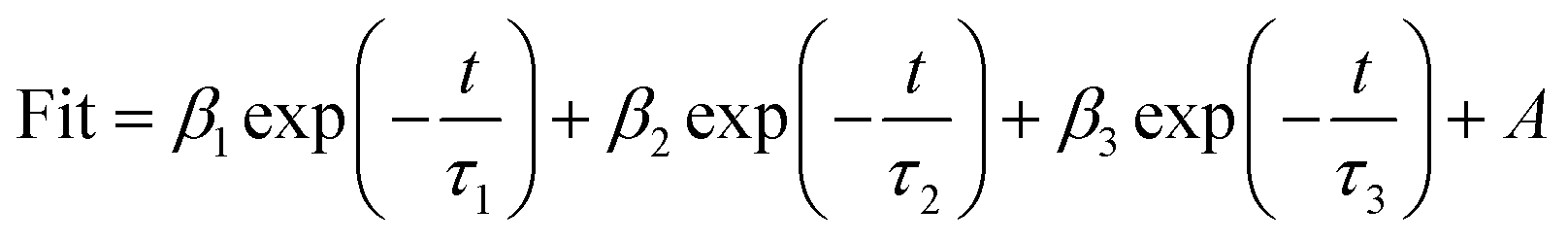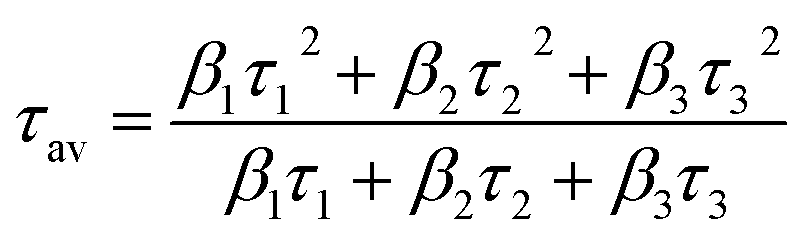DOI:
10.1039/D1RA08177B
(Paper)
RSC Adv., 2022,
12, 2928-2937
Synergistic oxygen vacancy-rich CuO/visible light activation of peroxymonosulfate for degradation of rhodamine B: fast catalyst synthesis and degradation mechanism†
Received
8th November 2021
, Accepted 14th January 2022
First published on 21st January 2022
Abstract
This work outlines the synthesis of copper oxide nanoparticles (CuO-SC) loaded with a number of oxygen vacancies by a fast sodium citrate assisted precipitation method with no need of calcination. X-ray diffraction, scanning electron microscopy, UV-Vis diffuse reflectance spectroscopy, time-resolved fluorescence lifetime and electrochemical impedance spectra were used to characterize the as-synthesized nanocomposites. The results indicated that the CuO-SC nanoparticles had regular fusiform shape with high surface area, wide light harvesting window, fast charge transport and high carrier concentration. As a result, the catalytic activity of the CuO-SC/peroxymonosulfate (PMS)/visible light (Vis) system for the degradation of rhodamine B (RhB) was much higher than that of as-prepared CuO nano powder in the absence of sodium citrate. Almost 98.0% of the initial RhB dyes was decomposed in 20 min with 0.12 g L−1 PMS and 0.3 g L−1 catalyst. Meantime, it exhibited high catalytic stability with little deactivation after four runs and a wide application range of pH. Moreover, RhB can be readily degraded with backgrounds of Cl−, NO3−, SO42−, HCO3− and low concentration of humic acid in a CuO-SC/PMS/Vis system. Combined with the results of electron spin resonance paramagnetic spectroscopy, X-ray photoelectron spectroscopy and radical quenching experiments, holes, superoxide radicals and a small amount of sulfate radicals, hydroxyl radicals and singlet oxygen were involved in the CuO-SC/PMS/Vis system. Furthermore, a possible degradation mechanism based on the synergistic effect of radical reaction and non-radical reaction was proposed based on the above results.
1. Introduction
Wastewater pollution becomes more threatening to the natural ecological environment as human beings make more and more industrial production. Among the varied techniques for treating organic pollutants, advanced oxidation processes (AOPs) attracted much more attention as they are highly efficient, economical and could degrade pollutants completely. Compared with the traditional hydroxyl radical (˙OH) based AOPs, sulfate-radical (SO4˙−)-based AOPs using peroxymonosulfate (PMS) or persulfate (PS) as oxidants have become increasingly important in recent years as the sulfate radical has higher oxidation potential, longer life, and stable structure.1 Sulfate radical-based AOPs have been reported to effectively degrade dyes,2 bisphenol,3 emerging contaminants,4 etc. Although PMS can be activated by metal ions, UV light, ultrasonication, etc., the application of metal oxide heterogeneous catalysts has become a hot spot due to their high efficiency, no secondary pollution, no extra power consumption and a great variety of choice for different materials.1 Up to now, metal oxides such as Co3O4, MnO2, CuO, Fe3O4, NiO and so on have been found to be effective catalysts for activating PMS.5–8
Among the various heterogeneous PMS activators, CuO is considered as one of the most promising due to its low biological toxicity, easy accessibility and low cost. However, the catalytic efficiency of CuO is still lower than that of Co3O4. In order to further improve its catalytic performance, researchers tried to construct CuO composites with other transition metal oxides, load CuO on specific carriers or modulate CuO crystalline morphology. For instance, Khan et al. prepared CuO–Co3O4@MnO2 nanostructures and used them to catalyze the degradation of phenol with PMS.5 It was found that the composites not only exhibit much higher efficiency than pure CuO and pure Co3O4, but also significantly reduced the leakage of metal ions in the reaction. Recent studies loaded CuO onto carbon fibers,9 biochar10 or carbon framework.11 It was found that the carriers provided electrons to CuO or O2, thus accelerating the generation of strong oxidizing groups and improving its catalytic efficiency of activating PMS.9 Qin et al. synthesized spongelike porous CuO as an efficient PMS activator for degradation of Acid Orange 7, by overcoming the mass-transfer limitation.12 However, more simple and feasible strategies for improving CuO catalytic performance is still lacking.
From another hand, light-assisted PMS activation by heterogeneous catalysts is considered as a promising method for refractory organic pollutants degradation. Light irradiation benefits the formation of free radicals by trapping photo-generated electrons, reducing the recombination rate of holes and accelerate the regeneration of low-valence metal species.13 For instance, ZnFe2O4/PMS/visible light (Vis) system showed degradation efficiency of 100% at 80 min for Orange II, much higher than the summation of ZnFe2O4/PMS and ZnFe2O4/Vis.14 Lin et al. synthesized a magnetic cobalt–graphene nanocomposite by carbonizing a self-assembly of a cobalt-based metal–organic framework and the as-prepared catalyst showed high efficiency and long-term stability to activate PMS under UV irradiation.15 It is also reported that the introduction of light, especially UV irradiation, can improve the organic pollutants degradation by ZnO/PMS.16 As an effective heterogeneous catalyst for PMS, CuO is a p-type semiconductor with narrow bandgap. However, the light-assisted CuO activation for PMS has not been studied, although it is found that doping Cu into BiFeO3 magnetic nanoparticles can effectively enhance its degradation of 2-chlorophenol when activating PS under visible light.17
In recent years, oxygen vacancy was reported to play important roles in improving catalytic activity for activating PMS by enhancing water dissociation or O2 adsorption. Besides, oxygen vacancy can also modulate light absorption, charge transfer, etc.18 However, the introduction of oxygen vacancies usually needs complex preparation conditions, such as high-temperature, post-chemical reduction or solvothermal reaction. Li et al. constructed oxygen vacancies in CuO/CeO2 composites by hydrothermal-calcination method, which greatly improved their activity for activating PMS to degrade rhodamine B (RhB).19 In our previous studies, oxygen vacancy-rich NiO was prepared by solvothermal method using mixed water-ethylene glycol solvents and it was found that the NiO had high catalytic efficiency for activating PMS.8 In addition, oxygen vacancy-rich CuO catalysts were prepared for the first time by the simple precipitation method using PVP–EDTA dual surfactants, however, calcination process was still needed.6 In general, the above methods are not energy-saving and need special instruments such as high-pressure vessels. Thus, it is highly desirable to develop fast and simple methods for preparing oxygen vacancy-rich CuO catalysts.
In this work, oxygen vacancy-rich CuO was synthesized for the first time by a fast precipitation method (30 min) with no need of calcination, through the use of sodium citrate as a guidance additive. The prepared oxygen vacancy-rich CuO, for the first time, showed excellent catalytic properties for degrading RhB by PMS under visible light irradiation. The operating parameters such as pH, catalyst dosage, PMS concentration on the degradation efficiency were systematically investigated. The role of oxygen vacancy on the properties of catalyst was probed by the method of photoluminescence (PL) spectroscopy, electrochemical impedance spectra (EIS), band structure analysis, electron spin resonance paramagnetic spectrometer (ESR) and photocurrent measurements. Based on the results of X-ray photoelectron spectroscopy (XPS), ESR and radical quenching experiments, the reaction mechanism was finally discussed.
2. Experiments
2.1 Material and chemicals
Copper sulfate pentahydrate (CuSO4·5H2O), sodium hydroxide (NaOH), sodium citrate, ethanol (EtOH), tert-butanol (TBA), 1,4-benzoquinone (BQ), ethylenediaminetetraacetic acid disodium salt (EDTA-2Na), sodium azide (NaN3), sodium chloride (NaCl), sodium sulfate (Na2SO4), sodium nitrate (NaNO3), sodium hydrogen carbonate (NaHCO3) and humic acid were purchased from Sinopharm Chemical Reagent Co., Ltd. PMS, carbon black, N-methylpyrrolidone (NMP), poly(vinylidene fluoride) (PVDF), RhB were purchased from Aladdin (Shanghai, China). All the chemicals were analytical grade and used without further purification. The water in this experiment is deionized water.
2.2 Preparation of CuO samples
0.03 mol CuSO4·5H2O and 0.24 g sodium citrate were dissolved in 275 mL water by stirring for 10 min. When the solution temperature was raised to 80 °C, 25 mL solution with 0.18 mol NaOH was added dropwise. After 20 min of reaction, the temperature was raised to 100 °C and maintained for 10 min until the end of reaction. The precipitation was collected by filter, washed with water and alcohol, and finally dried at 60 °C for 24 hours, which was marked as CuO-SC.
As control group, sample was prepared by similar procedures but without sodium citrate during synthesis process, which was recorded as CuO-B.
2.3 Degradation experiments
Experiments were conducted in a visible light photocatalytic reactor (visible light from a 300 W Xe lamp with a UV cut-off filter) with a 50 mL quartz tube containing 25 mL RhB solution at a constant stirring rate. Typically, 3 mg PMS was dissolved in 25 mL RhB solution (20 mg L−1) and the initial solution pH was adjusted to 7. Then 7.5 mg catalyst was added to initiate the catalytic oxidation reaction. During the reaction process, 1.5 mL mixture was extracted at regular intervals and filtered immediately using a 0.22 μm syringe filter. The resulting supernatants were obtained and the residual RhB concentration was analyzed by a 725 N UV-Vis spectrophotometer at 532.5 nm. Chemical oxygen demand (COD) was measured by potassium dichromate method on HACH DRB200 digester and HACH DR2800 multi parameter water quality analyzer. For comparison, the catalytic tests were conducted without adding the catalyst or PMS under the same conditions.
The effect of pH variation (5.0–10.0), CuO-SC dosages (0.2–0.6 g L−1) and PMS/RhB weight ratio (4–9) on catalytic performance was evaluated. RhB degradation with backgrounds of Cl−, NO3−, SO42−, HCO3− and humic acid was also determined. Then, 10 mM EtOH, 10 mM TBA, 6 mM BQ, 6 mM EDTA-2Na and 6 mM NaN3 were added to the reaction solution and the changes in the dye degradation rate were recorded.
During the recycling experiment, the catalysts were collected by vacuum filtration and thoroughly washed with distilled water and ethanol after each recycle, then dried at 60 °C for 24 hours.
2.4 Characterization
The crystal structure of the samples was analyzed by X-ray Diffraction Spectroscopy (XRD, Bruker advance D8, Germany, Morphologies) and their morphologies were analyzed with Field-Emission Scanning Electron Microscopy (FESEM, Megallan 400). The valence analysis of the elements on the catalyst's surface was detected by XPS (Thermo Scientific K-Alpha). Specific surface area and pore size distribution were analyzed by Brunauer–Emmett–Teller (BET, ASAP 2460). The optical properties were investigated by UV-Vis diffuse reflectance spectra (DRS, Shimadzu 3600Plus). O2 temperature-programmed desorption (O2-TPD) was conducted on Chembet TPD (Quantachrome). The PL spectra were recorded with an Edinburgh (FLS1000) spectrofluorometer, measured at room temperature under excitation at 355 nm. The time-resolved fluorescence lifetime (TRFL) was obtained at an excitation wavelength of 375 nm and an emission wavelength of 435 nm. An ESR analyzer (Bruker EMXnano) was used to detect the generated reactive oxygen species.
EIS was carried out on an electrochemical workstation (Solartron Analytical 1470E Cell Test System) with standard three electrodes. The applied potential was fixed at the open circuit voltage and the amplitude was controlled at 10 mV, with the frequency range of 0.1 to 105 Hz. The surface of conductive glass was covered with 20 μL of suspension composed of 16 mg catalyst, 2 mg carbon black, 2 mg PVDF and 0.5 mL NMP and used as working electrode. The test was taken under the visible light illumination using LED lamp. The electrolyte was 0.1 mol L−1 Na2SO4 solution. Ag/AgCl electrode and platinum plates were used as reference electrode and counter electrode, respectively. The photocurrent–voltage characteristic curve was obtained by measuring the photocurrent at a scan rate of 50 mV s−1 between −0.4 and 0.4 V vs. Ag/AgCl. The Mott–Schottky measurement was made at 1000 Hz.
3. Results and discussion
3.1 Synthesis and characterization of samples
3.1.1 Crystal structure and morphology. Fig. S1† shows the XRD patterns of the prepared samples. All reflection peaks can be indexed as monoclinic CuO (PDF card no. 80-1916). No obvious difference and other peaks are observed, indicating that high purity samples were successfully obtained. Fig. 1 present typical SEM images of CuO-B and CuO-SC, respectively. In the absence of sodium citrate during the preparation process, the CuO-B (Fig. 1a) is non-uniform elongated flake shape with length of 300–700 nm and width of 50–90 nm. Compared with CuO-B, CuO-SC (Fig. 1b) is much more regular and homogeneous in the presence of sodium citrate. It is fusiform shape with length of 150–250 nm and width of 20–30 nm (Fig. 1c). Apparently, sodium citrate was found to be an important structure directing agent controlling the particle size and morphology of CuO-SC.
 |
| | Fig. 1 SEM images of (a) CuO-B and (b and c) CuO-SC. The scales are 200 nm. | |
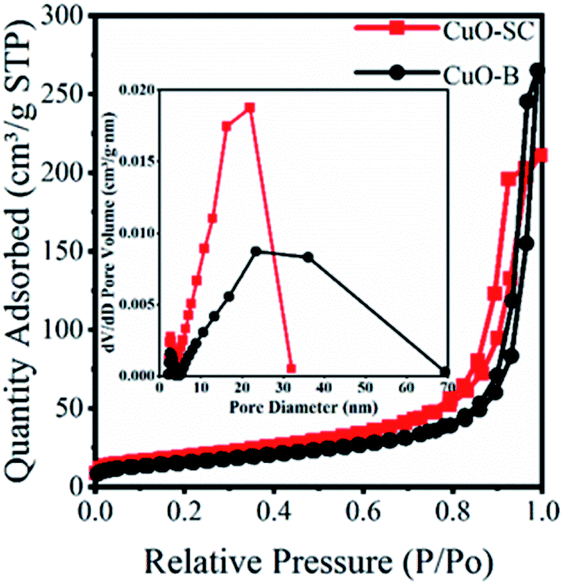
3.1.2 Textural properties. The surface areas and pore size distributions were investigated by N2 adsorption–desorption isotherms. As shown in Fig. 2, all samples exhibit type IV isotherms with H3 hysteresis with loop, meaning that the porosity is due to aggregation of particles with narrow slit-shaped pores. The pore size distribution profile in the inset of Fig. 2 shows a narrow distribution from 2 nm to 30 nm for CuO-SC and a wide distribution from 2 nm to 70 nm for CuO-B. Obviously, CuO-SC sample is mainly composed of mesoporous. And CuO-B contains both macroporous and mesoporous pores. The existence of macropores is negative for the increase of specific surface area. Therefore, the surface parameters calculated from BET measurements of CuO-B samples is 55 m2 g−1, which is lower than that of CuO-SC (70 m2 g−1). Fusiform shape CuO-SC samples with high surface areas is expected to supply plenty of active sites to contact with PMS and dye molecules under catalytic environment.
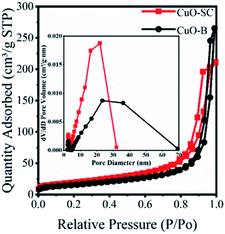 |
| | Fig. 2 BET of CuO samples and the inset is pore size distribution. | |
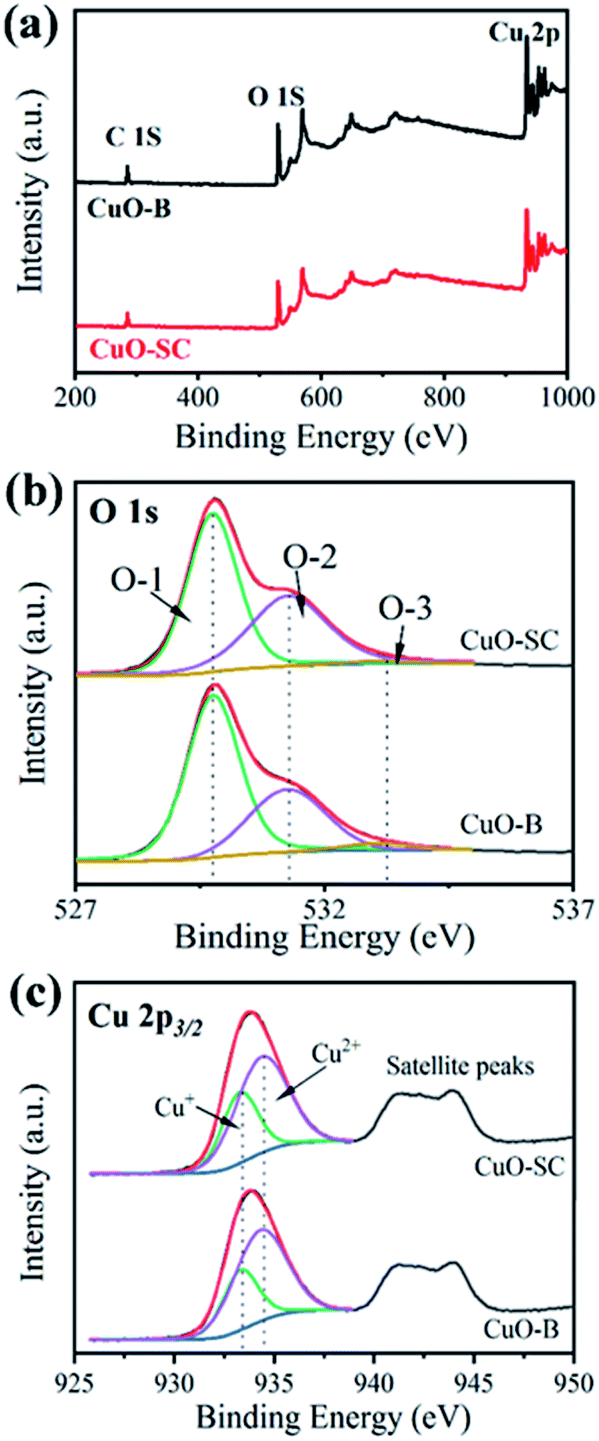
3.1.3 Surface properties. XPS measurements were taken to characterize their surface property discrepancy. The results are shown in Fig. 3. Fig. 3a presents survey spectrum referring to C 1s peak at 284.8 eV. It comprises peaks at 934 eV and 530 eV, which correspond to the characteristic peak of Cu 2p and O 1s, respectively. The high resolution XPS spectrum and fitting parameters for O 1s are shown in Fig. 3b and Table S1.† In Fig. 3b, the three peaks at 529.8 eV, 531.3 eV and 533.1 eV correspond to lattice oxygen (O-1), chemically adsorbed oxygen (O-2), and physically adsorbed oxygen (O-3), respectively. It is well known that the relative area of the O-2 peak is proportional to the percentage of surface oxygen vacancy.20 Table S1† shows that the content of O-2 in CuO-SC is 41.2%, in contrast to the 32.1% in CuO-B, implying that the CuO-SC contains more oxygen vacancies than CuO-B sample. Fig. 3c shows the high resolution XPS spectrum of Cu 2p. The fitting parameters are also shown in Table S1.† The peaks at 934.4 eV and 933.3 eV correspond to Cu2+ 2p3/2 and Cu+ 2p3/2, respectively. Compared with CuO-B, a higher ratio of Cu+/Cu2+ for CuO-SC means that there are more Cu+ species formed by the reduction function of sodium citrate on the surface of catalysts, which result in the creation of more oxygen vacancies to keep charge balance.
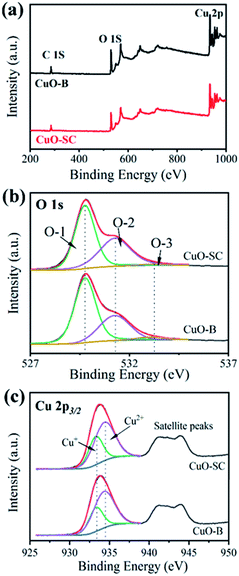 |
| | Fig. 3 X-ray photoelectron spectroscopy of (a) survey spectrum, (b) O1 s high resolution spectrum and (c) Cu 2p high resolution spectrum of samples. | |
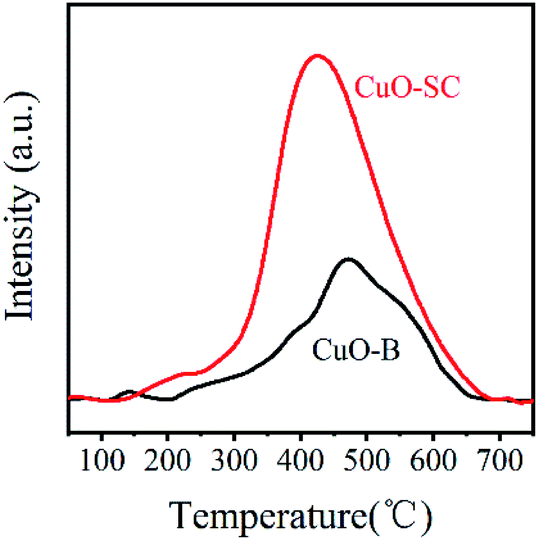
In order to further study the variation of oxygen vacancies on different CuO sample surfaces, O2-TPD tests were taken and the results are shown in Fig. 4. According to previous reports, chemically absorbed oxygen species (e.g. O− or O2−) formed by the chemisorbed O2 on CuO surface oxygen vacancies will be released at 300–500 °C.21 Clearly, the amount of surface adsorbed oxygen species in CuO-SC sample calculated by integrating area under the TPD curves is 0.24 mmol g−1, which is much higher than 0.11 mmol g−1 of CuO-B. Obviously, it contains more oxygen vacancies on the surface of CuO-SC, which is in agreement with XPS results.
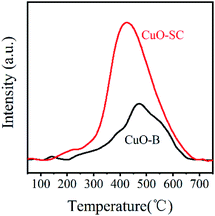 |
| | Fig. 4 Oxygen-temperature programmed desorption curves of samples. | |
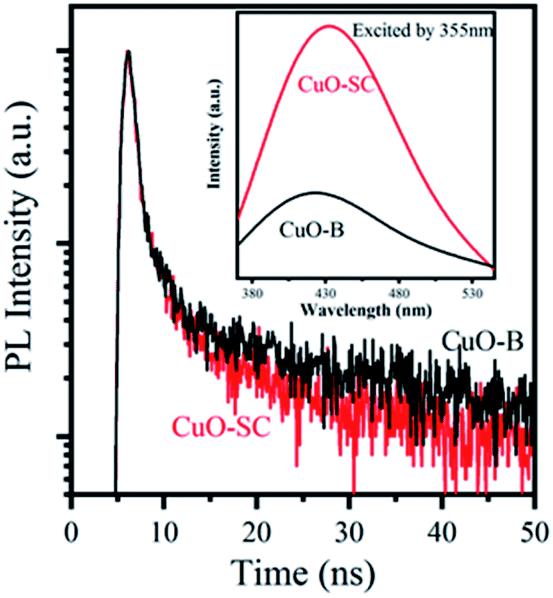
3.1.4 Optical and photo-electrochemical properties. As light activators, optical absorption property is crucial to catalytic activity. So UV-Vis DRS spectrum was measured to characterize the optical properties of the samples and their Tauc plot curves are shown in Fig. S2.† The band gaps of CuO-SC is estimated to be 1.65 eV (the absorption edge of 751 nm) and that of CuO-B is 1.71 eV (the absorption edge of 725 nm). The band gaps for both CuO-B and CuO-SC are larger than that reported for bulk CuO (Eg = 1.2 eV), which is due to the quantum confinement effect for nanostructured semiconductors. The result verifies that electrons and holes can be excited under visible light irradiation in CuO system. CuO occupied with plenty of oxygen vacancies can broaden light harvesting window and make use of much more solar energy, which plays an important role in catalytic reaction.PL were performed using an excitation wavelength of 355 nm in order to investigate the photogenerated charge carrier recombination. As shown in the inset of Fig. 5, the PL intensity of CuO-SC sample is higher than that of CuO-B from 380 to 530 nm because of the stronger recombination of much more excitons bounded to oxygen vacancies.22 The presence of much more oxygen vacancies in CuO-SC was confirmed by PL spectroscopy. The trapping effect of oxygen vacancies to electron can be further effectively studied with the help of TRFL measurements. The TRFL spectra were obtained at an excitation wavelength of 375 nm and an emission wavelength of 435 nm, shown in Fig. 5. It was fitted using the following tri-exponential function:
| |
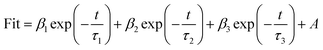 | (1) |
where
τ1,
τ2 and
τ3 are the decay time,
β1,
β2 and
β3 are the weighting parameter and
A is a constant. The average lifetimes (
τav) are calculated using
eqn (2):
| |
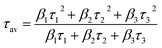 | (2) |
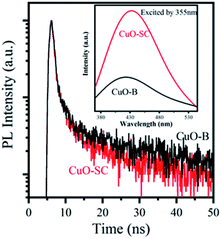 |
| | Fig. 5 Time-resolved fluorescence lifetime spectra (TRFL) of samples. The inset is PL emission spectra at excitation of λ = 355 nm. | |
Fitting parameters are shown in Table S2.† The τav of CuO-B and CuO-SC was 30.34 ns and 27.18 ns, respectively. This evidence of decrease in the τav for CuO-SC confirms that a separate energy level is generated, which effectively facilitates the trapping of photogenerated carriers.
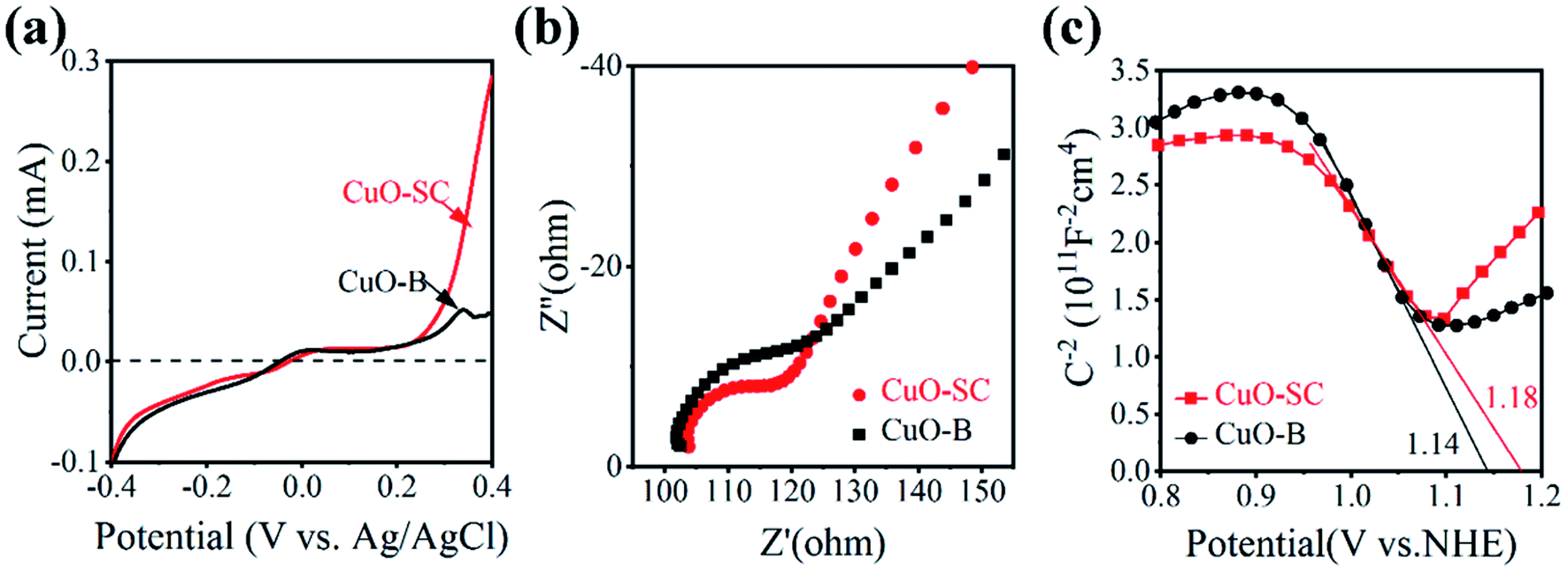
Besides the generation of carrier, efficient carrier transfer is also key factor for catalytic activity, which can be evaluated by electrochemical workstation. Fig. 6a is the obtained instantaneous current–voltage curves. It shows that CuO-SC can produce larger photocurrent than CuO-B, indicating that the former can more easily undergo electron transfer under light conditions. This conclusion can be further confirmed by the results of EIS. Fig. 6b shows the results under open circuit voltage conditions. The arc radius in the high frequency region is related to the charge transfer resistance. The larger semicircle diameter, the larger electrical impedance. It is clear that the diameter corresponding to CuO-SC is smaller than that to CuO-B, evidencing the smaller charge transfer resistance for CuO-SC. In order to obtain the number of carrier concentration, Mott–Schottky experiments were monitored and the results are shown in Fig. 6c. The negative slope of the curve confirms that CuO is P-type semiconductor. At the same time, the following M–S formula (eqn (3)) is used to calculate the carrier concentration (Nd) based on the slope of the curve.
| | |
Nd = (2/(eεrε0)) × [slop]−1
| (3) |
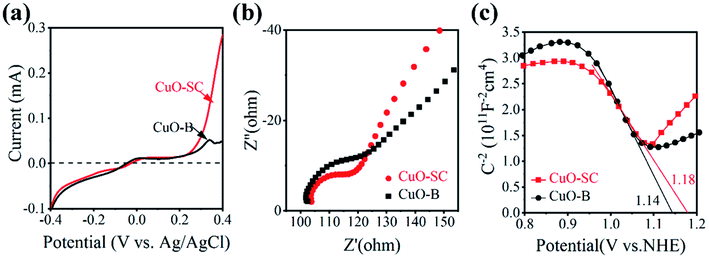 |
| | Fig. 6 (a) Instantaneous photocurrent–voltage curves under light condition, (b) electrical impedance spectrum under the light condition and (c) Mott–Schottky curves of the samples. | |
Among them, slop is calculated from the M–S curves, e is the electron charge (1.6 × 10−19 C), ε0 is the vacuum dielectric constant (8.85 × 10−12 F m−1) and εr is the relative permittivity (10.26 for CuO). The carrier concentration of CuO-SC and CuO-B calculated by eqn (3) are 10.7 × 1018 cm−3 and 7.9 × 1018 cm−3, respectively. These results prove again that the separation efficiency of the electron–hole pairs is largely promoted in the CuO-SC samples.
The above results prove that CuO samples synthesized with sodium citrate is superior to CuO-B in capability of charge transfer. It can be ascribed to high load of oxygen vacancies on the surface of CuO-SC. Oxygen vacancies would induce localized states, trap charge carriers efficiently and inhibit the recombination between electrons and holes.23 As a result, the number of electrons that effectively participate in the adsorption and reaction would be increased, which is beneficial for the catalytic process.
Generally, the charge transfer action of photocatalysts is related to their band structure. Thus, the corresponding band edge positions of both CuO-SC and CuO-B were estimated. The flat band position can be derived from the intersection of the straight-line part of the extended curve and the horizontal axis in Fig. 6c. For P-type semiconductors, flat band position is near to the valence band (VB). Thus, the position of VB for CuO-SC and CuO-B is 1.18 and 1.14 V vs. NHE (Normal Hydrogen Electrode), respectively. Furthermore, by analyzing these data combined with the band gaps achieved from Fig. S2,† the conduction band (CB) edge potentials of CuO-SC and CuO-B are estimated to be −0.47 and −0.57 V, respectively. According to these results, the band structure of samples is shown in Fig. S3.† It indicates that the CB potential is more negative than the redox potential O2/O2˙− (−0.046 V), meaning that samples are able to reduce the dissolved O2 to generate superoxide radicals (O2˙−).
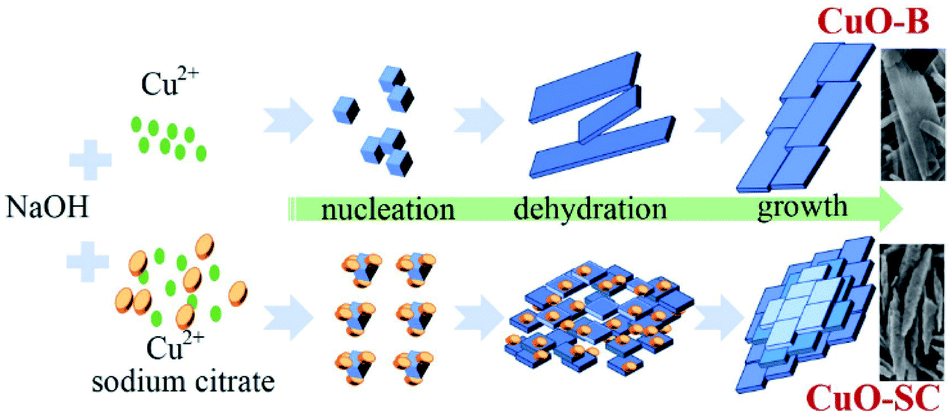
3.15 Formation mechanism. Although the formation mechanism is not yet fully understood, it is believed that the chelate effect of sodium citrate, ascribing to its carboxylate and hydroxyl groups, plays an important role during the formation of fusiform-shaped sample. As illustrated Fig. 7, in the synthesis process of CuO, Cu2+ ions reacted with OH− to form complex Cu(OH)42− firstly. Then Cu(OH)42− could decompose into Cu(OH)2 nuclei.24 Without the intervention of sodium citrate, these nuclei would grow into layer structure and form CuO-B through dehydration process finally. Under the help of chelate agent, sodium citrate could link its carboxylate and hydroxyl groups with Cu2+. Consequently, the decline of free metal ions would slow down the nucleation rate and crystal growth of precursors during the reaction, which would induce the aggregation of nanoparticles to form regular fusiform-shaped nanoassemblies. Similar modulation of the crystal shape by citrate ions were reported for NiCo2O4 and CoS2 systems.25,26
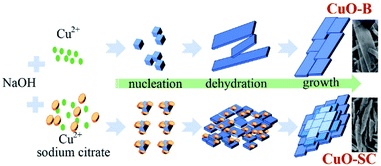 |
| | Fig. 7 Schematic illustration of the proposed growth mechanism for CuO samples. | |
The samples fast synthesized with sodium citrate can display high surface area and narrow mesoporous size distribution. More importantly, CuO-SC is occupied by a large number of oxygen vacancies. Previous reports have proved that the presence of oxygen vacancies on catalysts significantly improves catalytic performance through promoting the cleavage of the O–O bonds in PMS and forming the active oxidizing species.5,6 Therefore, oxygen vacancies located at CuO-SC are expected to act as active sites for catalysis. On one hand, it might advance the dissociative adsorption of O2 in water on CuO-SC surfaces where O2˙− are formed. On the other hand, oxygen vacancy might serve as electron trap, which is favorable for the separation of photoexcited electron (e−) and hole (h+), the formation of chemical bond between CuO-SC and PMS and the activation of PMS.7,8
3.2 Catalytic properties and mechanism of CuO-SC
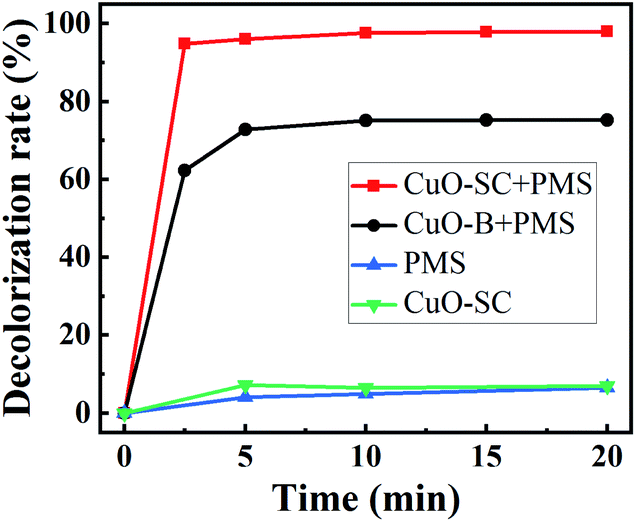
3.2.1 Catalytic activities. In order to confirm the advantage of CuO-SC, a typical degradation profile of RhB is given in Fig. 8. For comparison, the degradation of CuO-B and pure PMS was performed in the same condition. Under visible light irradiation, PMS itself could degrade RhB of only 6.4%. In the system of CuO-SC sample without PMS, a degradation efficiency of 7.0% is reached due to adsorption and photocatalysis under visible light. In great contrast, the degradation efficiency of RhB is sharply improved in the system of CuO/PMS. The degradation percentage for 2.5 min reaches 62.3% and 94.8% for CuO-B and CuO-SC, respectively. At 20 min, it increases to 75.3% and 98.0%, respectively. Fig. S4† shows the COD removal rates of CuO-B and CuO-SC. The result follows the same trend of Fig. 8. CuO-SC/PMS/Vis system not only provides higher RhB degradation rate but also possesses remarkable capacity of mineralization. Table 1 presents the degradation of RhB by PMS using different catalysts reported previously. In contrast to these results, CuO-SC shows much better and rapid activation efficiency, even at lower catalyst dosage and lower PMS concentration.
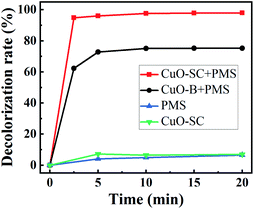 |
| | Fig. 8 Rhodamine B (RhB) degradation efficiency as a function of reaction time for different catalysts under visible light irradiation (reaction conditions: RhB concentration = 20 mg L−1, catalyst dosage = 0.3 g L−1, initial pH = 7, PMS/RhB weight ratio = 6). | |
Table 1 The degradation efficiency of RhB by PMS under different catalytic systems
| |
PMS (g L−1) |
Catalyst (g L−1) |
Initial RhB (mg L−1) |
Light condition |
Reaction time (min) |
Degradation efficiency |
Ref. |
| MnO2 |
0.25 |
0.25 |
20 |
No light |
15 |
<85% |
27 |
| BiVO4 |
∼0.6 |
0.5 |
10 |
Visible |
60 |
92.4% |
28 |
| BiFeO3 |
0.15 |
1 |
5 |
Visible |
40 |
97.5% |
29 |
| CuO-SC |
0.12 |
0.3 |
20 |
Visible |
2.5 |
94.8% |
Current work |
| CuO-SC |
0.12 |
0.3 |
20 |
Visible |
20 |
98.0% |
Current work |
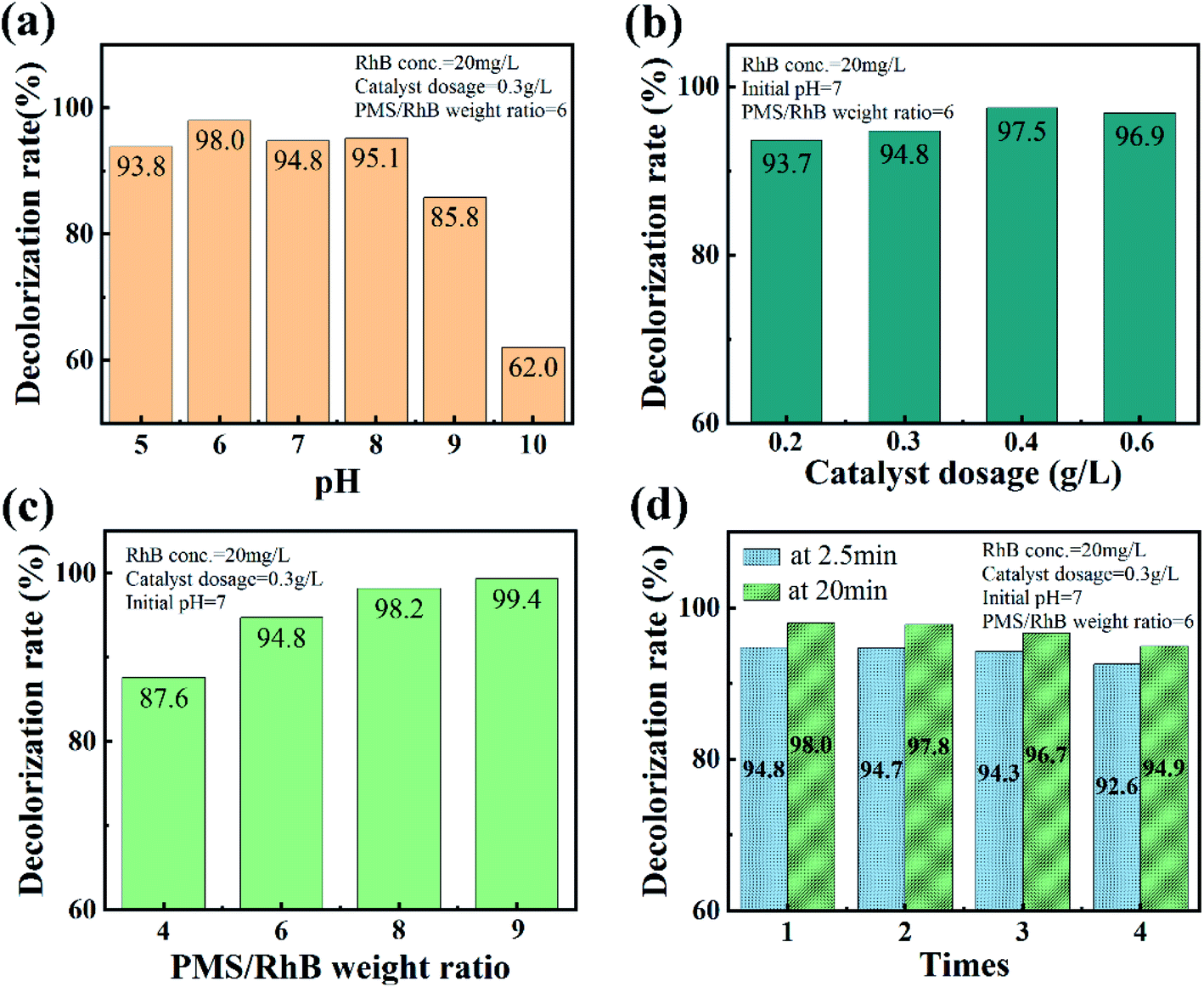
The effect of initial pH in the CuO-SC/PMS/Vis system on the degradation of RhB is shown in Fig. 9a. In the pH range of 5.0–8.0, CuO-SC reaches a 2.5 min-degradation efficiency higher than 90%, while decreases at pH higher than 9.0. It might be ascribed to the self-dissociation of PMS in alkaline medium. Obviously, the advantage of the CuO-SC/PMS/Vis system in degrading RhB can be maintained in wide pH range. Furthermore, the activation efficiency could be further enhanced by increasing the amount of catalyst and PMS, as shown in Fig. 9b and c. Based on the mechanism of activation, the enhancement can be attributed to the increase of contact opportunity between CuO-SC/PMS and RhB molecules. Besides, the stability and reusability of CuO-SC catalyst was verified by several cyclic degradation experiments. As shown in Fig. 9d, the degradation efficiency of RhB can still be maintained 92.6% at 2.5 min and 94.9% at 20 min after 4 cycles. Compared with other reported PMS system in Table 2, CuO-SC/PMS/Vis system shows a stronger stability.
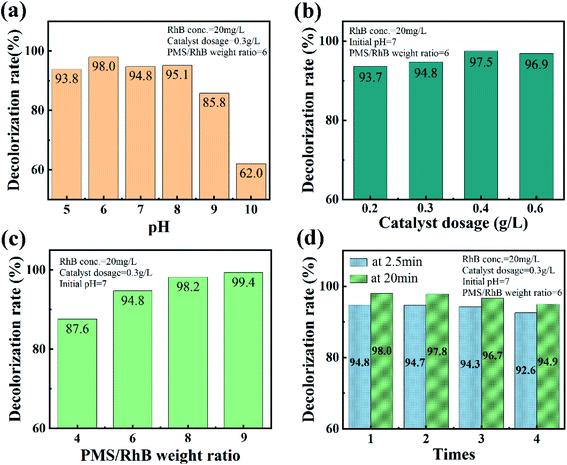 |
| | Fig. 9 Effects of (a) initial pH, (b) catalyst dosage and (c) PMS/RhB weight ratio on the catalytic degradation of RhB at 2.5 min in CuO-SC/PMS system under visible light irradiation. (d) Cycle test result graph of CuO-SC catalyst. | |
Table 2 Reaction cycle stability under various reaction systems
| Reaction system |
Cycles |
Degradation efficiency |
Ref. |
| CuO–Co3O4@MnO2/PMS–phenols |
4 |
84% |
5 |
| Fe3O4/PMS–RhB |
5 |
83% |
7 |
| CuFe2O4/PMS–bisphenol A |
3 |
80% |
30 |
| Mn2O3/PMS–RhB |
5 |
91.6% |
31 |
| CuO/PMS–RhB |
4 |
94.9% |
Current work |
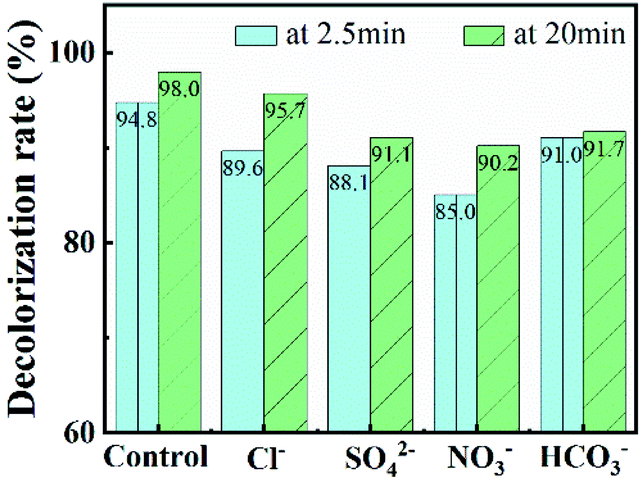
In further, various anions (Cl−, SO42−, NO3− and HCO3−) and natural organics were introduced into CuO-SC/PMS/Vis system to evaluate their effects on RhB degradation. Working as scavengers, Cl−, SO42−, NO3− and HCO3− can react with radicals (such as OH˙ and SO4˙−) and lower catalytic activities.32–34 As shown in Fig. 10, the degradation efficiency at 20 min of RhB is 95.7%, 91.1%, 90.2% and 91.7% in the presence of Cl−, SO42−, NO3− and HCO3−, respectively. Though all the degradation were suppressed, the inhibition effect is weak with the addition of 5 mM and all the degradation efficiency still maintained above 90%. Among them, Cl− provides the weakest inhibition. As reported previously, radicals scavenged by Cl− can be transformed into Cl˙ and Cl2˙−, which still have high redox potential to degradate RhB.32 As a result, a 95.7% degradation efficiency of RhB at 20 min was observed. Besides inorganic ions, the degradation of RhB would also be affected by natural organic matter, which would competitively react with radicals and decrease radicals number for degradation of RhB. Therefore, humic acid, as a common natural organic matter, was used to check the RhB degradation of CuO-SC/PMS/Vis system. The result is shown in Fig. S5.† With the addition of 10 mg L−1 humic acid, there are few effect on the degradation of RhB. When humic acid concentration increased to 20 mg L−1, the degradation was obviously suppressed. Obviously, natural organic matter with high concentration is adverse in CuO-SC/PMS/Vis system.
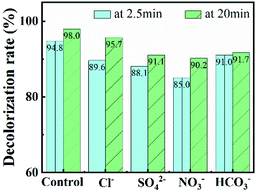 |
| | Fig. 10 Effects of co-existing inorganic ions on RhB removal (Cl− concentration = 5 mM, SO42− concentration = 5 mM, NO3− concentration = 5 mM, HCO3− concentration = 5 mM). | |
The above results indicate that regular fusiform-shaped CuO-SC catalysts, prepared in our easy and novel way, are superior because of excellent catalytic performance, exceptional stability and strong interference rejection to inorganic ions and natural organic matter with low concentration. In order to investigate the degradation mechanism of RhB in CuO-SC/PMS/Vis system, several crucial factors to the catalytic activity were investigated.
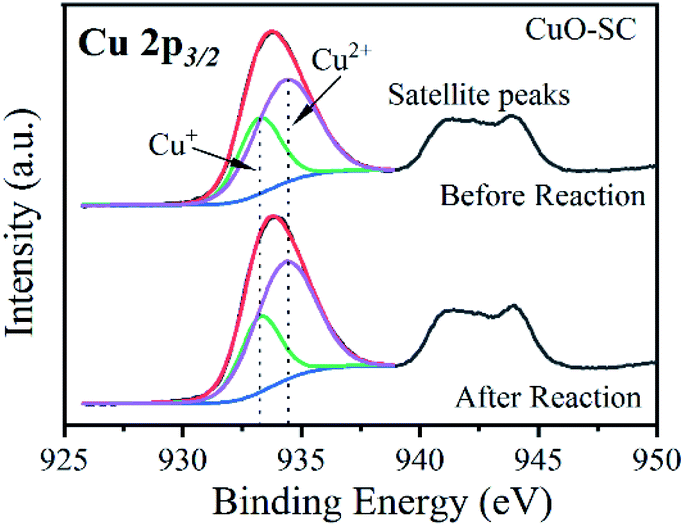
3.2.2 Catalytic mechanism. Fig. S6† presents the XRD and SEM of CuO-SC catalyst after four cyclic catalytic reaction. Compared with that before the reaction (Fig. S1† and 1b), crystal structure and morphology are almost unchanged except that there are some contaminants in Fig. S6b† deriving from residual reactants, which confirms the excellent stability of CuO-SC. XPS was used to investigate the difference of surface chemical states of CuO-SC before and after reaction. Fig. S7a† shows survey spectrum comprising C, O and Cu on the surface of all catalysts. In the high resolution XPS spectrum of Cu 2p (Fig. 11 and Table S3†), the proportion of Cu+ ions slightly decreases from 35.6% to 31.5% after reaction, while that of Cu2+ increases from 65.4% to 68.5%. Cu+ have been partially oxidized to Cu2+ by experiencing electron transfer, implying the generation of radicals (such as OH˙ and SO4˙−) in the process of reaction. Usually, the regeneration of Cu+ is the rate-determining step in heterogeneous PMS activation based on SO4˙− and OH˙. For CuO-SC, the proportion of Cu+ was still at a relatively high level after four cyclic reaction, which indicated that there were other active substances involved in the degradation of RhB. In the high resolution XPS spectrum of O1s (Fig. S7b and Table† S3), the percentage of the peaks at 533.1 eV for fresh and used samples is 1.3% and 11.2%, respectively. This increase results from physically adsorbed residual reactants, which is consistent with the result of SEM (Fig. S6b†). Meantime, after reaction, the ratio of O-2/O-1 is decrease a little for CuO-SC, which results from the decrease of percentage of Cu+.
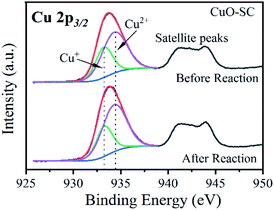 |
| | Fig. 11 Cu 2p high resolution X-ray photoelectron spectrum of CuO-SC before and after reaction. | |
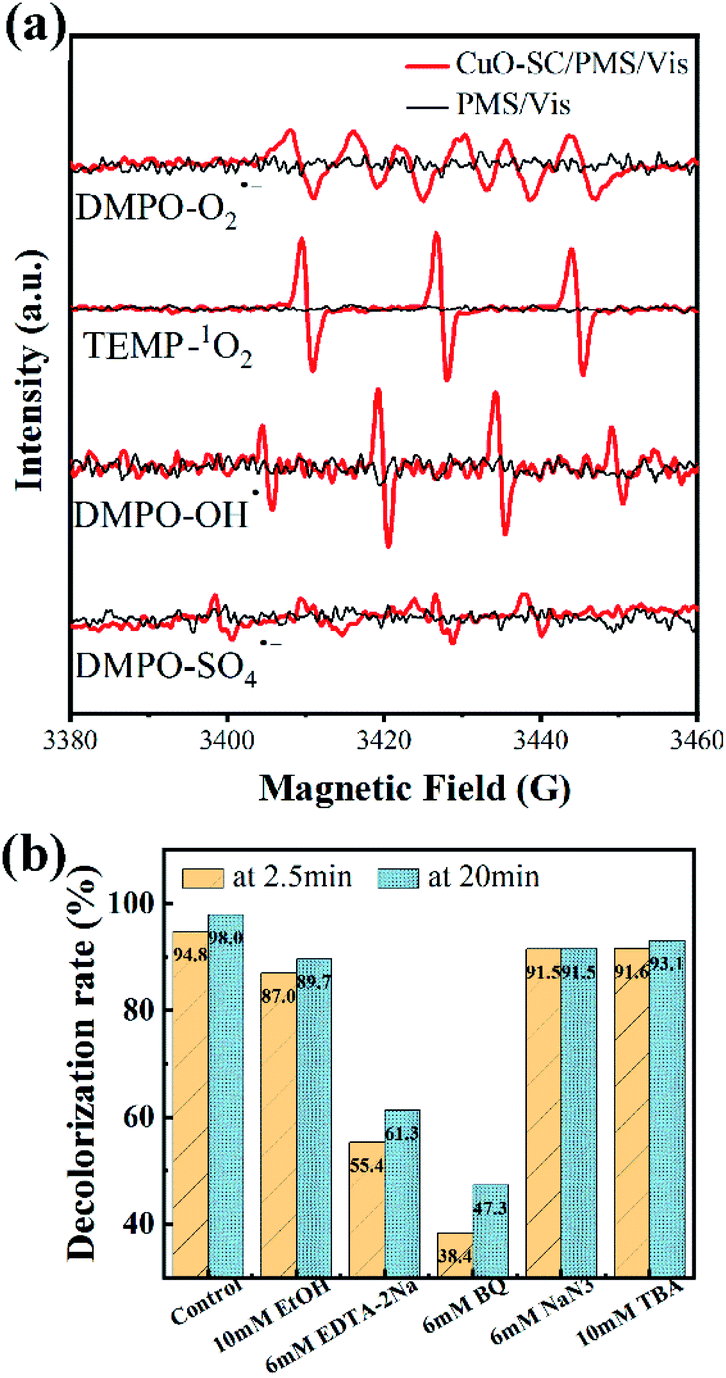
In order to further study the degradation mechanism in the CuO-SC/PMS/Vis system, ESR analysis with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) or 2,2,6,6-tetramethyl-4-piperidone (TEMP) as spin-trap agents was conducted. From Fig. 12a, all the characteristic peaks for PMS alone are not observed. While for CuO-SC/PMS, a strong 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet signal characteristic of TMPO-1O2, a 1:2:2:1 intensity ratio quarter pattern signal characteristic of DMPO–OH˙ and six characteristic peaks of the DMPO–O2˙− are observed. Meantime, weak signals of DMPO-SO4˙− are also detected. These results indicated that the CuO-SC/PMS/Vis system involved in many type of active species. Both the nonradical pathway (1O2) and radical pathway (O2˙−, OH˙ and SO4˙−) contribute to the RhB degradation.
:1:1 triplet signal characteristic of TMPO-1O2, a 1:2:2:1 intensity ratio quarter pattern signal characteristic of DMPO–OH˙ and six characteristic peaks of the DMPO–O2˙− are observed. Meantime, weak signals of DMPO-SO4˙− are also detected. These results indicated that the CuO-SC/PMS/Vis system involved in many type of active species. Both the nonradical pathway (1O2) and radical pathway (O2˙−, OH˙ and SO4˙−) contribute to the RhB degradation.
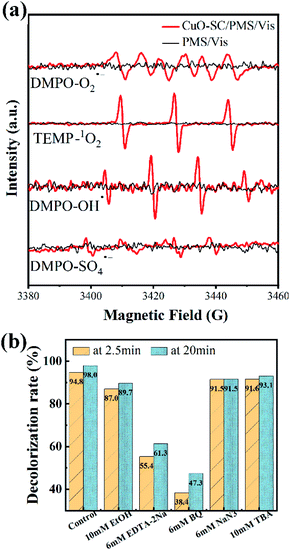 |
| | Fig. 12 (a) ESR signal detection for O2˙−, OH˙, 1O2 and SO4˙− (PMS dosage = 0.12 g L−1, catalyst dosage = 0.3 g L−1, DMPO concentration = 200 mM, TEMP concentration = 600 mM); (b) effects of various reactive species scavengers on RhB degradation at 2.5 and 20 min in CuO-SC/PMS/Vis system (reaction conditions: RhB concentration = 20 mg L−1, catalyst dosage = 0.3 g L−1, initial pH = 7, PMS/RhB weight ratio = 6). | |
To gain further insight into the contribution of different active species, quenching test was carried out. BQ, TBA, NaN3 and EDTA-2Na were used as the trapping agent for O2˙−, OH˙, singlet oxygen (1O2) and h+, respectively.35 In addition, EtOH was used to simultaneously capture OH˙ and SO4˙−.36 As shown in Fig. 12b, the degradation efficiency of RhB in the system is significantly reduced by BQ (from 98.0% to 47.3%) and EDTA-2Na (from 98.0% to 61.3%), indicating that O2˙− and h+ are the dominant active species. While the inhibition effect of TBA, EtOH and NaN3 is weak so that the degradation efficiency at 20 min of RhB is as high as 93.1%, 89.7% and 91.5%, respectively, indicating that there are a small percentage of OH˙, SO4˙− and 1O2 participated in the catalytic process.
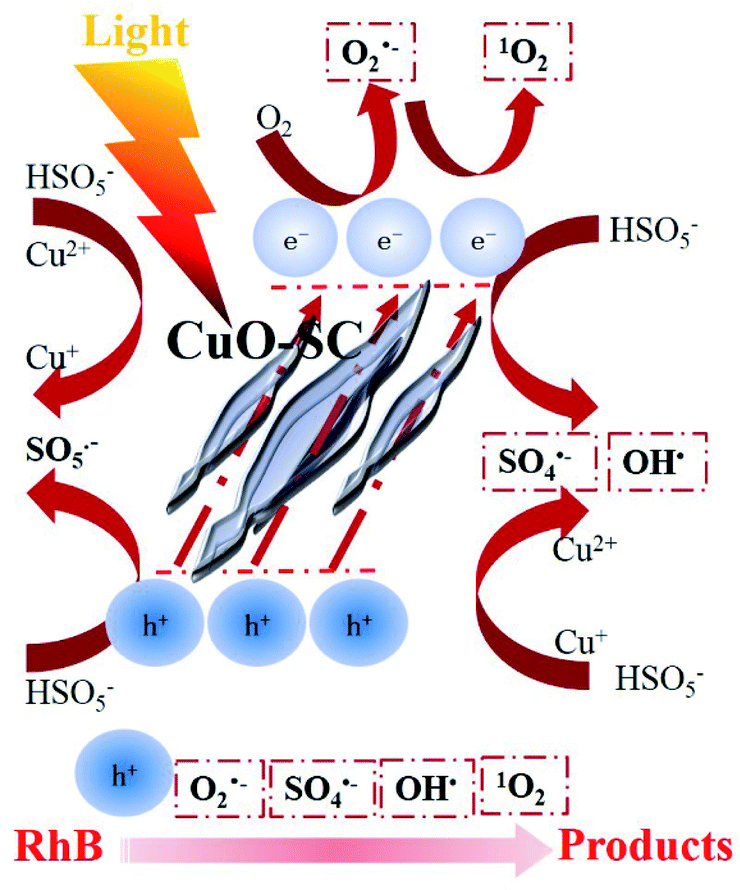
As dominant species, the generation of h+ derives from the well-known photoexcitation process (eqn (4)). At the same time, the generation of O2˙− derives from the reaction between photoelectron (e−) and adsorbed O2 over the oxygen vacancies (eqn (5)).37 Then, some O2˙− can react with H2O forming 1O2 (eqn (6)). The generation of SO4˙− and OH˙ is due to the electron transfer between Cu+ or e− and PMS (eqn (7)–(9)), as confirmed by Fig. 11. And the generated SO4˙− may be converted to OH˙ through eqn (10).
| | |
CuO-SC + hν → h+ + e−
| (4) |
| | |
2O2˙− + 2H2O → 1O2 + H2O2 + 2OH−
| (6) |
| | |
Cu+ + HSO5− → Cu2+ + SO4˙− + OH−
| (7) |
| | |
e− + HSO5− → SO4˙− + OH−
| (8) |
| | |
Cu+ + HSO5− → Cu2+ + SO42− + OH˙
| (9) |
| | |
SO4˙− + H2O → HSO4− + OH˙
| (10) |
| | |
Cu2+ + HSO5− → Cu+ + HSO5− + H+
| (11) |
| | |
h+ + HSO5− → SO5˙− + H+
| (12) |
| | |
2SO5˙− → 2SO4˙− + O2
| (13) |
| | |
h+/SO4˙−/O2˙−/OH˙/1O2 + RhB → intermediates → H2O + CO2 + …
| (14) |
Based on the aforementioned analysis, a rough catalytic degradation mechanism of RhB in the CuO-SC/PMS system under visible light irradiation is shown in Fig. 13. Firstly, e− and h+ are formed on the surface of CuO-SC under visible light irradiation. Holes can directly interact with RhB. The photoelectron can transfer to the oxygen vacancies on the surface of CuO-SC, reacting with oxygen vacancy-adsorbed oxygen to form O2˙− and 1O2. In addition, different valence of Cu on the surface of CuO-SC combined with PMS to produce SO4˙− and OH˙. Meanwhile, the loss of oxygen in the process (eqn (5)) could be replenished by the reaction of eqn (11)–(13).38 Finally, OH˙, 1O2, O2˙−, h+ and SO4˙− attack RhB molecules and the dye transforms to carbon dioxide, water and inorganic ions (eqn (14)). Overall, the degradation process is achieved by radical pathway (h+ and 1O2) and non-radical pathway (O2˙−, OH˙ and SO4˙−).
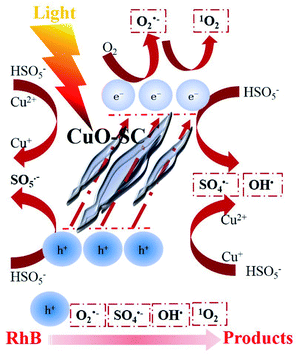 |
| | Fig. 13 Schematic illustration of the possible degradation mechanism of RhB in CuO-SC/PMS/Vis system. | |
CuO-SC nanoparticles exhibits a superior performance for RhB removal combined PMS and visible light irradiation. The advanced nature is mainly attributed to the following three aspects: (1) the regular shape provides high surface area, increasing the connection between catalyst and PMS/dye. (2) Plenty of oxygen vacancies on the surface of CuO-SC endow catalyst low carrier recombination and high charge carriers utilization, which play a determining role in the formation of species to degrade dye. (3) Despite the fact that oxygen vacancy-rich CuO-SC nanoparticles own remarkable advancement, the resultant preparation is extremely efficiency and convenient, providing unique insight into catalyst synthesis.
4. Conclusions
In conclusion, oxygen vacancy-rich CuO nanoparticles with regular fusiform shape were successfully synthesized via sodium citrate guidance strategy. The as-obtained CuO-SC exhibited extremely excellent catalytic behavior in the degradation of RhB. It showed 98.0% decolorization at 20 min, with 0.12 g L−1 PMS and 0.3 g L−1 catalyst under visible light irradiation. The degradation efficiency improved with the increase in CuO-SC and PMS dosage. While, it exhibited high catalytic stability with little deactivation after four runs and a wide application range of pH. According to quenching experiments, O2˙−, h+, OH˙, SO4˙− and 1O2 participated jointly in the catalytic process. While O2˙− and h+ were dominant radicals, whose formation was greatly enhanced by the presence of oxygen vacancies. High surface area, rich oxygen vacancies, wide light harvesting window, fast charge transport, and high carrier concentration worked together to create the remarkable catalytic activity of CuO-SC. This synthetic method might be extended to the production of a wide variety of oxygen vacancy-rich materials for other reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51878647, 61574148).
References
- W. D. Oh, Z. Dong and T. T. Lim, Appl. Catal., B, 2016, 160–201 Search PubMed.
- F. Ghanbari and N. Jafarzadeh, Res. Chem. Intermed., 2017, 43, 4623–4637 CrossRef CAS.
- X. Hao, W. Da and M. Jun, Appl. Catal., B, 2018, 238, 557–567 CrossRef.
- J. Wang and S. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- A. Khan, Z. Liao, Y. Liu, A. Jawad, J. Ifthikar and Z Chen, J. Hazard. Mater., 2017, 329, 262–271 CrossRef CAS PubMed.
- S. Sun, Y. Gu, Y. Wang and Y. Liu, Chem. Phys. Lett., 2020, 752, 137557 CrossRef CAS.
- G. Sheng, W. Hao and Z. Kun, Appl. Catal., B, 2020, 262, 118250 CrossRef.
- Y. Gu, S. Sun, Y. Liu, M. Dong and Q. Yang, ACS Omega, 2019, 4, 17672–17683 CrossRef CAS PubMed.
- S. Yang, L. Li, T. Xiao and D. Zheng, Appl. Surf. Sci., 2016, 383, 142–150 CrossRef CAS.
- Z. Li, D. Liu, W. Huang, X. Wei and W. Huang, Sci. Total Environ., 2020, 721, 137764 CrossRef CAS PubMed.
- D. Yang, P. Hong, Y. Hu, Y. Li, C. Wang, J. He, B. Sun, S. Zhu, L. Kong and J. Liu, Appl. Surf. Sci., 2021, 552, 149424 CrossRef CAS.
- Q. Qin, N. Qiao, Y. Liu and X. Wu, Appl. Surf. Sci., 2020, 521, 146479–146487 CrossRef CAS.
- Q. Yang, Y. Ma, F. Chen, F. Yao, J. Sun, S. Wang, K. Yi, L. Hou, X. Li and D. Wang, Chem. Eng. J., 2019, 378, 122149–122168 CrossRef CAS.
- K. X. Zhu, J. H. Wang, Y. J. Wang, C. Z. Jin and A. S. Ganeshraja, Catal. Sci. Technol., 2016, 6, 2296–2304 RSC.
- K. Y. A. Lin, F. K. Hsu and W. D. Lee, J. Mater. Chem. A, 2015, 3, 9480–9490 RSC.
- P. Shukl, I. Fatimah, S. B. Wang, H. M. Ang and M. O. Tadé, Catal. Today, 2010, 157, 410–414 CrossRef.
- T. Soltani and B. K. Lee, Chem. Eng. J., 2017, 313, 1258–1268 CrossRef CAS.
- G. Zhuang, Y. Chen, Z. Zhuang, Y. Yu and J. Yu, Sci. China Mater., 2020, 63, 2089–2118 CrossRef CAS.
- Z. Li, D. Liu, Y. Zhao, S. Li, X. Wei, F. Meng, W. Huang and Z. Lei, Chemosphere, 2019, 233, 549–558 CrossRef CAS PubMed.
- Y. Wang, Y. Lu, W. Zhan, Z. Xie, Q. Kuang and L. Zheng, J. Mater. Chem. A, 2015, 3, 12796–12803 RSC.
- R. X. Zhou, T. M. Yu and X. Y. Jiang, Appl. Surf. Sci., 1999, 148, 263–270 CrossRef CAS.
- A. Bhaumik, A. M. Shearin, R. Patel and K. Ghosh, Phys. Chem. Chem. Phys., 2014, 16, 11054 RSC.
- Y. Zhao, H. An, G. Dong, F. Jing and M. Jun, Chem. Eng. J., 2020, 388, 124371 CrossRef CAS.
- Z. Yang, J. Xu, W. Zhang, A. Liu and S. Tang, J. Solid State Chem., 2007, 180, 1390–1396 CrossRef CAS.
- M. Qin, D. Lan, G. Wu, X. Qiao and H. Wu, Appl. Surf. Sci., 2020, 504, 144480 CrossRef CAS.
- V. Singh, A. Tiwari and T. C. Nagaiah, J. Mater. Chem. A, 2018, 6, 22545–22554 RSC.
- S. Luo, L. Duan, B. Sun, M. Wei, X. Li and A. Xu, Appl. Catal., B, 2015, 164, 92–99 CrossRef CAS.
- Y. Liu, H. Guo, Y. Zhang, W. Tang, X. Cheng and H. Liu, Chem. Phys. Lett., 2016, 653, 101–107 CrossRef CAS.
- F. Chi, B. Song, B. Yang, Y. Lv, S. Ran and Q. Huo, RSC Adv., 2015, 5, 67412–67417 RSC.
- Y. Xu, J. Ai and H. Zhang, J. Hazard. Mater., 2016, 309, 87–96 CrossRef CAS PubMed.
- Y. Li, D. Li and S. Fan, Catal. Sci. Technol., 2020, 10, 864–875 RSC.
- W. D. Jiang, Y. Liu, J. Jiang, P. Wu, L. Feng and X. Q. Zhou, Aquat. Toxicol., 2015, 159, 245–255 CrossRef CAS PubMed.
- J. Sharma, I. M. Mishra and V. Kumar, J. Environ. Manage., 2015, 156, 266–275 CrossRef CAS PubMed.
- X. Ao and W. Liu, Chem. Eng. J., 2017, 313, 629–637 CrossRef CAS.
- Y. B. Zhou, Y. L. Zhang and X. M. Hu, Colloids Surf., A, 2020, 597, 124568 CrossRef CAS.
- T. J. Park, R. C. Pawer, S. Kang and C. S. Lee, RSC Adv., 2016, 6, 89944–89952 RSC.
- J. Bandara, I. Guasaquillo, P. Bowen, L. Soare and J. Kiwi, Langmuir, 2005, 21, 8554–8559 CrossRef CAS PubMed.
- F. J. Rivas, O. Gimeno and T. Borallho, Chem. Eng. J., 2012, 192, 326–333 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08177b |
| ‡ Yangqiao Liu and Qing Lan contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Qingfeng Yang*b
*a and
Qingfeng Yang*b