
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the mechanism of high-performance hydrogen evolution reaction on noble-metal-free (113)-faceted Ni3C: ab initio calculations†
Fuyun Hu‡
ab,
Jiahe Peng‡bc,
Wei Xiea and
Neng Li *bcd
*bcd
aSchool of Physics and Electronic-information Engineering, Huanggang Normal University, Huanggang 438000, China
bState Key Laboratory of Silicate Materials for Architectures, Research Center for Materials Genome Engineering, Wuhan University of Technology, Hubei 430070, P. R. China. E-mail: lineng@whut.edu.cn
cShenzhen Research Institute of Wuhan University of Technology, Shenzhen 518000, Guangdong, China
dState Center for International Cooperation on Designer Low-Carbon & Environmental Materials (CDLCEM), School of Materials Science and Engineering, Zhengzhou University, Zhengzhou 450001, Henan, China
First published on 4th January 2022
Abstract
To examine the reactivity of noble-metal-free Ni3C towards hydrogen evolution reaction (HER), we report a comprehensive first-principles density functional theory (DFT) study on the stability, geometric structure, electronic characteristics, and catalytic activity for HER on the Ni3C crystal (113) surfaces with different surface terminations, namely the C-rich and Ni-rich terminated surface of Ni3C (113). The results indicate that C-rich and some stoichiometric surfaces are thermodynamically stable. The bridge-site of C-rich Ni3C (113) is indispensable for HER because it not only displays improved electrocatalytic activity, but also possesses appropriate hydrogen adsorption energy, overpotential and robust stability. The ΔGH (0.02 eV) and overpotential obtained by C-rich Ni3C outperformed that obtained by Pt determined by computation (ΔGH = −0.07 eV). Thus, the bridge-sites of C-rich Ni3C (113) function as both excellent and stable active sites and adsorption/desorption sites. Increasing the density of active sites through doping or enlarging the surface area renders a prospective strategy to ameliorate the HER activity further. Overall, this study elucidates new insights into the surface properties of Ni3C for HER from water splitting and opens up a fascinating avenue to optimize the performance of solar energy conversion devices by synthesizing preferentially exposed catalyst facets.
Introduction
Energy crisis and environmental pollution have become serious problems that human society must face in the age of Anthropocene. In the war against energy security and climate change, this has attracted considerable attention to opt for renewable and clean energy sources.1–4 Because of the environmental friendliness, recycling utilization and high-energy capacity of hydrogen (H2) fuels, H2 has been considered as a green and potential new energy carrier over the past few decades.5 To date, hydrogen evolution reaction (HER) by water splitting, including electrocatalysis6 and photocatalysis,7,8 has been regarded as a pivotal technique to resolve the energy and environmental-related problems. At present, noble metals (e.g. Pt, Ag and Au) have been proven to be superior catalysts for HER. However, their high cost and scarcity severely limit their commercial applications with potential industrialization. In order to tackle these obstacles, searching for alternative catalysts composed of abundant elements for robust HER activity is actively pursued at this juncture. This includes the design of TiO2,9 g-C3N4,10 MXenes,11 transition metal chalcogenides,12 transition metal phosphides,13 metal–organic frameworks (MOFs),14,15 covalent organic frameworks (COFs)16 and so forth.For electrochemical water splitting, utilizing carbon-based nanomaterials17,18 and transition-metal dichalcogenides19 as electrocatalysts have spurred enormous interests due to their remarkably high catalytic activity6 and earth-abundance. In the recent years, nickel and its corresponding compounds have emerged as auspicious electrocatalysts,20 particularly nickel phosphide (NixPy)21 and nickel carbides.22 Co1−xNixP3 exhibits good OER activity with the Ni participation. The electronic structure can be effectively adjusted by Ni, indicating the great potential of Ni-based catalysts in electrocatalysis.23 Yan et al. dispersed uniform Ni3C nanodots in ultrathin N-doped carbon nanosheets and when doped with Fe, the Ni3C-based nanosheets exhibited outstanding electrocatalytic properties towards both HER and oxygen evolution reactions (OER).24 As for HER, Ni3C also exhibits great performance. The catalyst consisting of Ni nanoparticles with Ni3C nanosheets showed highly efficient overall water splitting.25 Moreover, combined with g-C3N4, the Ni3C cocatalyst/g-C3N4 catalyst showed high photocatalytic HER properties under visible light.26 In view of the unique traits and properties of Ni3C, it is of timely importance to demand a relentless pursuit to bridge the fundamental aspect of Ni3C at the molecular level towards the enhancement of functionality and HER activity.
Herein, we investigate the electronic characteristics and reactivity of Ni3C for the application in HER from the theoretical insight via first-principles density functional theory (DFT) calculations. Previous experiments have demonstrated that Ni3C exposed with the (113) facet is a highly stable surface.27 Sparked by the flourishing finding, we unveil the HER activity of (113)-faceted Ni3C by calculating the Gibbs free energy and adsorption energy of different active sites on the surface of Ni3C (113). Importantly, insights into the C-rich and Ni-rich terminated surface of Ni3C (113) for HER will be thoroughly unravelled. As a whole, this work casts favorable prospects for the potential utilization of earth-abundant Ni3C in the field of electrochemical water splitting, which will pave a new frontier in the materials science to engineer precious-metal-free electrocatalysts in experiments.
Computational methods
The CASTEP28 of the Materials Studio software (Accelrys Inc.) was employed for the quantum chemistry calculations. During the calculations, self-consistent periodic DFT was adopted to explore the electronic structure and catalytic activities on the facets. Ionic cores were represented by an ultrasoft pseudopotential. Perdew–Burke–Ernzerhof (PBE)29 approximation was selected as the generalized gradient approximation (GGA)30 method to calculate the exchange–correlation energy. The Broyden–Fletcher–Goldfarb–Shanno (BFGS)31 scheme was selected as the minimization algorithm. DFT-D3 correction was used for dispersion corrections. The energy cut-off was 380 eV and the SCF tolerance was 1.0 × 10−6 eV per atom. The optimization was completed when the energy, maximum force, maximum stress and maximum displacement were smaller than 5.0 × 10−6 eV per atom, 0.01 eV Å−1, 0.02 GPa and 5.0 × 10−4 Å, respectively.32 The gamma points only were set as k-point samplings during the calculations because there were no significant changes in the calculated energies for larger k-point mesh such as 2 × 2 × 1 and 3 × 3 × 1. Furthermore, at least four Ni layers were selected to reduce the dispersive error. The (113) surfaces with Ni rich and C rich terminations were built from the optimized Ni3C (space group 167, a = b = 4.553 Å, c = 12.920 Å) with a vacuum region of 15 Å (in Fig. S1, ESI†). The Gibbs free energy of the adsorption atomic hydrogen (ΔGH) is obtained by eqn (1):| ΔGH = ΔEH + ΔEZPE − TΔSH | (1) |
| ΔEH = E[Ni3C + H] − E[Ni3C] − ½E[H2] | (2) |
The exchange current at pH 0 can be calculated by eqn (3) and (4):34
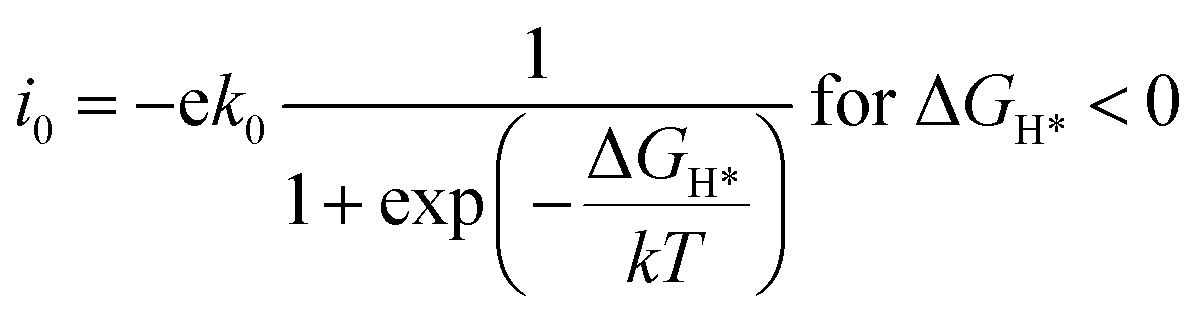 | (3) |
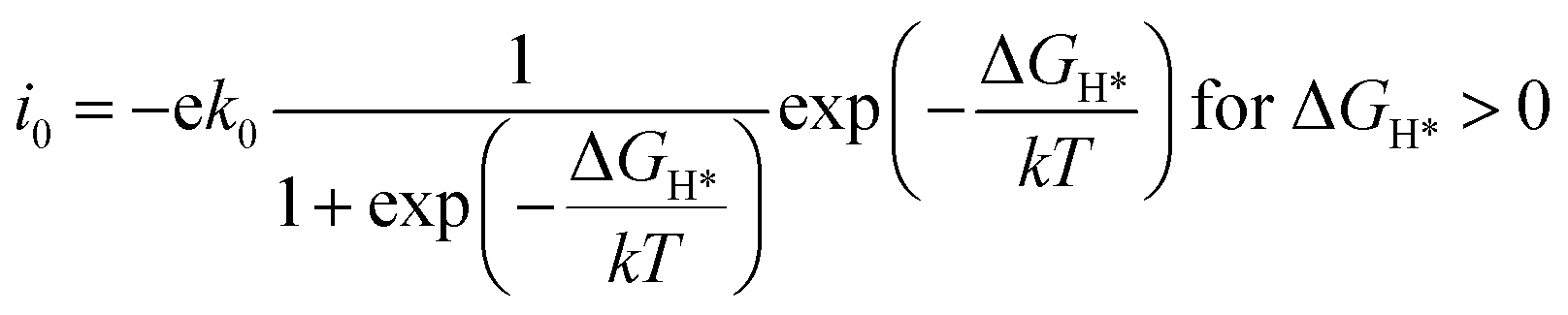 | (4) |
Furthermore, ab initio molecular dynamics (AIMD) simulations via the Vienna ab initio simulation package (VASP)35 were performed within the NVT ensemble at 400 K with a time step Δt = 1 fs and an overall time scale of 10 ps to assess the thermal stability of the Ni3C (113) structure.
Results and discussion
Ni3C (113) has been demonstrated as a stable surface in the experiment.27 With the same crystal surface index, the stable surface can be divided into two different terminations. One of the surfaces has carbon ratios larger than the other one, which is denoted as C-rich Ni3C (113). On the contrary, the higher surface nickel ratio represents Ni-rich Ni3C (113). The scaling factor is applied for the searching of optimised lattice parameters. The curve of the electronic energy vs. scaling factor is shown in Fig. S1.† Fig. 1 displays the top and side views of C-rich Ni3C (113) and Ni-rich Ni3C (113).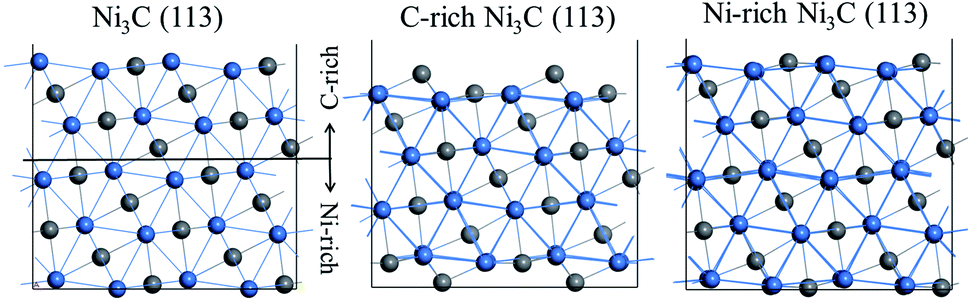 | ||
| Fig. 1 Model of Ni3C (113): (a) the terminated position of different surfaces. The side view of (b) C-rich Ni3C (113) and (c) Ni-rich Ni3C (113). | ||
In order to reveal the underlying mechanism of the HER activity of Ni3C and to investigate the electronic properties of Ni3C (113), the total and partial density of states (DOS) of C-rich Ni3C (113) and Ni-rich Ni3C (113) are depicted in Fig. 2. It is evident that the carrier density of C-rich Ni3C (113) and Ni-rich Ni3C (113) is continuous at the Fermi level, which is not equal to zero. This confirms that the two different terminated surfaces are metallic. Furthermore, the main contributions in the range from −3.75 to 1.25 eV originated from Ni 3d, and a minority from Ni 4p and C 2p orbitals. A broad and strong signal of the peak is observed from −3.75 to −0.5 eV for the Ni 3d orbital both in C-rich Ni3C (113) and Ni-rich Ni3C (113), but the intensity of the peak decreases rapidly near the Fermi level in which the free carrier density of Ni3C (113) under the Fermi level is reduced. In addition, owing to the interaction between Ni 3d and C 2p orbitals, a significant overlap of states appears from −7.5 to −5 eV, which indicates that the formation of Ni–C bonds is close and stable. In general, the difference between the DOS of C-rich Ni3C (113) and Ni-rich Ni3C (113) is not apparent.
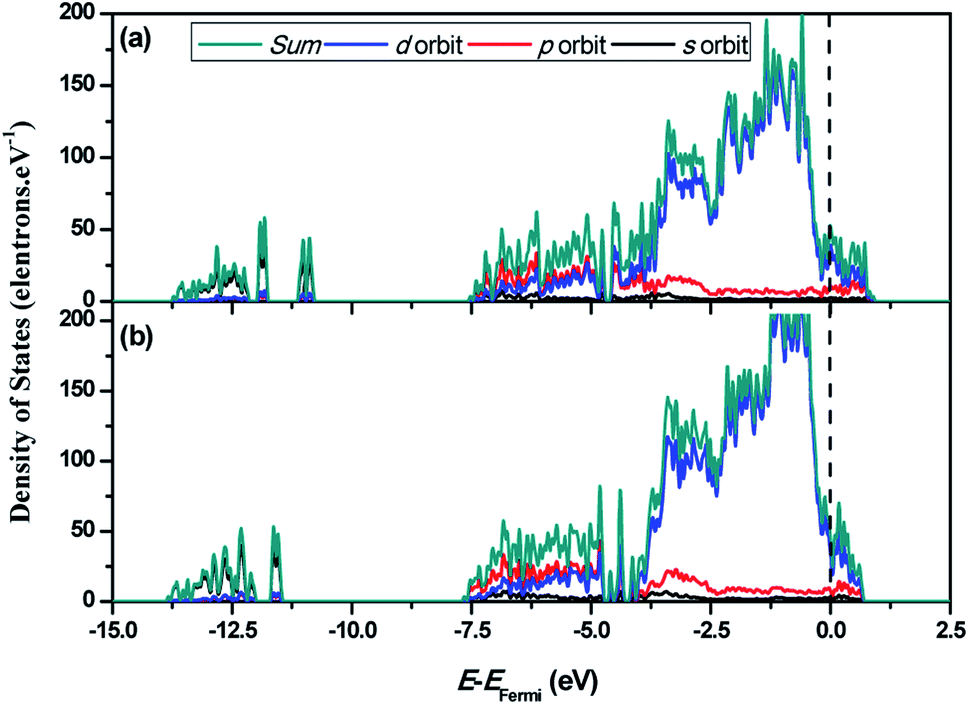 | ||
| Fig. 2 The calculated electronic characteristics of the Ni3C (113) surface with different terminations. (a) C-rich Ni3C (113) surface and (b) Ni-rich Ni3C (113) surface. | ||
Moreover, the work function of C-rich Ni3C (113) and Ni-rich Ni3C (113) is calculated to uncover the mechanism for HER (Fig. 3). The work function of two different terminated surfaces is calculated to be around 4.85 eV, which is relatively higher than the standard hydrogen electrode energy of 4.5 eV. Interestingly, the Fermi level of Ni3C is lower than the standard hydrogen electrode potential, rendering Ni3C (113) an appealing catalyst for HER.
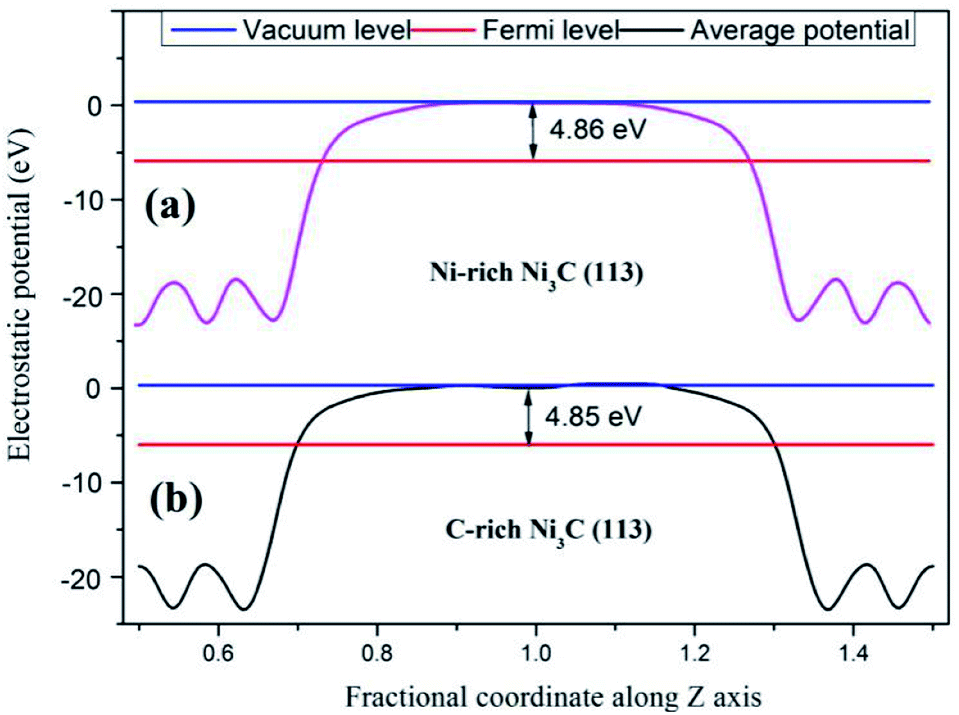 | ||
| Fig. 3 Work function of the Ni3C surface with different terminations. (a) Ni-rich Ni3C (113) surface and (b) C-rich Ni3C (113) surface. | ||
To clarify the origin of the HER catalytic activity of different facets, Gibbs free energies of hydrogen adsorption are calculated via DFT calculations. The Gibbs free energy of hydrogen adsorption (ΔGH) is one of the important criteria for evaluating the catalytic properties of HER. As a matter of fact, the optimal value for HER is ΔGH = 0. Alternatively, smaller |ΔGH| signifies better HER performance of the designed catalyst.
Fig. 4 illustrates the calculated free energy diagram for H2 evolution on different active surface sites. As shown in Fig. 4(a), the free energies of H2 evolution for Ni (111) and Pt (111) surfaces are labelled, and the values of ΔGH for Ni (111) and Pt (111) are consistent with the previous experiments and calculations.34,36 It is worth noting that although the Gibbs free energies of some active sites are close to zero, and their hydrogen adsorption energies (Eads) are greater than zero, inferring that the active site is not necessarily the stable adsorption site. Most of these stable adsorption sites are not catalytically active because they require a high energy for desorption of the adsorbed H*.
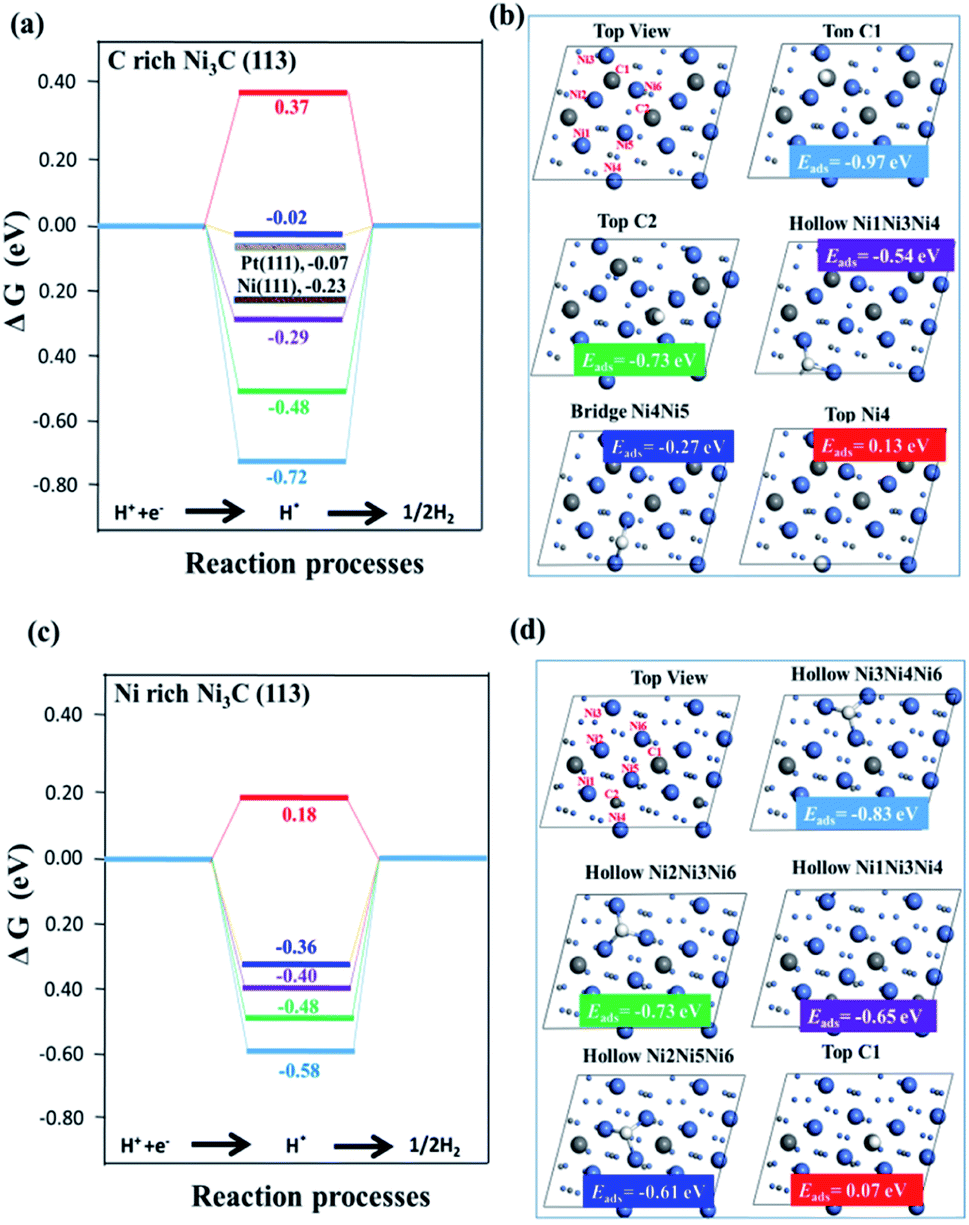 | ||
| Fig. 4 Free energy profiles for the H2 generation on different active sites of surfaces. (a and b) C-rich Ni3C (113) surface with Pt (111) and Ni (111) surfaces for comparison; (c and d) Ni-rich Ni3C (113) surface. White, grey and blue spheres represent H, C and Ni atoms, respectively. More details about the adsorption sites are described in Tables S1 and S2 (ESI†). | ||
For instance, the most stable adsorption position of C-rich Ni3C (113) is “Top C1” with an adsorption energy of −0.97 eV, while the |ΔGH| of “Top C1” is much higher than zero. Furthermore, by putting the focus on the “Bridge site of “Ni4Ni5”” (“Bridge Ni4Ni5”), the |ΔGH| of the “Bridge Ni4Ni5” site is 0.02 eV (very close to zero), highlighting that the “Bridge Ni4Ni5” site is an active site for HER. The hydrogen adsorption energies of the “Bridge Ni4Ni5” site is less than zero, manifesting that it is facile to capture protons and bond with hydrogen ions (H+) with the relative ease of desorbing H2. As for Ni-rich Ni3C (113), the most active site for HER is the “Top C1” site, and its Gibbs free energy is 0.18 eV.
However, the hydrogen adsorption energy of the “Top C1” site is greater than zero, revealing its difficulty for the adsorption of H+ ion in the “Top C1” site. As for the other sites on Ni-rich Ni3C (113), their Eads values are far less than zero, so it is hard for them to desorb the adsorbed H*. All in all, if the active site is a stable adsorption site, H* will continuously adsorb on the active sites, hence substantially boosting the H2 production rate. Nevertheless, when the stable adsorption site is not the active site, H* will first adsorb at the stable adsorption sites and then on the active sites, thus requiring a longer reaction pathway.
A plot of exchange current densities against ΔGH has a volcano shape in Fig. 5. As indicated in Fig. 5, there is a clear disparity between the exchange current densities on different Ni3C surfaces for H2 evolution. Compared to the Ni-rich Ni3C, the C-rich Ni3C (113) has much higher exchange current density and lower overpotential. Also, more than this, the plot of C-rich Ni3C (113) is much closer to the summit, which markedly outperforms the theoretically simulated Pt (111) and Ni (111).34,36 As such, the C-rich Ni3C (113) is an appropriate catalytic surface for HER.
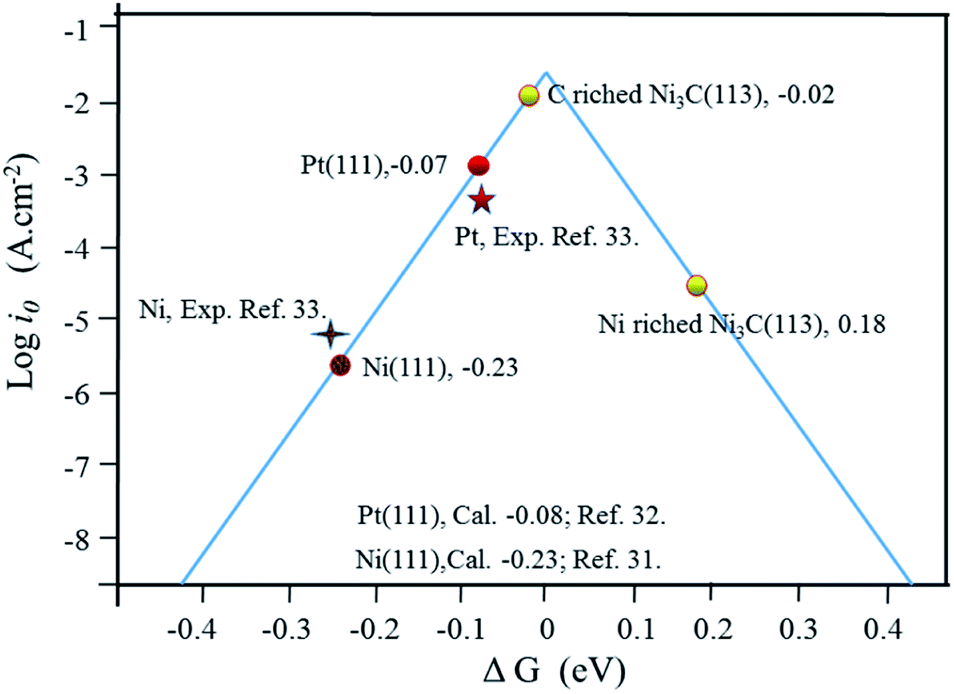 | ||
| Fig. 5 The exchange current for H2 evolution over different Ni3C surfaces, Pt (111) and Ni (111) plotted as a function of the calculated hydrogen chemisorption energy. The experimental and calculated data for Ni and Pt are also added.34,36,37 | ||
To further illustrate the stability of the structures in our calculations, AIMD simulations are performed at 400 K for 10 ps to test the stability of Ni3C. As shown in Fig. 6(a and b), the total energy and temperature oscillate near the initial condition and the geometric structures of Ni3C are preserved well within 10 ps.
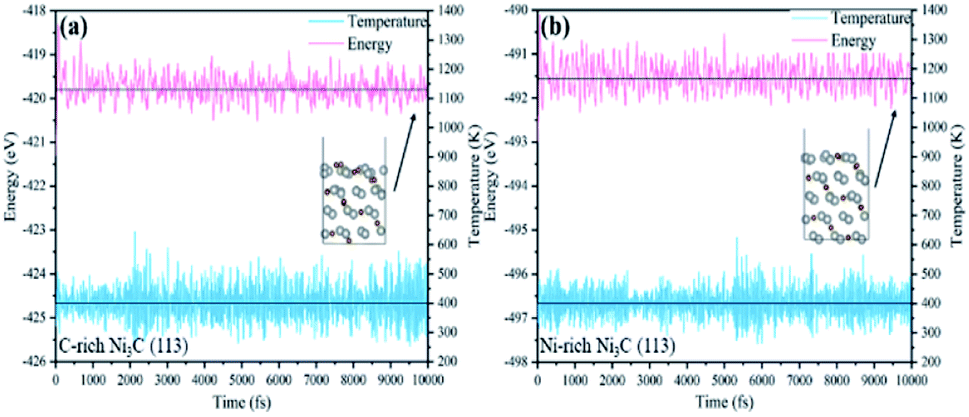 | ||
| Fig. 6 Variations of energy and temperature versus the AIMD simulation time for (a) C-rich Ni3C (113) and (b) Ni-rich Ni3C (113). The AIMD simulation lasts for 10 ps at 400 K. | ||
Conclusions
In summary, we have calculated the electronic properties and HER catalytic activity for both C-rich Ni3C (113) and Ni-rich Ni3C (113). Through calculation and analysis, C-rich Ni3C (113) endows excellent adsorption and active sites, and importantly, the Gibbs free energy of the “Bridge Ni4Ni5” site records a value of 0.02 eV, which is closer to zero than Pt (111). In addition, the overpotential of C-rich Ni3C (113) is smaller than that of Pt (111), which demonstrates that C-rich Ni3C (113) is an ideal surface for superior HER. However, the catalytic activity is not only related to these conditions, and further considerations on the number of active sites of the catalytic surface will be examined in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Fund for Distinguished Young Scholars of Hubei Province (No. 2020CFA087); the Fok Ying-Tong Education Foundation for Young Teachers in the Higher Education Institutions of China (No. 161008); the Basic Research Program of Shenzhen (No. JCYJ20190809120015163); the Central Government Guided Local Science and Technology Development Special Fund Project (2021Szvup106); the Overseas Expertise Introduction Project for Discipline Innovation of China (No. B18038), and Fundamental Research Funds for the Central Universities (2022WUT). We gratefully acknowledge HZWTECH for providing computation facilities.Notes and references
- X. Chen, N. Li, Z. Kong, W. J. Ong and X. Zhao, Mater. Horiz., 2017, 5, 9–27 RSC.
- N. Li, X. Z. Chen, W. J. Ong, D. R. MacFarlane, X. J. Zhao, A. K. Cheetham and C. H. Sun, ACS Nano, 2017, 11, 10825–10833 CrossRef CAS.
- W. J. Ong, L. K. Putri, Y. C. Tan, L. L. Tan, N. Li, H. N. Yun, X. Wen and S. P. Chai, Nano Res., 2017, 10, 1673–1696 CrossRef CAS.
- Z. Zeng, X. Chen, K. Weng, Y. Wu, P. Zhang, J. Jiang and N. Li, npj Comput. Mater., 2021, 7, 80 CrossRef CAS.
- J. Jiang, Y. Zou, A. Arrame, F. Li, J. Wang, J. Zou and N. Li, J. Mater. Chem. A, 2021, 9, 24195–24214 RSC.
- J. X. Ge, J. Hu, Y. T. Zhu, Z. Zeb, D. J. Zang, Z. X. Qin, Y. C. Huang, J. W. Zhang and Y. G. Wei, Acta Phys.-Chim. Sin., 2020, 36(1), 1906063 Search PubMed.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS.
- T. Su, Q. Shao, Z. Qin, Z. Guo and Z. Wu, ACS Catal., 2018, 8, 2253–2276 CrossRef CAS.
- W. J. Ong, L. L. Tan, S. P. Chai, S. T. Yong and A. R. Mohamed, Chemsuschem, 2014, 7, 690–719 CrossRef CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS.
- N. K. Chaudhari, H. Jin, B. Kim, S. B. Du, H. J. Sang and K. Lee, J. Mater. Chem. A, 2018, 6, 1865 RSC.
- D. Zeng, P. Wu, W.-J. Ong, B. Tang, M. Wu, H. Zheng, Y. Chen and D.-L. Peng, Appl. Catal., B, 2018, 233, 26–34 CrossRef CAS.
- D. Zeng, W. Xu, W.-J. Ong, J. Xu, H. Ren, Y. Chen, H. Zheng and D.-L. Peng, Appl. Catal., B, 2018, 221, 47–55 CrossRef CAS.
- H. Zhang, J. Nai, L. Yu and X. W. Lou, Joule, 2017, 1, 77–107 CrossRef CAS.
- H. B. Wu and X. Lou, Sci. Adv., 2017, 3, eaap9252 CrossRef.
- T. Banerjee, K. Gottschling, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CrossRef CAS.
- Y. P. Zhu, C. Guo, Y. Zheng and S. Z. Qiao, Acc. Chem. Res., 2017, 50, 915–923 CrossRef CAS.
- L. Zhang, J. Xiao, H. Wang and M. Shao, ACS Catal., 2017, 7, 7855–7865 CrossRef CAS.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 0105 CrossRef CAS.
- J. Lvlv, L. Cuncai, C. Zuofeng, H. Zhipeng and Z. Chi, Adv. Mater., 2018, 30, 1705653 CrossRef.
- H. Sun, X. Xu, Z. Yan, X. Chen, F. Cheng, P. S. Weiss and J. Chen, Chem. Mater., 2017, 29, 8539–8547 CrossRef CAS.
- Q. Qin, J. Hao and W. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 17827–17834 CrossRef CAS.
- Y.-C. Zhang, C. Han, J. Gao, L. Pan, J. Wu, X.-D. Zhu and J.-J. Zou, ACS Catal., 2021, 11, 12485–12509 CrossRef CAS.
- Q. Yan, H. Fan, H. Yu, Y. Zhang, Y. Zheng, Z. Dai, Y. Luo, B. Li and Y. Zong, Angew. Chem., 2017, 56, 12566 CrossRef.
- P. Wang, R. Qin, P. Ji, Z. Pu, J. Zhu, C. Lin, Y. Zhao, H. Tang, W. Li and S. Mu, Small, 2020, 16, 2001642 CrossRef CAS.
- K. He, J. Xie, Z.-Q. Liu, N. Li, X. Chen, J. Hu and X. Li, J. Mater. Chem. A, 2018, 6, 13110–13122 RSC.
- K. He, J. Xie, Z.-Q. Liu, N. Li, X. Chen, J. Hu and X. Li, J. Mater. Chem. A, 2018, 6, 13110–13122 RSC.
- M. D. Segall, J. D. L. Philip, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B, 1992, 46, 6671–6687 CrossRef CAS.
- J. D. Head and M. C. Zerner, Chem. Phys. Lett., 1985, 122, 264–270 CrossRef CAS.
- J. Hu, X. Zhao, W. Chen, H. Su and Z. Chen, J. Mater. Chem. C, 2017, 121, 18702–18709 CAS.
- Q. Tang and D. Jiang, ACS Catal., 2016, 6, 4953–4961 CrossRef CAS.
- J. K. Noerskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Cheminform, 2005, 152, J23–J26 Search PubMed.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS.
- T. L. Tan, L. L. Wang, D. D. Johnson and K. Bai, J. Mater. Chem. C, 2016, 117, 22696–22704 Search PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Stable adsorption structures of H on the C-rich and Ni-rich Ni3C (113) surface. See DOI: 10.1039/d1ra07448b |
| ‡ Equal contribution on this work. |
| This journal is © The Royal Society of Chemistry 2022 |