
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of mixed transition metal (Co, Mn, and Cu) oxide electrocatalysts anchored on different carbon supports for robust oxygen reduction reaction in neutral media†
Dena Z. Khatera,
R. S. Amina,
Mohamed Mahmoudb and
K. M. El-Khatib *a
*a
aChemical Engineering & Pilot Plant Department, Engineering Research Institute, National Research Centre, 33 El-Buhouth St., Dokki, Cairo 12311, Egypt. E-mail: kamelced@hotmail.com
bWater Pollution Research Department, National Research Centre, 33 El-Buhouth St., Dokki, Cairo 12311, Egypt
First published on 14th January 2022
Abstract
Oxygen reduction reaction (ORR) remains a pivotal factor in assessing the overall efficiency of energy conversion and storage technologies. A promising family of ORR electrocatalysts is mixed transition-metal oxides (MTMOs), which have recently gained a growing research interest. In this study, we developed MTMOs with different compositions (designated as AxB3−xO4; A = Cu, B = Co or Mn) anchored on two different carbon supports (activated carbon Vulcan XC-72 (AC) and graphene (G)) for catalyzing ORR in neutral media. Four different MTMO electrocatalysts (i.e., MnO2–CuO/AC, CoO–CuO/AC, CoO–CuO/G, and MnO2–CuO/G) were synthesized by a simple and scalable co-precipitation method. We documented the morphology and electrocatalytic properties of MTMO electrocatalysts using transmission and scanning electron microscopy, X-ray diffraction (XRD), X-ray photoelectron spectrometer (XPS), energy dispersive X-ray (EDX), and electrochemical techniques. Generally, MTMOs exhibited remarkably high ORR electrocatalytic activity with MTMOs anchored on an activated carbon support outperforming their respective MTMOs anchored on a graphene support, highlighting the importance of the catalyst support in determining the overall ORR activity of electrocatalysts. MnO2–CuO/AC has the highest diffusion limiting current density (j) value of 4.2 mA cm−2 at −600 mV (vs. SHE), which is ∼1.1–1.7-fold higher than other tested electrocatalysts (i.e., 3.9, 3.5, and 2.7 mA cm−2 for CoO–CuO/AC, CoO–CuO/G, and MnO2–CuO/G, respectively), and slightly lower than Pt/C (5.1 mA cm−2) at the same potential value. Moreover, all electrocatalysts exhibited good linearity and parallelism of the Koutechy–Levich (K–L) plots, suggesting that ORR followed first-order reaction kinetics with the number of electrons involved being close to four. Benefiting from their remarkable ORR electrochemical activities and low cost, our results reveal that non-precious MTMOs are efficient enough to replace expensive Pt for broad applications in energy conversion and electrocatalysis in neutral media, such as microbial fuel cells.
Introduction
Over the past decades, there has been an increasing demand for developing alternative environmentally-friendly, sustainable energy conversion and storage technologies.1,2 Fuel cell-based technologies represent a promising option to generate a wide range of valuable products (e.g., electric current and hydrogen). However, the overall performance of these fuel-cell systems, especially in neutral media (i.e., pH ≈ 7.0), is highly dependent on the electrocatalytic activity of the used catalysts for oxygen reduction reaction (ORR), which plays a crucial role in assessing the overall efficiency of energy conversion and storage technologies.3,4 In this context, the cathodic electrochemical reduction of oxygen (O2) in microbial fuel cells (MFCs) (as shown in eqn (1)) has recently gained a remarkable interest, owing to the capability of coupling electrochemical ORR with biological organic matter oxidation to generate an electric current.5–7| O2 + 2H2O + 4e− → 4OH− | (1) |
In a typical MFC, anaerobic consortia, including electroactive bacteria, covert complex organic waste streams, which are commonly found in wastewater, through a cascade of bioelectrochemical reactions under strictly anaerobic conditions into carbon dioxide, electrons, and protons in the anode chamber. The resulting electrons are transferred via the external circuit, while the protons pass through the membrane, into the cathode chamber. In the cathode chamber, efficient ORR electrocatalysts are usually required to reduce O2.8,9 Although Pt-based catalysts (as well as other precious metals) have been widely used as cathodic catalysts for ORR to minimize the cathodic overpotential and increase the catalytic current densities; their high cost, weak stability, and scarcity limit their application.5,10,11 In addition, the sluggish kinetics of ORR in neutral pH with complex multiple electron transfer reactions entails a much higher cathodic overpotential compared to ORR in acidic and alkaline media.12 Therefore, extensive efforts have been made to explore cost-effective, non-precious ORR electrocatalysts that can efficiently replace noble metal-based electrocatalysts.13–18
Among various alternatives, mixed transition metal oxides (MTMOs)-based electrocatalysts have gained growing interest for ORR. MTMOs are a mixture of transition metal oxides (TMOs) with different structural properties, leading to much higher electrical conductivity compared to that of a single TMO. The hallmark of MTMOs is the presence of multiple cation valences in their structures. This donor–acceptor chemisorption configuration enables strong interaction with molecular O2, which would facilitate the O–O bond splitting and improve the ORR kinetics by decreasing the activation energy required for electron transfer among cations in the system.19 Therefore, MTMOs have been widely used as a potential candidate to replace Pt-based electrocatalysts owing to their high catalytic efficiency, relatively low cost, thermodynamic stability, and availability.20,21
In addition, the nanometer-scale distribution of MTMOs on the electrode surface seems to be a prevailing factor for achieving high ORR activity through increasing the electroactive surface area and sites.22 Therefore, developing MTMOs with rich electroactive surface area as efficient ORR electrocatalysts remains a challenge. It requires tailoring and fine-tuning of the nanostructure's geometrical morphologies to improve their inherent electrical conductivity and electron transport.23 One strategy to maximize the electrocatalytic activity of the nanoscale MTMOs electrocatalysts is to embed them in a conductive catalyst support matrix, such as activated carbon and graphene-based materials. These materials have a high specific surface area, suitable functional groups, excellent thermal/chemical stability, and high electrical conductivity.24,25 The improved electrocatalytic performance of the composite might be due to one or a combination of the following factors: (1) the capacity of electron acceptance of the nitrogen and oxygen atoms that polarize in the framework of the adjacent metal atom and (2) the high surfaces area of the support, which a enhance electron transfer during electrochemical reactions. Therefore, a rational design of MTMO electrocatalysts with modified surface structure and high electrical conductivity through cationic substitution is an important factor in enhancing the ORR efficiency and activity. The other factor is anchoring them on carbon support with good electrical conductivity and large surface area,26–30 which, in turn, would provide a major boost for microbial fuel cells towards their potential scaling-up.
Yet, to the best of our knowledge, there is no mechanistic understanding of how binary MTMOs would facilitate ORR in neutral media (phosphate buffer electrolyte; pH = 7.2) compared to benchmark Pt/C. Taking into consideration the superior activity of binary MTMOs for ORR, we proposed a strategy for enhancing ORR activity in neutral media by co-precipitate Cu oxide with different TMOs (cobalt and manganese oxides) anchored on two different carbon supports (activated carbon Vulcan XC-72 (AC) and graphene (G)) (i.e., MnO2–CuO/G, MnO2–CuO/AC, CoO–CuO/G and CoO–CuO/AC). The effect of carbon supports type on the structure, morphology, and electrochemical activity of the MTMO electrocatalysts was investigated.
Materials and methods
Chemicals
All chemicals were obtained from Sigma Aldrich (Steinheim, Germany) and Fuel Cell Store Inc. (College Station, TX, USA), and were used without further purification. The only exception was the functionalization of carbon Vulcan XC-72 R, which was treated and purified in a mixture of 8.0 M HNO3 and 8.0 M H2SO4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) in an ultrasonic bath for 4 h. The product was then filtered and rinsed several times with DI water before drying at 80 °C for 6 h.
:1, v/v) in an ultrasonic bath for 4 h. The product was then filtered and rinsed several times with DI water before drying at 80 °C for 6 h.
Synthesis of MTMOs
MTMOs electrocatalysts were prepared by co-precipitation method of desired amounts of metal salts precursors (i.e. Co(NO3)2·6H2O, Cu(NO3)2·6H2O, and Mn(NO3)2·6H2O). The MTMOs were deposited onto two catalyst supports (i.e., activated carbon Vulcan XC-72R (AC) and graphene (G)). The corresponding mass ratios of the MTMOs were adjusted at 10 wt% to 90 wt% of AC or G. In a typical experiment, 0.35 mM Cu (NO3)2·6H2O was added to either 0.35 mM Co(NO3)2·6H2O or 0.35 mM Mn(NO3)2·xH2O. Then, 500 mg of catalyst supports were added to the mixture. The pH of the resulting solution was then adjusted to approximately 10, and vigorously mixed for 3 h to form a homogenous solution. Afterward, the precipitates were collected and washed several times with water and ethanol before drying at 80 °C for 6 h in air. Finally, the powder was then calcined at 300 °C for 3 h. Accordingly, four MTMOs electrocatalysts were prepared, namely MnO2–CuO/G, MnO2–CuO/AC, CoO–CuO/G, and CoO–CuO/AC. The schematic view of the experimental setup is illustrated in Fig. S1.†Physical characterization of the MTMOs
X-ray diffraction analysis (XRD) was performed to identify the crystal structure, lattice composition, and distinctive crystallite size using a RIGAKU-D/MAX-PC 2500 X-ray diffractometer fitted with Ni-filtered Cu Kα as the radiation source (λ = 0.154056 nm). XRD data were indexed using the X-Cell Materials Studio (Accelrys) software suites Reflex module. High-resolution transmission electron microscope (HRTEM; JEOL-JEM 2010) and field-emission scanning electron microscopy (FESEM; Quanta Feg 250) were used to explore the microstructure morphologies and particle size at an accelerating voltage of 160 kV and 20 kV, respectively. Data analysis for image processing and particle size measurement was performed using Gatan software. Elemental composition was determined using an energy-dispersive X-ray spectroscopy instrument (Thermo Jarrell-Ash Corporation, Franklin, MA, USA). We used X-ray photoelectron spectra (XPS) to determine the electronic interaction nature at the particle-on-carbon interfaces for the MTMOs electrocatalysts and the surface elemental composition using by XPS (PH1-5700 ESCA system, USA) that equipped with a hemispherical analyzer and an aluminum anode (monochromatic Al Ka 1.4866 keV) as an excitation source (at 12–14 kV and 10–20 mA). The system was operated under retarding model with a binding energy of 20–980 eV. High-resolution elemental analysis was performed on the Cu 2p (935–960 eV), Mn 2p (660–630 eV) and Co 2p (770–800 eV) regions with a 20 eV pass energy, a 0.1 eV step, and an 800 ms dwell time. All absolute energies were calibrated relative to copper, manganese, cobalt, and graphite. Then, the curve fitting of C 1s was performed by using a standard Gaussian–Lorentzian peak line shape fit with a Shirley background correlation. The peaks for the element were calibrated concerning the position of the C 1s line of carbon (284.6 eV).Electrocatalytic measurements and calculations
The electrochemical measurements were performed using a VoltaLab electrochemical workstation and a rotating disk electrode (RDE; CTV 101 speed control unit) connected to a personal computer. In a conventional three-electrode electrochemical cell setup, an Ag/AgCl electrode (Metrohm, Switzerland) and platinum (Pt) wire (Metrohm, Switzerland) were used as a reference and counter electrodes, respectively. The working electrodes were the as-prepared electrocatalysts deposited as an ultra-thin layer over a glassy carbon (GC) disk electrode, which was a part of the RDE. Before catalyst deposition, the GC electrode (with a geometrical surface area of 0.196 cm2) was first polished mechanically using 0.05 μm alumina slurry and a soft cloth to obtain a mirror-like surface, and then washed with deionized water and acetone. We prepared the catalyst ink by mixing 2.5 mg of prepared electrocatalysts with 0.5 mL of isopropanol and 15 μL of 5% Nafion solution. After homogenization by ultrasonication for 15 min, 5 μL of the ink was drop cast onto the GC electrode. The electrocatalyst thin film was left to dry at room temperature for one day before electrochemical measurements. The mass loading of dried electrocatalyst was about 0.12 mg cm−2.Linear sweep voltammetry (LSV) was conducted in a 50 mM O2-saturated phosphate buffer electrolyte (PBS; pH = 7.2) with a scan rate of 10 mV s−1 in a potential range of −1 to +1 V (vs. Ag/AgCl). In addition, for the RDE experiments, LSV was conducted at different rotations ranging from 400 to 2400 rpm. We reported all potentials versus the standard hydrogen electrode (SHE) (eqn (2)).31
| E (vs. SHE) = E (vs. Ag/AgCl) + 0.197 V | (2) |
The kinetic parameters and the number of electrons (n) involved in the oxygen reduction process were estimated from the slope of the fitting line (j−1 vs. ω−1/2) according to the Koutechy–Levich equation (eqn (3)).32,33
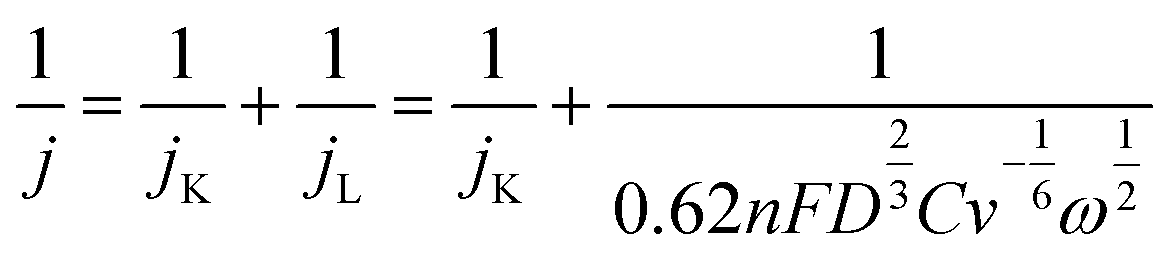 | (3) |
485C mol−1); D and C are the diffusion coefficient (1.9 × 10−5 cm2 s−1) and the bulk concentration (1.117 × 10−6 mol mL−1) of O2, respectively; and v is the kinetic viscosity of the electrolyte (0.01073 cm2 s−1) in PBS.34
Tafel slopes were calculated based on the LSV curves by plotting overpotential against logj according to the Tafel equation as follows:
|
η = a + blogj
| (4) |
We also calculated the exchange current density according to the following relation:
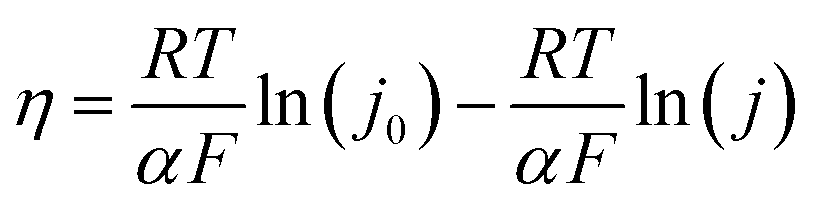 | (5) |
We estimated the real electrochemical surface area (ECSA), which is an important factor in normalizing intrinsic activities of different electrocatalysts to the same number of reactive surface sites. The ECSA cannot be estimated using the adsorption/desorption peaks of hydrogen, the ECSA of the CuO-based electrocatalysts is calculated according to the charge of CuO by the following equation:
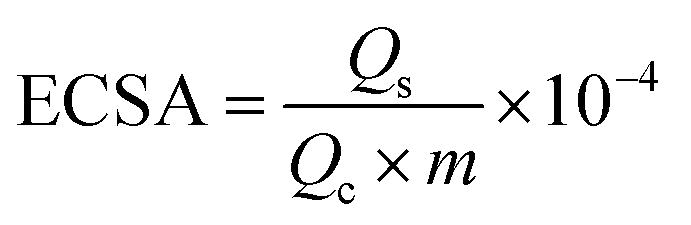 | (6) |
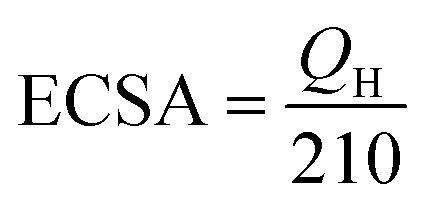 | (7) |
Results and discussion
Physicochemical properties of the MTMOs
The phases and the resulting composites' crystalline structure of all electrocatalysts were examined using XRD analysis as illustrated in Fig. 1. It could be observed that there was a broad peak at a 2θ value of ∼26° corresponding to the (002) plane of the graphitic structure for both activated carbon and graphene.37,38 Additionally, the presence of small broad peaks could be noticeably identified from all the XRD patterns at 2θ values of ∼35.6°, 38.7°, 48.8°, and 58.4° that were attributed to the (111), (200), (202), and (202) planes of CuO, respectively (JCPDS. 48-1548).39 Regarding the XRD patterns of MnO2–CuO electrocatalysts that were supported on graphene and activated carbon, it could be noticed that there was a peak at a 2θ value of 42.8°, which was assigned to the (400) plane of MnO2 (JCPDS. 24-0734).40,41 Moreover, the XRD pattern of CoO–CuO electrocatalysts showed the presence of three well-defined diffraction peaks at ∼35.3°, 38.6°, and 42.6° assigned to (222), (400), and (422) crystal planes of CoO, respectively (JCPDS. 74-2392).39,42 In addition, in the case of CoO–CuO/AC, we observed four diffraction peaks at 2θ values of 36.4°, 36.8°, 42.3°, and 61.6°, which were indexed to the (220), (311), (422) and (440) planes of CoO, respectively (JCPDS. 74-2392).43–45 It could be observed that the presence of weak intensity of different MTMOs owing to their low percentage in the nanocomposites (10%). In addition, the nature of the electrocatalysts was highly affected by changing the type of carbon support.46,47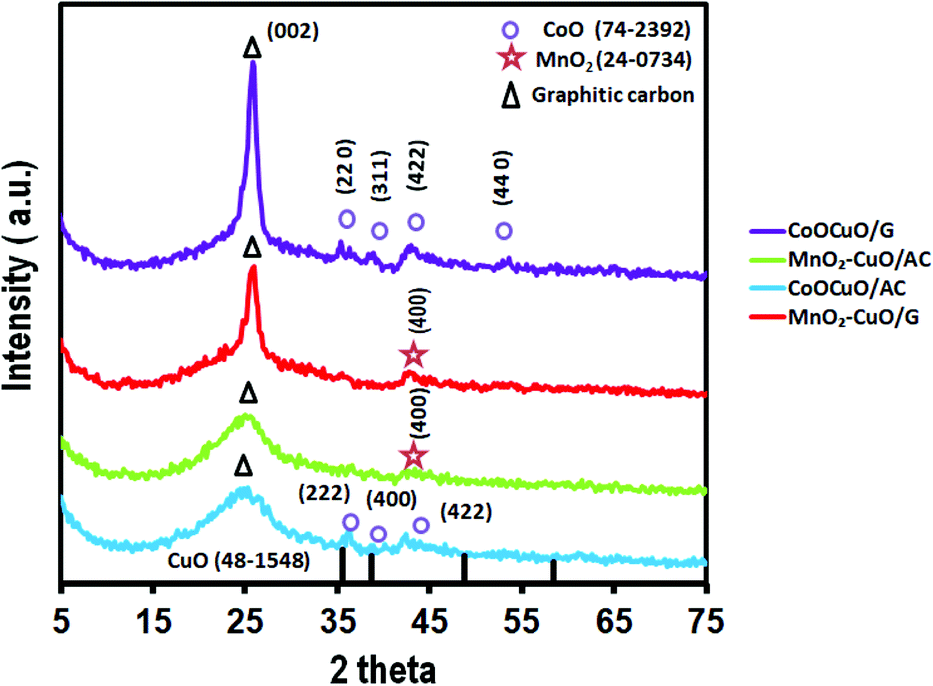 | ||
| Fig. 1 XRD patterns of MnO2–CuO/AC, CoO–CuO/AC, MnO2–CuO/G, and CoO–CuO/G electrocatalysts. | ||
TEM images illustrate the microstructure and the distribution of differently prepared electrocatalysts on the activated carbon or graphene surface as shown in Fig. 2. We observed that all MTMO electrocatalysts exhibited similar morphological and structural features and MTMOs were uniformly distributed and encapsulated within graphitic carbonaceous supports. In the case of MTMOs electrocatalysts anchored on AC, we noticed the presence of an amorphous carbon structure with rounded dark spots corresponding to metallic particles, which were uniformly dispersed on activated carbon lattice. However, MTMOs electrocatalysts anchored on G exhibited different features, in which there was a large amount of small spherically-shaped of MTMOs that were uniform in size and well dispersed with random agglomeration and heavily deposited on the graphene surface.48
 | ||
| Fig. 2 TEM images of (a) MnO2–CuO/AC, (b) CoO–CuO/AC, (c) MnO2–CuO/G, and (d) CoO–CuO/G electrocatalysts (images were taken under different magnifications). | ||
Our results were in agreement with the previous studies. For example, Sanaz Tajik et al.49 reported that MTMOs (i.e., FeCo2O4, MnCo2O4, and ZnCo2O4) possess distinct nanostructures, such as tetragonal, spherical nanoparticles, and hexagonal nanosheets. In another study, Nikolova et al.50 observed that the crystal sizes and surface areas of CuxCo3–xO4 electrocatalyst largely vary according to the precursor used. Although all CuxCo3–xO4 electrocatalysts exhibited a spinel structure, CuxCo3–xO4 prepared from carbonate precursors had a catalyst with smaller crystal sizes and higher surface area compared to the use of nitrate precursors.
To characterize the elemental structure of the MTMOs along with carbon supports, EDX analyses were performed during FESEM observation and the corresponding spectra and their mapping distribution of carbon, oxygen, manganese cobalt, and copper atoms were illustrated in Fig. 3 and Fig. S2,† while the weight and atomic ratios were shown in Table 1. Our results reveal that all metallic nanoparticles were well anchored on the surface of carbon supports. The total metal loadings were comparable with the calculated values, suggesting an efficient deposition of Cu, Mn, Co, and O on C during the co-precipitation method.
 | ||
| Fig. 3 FESEM and elemental mapping analysis of (a) CoO–CuO/AC, (b) MnO2–CuO/AC, (c) CoO–CuO/G, (d) MnO2–CuO/G. | ||
| Electrocatalyst | C | O | Cu | Mn | Co | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Wt% | Atomic% | Wt% | Atomic% | Wt% | Atomic% | Wt% | Atomic% | Wt% | Atomic% | |
| CoO–CuO/AC | 80.41 | 88.74 | 11.5 | 9.53 | 5.28 | 1.1 | — | — | 2.81 | 0.63 |
| MnO2–CuO/AC | 69.6 | 79.65 | 21.3 | 18.33 | 7.39 | 1.6 | 1.68 | 0.42 | — | — |
| CoO–CuO/G | 87.44 | 91.44 | 10.3 | 8.11 | 1.48 | 0.29 | — | — | 0.74 | 0.16 |
| MnO2–CuO/G | 71.25 | 82.76 | 16.5 | 14.44 | 8.45 | 1.85 | 3.75 | 0.95 | — | — |
The prepared MTMOs electrocatalysts in this study were further analyzed by XPS measurements to exploit the chemical bonding and the elemental composition. The chemical nature of AC and G surfaces as well as the coexistence of the oxygen-containing functioning groups were also examined. Fig. 4 and S3–S5† display the XPS spectra of MnO2–CuO/AC, CoO–CuO/AC, MnO2–CuO/G, and CoO–CuO/G. It was clear that the Cu, Co, and Mn were fitted with 2p spin–orbital interfaces, which could be deconvoluted into two doublets subsequent to the 2p1/2 and 2p3/2 along with satellites (sat.). The Cu 2p XPS spectra prominent peaks centered at about 933.52 and 950.48 eV, which is attributed to the Cu 2p3/2 and Cu 2p1/2, respectively.51 The characteristic bands for the oxidation state of the Cu corresponding to CuO (at ∼944.1 and ∼953.72 eV) and the zero-valent states of Cu (Cu0) (at ∼933.45 eV) were observed. Furthermore, it could be observed that the presence of 3 peaks at 943.58, 940.88, and 949.9 eV in Cu 2p3/2 doublet as well as the presence of 2 peaks at 961.76 and 962.04 eV in Cu 2p1/2 that corresponding to CuOsat. for all prepared MTMOs, indicating that the CuO phase exists in the electrocatalysts.52 The Cu 2p peak profile was similar to that reported for the CuCo2O4.36
 | ||
| Fig. 4 (a) XPS survey spectrum of MnO2–CuO/AC and high-resolution XPS spectra of (b) Mn 2p, (c) Cu 2p, (d) C 1s, and (e) O 1s. | ||
Furthermore, the high-resolution XPS spectrum of Mn 2p exhibits two peaks centered at 641.68 and 652.33 eV, which corresponds to Mn's spin–orbit splitting doublet states of 2p3/2 and 2p1/2, respectively. These binding energy manifests the selective formation of MnO2 free from other Mn oxidation states in appreciable amounts.52–55 In the case of MnO2–CuO/G, we observed the emergence of a distinctive peak at a binding energy of 640.4 eV that is well correlated with the zero-valent state of Mn. While in the case of MnO2–CuO/AC, there were two distinctive peaks at 645.68 and 648.85 eV that is correlated with MnO2 sat. peaks.56 It is obvious that the Co 2p3/2–2p1/2 spin–orbit splitting characteristic peaks of CoO were emerged at a binding energy of 782.92 eV (2p3/2), 794.71 eV (2p1/2), 796.93 eV (2p1/2), and 802.55 eV (2p1/2). Moreover, there was a CoO sat. peak at a binding energy of 786.04 and 789.96 eV as well as another peak at a binding energy of 780.73 eV could be assigned to Co metal.12,36,57
The C1s XPS spectrum could be fitted with adventitious carbon as C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at a binding energy of 284.23 and 285.57 eV; C–O at a binding energy of 286.23 eV; CO at 287.73, 288.53, and 289.15 eV; COOH at a binding energy of 290.69 eV; and secondary carbon C-COx/C at a binding energy of 285.57 eV.58,59 Significantly, the O 1s spectral regions at 529.49–533.68 eV were showed a strong feature that could be related to OH, CO, and COOH that assists as a nucleus for MTMOs structure on the surface of G or AC.
C at a binding energy of 284.23 and 285.57 eV; C–O at a binding energy of 286.23 eV; CO at 287.73, 288.53, and 289.15 eV; COOH at a binding energy of 290.69 eV; and secondary carbon C-COx/C at a binding energy of 285.57 eV.58,59 Significantly, the O 1s spectral regions at 529.49–533.68 eV were showed a strong feature that could be related to OH, CO, and COOH that assists as a nucleus for MTMOs structure on the surface of G or AC.
The detailed scan surveys (0–1400 eV) reveal that there are characteristic peaks of carbon, oxygen, copper, manganese, and cobalt elements as the main species in the as-prepared MTMOs electrocatalyst. Our XPS findings depict that all MTMOs electrocatalysts were successfully prepared and composed of two TMOs anchored onto both carbon supports (i.e. AC and G) by the co-precipitation method. The existence of mixed-valence metal cations in the as-synthesized MTMOs samples was advantageous toward neutral media applications. In addition, the electrocatalysts were successfully prepared and the metal transition oxides were (CuO, MnO2, and CoO). All of these results were correlated with the previously-mentioned XRD patterns (Fig. 1).
Electrochemical evaluation of MTMO
Fig. 5a displays the evaluation of the electrocatalytic activity of all prepared electrocatalysts towards ORR using LSV in 50 mM O2-saturated PBS electrolyte at a scan rate of 10 mV s−1 and a rotation rate of 1600 rpm. Pt/C (30 wt%, E-Tek) was also examined as a reference under the same conditions. MnO2–CuO/AC had the highest diffusion limiting current density value of 4.2 mA cm−2 at −600 mV (vs. SHE) among the other tested electrocatalysts. This current density value was ∼1.1–1.7-fold higher than other tested electrocatalysts (i.e., 3.9, 3.5, and 2.7 mA cm−2 for CoO–Cu O/AC, CoO–CuO/G, and MnO2–CuO/G, respectively), and slightly lower than Pt/C (5.1 mA cm−2) at the same potential value (Table 2). Compared with Pt/C electrocatalyst, the as-prepared MTMOs exhibited lower onset potential (Eonset). Interestingly, MTMOs anchored on AC had slightly higher Eonset than MTMOs/G electrocatalysts, suggesting a superior ORR electrocatalytic activity of activated carbon-based electrocatalysts. Half-wave potential (E1/2), which is another important factor to evaluate the catalytic activities of MTMOs, were 180, −43, −80, −242, and −284 mV for Pt/C, CoO–CuO/AC, MnO2–CuO/AC, MnO2–CuO/G, and CoO–CuO/G, respectively.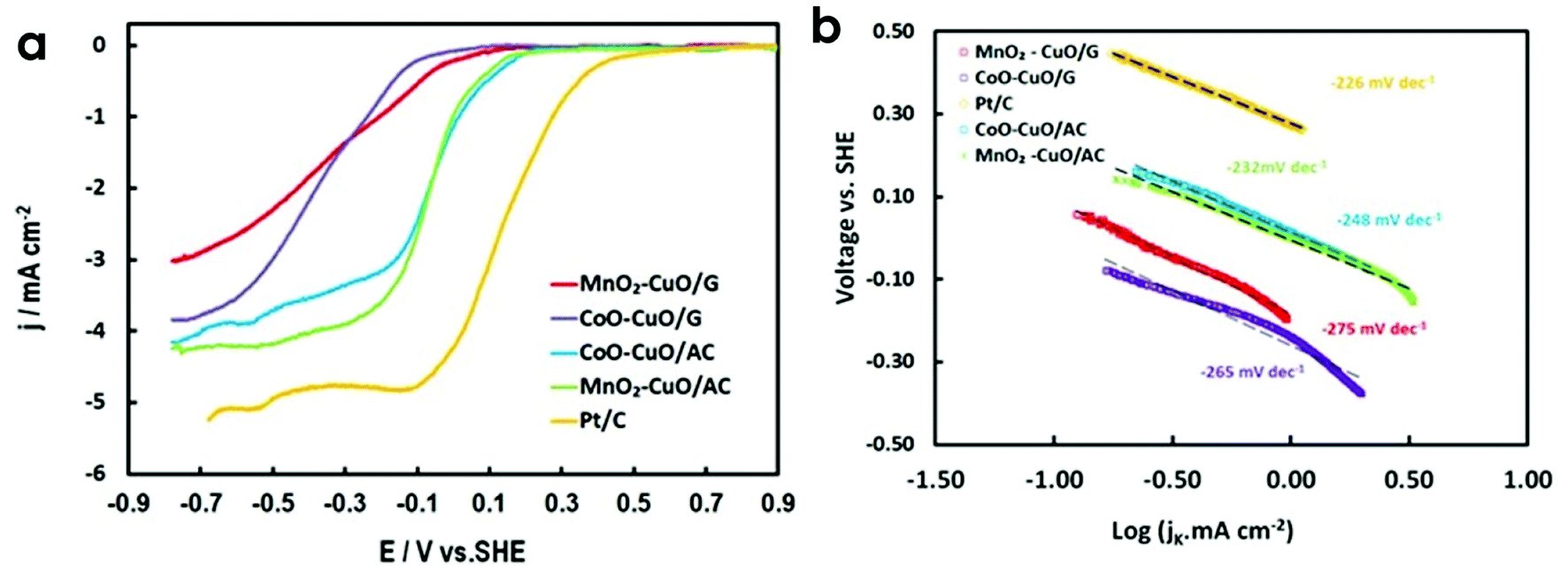 | ||
| Fig. 5 (a) ORR polarization curves of MnO2–CuO/AC, CoO–CuO/AC, MnO2–CuO/G and CoO–CuO/G and Pt/C electrocatalysts at a scan rate of 10 mV s−1 and a rotation rate of 1600 rpm in 50 mM O2-saturated PBS at, (b) the corresponding Tafel plots. | ||
| Electrocatalysts | Eonset (mV) | E1/2 (mV) | OCV (V) | j (mA cm−2) | n | jo (A cm−2) at −600 mV (vs. SHE) | b (mV dec−1) | ECSA (m2 g−1) |
|---|---|---|---|---|---|---|---|---|
| Pt/C | 416 | 180 | 0.986 | 5.1 | 4 | 2.5 × 10−5 | 235 | 19.37 |
| MnO2–CuO/AC | 220 | - 43 | 1.08 | 4.2 | 3.68 | 2.48 × 10−5 | 216 | 8.08 |
| CoO–CuO/AC | 223 | - 80 | 1.003 | 3.9 | 3.49 | 2.49 × 10−5 | 248 | 9.53 |
| MnO2–CuO/G | 130 | −242 | 1.079 | 3.5 | 3.2 | 2.46 × 10−5 | 275 | 7.4 |
| CoO–CuO/G | 89 | −284 | 0.897 | 2.7 | 3.02 | 2.45 × 10−5 | 208 | 3.18 |
Our results revealed that the activated carbon-supported electrocatalysts exhibited higher ORR performance compared to graphene-supported electrocatalysts. Thus, the nature of carbon supports can significantly affect the catalytic activity of the MnO2, CuO and CoO. The most likely reasons for this superior activity were the synergistic effect arising from the interfacial TMOs bond formation and the carbon layer that activate the sites for ORR.60 The beneficial effect of carbon supports to improve the electrical conductivity of MTMOs or both; leading to enhance the electron transfer and accelerate the ORR kinetics, excellent thermal, mechanical, electrical, and optical properties. The porosity of AC is higher than that of graphene5,61 with more complex surface chemistry that contains an abundance of reduced oxygen and increased nitrogen functionalities, which could be customized to enhance MTMOs performance for ORR in neutral media.62,63
Recently, Hazarika et al.64 synthesized mesoporous cubic Mn2O3 electrocatalyst supported on AC (Vulcan XC 72-R) and tested it for both ORR and oxygen evolution reaction (OER). They showed that the Mn2O3/AC exhibited higher ORR activity compared to the commercially available Pt/C and Pd/C electrocatalysts. However, Mn2O3 without carbon support shows less ORR activity compared to Mn2O3/C, Pt/C, and Pd/C.
Furthermore, we performed a linear fitting of the Tafel plots for all MTMO electrocatalysts to compare the electrocatalytic activity and reaction mechanism of MTMOs activities that provide information associated with the rate-determining steps.65 From the corresponding Tafel plots as illustrated in Fig. 5b, it could be observed that the Tafel slope for Pt/C (i.e., 226 mV dec−1) was comparable with those of MnO2–CuO and CoO–CuO supported on activated carbon (i.e., 232 and 248 mV dec−1, respectively). Tafel slopes of other electrocatalysts supported on graphene (MnO2–CuO and CoO–CuO) were 275 and 265 mV dec−1, respectively. It could be noticed that the electrocatalysts were displayed very similar slope values and kinetic current densities, especially in the high overpotential region. Tafel slope values imply that the first charge-transfer step was likely to be the rate-determining step. This verifies the superior ORR catalytic properties of MTMOs. Table 2 demonstrates that activated carbon-based MTMOs exhibited lower overpotential (η) (at 1 mA cm−2) compared to graphene-based electrocatalysts. Generally, Tafel slopes are dependent on the overpotential, type, and concentration of used electrolytes. For instance, it was observed that Pt/C cathode exhibited different Tafel slopes in acidic media (i.e., 76–77 mV dec−1 in 0.1 M HClO466 and 120–130 mV dec−1 in 0.05–0.5 M H2SO4)67 and alkaline media (i.e., 265 mV dec−1 in 0.1 M KOH).68 While in neutral media, relatively higher Tafel slopes are often observed.69–71
We also estimated j0 for all tested electrocatalysts and the results were shown in Table 2. Consistent with LSV results, Pt/C had the highest j0 (i.e., 2.5 × 10−5 A cm−2) compared to the as-prepared MTMOs (i.e., 2.48 × 10−5, 2.49 × 10−5, 2.46 × 10−5, and 2.45 × 10−5 A cm−2 for MnO2–CuO/AC, CoO–CuO/AC, MnO2–CuO/G, and CoO–CuO/G, respectively). These observations underscore the key role played by the AC and G supports indicating that the mechanism of ORR can be modified by used support and indicates electron transfer as the rate-determining step.
The superiority of the MTMOs towards ORR was basically due to the active role of both the transition metals to reduce the O2-dissociation energy. The dissociation of O2− can be the intermediate species for ORR as reported by Adzic and co-workers.72 Therefore, as elementary steps, the theoretical kinetic description in this study was illustrated as the following associative mechanism:73
The first step in the overall reaction path is the adsorption of O2 on the prepared MTMO electrocatalysts surface (eqn (8)). Following the initial adsorption step, the protonation of O2 on the active sites of MTMOs represents the rate-limiting step of ORR (eqn (9)–(11)).31
| M + O2 ↔ MO2(ad), | (8) |
| MO2(ad) + e− ↔ MO2(ad)−, | (9) |
| MO2(ad)− + H2O ↔ MO2H(ad) + OH(ad)−, | (10) |
| MO2H(ad) + e− ↔ MO(ad) + OH(ad)− or MO2H(ad) + e− ↔ MOOH(ad)− | (11) |
We could conclude that although the four electrocatalysts had the same mixed metals they had different surface characteristics, as indicated by the limiting current densities, according to functionalization of the AC and G that introduced different electronic interactions with O2 during the ORR.
There were obvious performance differences between the MTMOs electrocatalysts in 50 mM O2-saturated PBS at different electrode rotation speeds (i.e., 400 to 2400 rpm) as shown in Fig. 6(a–e). It could be noticed that there was an extrusive relation between cathodic current values, the rate of electrode rotation, and the potential scan rate. Furthermore, the current densities were gradually improved, implying an increase in the mass transfer of O2 from the bulk solution to the electrode surface. This behavior could be explained by the fact that; higher rotation speeds lead to faster oxygen flux at the electrode surface. As a result, generating higher currents and lower potential values were obtained. This indicated the presence of a controlled kinetics process.37 ORR generally occurs through two different mechanisms: (1) a 4-electron reduction pathway, in which O2 was directly reduced to produce H2O (eqn (12)) and/or (2) a 2-electron reduction pathway, which involves the production of hydrogen peroxide as an intermediate that undergoes electro-reduction to produce H2O (eqn (13) and (14)):3
| O2 + 4H+ + 4e− → H2O, E0 = 1.23 V (vs. SHE) | (12) |
| O2 + 2H+ + 2e− → H2O2, E0 = 0.70 V (vs. SHE) | (13) |
| H2O2 + 2H+ + 2e− → H2O, E0 = 1.78 V (vs. SHE) | (14) |
 | ||
| Fig. 6 ORR polarization curves of (a) MnO2–CuO/G, (b) MnO2–CuO/AC, (c) CoO–CuO/AC, (d) CoO–CuO/G, and (e) Pt/C electrocatalysts at different rotation rates. (f) The Koutecky–Levich plot of MTMOs at 0.5 V (vs. SHE). | ||
Ideal ORR electrocatalysts should catalyze O2 reduction in a direct 4-electron reduction process rather than in an indirect 2-electron reduction process. The latter process leads to a high overpotential and limits the overall electrode efficiency, resulting in much lower ORR efficiency.74,75 Here, the RDE measurements were used to investigate the electrochemical ORR kinetics and the number of transferred electrons involved in ORR. The corresponding K–L plot slope was determined by performing LSVs in 50 mM O2-saturated PBS at different rotation speeds (i.e., 400 to 2400 rpm). Fig. 6f shows the K–L plots for all electrocatalysts at −500 mV vs. SHE. It could be anticipated from the fitting of the K–L plot that it was distinctly parallel and linear. This behavior suggests an improved electrocatalytic behavior towards ORR (kinetically more facile) with a similar number of electrons involved in ORR (n) and first-order kinetics reaction with respect to O2 concentrations.76–78 The calculated number of electrons involved in ORR (n) for Pt/C, MnO2–CuO/AC, CoO–CuO/AC, MnO2–CuO/G, and CoO–CuO/G electrocatalysts were 4, 3.68, 3.49, 3.2, and 3.02, respectively. It is suggesting that electrocatalytic ORR catalyzed by the prepared MTMOs proceeded via a direct, one-step 4-electron transfer process with respect to molecular oxygen in the case of MnO2–CuO/AC and CoO–CuO/AC. Meanwhile, MnO2–CuO/G and CoO–CuO/G electrocatalysts seem to catalyze O2 reduction via two co-existing pathways: 2-electron and 4-electron ORR pathways. All of these observations confirm that the non-precious MTMOs were efficient enough to replace expensive Pt for broad applications in energy conversion and electrocatalysis in neutral media, owing to their availability, low cost, and they were comparable to Pt/C in terms of electrochemical activities towards ORR, the presence of variable oxidation states combined with high electrical conductivity, plentiful design and they were also higher more stable in microbial environments.79,80
Furthermore, our results indicate that the superiority of MTMOs towards ORR was due to the active role of both transition mixed metal oxides composites (i.e. Cu oxide as a post-TMO to the other TMOs (Co and Mn oxides)) to reduce the O2-dissociation energy. In this point of view, one O atom fills the surface vacancy of the transition metal having a higher affinity from electrons, whereas the other O atom binds with the second transition metal atom. As a result, enhanced ORR can be anticipated in the MTMOs cathode. Consequently, MTMOs can efficiently reduce hydrogen peroxide intermediate, resulting in high ORR efficiency as indicated from high current density associated with more positive Eonset potential.75
Recently, MTMOs electrocatalysts in neutral media have got much attention in recent years due to their promising performances in the field of energy storage. The coupling of two metal species could render the MTMOs with rich redox reactions and improved electronic conductivity, which are beneficial to electrochemical energy storage applications.49,81 They also have significantly higher electrical conductivity than single TMOs. The unique property of TMOs that facilitates better ORR performance is the presence of variable oxidation states and its ability to mix well into one material. Moreover, TMOs are commercially affordable due to their low price and high abundance which makes them very likely to be used as electrocatalysts for ORR.80 Table 3 summarizes the experimental results of different MTMOs-based electrocatalysts in neutral media. It showed the performance of different MTMOs in neutral media that were comparable to our MTMOs electrocatalysts. For example, Roche and Scott71 prepared Ni–MnOx/Vulcan by chemical deposition method onto various high specific surface area carbons. Ni–MnOx/Vulcan was found to be promising as an alternative, non-noble catalyst for the ORR in neutral media. The current density value for Ni–MnOx/Vulcan was 1.3 mA cm−2 and that of Pt/C Vulcan (E-Tek) was 18 mA cm−2 under the same condition. Furthermore, Gong et al.20 fabricated a new type of binder-free gas diffusion electrode made of cobalt oxide (Co3O4) micro-particles directly grown on stainless steel mesh (SSM) by using an ammonia-evaporation-induced method. They showed that the SSM/Co3O4 demonstrated improved performances in terms of electrocatalytic activity toward ORR in pH-neutral solution (1.75 mA), which was comparable with conventional carbon-supported platinum catalyst (2 mA).
| MTMOs Electrocatalysts | Preparation method | Eonset potential (V) | Current density (mA cm−2) | Ref. |
|---|---|---|---|---|
| MnO2–CuO/AC | Co-precipitation | 0.22 | 4.2 | This study |
| AuNPs-GC | Different combinations of reducing, stabilizing agents and solvent | −0.1 | 1600 (μA cm−2) | 3 |
| NiMn2O4 | Co-precipitation method | ∼−0.1 | 2.47 mA | 82 |
| Ni–MnOx/Monarch | Chemical deposition | 0.33 | 1.3 | 71 |
| AgNPs/rGO | Facile coreduction | −0.2 | 96 (μA) | 1 |
| SSM/Co3O4 | Ammonia-evaporation-induced method | 0.1 | 1.75 (mA) | 20 |
| Cu/Co based oxides (Cu0.15Co2.84O4) | Thermal decomposition method | ∼−0.1 | ∼2.7 | 83 |
The blocking part of the active surface area of electrocatalysts in the PBS according to the adsorption of –HPO42−, –H2PO4, –HPO43− onto the catalyst surface that was led to activity attenuation.33,71 Therefore, for measuring the specific ECSA, the CV's profiles analysis of the MTMOs was recorded in 0.5 M N2-saturated H2SO4 electrolyte within a potential window of −0.2 to 1.8 V (versus SHE) at a scan rate of 50 mV s−1 as shown in Fig. 7. It could be observed that there were two major broad peaks centered at 0.7 V. In addition, it was followed by a reversible process centered at about 0.6 V for all MTMOs. The calculated ECSA from the hydrogen adsorption region (0.2–0.8 V) were 19.37, 8.08, 9.53, 7.4, and 3.18 m2 g−1 for Pt/C, MnO2–CuO/AC, CoO–CuO/AC, MnO2–CuO/G, and CoO–CuO/G, respectively. The slight decrease of the ECSA for graphene-based catalysts was due to the lack of full utilization of activities in the thick layer of electrocatalysts.
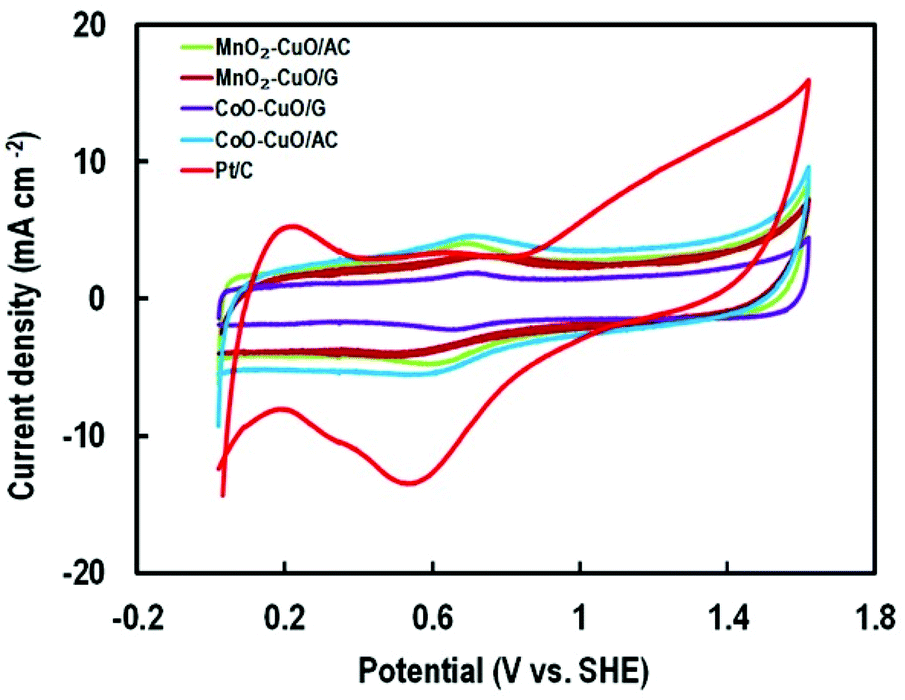 | ||
| Fig. 7 CV plots of MTMOs and Pt/C at a sweep rate of 50 mV s−1 in 0.5 M H2SO4. | ||
Conclusion
In summary, we developed highly efficient, non-precious MTMO electrocatalysts anchored on carbon supports for ORR in neutral media. MTMOs, which were prepared by co-precipitation of Cu oxide with either Mn or Co oxides, were anchored in two different carbon supports (i.e., graphene and activated carbon). Compared to the commercial Pt/C electrocatalyst, all prepared electrocatalysts exhibited excellent electrocatalytic ORR activity. This is attributed to the synergistic coupling effect of two transition metal oxides with graphene or activated carbon as well as the high electrical conductivity and specific surface area of the resulting MTMOs.It was obvious that MnO2–CuO/AC has the highest current density values of 4.2 mA cm−2 at −600 mV among the other tested electrocatalysts (i.e., 3.9, 3.5, and 2.7 mA cm−2 for CoO–Cu O/AC, CoO–CuO/G, and MnO2–CuO/G, respectively). In addition, it had a lower current density value than that of Pt/C (5.1 mA cm−2) at the same potential value. We noticed that all electrocatalysts exhibited good linearity and parallelism of the polarization plots, suggesting that ORR followed first-order reaction kinetics with the number of electrons involved close to four.
Our results indicate that the superiority of MTMOs towards ORR was due to the active role of both transition mixed metal oxides composites (i.e. Cu oxide as a post-TMO to the other TMOs (Co and Mn oxides)) to reduce the O2-dissociation energy. This significantly enhanced the ORR electrochemical activity, which was comparable to benchmark Pt/C cathode. Therefore, our MTMOs are efficient enough to replace expensive electrocatalysts (e.g., Pt) for broad applications in energy conversion and electrocatalysis in neutral media, such as microbial fuel cells.
Funding
We acknowledge the National Research Centre, Egypt, for providing funding.Availability of data and material
The online version contains supplementary material.†Author contributions
Dena Z. Khater: conceptualization, methodology, investigation, writing – original draft, visualization. R. S. Amin: methodology, investigation, data curation, writing – review & editing. Mohamed Mahmoud: methodology, investigation, data curation, visualization, writing – review & editing. K. M. El-Khatib: conceptualization, methodology, investigation, data curation, writing – review & editing, visualization, supervision.Conflicts of interest
There are no conflicts to declare.References
- H. Sun, K. Xu, G. Lu, H. Lv and Z. Liu, IEEE Trans. Nanotechnol., 2014, 13, 789–794 CAS.
- J. Chattopadhyay, T. Sankar, R. Srivastava and A. C. Singh, Electrochim. Acta, 2015, 167, 429–438 CrossRef CAS.
- G. Gotti, D. Evrard, K. Fajerwerg and P. Gros, J. Solid State Electrochem., 2016, 20, 1539–1550 CrossRef CAS.
- C. Goswami, K. K. Hazarika and P. Bharali, Mater. Sci. Energy Technol., 2018, 1, 117–128 Search PubMed.
- P. Dange, N. Savla, S. Pandit, R. Bobba and S. P. Jung, J. Renewable Mater., 2022, 10(3), 561–589 Search PubMed.
- F. Ando, T. Gunji, T. Tanabe, I. Fukano, D. Abrun, J. Wu, T. Ohsaka and F. Matsumoto, ACS Catal., 2021, 11, 9317–9933 CrossRef CAS.
- M. Mahmoud, T. A. Gad-Allah and K. M. El-Khatib, Bioresour. Technol., 2011, 102, 10459–10464 CrossRef CAS PubMed.
- K. M. El-Khatib and M. Mahmoud, Int. J. Hydrogen Energy, 2020, 45, 32413–32422 CrossRef.
- Z. H. W.-W. Li and H.-Q. Yu, Energy Environ. Sci., 2014, 7, 911–924 RSC.
- M. Mashkour, M. Rahimnejad, F. Raouf and N. Navidjouy, Biofuel Res. J., 2021, 30, 1400–1416 CrossRef.
- H. Oh, J. Oh, W. Hee, H. Kim and H. Kim, Int. J. Hydrogen Energy, 2011, 36, 8181–8186 CrossRef CAS.
- S. K. Bikkarolla and P. Papakonstantinou, J. Power Sources, 2015, 281, 243–251 CrossRef CAS.
- X. W. Liu, W. W. Li and H. Q. Yu, Chem. Soc. Rev., 2014, 43, 7718–7745 RSC.
- M. Kodali, R. Gokhale, C. Santoro, A. Serov, K. Artyushkova and P. Atanassov, Mater. Horiz., 2017, 164, H3041–H3046 CAS.
- H. Yuan, Y. Hou, I. M. Abu-Reesh, J. Chen and Z. He, Mater. Horiz., 2016, 3, 382–401 RSC.
- Y. Qiao, S. Bao and C. M. Li, Energy Environ. Sci., 2010, 3, 544–553 RSC.
- M. Mustakeem, Mater. Renewable Sustainable Energy, 2015, 4, 1–11 CrossRef.
- D. Z. Khater, R. S. Amin, A. E. Fetohi, K. M. El-Khatib and M. Mahmoud, Sustainable Energy Fuels, 2022 10.1039/D1SE01479J , in press.
- F. H. B. Lima, J. F. R. De Castro and E. A. Ticianelli, J. Power Sources, 2006, 161, 806–812 CrossRef CAS.
- X. Gong, S. You, X. Wang, J. Zhang, Y. Gan and N. Ren, Biosens. Bioelectron., 2014, 55, 237–241 CrossRef CAS PubMed.
- K. Ben, W. Ramli, W. Daud and M. Ghasemi, Int. J. Hydrogen Energy, 2014, 39, 4870–4883 CrossRef.
- P. C. M. Hamdani and R. N. Singh, Int. J. Electrochem. Sci., 2010, 5, 556–577 Search PubMed.
- X. W. Lou, C. Yuan, H. B. Wu and Y. Xie, Angew. Chem., Int. Ed., 2014, 53, 1488–1504 CrossRef PubMed.
- J. Yang, X. Shen, Z. Ji, H. Zhou, G. Zhu and K. Chen, Appl. Surf. Sci., 2014, 316, 575–581 CrossRef CAS.
- K. Lee, L. Zhang, H. Lui, R. Hui, Z. Shi and J. J. Zhang, Electrochim. Acta, 2009, 54, 4704–4711 CrossRef CAS.
- Z. Wang, C. Cao, Y. Zheng, S. Chen and F. Zhao, ChemElectroChem, 2014, 1813–1821 CrossRef CAS.
- B. Sompalli, F. T. Wagner, H. A. Gasteiger and S. S. Kocha, Appl. Catal., B, 2005, 56, 9–35 CrossRef.
- J. L. D. Chen and L. Tang, Chem. Soc. Rev., 2010, 39, 3157–3175 RSC.
- Z. Qu, W. Huang, S. Zhou, H. Zheng, X. Liu and M. Cheng, et al., J. Catal., 2005, 234, 33–36 CrossRef CAS.
- A. S. A. D. Sebastián, A. García-Ruíz, I. Suelves, R. Moliner, M. J. Lázaro, V. Baglio and A. Stassi, Appl. Catal., B, 2012, 116, 269–275 CrossRef.
- T. Noori, B. R. Tiwari, M. M. Ghangrekar and B. Min, Sustainable Energy Fuels, 2019, 3, 3430–3440 RSC.
- T. Stephen, T. Andrew and D. C. Johnson, Electroanalysis, 2002, 14, 165–171 CrossRef.
- X. Gong, S. You, X. Wang, Y. Gan, R. Zhang and N. Ren, J. Power Sources, 2013, 225, 330–337 CrossRef CAS.
- S. Rojas-carbonell, C. Santoro, A. Serov and P. Atanassov, Electrochem. Commun., 2017, 75, 38–42 CrossRef CAS.
- S. Trasatti and A. Petri, Pure Appl. Chem., 1991, 63, 711–734 CAS.
- M. De Koninck, S. Poirier and B. Marsan, J. Electrochem. Soc., 2006, 153, A2103–A2110 CrossRef CAS.
- F. Papiya, A. Nandy, S. Mondal and P. Paban, Electrochim. Acta, 2017, 254, 1–13 CrossRef CAS.
- J. H. L. M. Jana, S. Saha, P. Khanra, N. C. Murmu, S. K. Srivastava and T. Kuila, Mater. Sci. Eng. B, 2014, 186, 33–40 CrossRef.
- W.-D. Zhang and L.-C. Jiang, Biosens. Bioelectron., 2010, 25, 1402–1407 CrossRef PubMed.
- X. Q. J.-L. Liu and L.-Z. Fan, Electrochim. Acta, 2012, 66, 302–305 CrossRef.
- B.-L. H. Y. Zhang, G.-Y. Li, Y. Lv, L.-Z. Wang, A.-Q. Zhang and Y.-H. Song, Int. J. Hydrogen Energy, 2011, 36, 11760–11766 CrossRef.
- S. W. Wang W, L. Zhang, S. Tong and X. Li, Biosens. Bioelectron., 2009, 25, 708–714 CrossRef PubMed.
- F. B. Zhang X, G. Wang, W. Zhang and Y. Wei, Biosens. Bioelectron., 2009, 24, 3395–3398 CrossRef PubMed.
- W. Y. C. Wang, P. Shi, X. Cai, Q. Xu, X. Zhou and X. Zhou, J. Phys. Chem. C, 2016, 120, 336–344 CrossRef.
- A. M. Ghazalah Allaedini, J. Nanostruct. Chem., 2013, 1–16 Search PubMed.
- Y.-B. Kim, M. S. Ahmed and B. Choi, Sci. Rep., 2018, 8, 2543–2553 CrossRef PubMed.
- M. Ghasemi, W. Ramli, W. Daud, M. Rahimnejad and M. Rezayi, Int. J. Hydrogen Energy, 2013, 1–8 Search PubMed.
- L. Lan, L. Shuxin, W. Hui and Y. Ping, Environ. Eng. Sci., 2018, 35, 1–11 CrossRef.
- S. Tajik, D. P. Dubal and P. Gomez-romero, Int. J. Hydrogen Energy, 2017, 42, 12384–12395 CrossRef CAS.
- D. S. V. Nikolova, P. Iliev, K. Petrov, T. Vitanov, E. Zhecheva, R. Stoyanova and I. Valov, J. Power Sources, 2008, 185, 727–733 CrossRef.
- H. Zou, S. Chen, Z. Liu and W. Lin, Powder Technol., 2011, 207, 238–244 CrossRef CAS.
- K. Qian, Z. Qian, Q. Hua, Z. Jiang and W. Huang, Appl. Surf. Sci., 2013, 273, 357–363 CrossRef CAS.
- D. Briggs and M. P. Seah, Practical Surface Analysis: Auger and X-Ray Photoelectron Spectroscopy, 1990 Search PubMed.
- http://srdata.nist.gov/xps.
- D. B. L. Bigiani, D. Zappa, E. Comini, C. Maccato and A. Gasparotto, J. Nanosci. Nanotechnol., 2020, 20, 3025 CrossRef PubMed.
- E. S. Ilton, J. E. Post, P. J. Heaney, F. T. Ling and S. N. Kerisit, Appl. Surf. Sci., 2016, 366, 475–485 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson, R. St and C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- C. M. Bigiani, D. Barreca and A. Gasparotto, Surf. Sci. Spectra, 2018, 25, 014003 CrossRef.
- D. F. Gri, L. Bigiani, A. Gasparotto and C. Maccato, Surf. Sci. Spectra, 2018, 25, 024004 CrossRef.
- Z. H. C. Shenghai, S. Liping, K. Fanhao and H. Lihua, J. Power Sources, 2019, 430, 25–31 CrossRef.
- G. Wang, M. Yu, K. Xie, R. Zhao, Y. Fu and T. Chen, J. Power Sources, 2019, 438, 227002 CrossRef CAS.
- F. Zhang, S. Cheng, D. Pant, G. Van Bogaert and B. E. Logan, Electrochem. Commun., 2009, 11, 2177–2179 CrossRef CAS.
- V. J. Watson, C. N. Delgado and B. E. Logan, J. Power Sources, 2013, 756, 756–761 CrossRef.
- P. B. K. K. Hazarika, C. Goswami, H. Saikia and B. J. Borah, Mol. Catal., 2018, 451, 153–160 CrossRef.
- T. Shinagawa, A. T. Garcia-esparza and K. Takanabe, Nat. Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- N. M. Markovic, H. A. Gasteiger, B. N. Grgur and P. N. Ross, J. Electroanal. Chem., 1999, 467, 157 CrossRef CAS.
- M. D. Macia, J. M. Campina, E. Herrero and J. M. Feliu, J. Electroanal. Chem., 2004, 564, 141 CrossRef CAS.
- N. M. Markovic, H. A. Gasteiger and P. N. Ross, J. Phys. Chem., 1996, 100, 6715 CrossRef.
- S. Strbac, Electrochim. Acta, 2011, 56, 1597 CrossRef CAS.
- B. Liu and A. J. Bard, J. Phys. Chem. B, 2002, 106, 12801 CrossRef CAS.
- I. Scott and K. Roche, J. Appl. Electrochem., 2009, 39, 197–204 CrossRef.
- M.-H. Shao, P. Liu and R. R. Adzic, J. Am. Chem. Soc., 2006, 128, 7408 CrossRef CAS PubMed.
- A. Holewinski and S. Linic, J. Electrochem. Soc., 2012, 159, H864 CrossRef CAS.
- D. Thompsett, Catalyst for the proton exchange membrane fuel cell. CRC Press LLC, 2003 Search PubMed.
- P. K. S. S. Yin, M. Cai and C. Wang, Energy Environ. Sci., 2011, 4, 558–563 RSC.
- A.-J. W. L.-L. He, P. Song, J.-J. Feng, W.-H. Huang and Q.-L. Wang, Electrochim. Acta, 2015, 176, 86–95 CrossRef.
- S. J. M. Yun and M. S. Ahmed, J. Power Sources, 2015, 293, 380–387 CrossRef.
- Z. D. Y. Zheng, S. Zhao, S. Liu, H. Yin, Y.-Y. Chen, J. Bao and M. Han, ACS Appl. Mater. Interfaces, 2015, 7, 5347–5357 CrossRef PubMed.
- Y. Xue, S. Sun, Q. Wang, Z. Dong and Z. Liu, J. Mater .Chem. A, 2018, 6, 10595–10626 RSC.
- P. B. C. Goswami and K. K. Hazarika, Mater. Sci. Energy Technol., 2018, 1, 117–128 Search PubMed.
- R. J. Toh, Z. Sofe and M. Pumera, ChemPhysChem, 2015, 16, 3527–3531 CrossRef CAS PubMed.
- V. M. Ortiz-martínez, K. Touati, M. J. Salar-garcía and F. J. Hernández-fernández, Biochem. Eng. J., 2019, 151, 107310 CrossRef.
- K. Touati, F. Belhoucine and A. A. Berrabbah, Energy, 2016, 113, 1241e1249 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07721j |
| This journal is © The Royal Society of Chemistry 2022 |