Lepadiformine and Related Compounds, Bull. Chem. Soc. Jpn., 2003, 76, 2059 CrossRef CAS.
- (a) K.-J. Xiao, J.-M. Luo, K.-Y. Ye, Y. Wang and P.-Q. Huang, Direct, One-pot Sequential Reductive Alkylation of Lactams/Amides with Grignard and Organolithium Reagents through Lactam/Amide Activation, Angew. Chem., Int. Ed., 2010, 49, 3037 CrossRef CAS; (b) K.-J. Xiao, Y. Wang, K.-Y. Ye and P.-Q. Huang, Versatile One-pot Reductive Alkylation of Lactams/Amides via Amide Activation: Application to the Concise Syntheses of Bioactive Alkaloids (±)-Bgugaine, (±)-Coniine, (+)-Preussin, and (–)-Cassine, Chem. – Eur. J., 2010, 16, 12792 CrossRef CAS; (c) W. Ou and P.-Q. Huang, Amides as Surrogates of Aldehydes for C-C Bond Formation: Amide-Based Direct Knoevenagel-Type Condensation Reaction and Related Reactions, Sci. China: Chem., 2020, 63, 11 CrossRef CAS; (d) W. Ou, G.-S. Lu, D. An, F. Han and P.-Q. Huang, Two-Step Catalytic Transformation of N-Benzyllactams to Alkaloids (±)-Solenopsin, (±)-Solenopsin A, and (+)-Julifloridine, Eur. J. Org. Chem., 2020, 52 CrossRef CAS; (e) X.-Z. Huang, L.-H. Gao and P.-Q. Huang, Enantioselective Total Syntheses of (+)-Stemofoline and Three Congeners Based on a Biogenetic Hypothesis, Nat. Commun., 2020, 11, 5314 CrossRef CAS PubMed; (f) D.-H. Chen, W.-T. Sun, C.-J. Zhu, G.-S. Lu, D.-P. Wu, A.-E. Wang and P.-Q. Huang, Enantioselective Reductive Cyanation and Phosphonylation of Secondary Amides by Iridium and Chiral Thiourea Sequential Catalysis, Angew. Chem., Int. Ed., 2021, 60, 8827 CrossRef CAS.
- A. Hager, N. Vrielink, D. Hager, J. Lefranc and D. Trauner, Synthetic Approaches towards Alkaloids Bearing α-Tertiary Amines, Nat. Prod. Rep., 2016, 33, 491 RSC.
- For other methods, see: (a) K. Shirokane, Y. Kurosaki, T. Sato and N. Chida, A Direct Entry to Substituted N-Methoxyamines from N-Methoxyamides via N-Oxyiminium Ions, Angew. Chem., Int. Ed., 2010, 49, 6369 CrossRef CAS PubMed; (b) G. Vincent, R. Guillot and C. Kouklovsky, Stereodivergent Synthesis of Substituted N,O-Containing Bicyclic Compounds by Sequential Addition of Nucleophiles to N-Alkoxybicyclolactams, Angew. Chem., Int. Ed., 2011, 50, 1350 CrossRef CAS; (c) S. Hiraoka, T. Matsumoto, K. Matsuzaka, T. Sato and N. Chida, Approach to Fully Substituted Cyclic Nitrones from N-Hydroxylactam Derivatives: Development and Application to the Total Synthesis of Cylindricine C, Angew. Chem., Int. Ed., 2019, 58, 4381 CrossRef CAS; (d) S. Yamamoto, Y. Komiya, A. Kobayashi, R. Minamikawa, T. Oishi, T. Sato and N. Chida, Asymmetric Total Synthesis of Fasicularin by Chiral N-Alkoxyamide Strategy, Org. Lett., 2019, 21, 1868 CrossRef CAS PubMed.
- (a) A. J. Blackman, C. Li, D. C. R. Hockless, B. W. Skelton and A. H. White, Cylindricines A and B, Novel Alkaloids from the Ascidian Clavelina Cylindrica, Tetrahedron, 1993, 49, 8645 CrossRef CAS; (b) C. Li and A. J. Blackman, Cylindricines C-G, Perhydropyrrolo[2,1-j]quinolin-7-one Alkaloids from the Ascidian Clavelina Cylindrica, Aust. J. Chem., 1994, 47, 1355 CrossRef CAS; (c) C. Li and A. J. Blackman, Cylindricines H-K, Novel Alkaloids from the Ascidian Clavelina Cylindrica, Aust. J. Chem., 1995, 48, 955 CrossRef CAS.
- For reviews, see: (a) P. Schar, S. Cren and P. Renaud, Tricyclic Marine Alkaloids: Synthetic Approaches to Cylindricines, Lepadiformine, and Fasicularin, Chimia, 2006, 60, 131 CrossRef CAS; (b) S. M. Weinreb, Studies on Total Synthesis of the Cylindricine/Fasicularin/Lepadiformine Family of Tricyclic Marine Alkaloids, Chem. Rev., 2006, 106, 2531 CrossRef CAS PubMed; (c) A. Kaga and S. Chiba, Synthesis of Tricyclic Marine Alkaloids, Cylindricines, Lepadiformines, Fasicularin, and Polycitorols: A Recent Update, Synthesis, 2018, 685 CAS.
- For racemic synthesis of cylindricines, see: (a) B. B. Snider and T. Liu, Synthesis of (±)-Cylindricines A, D, and E, J. Org. Chem., 1997, 62, 5630 CrossRef CAS; (b) K. M. Werner, J. M. de los Santos, S. M. Weinreb and M. Shang, An Intramolecular Nitrone−Olefin Dipolar Cycloaddition-Based Approach to Total Synthesis of the Cylindricine and Lepadiformine Marine Alkaloids, J. Org. Chem., 1999, 64, 4865 CrossRef CAS; (c) J. F. Liu and C. H. Heathcock, Total Synthesis of (±)-Cylindricines A and B, J. Org. Chem., 1999, 64, 8263 CrossRef CAS; (d) G. Lapointe, K. Schenk and P. Renaud, Total Synthesis of (±)-Cylindricine C, Org. Lett., 2011, 13, 4774 CrossRef CAS PubMed; (e) T. J. Donohoe, P. M. Brian, G. C. Hargaden and T. J. C. O'Riordan, Synthesis of Cylindricine C and a Formal Synthesis of Cylindricine A, Tetrahedron, 2010, 66, 6411 CrossRef CAS; (f) A. C. Flick, M. J. A. Caballero and A. Padwa, Utilization of a Michael Addition: Dipolar Cycloaddition Cascade for the Synthesis of (±)-Cylindricine C, Org. Lett., 2008, 10, 1871 CrossRef CAS PubMed; (g) A. C. Flick, M. J. A. Caballero and A. Padwa, A Conjugate Addition/Dipolar-Cycloaddition Cascade Sequence for the Synthesis of (±)-Cylindricine C, Tetrahedron, 2010, 66, 3643 CrossRef CAS. For synthetic studies, see: (h) T. Taniguchi, O. Tamura, M. Uchiyama, O. Muraoka, G. Tanabe and H. Ishibashi, Concise Synthesis of the Tricyclic Skeleton of Cylindricines Using a Radical Cascade Involving 6-Endo Selective Cyclization, Synlett, 2005, 1179 CAS; (i) J. Zou, D. W. Cho and P. S. Mariano, A Concise, Metathesis Based Approach to Construction of the Lepadiformine/Cylindricine Tricyclic Framework, Tetrahedron, 2010, 66, 5955 CrossRef CAS.
- (a) G. A. Molander and M. Rönn, Total Synthesis of (−)-Cylindricine C, J. Org. Chem., 1999, 64, 5183 CrossRef CAS; (b) B. M. Trost and M. T. Rudd, Chemoselectivity of the Ruthenium-Catalyzed Hydrative Diyne Cyclization: Total Synthesis of (+)-Cylindricine C, D, and E, Org. Lett., 2003, 5, 4599 CrossRef CAS; (c) T. Arai, H. Abe, S. Aoyagi and C. Kibayashi, Total Synthesis of (+)-Cylindricine C, Tetrahedron Lett., 2004, 45, 5921 CrossRef CAS; (d) S. Canesi, D. Bouchu and M. Ciufolini, Fully Stereocontrolled Total Syntheses of (−)-Cylindricine C and (−)-2-Epicylindricine C: A Departure in Sulfonamide Chemistry, Angew. Chem., Int. Ed., 2004, 43, 4336 CrossRef CAS; (e) J. Liu, R. P. Hsung and S. D. Peters, Total Syntheses of (+)-Cylindricines C−E and (−)-Lepadiformine through a Common Intermediate Derived from an aza-Prins Cyclization and Wharton's Rearrangement, Org. Lett., 2004, 6, 3989 CrossRef CAS PubMed; (f) H. Abe, S. Aoyagi and C. Kibayashi, Total Synthesis of the Tricyclic Marine Alkaloids (−)-Lepadiformine, (+)-Cylindricine C, and (−)-Fasicularin via a Common Intermediate Formed by Formic Acid-Induced Intramolecular Conjugate Azaspirocyclization, J. Am. Chem. Soc., 2005, 127, 1473 CrossRef CAS PubMed; (g) J. Liu, J. J. Swidorski, S. D. Peters and R. P. Hsung, An N-Acyliminium Cyclization Approach to a Total Synthesis of (+)-Cylindricine C, J. Org. Chem., 2005, 70, 3898 CrossRef CAS; (h) J. J. Swidorski, J. Wang and R. P. Hsung, A Concise Total Synthesis of (−)-Cylindricine C through a Stereoselective Intramolecular Aza-[3+3] Annulation Strategy, Org. Lett., 2006, 8, 777 CrossRef CAS PubMed; (i) J. Wang, J. J. Swidorski, N. Sydorenko, R. P. Hsung, H. A. Coverdale, J. M. Kuyava and J. Liu, Aza-[3+3] Annulations. Part 6. Total Synthesis of Putative (−)-Lepadiformine and (−)-Cylindricine C, Heterocycles, 2006, 70, 423 CrossRef CAS; (j) T. Shibuguchi, H. Mihara, A. Kuramochi, S. Sakuraba, T. Ohshima and M. Shibasaki, Short Synthesis of (+)-Cylindricine C by Using a Catalytic Asymmetric Michael Reaction with a Two-Center Organocatalyst, Angew. Chem., Int. Ed., 2006, 45, 4635 CrossRef CAS; (k) T. Shibuguchi, H. Mihara, A. Kuramochi, T. Ohshima and M. Shibasaki, Catalytic Asymmetric Phase-Transfer Michael Reaction and Mannich-Type Reaction of Glycine Schiff Bases with Tartrate-Derived Diammonium Salts, Chem. - Asian J., 2007, 2, 794 CrossRef CAS; (l) H. Mihara, T. Shibuguchi, A. Kuramochi, T. Ohshima and M. Shibasaki, Short Synthesis of (+)-Cylindricine C and Formal Total Synthesis of (−)-Lepadiformine, Heterocycles, 2007, 72, 421 CrossRef CAS; (m) G. Pandey and V. Janakiram, Aza-Quaternary Scaffolds from Selective Bond Cleavage of Bridgehead-Substituted 7-Azabicyclo[2.2.1]heptane: Total Synthesis of (+)-Cylindricines C–E and (−)-Lepadiformin A, Chem. – Eur. J., 2015, 21, 13120 CrossRef CAS PubMed; (n) Ref. 6c. For enantioselective synthetic studies, see: ; (o) M. C. Bagley and W. Oppolzer, Asymmetric Synthesis of 1-Azaspiro[4.5]decanes via Intramolecular Dipolar Cycloaddition of Nitrones Containing the Bornane-10,2-sultam Chiral Auxiliary, Tetrahedron: Asymmetry, 2000, 11, 2625 CrossRef CAS; (p) W. Oppolzer and C. G. Bochet, Asymmetric Synthesis of the Spirocyclic Core of the Cylindricine-type Alkaloids, Tetrahedron: Asymmetry, 2000, 11, 4761 CrossRef CAS; (q) X.-M. Zhang, M. Wang, Y.-Q. Tu, C.-A. Fan, Y.-J. Jiang, S.-Y. Zhang and F.-M. Zhang, Prolinamide/PPTS-Catalyzed Hajos-Parrish Annulation: Efficient Approach to the Tricyclic Core of Cylindricine-Type Alkaloids, Synlett, 2008, 2831 CAS; (r) D. M. Dalton and T. Rovis, Catalytic, Asymmetric Indolizidinone Aza-Quaternary Stereocenter Synthesis: Expedient Synthesis of the Cylindricine Alkaloids Core, Org. Lett., 2013, 15, 4915 CrossRef CAS; (s) K. Srinivas, N. Singh, D. Das and D. Koley, Organocatalytic, Asymmetric Synthesis of Aza-Quaternary Center of Izidine Alkaloids: Synthesis of (−)-Tricyclic Skeleton of Cylindricine, Org. Lett., 2017, 19, 274 CrossRef CAS PubMed.
- (a) J.-F. Wu and P.-Q. Huang, Concise, Enantioselective Total Syntheses of both Proposed and Revised Structures of (−)-Versiquinazoline H, Chin. Chem. Lett., 2020, 31, 61 CrossRef CAS; (b) P.-Q. Huang, Procedure-Economical, Enantioselective Total Syntheses of Polycyclic Natural Products and Analogs Containing a 3a-Hydroxyhexahydropyrrolo[2,3-b]indole-2-carboxylic Acid Residue, Synlett, 2020, 1681 CrossRef CAS.
- (a) J. F. Biard, S. Guyot, C. Roussakis, J. F. Verbist, J. Vercauteren, J. F. Weber and K. Boukef, Lepadiformine, a New Marine Cytotoxic Alkaloid from Clavelina Lepadiformis Müller, Tetrahedron Lett., 1994, 35, 2691 CrossRef CAS; (b) M.-P. Sauviat, J. Vercauteren, N. Grimaud, M. Jugé, M. Nabil, J.-Y. Petit and J.-F. Biard, Sensitivity of Cardiac Background Inward Rectifying K+ Outward Current (IK1) to the Alkaloids Lepadiformines A, B, and C, J. Nat. Prod., 2006, 69, 558 CrossRef CAS.
- A. D. Patil, A. J. Freyer, R. Reichwein, B. Carte, L. B. Killmer, L. Faucette, R. K. Johnson and D. J. Faulkner, Fasicularin, a Novel Tricyclic Alkaloid from the Ascidian Nephteis Fasicularis with Selective Activity against a DNA Repair-deficient Organism, Tetrahedron Lett., 1997, 38, 363 CrossRef CAS.
- H. Abe, S. Aoyagi and C. Kibayashi, Total Synthesis of the Natural Enantiomer of (−)-Lepadiformine and Determination of Its Absolute Stereochemistry, Angew. Chem., Int. Ed., 2002, 41, 3017 CrossRef CAS.
- S. M. Weinreb, Lepadiformine: A Case Study of the Value of Total Synthesis in Natural Product Structure Elucidation, Acc. Chem. Res., 2003, 36, 59 CrossRef CAS.
- (a) S. Nahm and S. M. Weinreb, N-Methoxy-N-Methylamides as Effective Acylating Agents, Tetrahedron Lett., 1981, 22, 3815 CrossRef CAS; (b) For a review, see: S. Balasubramaniam and I. S. Aidhen, The Growing Synthetic Utility of the Weinreb Amide, Synthesis, 2008, 3707 CAS.
- L. J. Breña-Valle, R. C. Sánchez and R. Cruz-Almanza, Diastereoselective Alkylation of 1-Benzyl-(5S)-Substituted 2-Pyrrolidinones, Tetrahedron: Asymmetry, 1996, 7, 1019 CrossRef.
- Q. He, J.-L. Ye, F.-F. Xu, H. Geng, T.-T. Chen, H. Chen and P.-Q. Huang, Tf2O/TTBP (2,4,6-tri-tert-Butylpyrimidine): An Alternative Amide Activation System for the Direct Transformations of Both Tertiary and Secondary Amides, J. Org. Chem., 2021, 86 DOI:10.1021/acs.joc.1c01572.
- A. R. Prosser and D. C. Liotta, One-pot Transformation of Esters to Analytically Pure Ketones: Methodology and Application in Process Development, Tetrahedron Lett., 2015, 56, 3005 CrossRef CAS.
- Step count is on the basis of the number of extractive/chromatographic separation involved.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qo01408k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2022 |