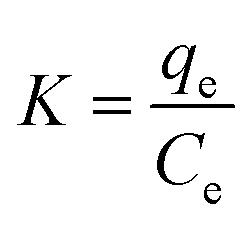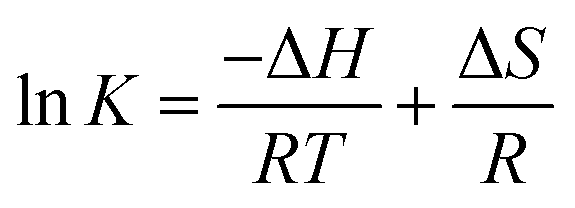DOI:
10.1039/D1RA08325B
(Paper)
RSC Adv., 2022,
12, 1885-1896
Efficient removal of methylene blue by activated hydrochar prepared by hydrothermal carbonization and NaOH activation of sugarcane bagasse and phosphoric acid
Received
13th November 2021
, Accepted 3rd January 2022
First published on 12th January 2022
Abstract
Activated-hydrochar (AHC) derived from sugarcane bagasse was synthesized by hydrothermal carbonization (HTC) using phosphoric acid and sodium hydroxide (NaOH) as activators. The properties of AHC were systematically characterized by elemental analysis, BET, SEM, FTIR, XPS and zeta potential, and applied to evaluate the adsorption ability of methylene blue (MB) by batch adsorption tests. The MB adsorption isotherm and kinetics of AHC were well described by the Langmuir model and pseudo-second-order kinetic model. Characteristic analysis suggested electrostatic attraction, hydrogen bonding and π–π interactions were the main contributors to MB adsorption. Analysis of mass transfer mechanisms demonstrated the adsorption process towards MB by AHC involved intra-particle diffusion to some extent. Thermodynamic studies indicated MB adsorption was an endothermic, spontaneous process associated with a disorder increase at the solid–liquid interface. The maximum adsorption capacity of AHC for MB was 357.14 mg g−1 at 303 K. Thus, the combination of HTC in phosphoric acid and NaOH activation offered a facile, green and economical alternative for conversion of sugarcane bagasse into efficient adsorbents used in wastewater treatment.
1. Introduction
Organic dyes are common contaminants in wastewater originating from the fields of textiles, plastics, paper, cosmetics, food processing and other industries.1,2 Considering the highly toxic, carcinogenic, mutagenic and teratogenic nature of organic dyes, their direct release into the environment can pose serious environmental issues as well as health threats to humans and other organisms.3 Thus, efficient and economical removal of dyes before the discharge of dye wastewater is necessary. So far, numerous techniques including catalytic degradation, membrane separation, adsorption, oxidation and flocculation have been utilized in the region of wastewater treatment.4–8 Adsorption stands out as the most promising technique based on its great efficiency, simplicity and inexpensiveness.9,10 However, some efficient adsorbents, such as activated carbon, resin and bentonite, restricted by the high manufacturing cost or complicated processes are difficult to be applied in massive wastewater treatment.11 Hence, new adsorbents with high efficiency, low cost and simple procedures are urgently to be developed. Given the scarcity of resources, most studies have focused on fabricating adsorbent materials using waste biomass for dye wastewater treatment.12,13
Waste biomass can be directly converted by hydrothermal carbonization (HTC) technology into a carbon-rich material, known as hydrochar, under moderate temperatures (180–350 °C) and autogenous pressures for several hours.14–16 Hydrochar is recognized as an efficient alternative adsorbent for organic dye removal from wastewater owing to its good porosity structure, well-developed surface area and substantial oxygen-containing functional groups (hydroxyl, phenolic, carbonyl, or carboxylic) on the surface.16,17 However, hydrochar prepared by HTC has a low surface area and deficient strong binding sites that limit its adsorption performance. Therefore, it is imperative to modify or activate the hydrochar to increase its adsorption ability. Except for feedstock nature and HTC processing conditions, the incorporation of chemicals especially acids can also improve the physicochemical properties of hydrochar. The added acid may act as catalyst of hydrolysis reaction, which is conducive to enhancing active surface area and functional groups of hydrochar.18–20 The addition of phosphoric acid to the HTC of biomass has been reported to promote the formation of large surface areas and acidic functional groups of hydrochar.21,22 Therefore, mixing with waste biomass and phosphoric acid to prepare hydrochar, intending to enhance the adsorption capacity seems to be highly feasible.
Chemical activation has been widely adopted to improve the adsorption capacity of hydrochar. Cold alkali activation is an efficient and simple activation method linked with the formation of oxygenated functional groups on the surface of hydrochar.23 The alkaline activated hydrochar has higher oxygenated functional group content and adsorption capacities for removing cationic pollutants compared to unactivated hydrochar.24,25 Previously, the maximum adsorption capacity of sewage sludge activated hydrochar prepared by KOH cold alkali activation was increased up to 247.06 mg g−1.24 The adsorption capacities of hydrochar for Cd2+ from different feedstocks (sawdust, wheat straw, and corn stalk) increased by 2–3 times after cold KOH activation.25 It has been reported that NaOH has a stronger the ability to form oxygenated functional groups on the surface and is more eco-friendly than KOH.26 However, few studies have focused on the use of chemically-activated hydrochar with NaOH to improve the adsorption capacity and evaluate its applicability for MB removal from wastewater. Therefore, NaOH was employed to activate hydrochar prepared by HTC in phosphoric acid environment by cold activation method in this study.
Sugarcane bagasse (SCB) is a typical waste biomass generated after sugarcane milling, comprised mainly of cellulose (40–45%), hemicelluloses (30–35%) and lignin (20–30%).27,28 These polysaccharides play an important role in framing the carbon skeleton of hydrochar with functional groups.19 Hence reuse of sugarcane bagasse for producing hydrochar as adsorbent materials to eradicate water pollutants seems to be prospective. In this work, the NaOH-activated hydrochar from SCB and phosphoric acid was synthesized and assessed its applicability for MB removal from aqueous solution. The overarching aims were to (i) prepare two hydrochars by HTC of sugarcane bagasse in phosphoric acid medium (labeled as HC) and followed by NaOH activation (labeled as AHC); (ii) characterize both hydrochars and compare the MB adsorbing ability; (iii) study the mechanism of adsorbing MB onto AHC.
2. Materials and methods
2.1. Materials and chemicals
SCB powder with diameter below 100 mesh was kindly provided by Guangxi Fengtang Biochemical Co., Ltd. (Liuzhou, China). NaOH (AR), HCl (AR), H3PO4 (AR) and MB (C16H18ClN3S·3H2O, Ind) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). All of the chemical solutions were prepared based on deionized water. A stock solution (1000 mg L−1) was prepared directly by dissolving MB (1.0000 g) in deionized water and then diluted to the desired concentrations.
2.2. Preparation of HC and AHC
Firstly, SCB powder and 1.5 M phosphoric acid solution at a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19 were added to a 100 mL Teflon autoclave. The reactor was heated at 513 K for 10 h and then cooled to ambient temperature in air atmosphere. Using vacuum filtration, the solids were retained, then washed repeatedly with deionized water. The raw hydrochar was obtained by drying the solids overnight at 373 K, which was named as HC. Subsequently, HC (2.5 g) was immersed in 1.5 M 50 mL NaOH solution, stirred for 2 h at ambient temperature to obtain activated hydrochar, neutralized by washing with deionized water and oven-dried at 373 K overnight. The activated hydrochar was henceforth denoted as AHC.
:19 were added to a 100 mL Teflon autoclave. The reactor was heated at 513 K for 10 h and then cooled to ambient temperature in air atmosphere. Using vacuum filtration, the solids were retained, then washed repeatedly with deionized water. The raw hydrochar was obtained by drying the solids overnight at 373 K, which was named as HC. Subsequently, HC (2.5 g) was immersed in 1.5 M 50 mL NaOH solution, stirred for 2 h at ambient temperature to obtain activated hydrochar, neutralized by washing with deionized water and oven-dried at 373 K overnight. The activated hydrochar was henceforth denoted as AHC.
2.3. Characterization
The elemental compositions of SCB, HC and AHC (C, H, O and N) was analyzed by Elemental analyzer (Vario EL, Elementar, Germany). The surface morphology of the samples was observed using an scanning electron microscopy (SEM; Phenom Pro, Phenom, Holland) at 5000× magnification. The chemical functional groups were characterized by Fourier transform infrared (FT-IR; IRTracer-100, Shimadzu, Japan). The X-ray photoelectron spectroscopy (XPS; K-alpha, Thermo Fisher Scientific, America) was used to analyse the surface chemical composition. All XPS spectra were calibrated by referencing the C 1s peak at 284.6 eV. N2 adsorption–desorption was used to test the specific surface area by employing a surface-area and pore-size analyzer (ASAP 2460, Atlanta, America) at 77 K using the Brunauer–Emmett–Teller (BET) theory as the test method. The sample pore size and pore volume were determined using the Barrett–Joyner–Halenda (BJH) formula. The surface charge of the AHC was measured by a zeta-potential analyzer (NanoBrook Omni, Brookhaven, America).
2.4. Batch adsorption experiments
The adsorption experiments were performed by batch experiments. A 100 mL conical flask containing 40 mg of AHC and 50 mL of MB solution at specific concentrations was shaken in a constant-temperature incubator (ZQZY-85CN, LUBEX, China) with constant speed of 150 rpm for a given time at predetermined temperatures. After reaching the pre-set time, the solution was filtered by using a 0.45 μm membrane. Then, the MB concentration in the filtrate was measured from the calibration curve of MB solutions at a maximum adsorption wavelength of 665 nm by using a spectrophotometer (UV-2550, Shimadzu, Japan) after appropriate dilution.29
Adsorption isotherm assays were executed using a series of MB solutions with initial concentrations ranging from 150 to 450 mg L−1 at 303 K, 313 K, and 323 K for 24 h, respectively. The adsorption capacity of the AHC for MB at equilibrium was defined by eqn (1):11
| |
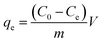 | (1) |
where
qe (mg g
−1) was the adsorption capacity of AHC onto MB at equilibrium;
C0 (mg L
−1) and
Ce (mg L
−1) represented the initial and equilibrium MB concentrations, respectively;
V (L) and
m (g) meant the solution volume and adsorbent dosage, respectively.
The kinetics of MB adsorption onto AHC were determined at concentrations of 200, 250 and 300 mg L−1 at 303 K and at different time intervals. The adsorption capacity of the AHC for MB at time t was calculated using eqn (2):11
| |
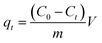 | (2) |
where
qt (mg g
−1) was the adsorption capacity at time
t (min);
C0 (mg L
−1) and
Ct (mg L
−1) were the concentration of MB initially and at time
t, respectively;
V (L) and
m (g) meant the solution volume and adsorbent dosage, respectively.
The effect of solution pH on the adsorption capacity of AHC was investigated in the pH range of 2.0–11.0 for 300 mg L−1 MB solution adjusted by drops of 0.1 M HCl or NaOH solutions. The suspensions were shaken at 303 K and 150 rpm for 24 h. All batch experiments were performed in triplicate, and the parameters were fitted by Origin Pro 2018 software.
3. Results and discussion
3.1. Characterizations
3.1.1. Elemental analysis. Table 1 depicted the elemental compositions and atomic ratios of SCB, HC and AHC. The nitrogen content in the samples were slightly affected by either HTC or activation treatment. HC had higher C content and lower H and O contents relative to SCB, which was attributed to the decarboxylation and dehydration reactions during the HTC process and the catalysis of phosphoric acid on hydrolysis carbonization of biomass.30,31 After NaOH activation, the C content in HC declined from 69.66% to 65.27%, and the H and O contents increased from 5.16% and 24.44% to 5.57% and 29.01%, respectively. These results were probably achieved due to NaOH activation, which promoted the dissociation of weak bonds and ionization of phenol and humus, leading to the loss of surface functional groups and an increase in oxygen-containing functional groups.32,33
Table 1 Elemental compositions and atomic ratios of SCB, HC and AHC
| Samples |
Elemental compositions (%) |
Atomic ratio |
| C |
H |
N |
Oa |
H/C |
O/C |
(O + N)/C |
| O = 100 − (C + H + N). |
| SCB |
45.06 |
6.31 |
0.60 |
48.03 |
1.680 |
0.799 |
1.080 |
| HC |
69.66 |
5.16 |
0.73 |
24.44 |
0.889 |
0.263 |
0.361 |
| AHC |
65.27 |
5.57 |
0.65 |
29.01 |
1.024 |
0.333 |
0.454 |
The atomic ratios of H/C and O/C were related to the contents of hydroxyl, carboxyl, and carbonyl groups. The HTC was usually accompanied by deoxygenation, dehydrogenation, and polycondensation, which were responsible for the decrease in H/C and O/C.34 The atomic ratios of H/C and (O + N)/C reflected the aromaticity and polarity of the biomass materials. The higher H/C and (O + N)/C ratios of HC compared to SCB indicated the aromaticity was enhanced and the polarity was weakened after HTC using phosphoric acid as the reaction medium.35 Moreover, the O/C ratio of AHC increased upon NaOH activation, suggesting more oxygen-containing functional groups were generated on the surface of the AHC, which was conducive to improving its adsorption capacities.
3.1.2. BET analysis. The N2 adsorption–desorption isotherms and pore size distribution curves of HC and AHC were presented in Fig. 1. Although variations existed in the adsorption behaviors, the isothermal curves of HC and AHC were similar, indicating NaOH activation treatment did not change the pore type. According to the International Union of Pure and Applied Chemistry (IUPAC) classification, the isotherms were equivalent to Type-IV isotherms, which was an indicative of the presence of a mesoporous structure and abundant slit pores on the surfaces.6 The isotherm profiles obtained herein were similar to those reported by Jais et al.36 However, the increase in relative pressure (p/p0) promoted the adsorption capacity of AHC, which was significantly greater than that of HC, suggesting that AHC had a richer pore structure than HC. A type H3 hysteresis loop was clearly observed in the isotherms of both HC and AHC, which revealed that their pore networks were composed of macropores that were not completely filled with pore condensate.37 As shown in Fig. 1b, the pore size distribution curves of HC and AHC showed multimodal distribution patterns. However, the latter seemed to have a larger pore size and a greater number of large pores.
 |
| | Fig. 1 (a) N2 adsorption–desorption isotherms and (b) pore size distributions of HC and AHC. | |
As summarized in Table 2, the BET surface area, total pore volume and average pore size for HC were 7.845 m2 g−1, 0.060 m3 g−1 and 6.869 nm, which proved that HC produced by HTC in phosphoric acid medium had a good porosity and surface area. After treatment with NaOH, AHC revealed a higher value in BET surface area, total pore volume and average pore size with 15.340 m2 g−1, 0.084 m3 g−1 and 7.325 nm, respectively. The same result was observed for cold alkali modification of other hydrochars.38,39 This was because that the ash and organic compounds contained in HC were transferred to the liquid phase and washed off during the NaOH treatment. Moreover, the interactions between NaOH and carbon during cold alkali activation were also helpful for the formation of high specific surface area and well-developed porosity. As mentioned by Martins et al., the possible activation mechanisms were summarized as follow:40
| 6NaOH + 2C → 2Na + 2Na2CO3 + 3H2 |
Table 2 Surface areas, total pore volumes and average pore sizes of SCB, HC and AHC
| Samples |
BET surface area (m2 g−1) |
Total pore volume (cm3 g−1) |
Average pore size (nm) |
| HC |
7.845 |
0.060 |
6.869 |
| AHC |
15.340 |
0.084 |
7.325 |
The reaction of NaOH with carbon could form sodium carbonate and was capable of reducing Na+ and hydroxyl anion cation to metallic sodium H2, respectively. The sodium embedded in the partially crystallized carbon layer and the release of H2 promoted the generation of micropore structure. In addition, CO2 and CO produced from Na2CO3 decomposition and CO2 reduction diffused out from the interior of AHC, which further improved the formation of micro/mesopores and specific surface area in the AHC. Therefore, cold alkali activation with NaOH helped to clear up partially blocked pores and increase the porosity of the AHC, which led to a rise in activated surface area as well as adsorption capacity.
3.1.3. SEM analysis. The SEM images of SCB, HC, AHC and AHC loaded on MB were distinctly different in Fig. 2. The SCB in Fig. 2a had a relatively smooth and flat surface showing less porosity. In contrast, the appearance of HC was rough with numerous homogeneous pores distributed on the surface, although some pores were covered and clogged by impurities (Fig. 2b). The pore blockages were ascribed to the condensation of hydrocarbon on the surface during the HTC process.41 Many smooth, mono-disperse carbon microspheres formed by the thermal polycondensation of hemicellulose, cellulose and partial lignin were also observed on the surface of HC.42 A comparison of HC and AHC showed that the latter was rougher and had more cracks and channels on the surface (Fig. 2b and c). Some of the impurities that blocked the pores were decomposed or volatilized during NaOH activation, thus removing the impurities from the cracks and pores.43 These cracks and channels provided a larger contact surface and more binding sites which facilitated the adsorption of MB into the interior surface of AHC. The morphology of AHC/MB after MB adsorption was displayed in Fig. 2d. The surfaces of the carbon microspheres and reticulate structure were apparently covered with a layer of MB molecules, which reflected the efficient adsorption of MB by AHC.
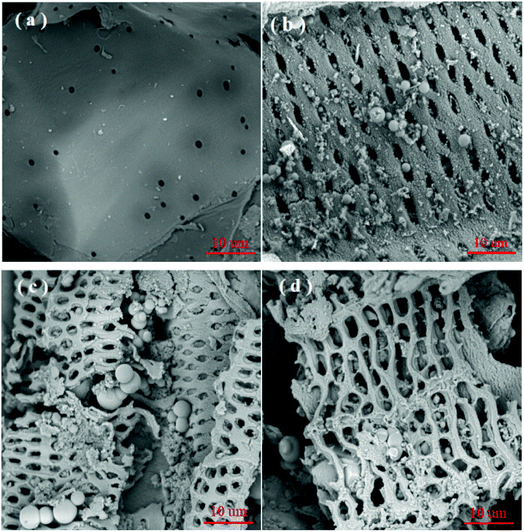 |
| | Fig. 2 SEM analysis for (a) SCB, (b) HC, (c) AHC and (d) AHC after MB adsorption at magnification = 5000×. | |
3.1.4. FT-IR analysis. FT-IR spectra demonstrated that HC contained abundant oxygen-containing functional groups under HTC in the presence of phosphoric acid (Fig. 3). A broad peak around 3420 cm−1 represented O–H stretching vibrations of hydroxyl or carboxyl groups, which appeared that the O–H groups originally in hemicellulose, cellulose, and lignin were not completely degraded during the HTC process.39,44 The peaks at 3000–2800 cm−1 were ascribed to asymmetric and symmetric of aliphatic C–H. The bands at 1697 cm−1 represented the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in the carboxyl and aldehyde groups. The stretching vibration peaks of CC and CO from the aromatic rings were at 1608 and 1510 cm−1, respectively. The peaks at 1265–1215 cm−1 were associated with the stretching vibrations of aromatic C–O and phenolic C–OH bonds.16 The bands at 1145–1027 cm−1 were assigned to the PO stretching mode in polyphosphate and phosphate bonds.45 The characteristic peaks at 830–540 cm−1 belonged to the out-of-plane aromatic C–H bending vibrations. After NaOH activation, the peak intensities of AHC at 3410 and 1697 cm−1 increased dramatically, which showed a larger amount of hydroxyl and carboxyl groups on the surface of AHC than HC. The oxygen-containing functional groups could serve as proton donors, so that the increase in oxygen-containing functional groups indicated that there were the presence of more protonated groups, which could interact with cationic MB to enhance the adsorption capacity of AHC.39
O bonds in the carboxyl and aldehyde groups. The stretching vibration peaks of CC and CO from the aromatic rings were at 1608 and 1510 cm−1, respectively. The peaks at 1265–1215 cm−1 were associated with the stretching vibrations of aromatic C–O and phenolic C–OH bonds.16 The bands at 1145–1027 cm−1 were assigned to the PO stretching mode in polyphosphate and phosphate bonds.45 The characteristic peaks at 830–540 cm−1 belonged to the out-of-plane aromatic C–H bending vibrations. After NaOH activation, the peak intensities of AHC at 3410 and 1697 cm−1 increased dramatically, which showed a larger amount of hydroxyl and carboxyl groups on the surface of AHC than HC. The oxygen-containing functional groups could serve as proton donors, so that the increase in oxygen-containing functional groups indicated that there were the presence of more protonated groups, which could interact with cationic MB to enhance the adsorption capacity of AHC.39
 |
| | Fig. 3 FT-IR spectra of SCB, HC, AHC as well as AHC following MB adsorption. | |
The characteristic peaks of MB appeared at 1597, 1386, 1328 and 883 cm−1 after adsorption, which manifested MB was successfully adsorbed on the AHC surface.11 The O–H stretching vibration of AHC and CN stretching vibration in MB shifted from 3410 and 1589 cm−1 to 3398 and 1597 cm−1, respectively. This result authenticated the existence of hydrogen bond interactions between the N atoms of phenothiazine in MB and C–OH of AHC.46,47 The C–N stretching vibration in the –N(CH3)2+ group and methyl symmetric deformation migrated from 1389 and 1319 cm−1 to 1386 and 1328 cm−1, which confirmed the electrostatic interaction between the AHC and the –N(CH3)2+ group of MB.46 The out-of-plane bending vibration of aromatic C–H group at 826 shifted to 883 cm−1, which suggested π–π interaction also participated in adsorption.11
3.1.5. XPS analysis. The surface chemical compositions of HC, AHC and AHC after MB adsorption were analyzed by XPS (Fig. 4). The XPS spectra of HC and AHC showed that major elemental peaks (eV) at 284.60/284.60 (C 1s), 401.21/399.97 (N 1s), 532.74/532.33 (O 1s) and 1071.43 (Na 1s) (Fig. 4a and d).48 As shown in Fig. 4b and e, the C 1s spectra of HC and AHC could be deconvoluted into three peaks at 284.52/284.56 eV (C–C/CC), 285.77/286.07 eV (C–O) and 288.81/288.56 eV (OC–O), respectively.49 The C 1s spectra revealed that AHC contained more C–C/CC (72.86% vs. 63.44%) and OC–O (10.00% vs. 7.64%) and less C–O (17.14% vs. 28.92%) than HC, which indicated the relative ratio of OC–O rose by 30% after NaOH activation. Meanwhile, the peaks at 531.88/531.87 eV, 533.03/533.03 eV and 533.72/533.41 eV indicated CO, C–O and OC–O for HC and AHC (Fig. 4c and f), respectively.50 The quantitative area analysis of O 1s spectra also suggested the relative percentage of CO and OC–O increased, while that of C–O decreased in AHC compared with HC. The above analyses convinced that NaOH activation treatment increased the carboxyl content on the surface of AHC, which contrariwise was beneficial for improving its MB adsorption capacity.
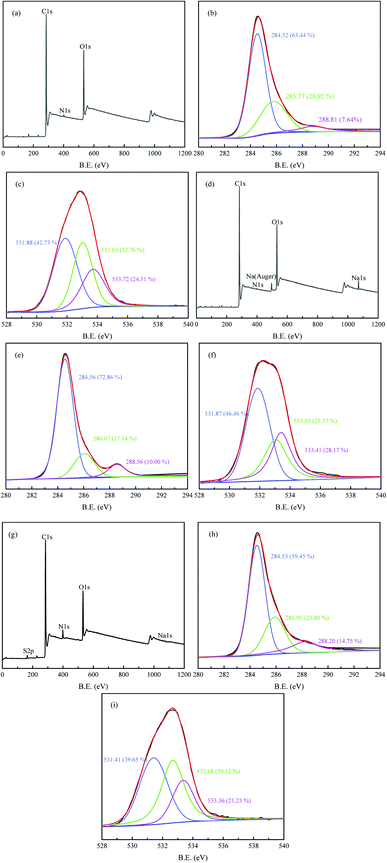 |
| | Fig. 4 XPS spectra of HC (a) AHC (d) AHC after MB adsorption (g); high-resolution C 1s XPS spectra (b, e and h) and O 1s (c, f and i) of HC/AHC/AHC after MB adsorption. | |
After MB adsorption, a new elemental peak emerged around 164.26 eV (S 2p) and the content of N increased while the peak intensity of Na 1s decreased in the XPS survey of AHC/MB (Fig. 4h), which demonstrated the successful adsorption of MB onto AHC and ion exchange between sodium ion and MB involved in the adsorption process. The binding energies of C 1s and O 1s changed obviously after MB adsorption in AHC/MB, confirming that the oxygen-containing functional groups participated in the adsorption, which conformed well with the FT-IR analysis.
3.2. Adsorption ability comparison of SCB, HC and AHC
The adsorption performances of SCB, HC and AHC were investigated. Their adsorption capacities for MB were 32.15, 116.65 and 334.74 mg g−1, respectively (Fig. 5). It was obvious that the MB adsorption ability of HC at 303 K was higher than that of SCB, because the HTC process in phosphoric acid medium greatly improved the aromaticity and porosity of HC. What's more, the adsorption capacity of AHC for MB was almost three times that of HC because of the further rise in BET surface area, pore volume and functional groups of AHC after NaOH activation, as mentioned in the BET and FT-IR analyses. Hence, cold alkali activation using NaOH is a promising activation method for improving the adsorption capacity of hydrochar.
 |
| | Fig. 5 Comparison of MB adsorption capacity of SCB, HC and AHC (C0 = 300 mg L−1, m = 40 mg, V = 50 mL, pH = 7, T = 303 K, t = 24 h). | |
3.3. Adsorption isotherms
The MB adsorption isotherms of AHC at three different temperatures were illustrated in Fig. 6a. The adsorption quantities (qe) increased quickly at low Ce values while the increase in qe slowed down at higher values of Ce, and ultimately reached an adsorption plateau. The adsorption capacities of AHC increased with the temperature increment, suggesting the endothermic nature of the adsorption process.
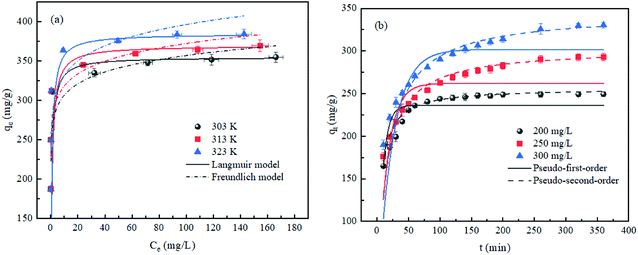 |
| | Fig. 6 (a) MB adsorption isotherms of AHC and Langmuir and Freundlich fitting curves (C0 = 150–450 mg L−1, m = 40 mg, V = 50 mL, pH = 7, T = 303–323 K, t = 24 h). (b) MB adsorption kinetics of AHC and pseudo-first/second-order kinetic fitting curves (C0 = 200–300 mg L−1, m = 40 mg, V = 50 mL, pH = 7, T = 303 K). | |
To investigate the relevant mechanisms of adsorption at equilibrium, Langmuir (eqn (3)) and Freundlich (eqn (4)) isotherm models were adopted to fit the experimental data. The Langmuir model simulated the homogenous monolayer adsorption behavior, whereas the Freundlich model described the heterogeneous multi-layer adsorption behavior.51
| |
 | (3) |
| |
 | (4) |
where
qe (mg g
−1) stood for the equilibrium adsorption capacity of AHC;
qm (mg g
−1) denoted the estimated theoretical saturation adsorption capacity by Langmuir model;
KL (L mg
−1),
KF ((mg g
−1) (L mg
−1)
1/n) and
n were assigned to Langmuir constant, Freundlich constants used to assess the relative adsorption capacity and favorability of adsorption, respectively.
The corresponding parameters were listed in Table 3 and the fitting curves were displayed in Fig. 6a. The Langmuir model exhibited larger correlation coefficients (R2 > 0.999) than the Freundlich model (R2 < 0.822). Besides, the maximum adsorption capacities (qm) of MB calculated by the Langmuir model agreed well with the equilibrium adsorption data. Hence, the Langmuir model provided a better description of the adsorption process, indicating that the MB adsorption onto AHC was homogenous monolayer adsorption.47 The surface of AHC had a monolayer coverage of active sites, and each adsorption site was occupied by only one MB molecule, thus forming a homogeneous monolayer of adsorption layer.
Table 3 Isotherm parameters for MB adsorption by AHC
| Temperature (K) |
qe, exp (mg g−1) |
Langmuir model |
Freundlich model |
| qm (mg g−1) |
KL (L mg−1) |
R2 |
KF (mg1−1/n L1/n g−1) |
1/n |
R2 |
| 303 K |
354.79 |
357.14 |
1.642 |
0.9999 |
263.35 |
0.066 |
0.8222 |
| 313 K |
369.32 |
370.37 |
1.688 |
0.9998 |
272.08 |
0.068 |
0.7996 |
| 323 K |
384.20 |
384.62 |
2.889 |
0.9999 |
277.94 |
0.077 |
0.7633 |
The maximum adsorption capacity of AHC for MB was 357.14 mg g−1 at 303 K, which was greater than that of the majority of hydrochars derived from other biomass wastes in Table 4. The BET surface area of AHC retained significantly smaller than that of pyrolysis-activated hydrochars, because the channels in AHC were not opened completely at the relatively low activation temperature.53–55 However, AHC exhibited higher adsorption amount than most pyrolytic hydrochars, which might be due to the fact that activation at low temperature effectively avoided the decomposition of surface oxygen-containing functional groups caused by high temperature.58 It seemed that the effect of oxygen-containing functional groups on adsorption was more significant than that of surface area during MB adsorption. Compared with AHC, hydrochar modified with maleic anhydride demonstrated the best adsorption performance, which was ascribed to the strong affinity of the introduced carboxyl groups for MB.46 Nevertheless, NaOH-modified AHC had the special advantages of low activation temperature and environmental friendliness. Furthermore, HTC in the phosphoric acid medium introduced more acidic functional groups, thus improving the adsorption capacity. In comparison, AHC proved to be an effective and economical method for purifying MB-contaminated wastewater.
Table 4 Adsorption capability of hydrochars for MB in literatures
| Raw materials/modification method |
BET surface area (m2 g−1) |
Adsorption batch conditions |
qm, (mg g−1) |
Ref. |
| Bamboo/HTC with polyvinyl chloride |
4.080 |
50 mg/50 mL at 100–400 mg L−1 for 12 h |
234.46 |
48 |
| Pine wood/oxidated with oxone |
7.662 |
50 mg/15 mL at 0–400 mg L−1 for 2 h |
47.77 |
52 |
| Hickory/mixed with H3PO4 + pyrolysis at 600 °C for 1 h |
1436.000 |
50 mg/25 mL at 10–1000 mg L−1 for 24 h |
214.60 |
53 |
| Camellia oleifera seed shells/ground with KOH + pyrolysis at 650 °C for 2 h |
1882.406 |
20 mg/40 mL at 50–300 mg L−1 for 12 h |
511.80 |
54 |
| Coconut shell/impregnated with NaOH + pyrolysis at 600 °C for 1 h |
876.140 |
200 mg/200 mL at 25–250 mg L−1 for 24 h |
200.01 |
55 |
| Bamboo/modified with maleic anhydride at 140 °C for 6 h |
28.189 |
40 mg/50 mL at 500–1300 mg L−1 for 24 h |
1155.57 |
46 |
| Bamboo/modified with ZnCl2 |
29.600 |
50 mg/50 mL at 200 mg L−1 for 24 h |
47.30 |
56 |
| Orange peel/mixed with nitric acid for 4 h |
20.250 |
50 mg/25 mL at 100–1000 mg L−1 for 24 h |
107.12 |
57 |
| Bamboo/HTC with HCl + activated with NaOH |
26.249 |
40 mg/50 mL at 100–900 mg L−1 for 24 h |
657.89 |
16 |
| Sugarcane bagasse/HTC with H3PO4 + activated with NaOH |
15.340 |
40 mg/50 mL at 150–450 mg L−1 for 24 h |
357.14 |
This study |
3.4. Adsorption kinetics
The adsorption capacity of AHC for MB as a function of time were depicted in Fig. 6b. Based on the analysis of adsorption isotherms, the MB adsorption kinetics of AHC was studied by choosing three specific concentrations of 200, 250 and 300 mg L−1 as low, medium and high concentrations. The adsorption capacity was positively correlated with the MB concentration, which was ascribed to the increase in the driving force derived from the concentration difference of adsorbate between substrate solution and the surface of the AHC. The MB adsorption rate onto the AHC was fast during the initial 50 min and then gradually slowed down until reaching equilibrium. It was because that numerous active sites were available initially on AHC, but were gradually saturated as the adsorption progressed.16
The test data were fitted by two frequently-used kinetic models, the pseudo-first-order model (eqn (5)) and pseudo-second-order model (eqn (6)), for mathematically analysing the adsorption kinetics (Fig. 6b).51
| |
 | (6) |
where
qe (mg g
−1) and
qt (mg g
−1) denoted the MB adsorption capacity at equilibrium and time
t (min);
k1 (min
−1) and
k2 (g mg
−1 min
−1) were the pseudo-first-order and pseudo-second-order adsorption rate constants, respectively.
The correlation coefficients (R2) and fitting parameters were summarized in Table 5. The high R2 values (>0.943) from the pseudo-first-order and pseudo-second-order kinetic models suggested that chemical and physical adsorption occurred simultaneously during MB adsorption.59 However, the larger R2 (>0.999) of the pseudo-second-order kinetic model revealed the pseudo-second-order kinetic model was preferable to describe the adsorption kinetics and MB adsorption by AHC was dominated by chemisorption.47 Furthermore, the equilibrium adsorption quantity values (qe, cal.) calculated by the non-linear regression method were correlated with the experimental values (qe, exp.), which further confirmed the pseudo-second-order kinetic model better described the MB adsorption behaviors.
Table 5 Kinetic parameters for MB adsorption by AHC
| Concentration (mg L−1) |
qe, exp (mg g−1) |
Pseudo-first-order model |
Pseudo-second-order model |
| qe, cal (mg g−1) |
k1 (min−1) |
R2 |
qe, cal (mg g−1) |
k2 [g·(mg−1 min−1)] |
R2 |
| 200 |
249.37 |
236.30 |
0.0181 |
0.9431 |
256.41 |
7.042 × 10−4 |
0.9999 |
| 250 |
294.92 |
261.94 |
0.0081 |
0.9764 |
303.03 |
2.521 × 10−4 |
0.9995 |
| 300 |
330.49 |
301.83 |
0.0106 |
0.9901 |
344.83 |
1.676 × 10−4 |
0.9991 |
3.5. Mass transfer mechanisms
The diagram of qt versus t0.5 for MB adsorption by AHC was shown in Fig. 7. Adsorption is essentially a mass transfer process that the adsorbate diffuses from the liquid phase to the solid adsorbent. To determine the diffusion mechanism in MB adsorption process, a linear intra-particle diffusion (IPD) models (eqn (7)) deduced by Weber and Morris was applied to describe the mass transfer process.51Where qt (mg g−1) was the adsorption capacity of MB by AHC at time t (min); KIPD (mg g−1 min−0.5) was the IPD rate constant and c (mg g−1) was the intercept of the linear plot of qt versus t0.5.
 |
| | Fig. 7 Linear of intra-particle diffusion model for MB adsorption by AHC (C0 = 200–300 mg L−1, m = 40 mg, V = 50 mL, pH = 7, T = 303 K). | |
The relatively high R2 values of the IPD model indicated that MB adsorption onto the AHC was governed by intra-particle diffusion to some extent. Fig. 7 displayed a multi-level linear relationship deviating from the origin, revealing that the MB adsorptive process onto AHC was involved in multiple mechanisms. The first stage was mainly external surface adsorption (pore filling), where the adsorbates passed through the liquid film around the adsorbent. The second stage of linearity was internal diffusion, which involved the transportation of MB into the pores of the adsorbent by intra-particle diffusion and/or pore diffusion. The third stage was the final equilibrium stage.11 It was concluded that intra-particle diffusion and chemical adsorption affected the adsorption results (Table 6).
Table 6 The intra-particle diffusion parameters for MB adsorption by AHC
| Concentration mg L−1 |
KIPD (mg g−1 min−0.5) |
c (mg g−1) |
R2 |
| 200 |
4.3935 |
184.97 |
0.6552 |
| 250 |
6.8583 |
182.50 |
0.8797 |
| 300 |
8.1224 |
196.57 |
0.8996 |
3.6. Impacts of aqueous pH on MB adsorption
The MB can exist in aqueous solution in both the cationic form (MB+) and as undissociated molecules (MBo). The MB speciation curve was displayed in Fig. 8a. Obviously, the MBo predominated at pH < 3.8, both MBo (50%) and MB+ (50%) coexisted at pH = 3.8, and MB+ was the only species at pH > 6.0.
 |
| | Fig. 8 (a) Effect of pH on MB adsorption onto AHC (C0 = 300 mg L−1, m = 40 mg, V = 50 mL, pH = 7, T = 303 K, t = 24 h); and (b) the zeta potential of AHC. | |
Fig. 8b showed the impacts of initial aqueous pH (2.0–11.0) on MB adsorption capacity and zeta potential of AHC. The MB uptakes of AHC ascended sharply from 223.29 to 298.55 mg g−1 with aqueous pH from 2.0 to 5.0 and then remained generally unchanged at pH 6.0–8.0, followed by a continuous increase from pH 9.0 to 11.0. The zeta potential of AHC dropped from +2.43 to −34.12 mV within pH 2.0–11.0 and the point zero charge (pHpzc) of AHC was 2.2. Therefore, the surface of AHC was positively charged due to the protonation of functional groups at pH < pHpzc. On the contrary, the functional groups on the surface of AHC were deprotonated and became negatively charged at pH > pHpzc.
The relatively low adsorption capacities of AHC at pH < 2.2 could be explained by the fact that the MB existed almost as MBo and the surface of AHC was positive charged. The electrostatic repulsion between the small amount of dissociated MB+ and positively charged AHC inhibited MB adsorption.47,48 On the another hand, the N atoms on the phenothiazine were easy to protonate at low pH, which was also adverse to adsorbing MB by hydrogen bonding.47 With rising pH, the cationic MB+ was the dominant species of MB while the zeta potentials decreased and the charge of AHC surface became negative, which resulted in the gradual increase in adsorption capacity of AHC driven by strong electrostatic force.11 Meanwhile, hydrogen bonding was more likely to occur between N atoms on the phenothiazine in MB and C–OH of AHC.48 Hence, electrostatic attraction and hydrogen bonding were key mechanisms for MB removal by AHC. The π–π interaction also played a crucial role in the adsorption system, as AHC still exhibited a high adsorption ability at pH 2.
3.7. Thermodynamic parameters
Thermodynamic indices such as variations in Gibbs free energy (ΔG), enthalpy (ΔH) and entropy (ΔS) shown in Table 7 were computed by isothermal equation (eqn (9)) and the van't Hoff equation of lnK against 1/T (eqn (10)).60| |
 | (8) |
| | |
ΔG = −RTlnK
| (9) |
| |
 | (10) |
where K was thermodynamic equilibrium constant; T (K) and R (8.314 J mol−1 K−1) represented the absolute temperature and the universal gas constant, respectively; ΔG (kJ mol−1), ΔH (kJ mol−1), and ΔS (J mol−1 K−1) were the Gibbs free energy, enthalpy and entropy, respectively; the ΔH and ΔS parameters could be calculated from the slope and intercept of a plot of lnK versus 1/T.
Table 7 Thermodynamic parameters for MB adsorption on AHC
| Temperature (K) |
ΔG (kJ mol−1) |
ΔH (kJ mol−1) |
ΔS [J mol−1 K−1] |
R2 |
| 303 |
−5.91 |
53.77 |
196.00 |
0.9096 |
| 313 |
−6.95 |
| 323 |
−9.87 |
The ΔH of 53.77 kJ mol−1 and ΔS of 196.00 J mol−1 K−1 confirmed an endothermic procedure of MB adsorption onto AHC and an intensifying randomness of MB adsorption at solid–liquid interface.16 The negative ΔG (−5.91,-6.95 and −9.87 kJ mol−1) at tested temperatures reflected the MB adsorption by AHC was spontaneous and thermodynamically favorable.46 The decreased in ΔG with rising temperature showed the elevated temperatures increased the spontaneity of the reaction, which was more favorable to MB adsorption. The adsorption reaction was generally believed to proceed through physical adsorption (ΔG value, 0∼-20 kJ mol−1) or chemical adsorption (ΔG values, −80∼-400 kJ mol−1).39 The MB adsorption was a physical process on the basis of ΔG values, which was inconsistent with adsorption kinetics and isotherm analysis. One possible explanation for this discrepancy was that chemical and physical adsorption occurred simultaneously amid MB adsorption.
3.8. Proposed MB adsorption mechanisms
The proposed adsorption mechanism of MB onto AHC were presented in Fig. 9. MB was a cationic phenothiazine dye. According to the FT-IR results in Section 3.1.4, the surface of AHC contained –COOH, –OH, –CO and PO functional groups. When the pH of the aqueous solution was higher than the pHpzc of AHC, the hydroxyl or carboxyl groups were readily dehydrogenated and converted to hydroxyl or carboxyl anions, which could interact with MB cations. Moreover, the existence of π–π conjugation structure in the –CO and –OP groups made AHC generate great adsorption driving force towards MB. In Section 3.6, it has been stated that the adsorption capacity of AHC was promoted with an increment of pH. Theoretically, the increase in pH only affected the electrostatic interaction between the quaternary ammonium cations in MB and carboxyl anions in AHC, and hydrogen bonding between N atoms on the phenothiazine of MB and hydroxyl anions in AHC. But the other adsorption mechanisms, such as micropore filling, were unaffected. At the same time, the analysis of adsorption kinetics in Section 3.4 showed that the adsorption of MB by AHC fitted pseudo-second-order kinetic model, which revealed that chemisorption was the absolute predominant mechanism, namely the electrostatic attraction and hydrogen bonding were the dominant interaction mechanisms. Analysis of the adsorption isotherms (Section 3.3) demonstrated MB adsorption onto AHC followed a homogenous monolayer adsorption. The IPD model, which described the mass transfer mechanisms, revealed MB molecules diffused throughout the surface into the internal surface and filled pores of the AHC. Therefore, the entire adsorption process towards MB by AHC proceeded mostly by electrostatic attraction and hydrogen bonding supplemented with π–π interactions and intra-particle diffusion.
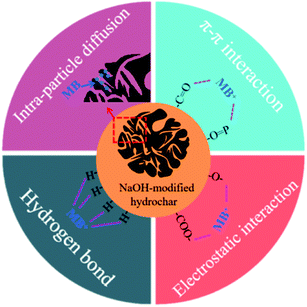 |
| | Fig. 9 Proposed mechanism for MB removal. | |
3.9. Regeneration of AHC
Regeneration experiments were conducted through adsorption–desorption assays to evaluate the reusability of AHC for MB adsorption from aqueous solution. In the desorption experiments, the MB-loaded AHC was eluted with 0.1 M HCl solution, filtered by rinsing with 0.1 M NaOH and deionized water, and then dried. Afterwards, the regenerated AHC was used in successive adsorption–desorption cycles. Fig. 10 presented that the regenerated AHC still maintained a high MB adsorption capacity by about 247.58 mg g−1 even after five cycles, which proved the reusability of AHC for MB adsorption.
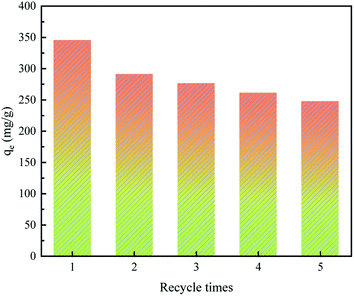 |
| | Fig. 10 Effects of number of regeneration cycles of AHC on MB adsorption performance (C0 = 300 mg L−1, m = 40 mg, V = 50 mL, pH = 7, T = 303 K, t = 24 h). | |
4. Conclusion
AHC from sugarcane bagasse via HTC in phosphoric acid medium followed by NaOH activation was satisfactorily applied to the adsorption of MB in aqueous solution. The characterizations confirmed that AHC prepared in the presence of phosphoric acid had high porosity and surface area, and some acidic functional groups were introduced into its surface. Furthermore, NaOH treatment enhanced the porosity structure and increased the oxygen-containing functional groups on the surface of the AHC. These two characteristics led to the high adsorption capacity of AHC for MB. The excellent adsorption performance of AHC for MB was mainly attributed to the electrostatic attraction, hydrogen bonding, π–π interaction and intra-particle diffusion arising from the functional groups and porous structure in AHC. Therefore, hydrochars prepared by HTC catalyzed by phosphoric acid followed by NaOH modification can be applied as high-efficiency adsorbents for the removal of cationic pollutants from wastewater.
Author contributions
Feng Zhou: investigation, data curation, formal analysis, writing-original draft, writing review & editing, visualization. Fangxue Hang: supervision, conceptualization, methodology, visualization, writing-review & editing. Zhiming Zhang: writing-original draft, writing review & editing, visualization. Peng Chen: conceptualization, methodology, visualization, writing-review & editing. Wei Lin: formal analysis, validation. Caifeng Xie: visualization, supervision, project administration, funding acquisition, writing-review & editing. Kai Li: supervision, conceptualization, methodology, formal analysis.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant number: 32160570), the Research Programs of the Science and Technology of Guangxi Zhuang Autonomous (Grant numbers: AA17204092, AB1795069 and AB16380011), and the Research Program of Agriculture Research System of China (CSRS-170502).
References
- V. Katheresan, J. Kansedo and S. Y. Lau, J. Environ. Chem. Eng., 2018, 6, 4676–4697 CrossRef CAS.
- A. Haji and M. Naebe, J. Cleaner Prod., 2020, 265, 121866 CrossRef CAS.
- Z. Li, H. Hanafy, L. Zhang, L. Sellaoui, M. S. Netto, M. L. S. Oliveira, M. K. Seliem, G. L. Dotto and A. Bonilla-Petriciolet, Chem. Eng. J., 2020, 388, 124263 CrossRef CAS.
- S. Iqbal, C. Zahoor, S. Musaddiq, M. Hussain, R. Begum, A. Irfan, M. Azam and Z. H. Farooqi, Ecotoxicol. Environ. Saf., 2020, 202, 110924 CrossRef CAS PubMed.
- H. Nawaz, M. Umar, A. Ullah, H. Razzaq, K. M. Zia and X. Q. Liu, J. Hazard. Mater., 2021, 403, 123587 CrossRef CAS PubMed.
- R. F. Wang, L. G. Deng, K. Li, X. J. Fan, W. Li and H. Q. Lu, Ceram. Int., 2020, 46, 27484–27492 CrossRef CAS.
- B. L. Alderete, J. da Silva, R. Godoi, F. R. da Silva, S. R. Taffarel and L. P. da Silva, Chemosphere, 2021, 263, 128291 CrossRef CAS PubMed.
- J. E. Gaayda, F. E. Titchou, R. Oukhrib, P. S. Yap, T. Q. Liu and M. Hamdani, J. Environ. Chem. Eng., 2021, 9, 106060 CrossRef.
- E. Santoso, R. Ediati, Y. Kusumawati, H. Bahruji, D. O. Sulistiono and D. Prasetyoko, Mater. Today Chem., 2020, 16, 100233 CrossRef CAS.
- M. M. Hassan and C. M. Carr, Chemosphere, 2021, 265, 129087 CrossRef CAS PubMed.
- B. Li, J. Q. Lv, J. Z. Guo, S. Y. Fu, M. Guo and P. Yang, Bioresour. Technol., 2019, 275, 360–367 CrossRef CAS PubMed.
- S. J. Zhou, L. J. Xia, Z. Fu, C. H. Zhang, X. Y. Duan and S. Y. Zhang, Carbohydr. Polym., 2021, 258, 117690 CrossRef CAS PubMed.
- S. Mishra, L. Cheng and A. Maiti, J. Environ. Chem. Eng., 2021, 9, 104901 CrossRef CAS.
- N. Khan, S. Mohan and P. Dinesha, J. Cleaner Prod., 2021, 288, 125629 CrossRef CAS.
- Z. Zhang, J. Yang, J. Qian, Y. Zhao, T. Wang and Y. Zhai, Bioresour. Technol., 2021, 324, 124686 CrossRef CAS PubMed.
- W. C. Qian, X. P. Luo, X. Wang, M. Guo and B. Li, Ecotoxicol. Environ. Saf., 2018, 157, 300–306 CrossRef CAS PubMed.
- X. Yu, S. Liu, G. Lin, Y. Yang, S. Zhang, H. Zhao, C. Zheng and X. Gao, Colloids Surf., A, 2020, 588, 124372 CrossRef CAS.
- F. L. Braghiroli, V. Fierro, J. Parmentier, L. Vidal, P. Gadonneix and A. Celzard, Ind. Crops Prod., 2015, 77, 364–374 CrossRef CAS.
- N. Zhou, H. Chen, J. Xi, D. Yao, Z. Zhou, Y. Tian and X. Lu, Bioresour. Technol., 2017, 232, 204–210 CrossRef CAS PubMed.
- M. T. Reza, E. Rottler, L. Herklotz and B. Wirth, Bioresour. Technol., 2015, 182, 336–344 CrossRef CAS PubMed.
- N. Zhou, H. G. Chen, Q. J. Feng, D. H. Yao, H. L. Chen, H. Y. Wang, Z. Zhou, H. Y. Li, Y. Tian and X. Y. Lu, J. Cleaner Prod., 2017, 165, 221–230 CrossRef CAS.
- Y. Liu, S. Ma and J. Chen, J. Cleaner Prod., 2018, 176, 187–195 CrossRef CAS.
- J. T. Petrović, M. D. Stojanović, J. V. Milojković, M. S. Petrović, T. D. Šoštarić, M. D. Laušević and M. L. Mihajlović, J. Environ. Manage., 2016, 182, 292–300 CrossRef PubMed.
- R. Ferrentino, R. Ceccato, V. Marchetti, G. Andreottola and L. Fiori, Appl. Sci., 2020, 10, 3445 CrossRef CAS.
- K. Sun, J. Tang, Y. Gong and H. Zhang, Environ. Sci. Pollut. Res., 2015, 22, 16640–16651 CrossRef CAS PubMed.
- A. J. Romero-Anaya, M. Ouzzine, M. A. Lillo-Ródenas and A. Linares-Solano, Carbon, 2014, 68, 296–307 CrossRef CAS.
- P. Buapeth, W. Watcharin, D. Dechtrirat and L. Chuenchom, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 515, 012003 CAS.
- R. Z. Khoo, W. S. Chow and H. Ismail, Cellulose, 2018, 25, 4303–4330 CrossRef CAS.
- T. H. Tran, A. H. Le, T. H. Pham, D. T. Nguyen, S. W. Chang and W. J. Chung, Sci. Total Environ., 2020, 725, 138325 CrossRef CAS PubMed.
- F. Liu and M. Guo, J. Mater. Sci., 2015, 50, 1624–1631 CrossRef CAS.
- Y. O. Donar, E. Çağlar and A. Sınağ, Fuel, 2016, 183, 366–372 CrossRef CAS.
- Y. Lin, P. Munroe, S. Joseph, R. Henderson and A. Ziolkowski, Chemosphere, 2012, 87, 151–157 CrossRef CAS PubMed.
- X. J. Zhang, L. Zhang and A. M. Li, J. Environ. Manage., 2018, 206, 989–998 CrossRef CAS PubMed.
- Y. Q. Lei, H. Q. Su and R. K. Tian, RSC Adv., 2016, 6, 107829–107835 RSC.
- X. M. Liu, C. Ma, F. Wu and B. Zhao, Environ. Sci. Technol., 2018, 41, 226–232 Search PubMed.
- F. M. Jais, C. Y. Chee, Z. Ismail and S. Ibrahim, J. Environ. Chem. Eng., 2021, 9, 104829 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso and J. Rouquerol, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- T. Zhang, X. Wu, X. Fan, D. C. W. Tsang, G. Li and Y. Shen, J. Environ. Manage., 2019, 236, 108–117 CrossRef CAS PubMed.
- T. Zhang, X. Wu, S. M. Shaheen, Q. Zhao, X. J. Liu and J. Rinklebe, Chem. Eng. J., 2020, 379, 122254 CrossRef CAS.
- A. C. Martins, O. Pezoti, A. L. Cazetta, K. C. Bedin, D. A. S. Yamazaki, G. F. G. Bandoch, T. Asefa, J. V. Visentainer and V. C. Almeida, Chem. Eng. J., 2015, 260, 291–299 CrossRef CAS.
- S. Masoumi and A. K. Dalai, J. Cleaner Prod., 2020, 263, 121427 CrossRef CAS.
- W. Yan, H. Zhang, K. Sheng, A. M. Mustafa and Y. Yu, Bioresour. Technol., 2017, 250, 806–811 CrossRef PubMed.
- L. Trakal, R. Šigut, H. Šillerová, D. Faturíková and M. Komárek, Arabian J. Chem., 2014, 7, 43–52 CrossRef CAS.
- G. Prasannamedha, P. S. Kumar, R. Mehala, T. J. Sharumitha and D. Surendhar, J. Hazard. Mater., 2021, 407, 124825 CrossRef CAS PubMed.
- N. V. Sych, S. I. Trofymenko, O. I. Poddubnaya, M. M. Tsyba, V. I. Sapsay and D. O. Klymchuk, Appl. Surf. Sci., 2012, 261, 75–82 CrossRef CAS.
- B. Li, J. Z. Guo, K. L. Lv and J. J. Fan, Environ. Pollut., 2019, 254, 113014 CrossRef CAS PubMed.
- B. W. Lv, H. Xu, J. Z. Guo, L. Q. Bai and B. Li, J. Hazard. Mater., 2022, 421, 126741 CrossRef CAS PubMed.
- H. Z. Li, Y. N. Zhang, J. Z. Guo, J. Q. Lv, W. W. Huan and B. Li, Bioresour. Technol., 2021, 337, 125442 CrossRef CAS PubMed.
- M. Ayiania, M. Smit, A. J. R. Hensley, L. Scudiero, J. S. McEwen and M. Garcia-Perez, Carbon, 2020, 162, 528–544 CrossRef CAS.
- Y. Xu, Z. Lou, P. Yi, J. Chen, X. Ma and Y. Wang, Bioresour. Technol., 2014, 172, 212–218 CrossRef CAS PubMed.
- Y. Xia, T. Yang, N. Zhu, D. Li, Z. Chen and Q. Lang, Bioresour. Technol., 2019, 288, 121593 CrossRef CAS PubMed.
- S. Madduri, I. Elsayed and E. B. Hassan, Chemosphere, 2020, 260, 127683 CrossRef CAS PubMed.
- J. Fang, B. Gao, A. Mosa and L. Zhan, Chem. Speciation Bioavailability, 2017, 29, 197–204 CrossRef CAS.
- H. Guo, C. Bi, C. Zeng, W. Ma, L. Yan and K. Li, J. Mol. Liq., 2018, 249, 629–636 CrossRef CAS.
- M. A. Islam, M. J. Ahmed, W. A. Khanday, M. Asif and B. H. Hameed, J. Environ. Manage., 2017, 203, 237–244 CrossRef CAS PubMed.
- F. Li, A. R. Zimmerman, X. Hu, Z. Yu, J. Huang and B. Gao, Chemosphere, 2020, 254, 126866 CrossRef CAS PubMed.
- D. H. Nguyen, H. N. Tran, H. P. Chao and C. C. Lin, Adsorpt. Sci. Technol., 2019, 37, 607–622 CrossRef CAS.
- X. M. Jian, X. Z. Zhuang, B. S. Li, X. W. Xu, Z. B. Wei, Y. P. Song and E. Jiang, Environ. Technol. Innovation, 2018, 10, 27–35 CrossRef.
- L. Y. Guo, H. Q. Lu, D. Rackemann, C. R. Shi, W. Li and K. Li K, Chem. Eng. J., 2021, 416, 129084 CrossRef CAS.
- Y. Shen, W. X. Ni and B. Li, ACS Omega, 2021, 6, 3202–3208 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc,
Zhiming Zhanga,
Peng Chena,
Lin Weia and
Caifeng Xie
abc,
Zhiming Zhanga,
Peng Chena,
Lin Weia and
Caifeng Xie