
In situ phosphonium-containing Lewis base-catalyzed 1,6-cyanation reaction: a facile way to obtain α-diaryl and α-triaryl acetonitriles†
Jian-Ping
Tan‡
ab,
Yuan
Chen‡
b,
Xiaoyu
Ren
*b,
Yumeng
Guo
a,
Bing
Yi
*a,
Hongkui
Zhang
b,
Guowei
Gao
b and
Tianli
Wang
 *b
*b
aHunan Province Key Laboratory of Environmental Catalysis and Waste Recycling, College of Materials and Chemical Engineering, Hunan Institute of Engineering, Xiangtan, 411104, P. R. China
bKey Laboratory of Green Chemistry & Technology of Ministry of Education, College of Chemistry, Sichuan University 29 Wangjiang Road, Chengdu 610064, P. R. China. E-mail: wangtl@scu.edu.cn
First published on 11th November 2021
Abstract
We present a phosphonium-containing catalyst generated in situ from phosphine and tert-butyl acrylate that serves as an unusual Lewis base catalyst. It was applied for the promotion of a remote 1,6-cyanation reaction of p-quinone methides and fuchsones employing trimethylsilyl cyanide as the cyanide source. A diverse range of α-diaryl and α-triaryl acetonitriles was obtained in high yields under mild reaction conditions with low catalyst loading (5 mol%). The practicality and utility of this protocol were demonstrated via the gram-scale preparation and facile elaboration of products. Mechanistic investigations (in situ NMR and ESI-MS analysis) were employed to characterize the active zwitterionic phosphonium intermediate, which was the “true” active catalyst.
In recent decades, organophosphine-participating nucleophilic catalysis has emerged as one of the most flourishing and attractive research field, and also as a powerful tool for the preparation of structurally diverse molecules.1 In a general mode of phosphine catalysis, the reaction between a nucleophilic phosphine and a suitable electrophile initially generates a zwitterionic species A, and then captures appropriate electrophiles such as aldehydes or imines,2 electron-deficient alkenes,3 or allenes4 to afford many useful products. Alternatively, the basic nature of such zwitterionic intermediates can be utilized to deprotonate pronucleophiles, leading to the Michael addition5 or γ-addition6 (Fig. 1a). Limited by this traditional activation mode, the application and development of organophosphine catalysis has been restricted. Interestingly, recent studies have shown that the zwitterion A generated in situ from phosphines and active electrophiles could be utilized as organic base catalysts for chemical synthesis. In 2008, Tian and colleagues reported a Henry reaction using an phosphonium salt in situ as a Brønsted base catalyst by combining triphenylphosphine and methyl acrylate.7 Zhao and coworkers established chiral phosphine-acrylate dual-reagent catalysis for many asymmetric transformations whereby the zwitterionic A generated in situ from a chiral amino acid-based phosphine and acrylate were rightfully utilized as Brønsted base catalysts.8 Recently, our research team also developed a novel and efficient in situ phosphonium-containing Brønsted base catalyst assembled with dipeptide-based phosphine and allenoate to enable a facile asymmetric decarboxylative Mannich reaction between cyclic ketimines and β-keto acids with excellent yields and enantioselectivities (Fig. 1b, left).9 The phosphine-acrylate dual-reagent from the work of Zhao and colleagues could also serve as a Lewis base catalyst to achieve enantioselective Strecker-type reactions.10 Accordingly, Wu and coworkers reported cyanosilylation of α,α-dialkoxy ketones by phosphine-acrylate dual-reagent catalysis.11 Recently, Zhang and Liu reported the 1,4-conjugate addition of β-trifluoromethyl enones with analogous strategy (Fig. 1b, right).12 Despite such impressive progress in this field, the novel catalytic model is underdeveloped and remains challenging. In particular, application of an in situ phosphonium-containing organic base catalyst to promote a remote 1,6-cyanation has not been reported. Thus, focusing on replenishing novel reactions and challenging substrates to achieve diverse organic synthesis by such in situ Lewis base catalysis is worthwhile.
 | ||
| Fig. 1 (a) General phosphine catalysis; (b) an in situ phosphonium catalyst serving as an organic base catalyst; and (c) an in situ phosphonium-containing Lewis base catalyst enabling a remote 1,6-cyanation reaction (this work). | ||
Conversely, α-diaryl and α-triaryl nitriles have emerged as essential scaffolds present in many pharmaceutical agents and biologically active molecules (Fig. 2).13 Accordingly, efforts have been directed toward the development of a new process to furnish these scaffolds.14 The direct cyanation reaction of p-quinone methides (p-QMs)15 is a straightforward and attractive way to construct such structural motifs.16 We have conducted considerable research on phosphonium-containing organocatalysis.9,17,18 We hypothesized that combining an organophosphine with a suitable acrylate for in situ formation of a phosphonium species might be employed as an impactful Lewis base catalyst to activate trimethylsilyl cyanide. That hypothesis was inspired by the natural affinity of silicon to oxygen or fluorine anions. Herein, we present an efficient and applicative methodology for preparing functionalized α-diaryl and α-triaryl nitrile molecules. In this strategy, an in situ phosphonium salt generated from tricyclohexylphosphine and tert-butyl acrylate was employed as a novel Lewis base catalyst for promoting 1,6-cyanation of p-QMs and fuchsones using trimethylsilyl cyanide (TMSCN) as a safe and convenient cyanide source (Fig. 1c).
 | ||
| Fig. 2 Representative bioactive molecules containing α-diaryl- or α-triaryl nitrile units. | ||
We began our studies by using PPh2Me (P1) and ethyl acrylate (E1) as catalyst partners to investigate the 1,6-cyanation reaction between p-QM (1a) and TMSCN (2a) in CHCl3 at room temperature (Table 1). Gratifyingly, the desired product 3a was isolated in 65% yield after 12 h (entry 1). Encouraged by this initial result, we next screened various phosphines with ethyl acrylate (E1) as the in situ phosphonium salt catalyst (entries 2–8). Finally, P(Cy)3 (P6) was found to be the most effective one, and gave the desired product in 94% yield (entry 6). PAr3-type phosphines could not initiate this reaction likely due to the low activity of phosphine itself (entries 7 and 8). Then, we fixed P6 as the phosphine moiety to evaluate different electrophiles (E) for this transformation (entries 9–11). Tert-butyl acrylate (E3) was the best partner, offering the desired product in 98% yield (entry 10). Besides, we investigated allenoate E4 as the electrophile partner combined with phosphine P6 as the in situ catalyst for this reaction, but a product was not obtained (entry 11). This may have been because this in situ phosphonium species generated from phosphine and allenoate was inclined to be π34 carbon anion rather than enolate oxygen anion, so it may have been unable to act as a Lewis base catalyst.9 In addition, we investigated the solvent type but this did not elicit a better result (entries 12–15). Upon reducing the catalyst load to 5 mol%, the model reaction was also completed smoothly after 24 h, and afforded the target product without loss of yield (entry 16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | P | E | Solvent | Time (h) | Yield (%) |
| a Reaction conditions: 1a (0.10 mmol), 2a (0.2 mmol), P (10 mol%), and E (10 mol%) in CHCl3 (1.0 mL) at room temperature. b Yields of isolated products of 3a. c 5 mol% P6 and E3 were used. | |||||
| 1 | P1 | E1 | CHCl3 | 12 | 65 |
| 2 | P2 | E1 | CHCl3 | 12 | 76 |
| 3 | P3 | E1 | CHCl3 | 12 | 74 |
| 4 | P4 | E1 | CHCl3 | 12 | 32 |
| 5 | P5 | E1 | CHCl3 | 12 | 86 |
| 6 | P6 | E1 | CHCl3 | 12 | 94 |
| 7 | P7 | E1 | CHCl3 | 12 | Trace |
| 8 | P8 | E1 | CHCl3 | 12 | Trace |
| 9 | P6 | E2 | CHCl3 | 12 | 55 |
| 10 | P6 | E3 | CHCl 3 | 12 | 98 |
| 11 | P6 | E4 | CHCl3 | 12 | NR |
| 12 | P6 | E3 | CH2Cl2 | 12 | 89 |
| 13 | P6 | E3 | Et2O | 12 | 91 |
| 14 | P6 | E3 | Hexane | 12 | 88 |
| 15 | P6 | E3 | Toluene | 12 | 75 |
| 16c | P6 | E3 | CHCl3 | 24 | 98 |

With the optimal reaction conditions in hand, the scopes of p-QMs for synthesis of α-diaryl acetonitriles were investigated (Table 2). In general, p-QMs bearing electron-donating groups (R = Me, OMe) on the phenyl ring regardless of the position were perfectly compatible with the reaction conditions, and afforded the desired cyanation products in high yields (92–99%). Halogenated substituents at the ortho, meta, or para position of the phenyl ring could be excellent substrates and afford the corresponding products in high yields (88–93%). Substrates bearing an electron-withdrawing group (R = CN, CF3) were also well tolerated to the reaction conditions, and furnished the corresponding α-diaryl nitriles in 92% yield (3q) and 86% yield (3r), respectively. Furthermore, disubstitution on the benzene ring of p-QMs gave the corresponding products (3s) in 99% yield and (3t) 98% yield, respectively. To our delight, heterocyclic-substituted p-QMs such as 1-naphthyl and thienyl were also amenable to this protocol, and delivered the corresponding α-diaryl acetonitriles in 84–89% yields (3u–3v). Notably, p-QMs bearing two isopropyl groups at the ortho position were also well tolerated, and afforded the product (3w) in 85% yield with prolongation of the reaction time to 32 h. When methyl-substituted p-QMs were used as the substrate, the desired product (3x) was not obtained due to the instability of the substrate.

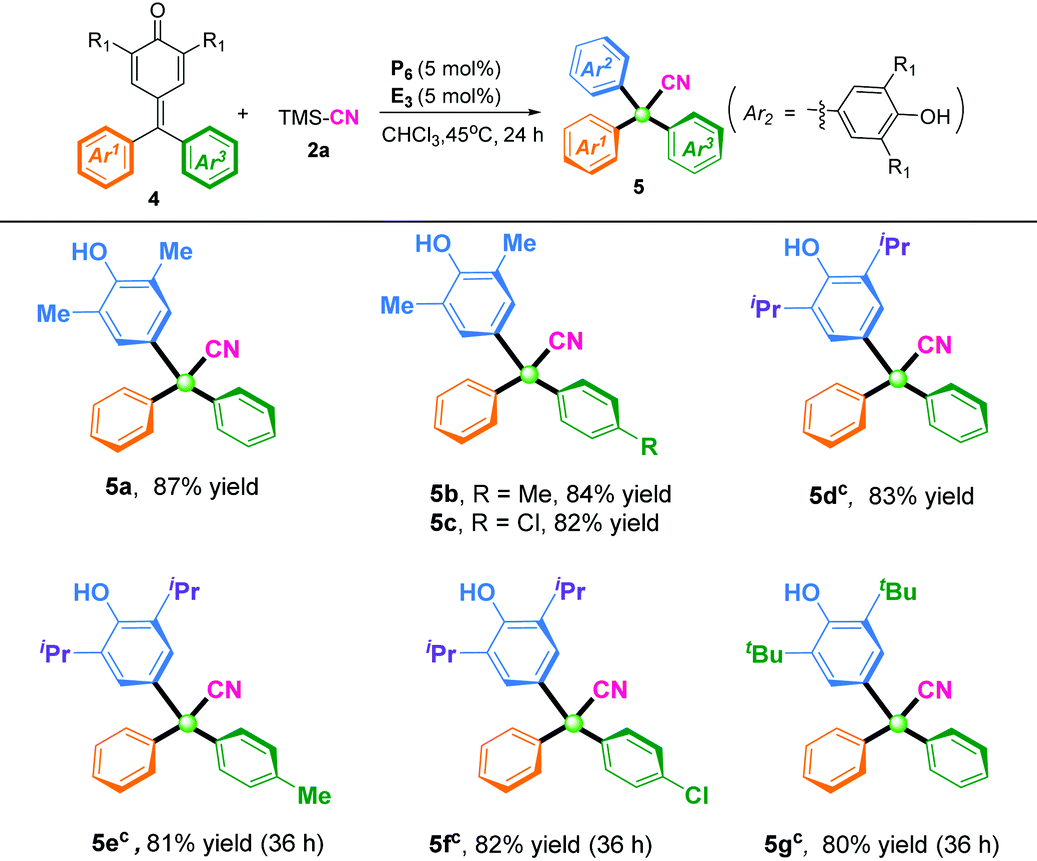
Based on successful implementation of direct cyanation of p-QMs with TMSCN for preparing α-diaryl nitriles, we used this 1,6-cyanation reaction of fuchsones towards constructing fully substituted α-triaryl acetonitriles. This is a big challenge because of the steric hindrance and low reactivity of fuchsone substrates. To overcome this difficulty, we adjusted slightly the reaction temperature to 45 °C. To our delight, all the fuchsones tested underwent this reaction efficiently, and produced the expected α-triaryl acetonitriles in moderate-to-good yields (Table 3). In general, fuchsones with a methyl substituent group at the ortho position, regardless of their electronic properties on the aromatic moiety (Ar3), were perfectly compatible with the reaction conditions, and delivered the corresponding α-triaryl nitrile products (5a–5c) in 82–87% yields. By increasing the catalyst loading to 10 mol%, isopropyl and tert-butyl substitutional fuchsones could also be suitable substrates, and the corresponding products bearing a quaternary carbon center (5d–5g) were obtained in 80–83% yields after 36 h.
To verify the utility and practicality of this protocol, the gram-scale reaction between p-QM 1f (3.5 mmol, 1.13 g) and TMSCN was undertaken, and gave the α-diaryl acetonitrile product 3f with 97% yield (1.23 g) under standard conditions. Besides, the fuchsone 4a could be accomplished under a large-scale reaction system to offer the quaternary-centered product 5a in 85% yield (Scheme 1A). Further transformations of product 3f were carried out. The CN group could be reduced readily to form primary amine 6a in 91% yield. The α-diaryl nitrile 3f could be oxidized by activated MnO2 to give the cyan-containing quinone 6b in 89% yield. Furthermore, a similar 1,6-cyanation reaction of product 6b by in situ phosphonium provided the dicyano-substituted compound 6c in 84% yield. Besides, the p-quinone methide 6b underwent a hydrophosphonylation reaction to give the phosphorus-containing cyanide 6d in 97% yield. Analogously, sulfur-containing cyanide 6e was obtained in 92% yield by a nucleophilic addition reaction of p-quinone methide 6b with thiophenol (Scheme 1B).
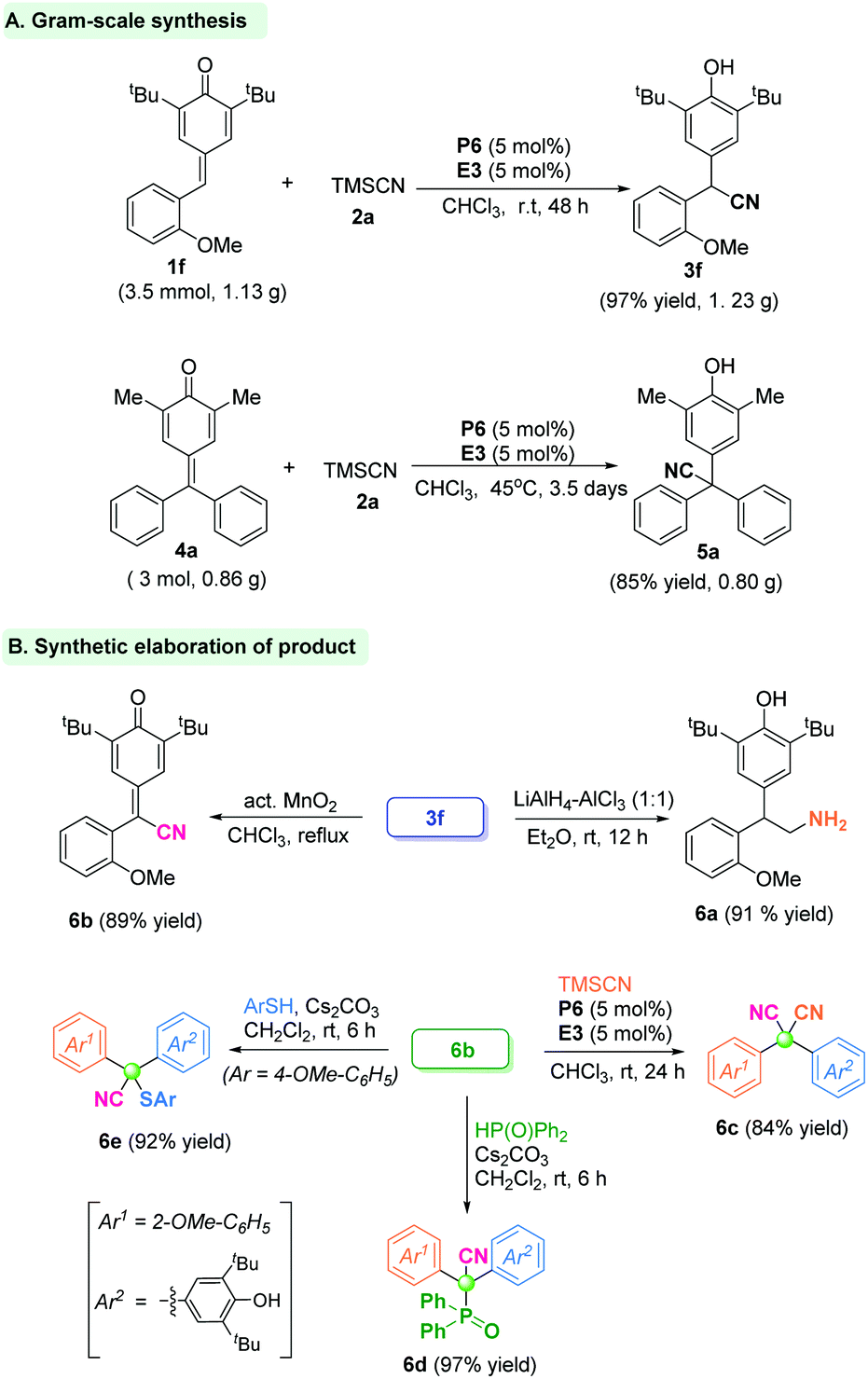 | ||
| Scheme 1 The gram-scale synthesis and synthetic elaboration of products. | ||
Then, a preliminary study on the enantioselective version of this 1,6-cyanation reaction was conducted. We first carried out the model reactions of p-QMs 1a or fuchsone 4b employing a dipeptide-based chiral organophosphine P9 with acrylate E3 to produce the in situ phosphonium salt catalyst (Table 4). Regrettably, both of the corresponding desired products (3a and 5b) were generated in low yields but with 0% ee. Afterwards, we prepared chiral acrylate E5. This was selected as a catalyst partner with P(Cy)3 for in situ formation of a chiral phosphonium catalyst to provide the enantioselectivity of this reaction. However, no chiral induction was shown. In addition, chiral phosphine (P9) and chiral acrylate (E5) were assembled together for this reaction, but promising results were not obtained (see ESI† for more details). We speculated that the long distance between the reaction site and chiral control site, as well as the flexible cyanion, may have hampered enantioselective control.
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Product | Yield (%) | ee (%) |
| 1 | P9 + E3 | 3a | 48 | 0 |
| 2 | P9 + E3 | 5b | 31 | 0 |
| 3 | P6 + E5 | 3a | 89 | 0 |
| 4 | P6 + E5 | 5b | 67 | 1 |
| 5 | P9 + E5 | 3a | 39 | 2 |
| 6 | P9 + E5 | 5b | 22 | 1 |

|

|
|||

Important control experiments were carried out to gain insight into the mechanism of this reaction. When the model substrates were used with only phosphine or tert-butyl acrylate, the reaction did not occur. This demonstrated the importance of phosphine and tert-butyl acrylate in this catalytic system. A Brønsted base, such as triethylamine, could not promote this reaction (Scheme 2A). We wished to further understand the role of an in situ-generated phosphonium catalyst from phosphine and tert-butyl acrylate. Hence, we employed electrospray ionization mass spectrometry (ESI-MS) and nuclear magnetic resonance (31P NMR) spectroscopy for characterization of active species. First, P(Cy)3 and tert-butyl acrylate generated a zwitterion Ain situ, which was tested by ESI-MS (ESI Fig. S2†), and 31P NMR was employed to track the reaction intermediate (Fig. 3). In particular, when P(Cy)3 and tert-butyl acrylate were added together, the expected zwitterion A was generated rapidly, and showed a new 31P NMR signal at δ = 32.78 ppm. Then, TMSCN was added and another new 31P NMR signal at δ = 32.95 ppm arose. We inferred that the oxygen anion of zwitterion A may act as a Lewis base to activate TMSCN, thereby likely forming a new species B (Scheme 2B).11
 | ||
| Scheme 2 Control experiments and in situ31P NMR studies. | ||
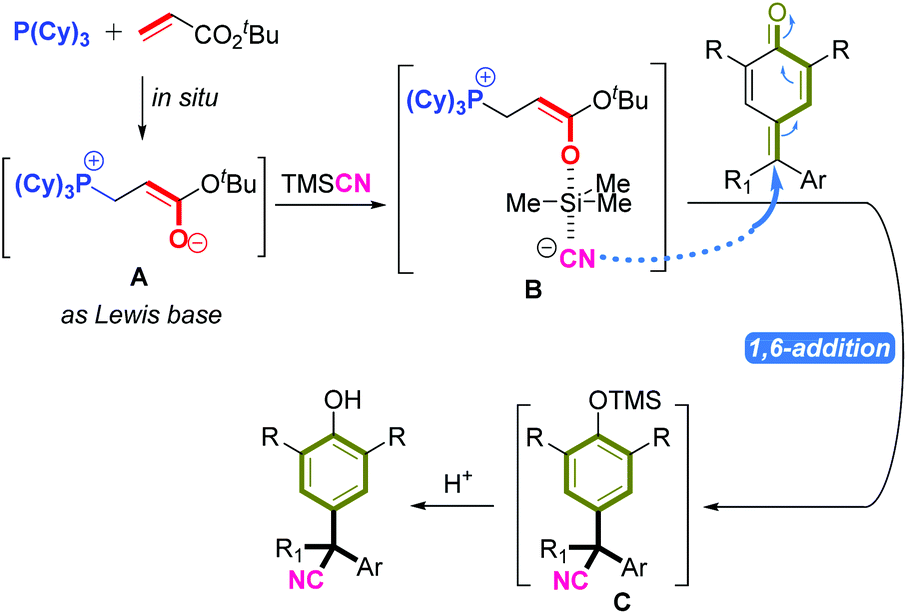 | ||
| Fig. 3 The proposed reaction mechanism. | ||
According to the mechanistic results stated above, we proposed a plausible reaction pathway for this 1,6-cyanation reaction (Fig. 3). First, the Michael-type addition of P(Cy)3 to tert-butyl acrylate generated zwitterion A rapidly, which acted as a Lewis base to activate TMSCN via species B. Then, the new zwitterion B released the active CN anion, which attacked the δ-carbon of p-QM or fuchsone to accomplish 1,6-addition/aromatization, thus affording the intermediate C that could be quenched further by acid to give the target product.
In summary, we demonstrated the synthesis of an efficient in situ phosphonium salt catalyst via combining P(Cy)3 and tert-butyl acrylate for the direct 1,6-cyanation reaction of p-QMs and fuchsones under mild reaction conditions and low catalyst loading. It provided facile access to a diverse range of α-diaryl and α-triaryl nitriles in good-to-excellent yields. Mechanistic studies showed that the zwitterion A generated in situ served as a Lewis base to activate the silicon of TMSCN. We hope that this elegant work strongly supplements the application of phosphine-acrylate catalysis. We also expect that this novel catalytic protocol might aid the construction of α-diaryl and α-triaryl nitrile scaffolds. Further development of asymmetric processes for such in situ phosphonium catalysis is ongoing in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Natural Science Foundation of China (21971165, 21921002, 21772035, 22101189), National Key R&D Program of China (2018YFA0903500), and National Natural Science Foundation of Hunan (2021JJ40150). We also acknowledge the comprehensive training platform of the Specialized Laboratory in the College of Chemistry at Sichuan University and the Analytical & Testing Center of Sichuan University for compound testing.Notes and references
- For selected Organophosphines catalysis reviews, see:
(a) Q.-Y. Zhao, Z. Lian, Y. Wei and M. Shi, Development of asymmetric phosphine-promoted annulations of allenes with electron-deficient olefins and imine, Chem. Commun., 2012, 48, 1724 RSC
; (b) T. Wang, X. Han, F. Zhong, W. Yao and Y. Lu, Amino acid-derived bifunctional phosphines for enantioselective transformations, Acc. Chem. Res., 2016, 49, 1369 CrossRef CAS PubMed
-
(a) V. Declerck, J. Martinez and F. Lamaty, aza-Baylis-Hillman reaction, Chem. Rev., 2009, 109, 1 CrossRef CAS PubMed
-
(a) S. Li, Y. Liu, B. Huang, T. Zhou, H. Tao, Y. Xiao, L. Liu and J. Zhang, Phosphine-catalyzed asymmetric intermolecular cross-vinylogous rauhut−currier reactions of vinyl ketones with para-Quinone Methides, ACS Catal., 2017, 7, 2805 CrossRef CAS
-
(a) Y. Fujiwara and G. C. Fu, Application of a new chiral phosphepine to the catalytic asymmetric synthesis of highly functionalized cyclopentenes that bear an array of heteroatom-substituted quaternary stereocenters, J. Am. Chem. Soc., 2011, 133, 12293 CrossRef CAS PubMed
- F. Zhong, X. Dou, X. Han, W. Yao, Q. Zhu, Y. Meng and Y. Lu, Chiral phosphine catalyzed asymmetric Michael addition of oxindoles, Angew. Chem., Int. Ed., 2013, 52, 943 CrossRef CAS
-
(a) Y. K. Chung and G. C. Fu, Phosphine-catalyzed enantioselective synthesis of oxygen heterocycles, Angew. Chem., Int. Ed., 2009, 48, 2225 CrossRef CAS PubMed
- X. Wang, F. Fang, C. Zhao and S.-K. Tian, Dual-reagent organocatalysis with a phosphine and electron-deficient alkene: application to the Henry reaction, Tetrahedron Lett., 2008, 49, 6442 CrossRef CAS
-
(a) H.-Y. Wang, K. Zhang, C.-W. Zheng, Z. Chai, D.-D. Cao, J.-X. Zhang and G. Zhao, Asymmetric dual-reagent catalysis: Mannich-type reactions catalyzed by ion pair, Angew. Chem., Int. Ed., 2015, 54, 1775 CrossRef CAS PubMed
- H. Zhang, C. Jiang, J.-P. Tan, H.-L. Hu, Y. Chen, X. Ren, H.-S. Zhang and T. Wang, Highly enantioselective construction of fully-substituted stereocenters enabled by in situ phosphonium-containin organocatalysis, ACS Catal., 2020, 10, 5698 CrossRef CAS
- H.-Y. Wang, C.-W. Zheng, Z. Chai, J.-X. Zhang and G. Zhao, Asymmetric cyanation of imines via dipeptide-derived organophosphine dual-reagent catalysis, Nat. Commun., 2016, 7, 12720 CrossRef
- Q.-W. Yu, L.-P. Wu, T.-C. Kang, J. Xie, F. Sha and X.-Y. Wu, Enantioselective cyanosilylation of α,α-Dialkoxy ketones by using phosphine–thiourea dual–reagent catalysis, Eur. J. Org. Chem., 2018, 3992 CrossRef CAS
- T. Zhou, X. Ji, J. Zhang and L. Liu, Phosphine-catalyzed conjugate cyanation of β-trifluoromethyl enones: access to α-trifluoromethyl γ-carbonyl nitriles, Org. Chem. Front., 2020, 7, 2644 RSC
-
(a) D. I. Lainé, H. Yan, H. Xie, R. S. Davis, J. Dufour, K. L. Widdowson, M. R. Palovich, Z. Wan, J. J. Foley, D. B. Schmidt, G. E. Hunsberger, M. Burman, A. M. Bacon, E. F. Webb, M. A. Luttmann, M. Salmon, H. M. Sarau, S. T. Umbrecht, P. S. Landis, B. J. Peck and J. Busch-Petersen, Design, synthesis and structure–activity relationship of N-substituted tropane muscarinic acetylcholine receptor antagonists, Bioorg. Med. Chem. Lett., 2012, 22, 3366 Search PubMed
- For selected review, see: G. Yan, Y. Zhang and J. Wang, Recent advances in the synthesis of aryl nitrile compounds, Adv. Synth. Catal., 2017, 359, 4068 CrossRef CAS
- For selected reviews on p-QMs and the references cited therein, see:
(a) P. Chauhan, U. Kaya and D. Enders, Advances in organocatalytic 1,6-addition reactions: enantioselective construction of remote stereogenic centers, Adv. Synth. Catal., 2017, 359, 888 CrossRef CAS
-
(a) P. Goswami, G. Singh and R. V. Anand, N-heterocyclic carbene catalyzed 1,6-conjugate addition of Me3Si-CN to para-Quinone Methides and fuchsones: access to α-arylated nitriles, Org. Lett., 2017, 19, 1982 CrossRef CAS PubMed
- G. Huang, X. Ren, C. Jiang, J.-H. Wu, G. Gao and T. Wang, Efficient synthesis of (E,)-2-nitromethylcinnamates via phosphine-catalyzed tandem α-addition and 1, 3-rearrangement, Org. Chem. Front., 2019, 6, 2872 RSC
- Selected examples, see:
(a) J.-P. Tan, P. Yu, J.-H. Wu, Y. Chen, J. Pan, C. Jiang, X. Ren, H.-S. Zhang and T. Wang, Bifunctional phosphonium salt directed enantioselective formal [4 + 1] annulation of hydroxyl-substituted Quinone Methides with α-halogenated ketones, Org. Lett., 2019, 21, 7298 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Complete experimental procedures and characterization data for the prepared compounds. See DOI: 10.1039/d1qo01501j |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2022 |