
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-assisted preparation of pyridinyl sulfonate esters from hydroxypyridines and sodium sulfinates†
Qian Lia,
Haibo Zhu *ab,
Yishuai Liua,
Liu Yanga,
Qiangwen Fan*ac,
Zongbo Xie*a and
Zhang-Gao Lea
*ab,
Yishuai Liua,
Liu Yanga,
Qiangwen Fan*ac,
Zongbo Xie*a and
Zhang-Gao Lea
aJiangxi Province Key Laboratory of Synthetic Chemistry, School of Chemistry, Biology and Material Science, East China University of Technology, 330013, Nanchang, China. E-mail: hbzhu@ecut.edu.cn; fanqw2019@ecut.edu.cn; zbxie@ecut.edu.cn
bJiangxi Province Key Laboratory of Polymer Micro/Nano Manufacturing and Devices, East China University of Technology, Nanchang, 330013, China
cJiangxi Key Laboratory for Mass Spectrometry and Instrumentation, East China University of Technology, Nanchang, 330013, China
First published on 20th January 2022
Abstract
An efficient and powerful copper-assisted method for the effective conversion of a broad range of hydroxypyridines and sodium sulfinates into their corresponding pyridinyl tosylates was developed. Key features of this base- and ligand-free protocol include using the cheap and readily available CuBr2 as a medium and the use of sodium sulfinates as formal sulfonylation reagents. A variety of functional pyridinyl tosylates could be formed with good yields, which can easily be converted into C–C and C–N bond-containing compounds.
Sulfonate esters are well-known ester compounds and important pharmaceutical ingredients (Fig. 1).1–3 As a bridge structure or ligand, they have special biological activities in medicinal chemistry, such as undesired thrombosis prevention and monoamine oxidase inhibitory activity.4–6 In addition, sulfonate ester groups play a unique role in coupling reactions due to their easy leaving ability.7–11 Traditional methods for the synthesis of sulfonate esters have usually focused on the esterification reaction of phenols or alcohols with sulfonyl chlorides, which requires the preparation of unstable and strongly corrosive sulfonyl chlorides in a low temperature system (down to −20 °C).12,13 Over the last decade, many chemists have become interested in direct sulfonylation reactions using various sulfonylation reagents,14,15 including sulfinic acids,16,17 sulfinates18,19 and sulfonyl hydrazides.20–22 Among them, the stable and readily available sodium sulfinate seems to be more attractive because it has demonstrated flexible reactivity, such as being nucleophilic, electrophilic, and a radical reagent by providing suitable reaction conditions.23–26 Therefore, it is highly desirable to design and develop a suitable low-cost and green approach to construct sulfonate ester building blocks.
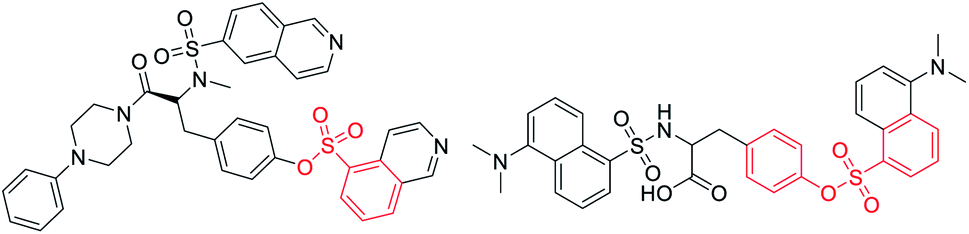 | ||
| Fig. 1 Representative biologically active sulfonate ester scaffolds. | ||
In 2016, Chiranjeevi and co-authors developed an efficient practical tosylation of phenols, employing the mild reagent [DMAPTs]+Cl− under base-free and chromatography-free conditions (Scheme 1a).27 Notably, Gao's group reported the synthesis of sulfonate esters induced by iodine through the cross-coupling reaction of sodium sulfinates and phenols (Scheme 1b).28 Compared with the reported methods using sulfonyl chlorides as starting materials, the present method appears to be more general and efficient for the synthesis of sulfonate esters. Electro-oxidation29,30 and transition-metal-catalyzed insertion of sulfur dioxide31–36 were also alternatives to generate arylsulfonate esters. An electro-oxidative sulfonylation of phenols with sodium arenesulfinates under mild reaction conditions was developed. This protocol showed a wide substrate scope and high functional group tolerance without external stoichiometric oxidants (Scheme 1c).37 Although great progress has been made in the preparation of sulfonate esters, the relevant studies are still in their infancy and a high efficiency protocol for nitrogen-containing heterocycle sulfonate esters is far from fully developed.
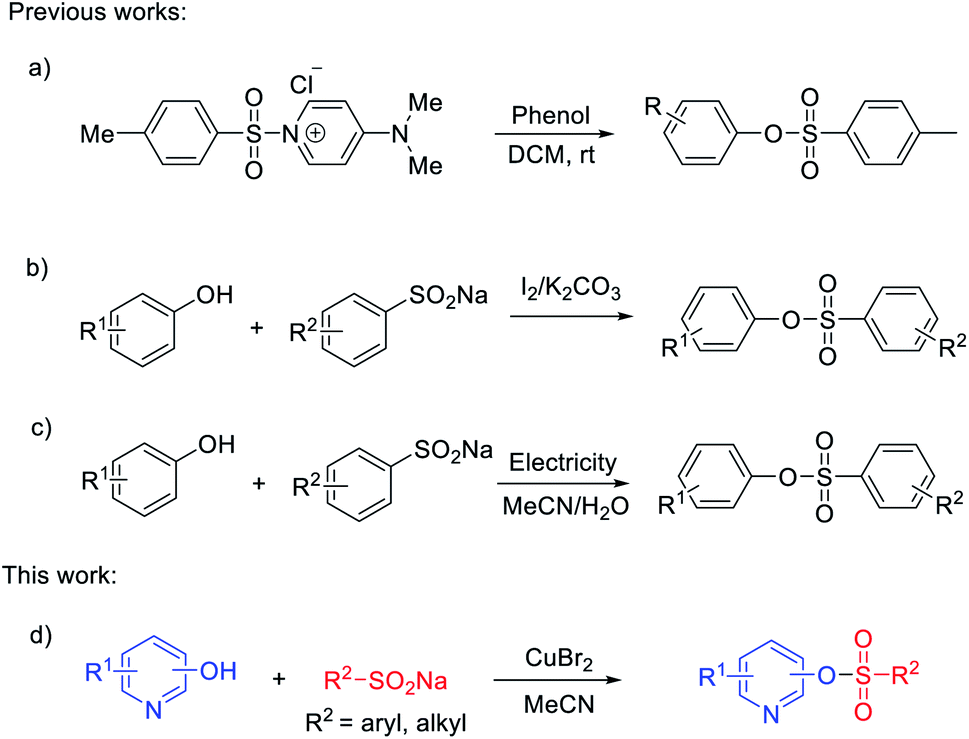 | ||
| Scheme 1 Methods for the synthesis of (hetero)arylsulfonate esters. | ||
On account of our strong interest in researching sulfonyl-containing compounds,19,38–41 herein, we report a facile, cost-effective and efficient approach using CuBr2 as a catalytic medium for the cross-coupling reaction of sodium sulfinates and hydroxypyridines to generally prepare a variety of functional pyridinyl sulfonate esters with good yields. The synthetic route has a simple operation and good functional group compatibility, and can afford a series of heterocyclic sulfonate esters with medium to high yields under mild conditions. To the best of our knowledge, such a route for the synthesis of pyridinyl sulfonate esters has not been reported to date.
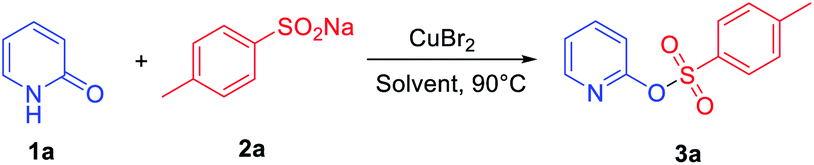
Our study was initiated by evaluating the model reaction between 2-pyridone 1a (0.2 mmol) and sodium sulfinate 2a (1.0 equiv.) with CuBr2 as the additive. After a 24 h reaction at 90 °C in acetonitrile (MeCN), the desired product 3a can be isolated in 24% yield (Table 1, entry 1). Various transition metal catalysts, including copper, nickel, iron salts, and especially palladium catalysts, were then investigated, and CuBr2 was the best additive for this transformation (see ESI for details, Table S1†). Further solvent screening indicated that MeCN is the best solvent; other solvents including polar and non-polar solvents, such as (dimethyl sulfoxide) DMSO, 1,4-dioxane, toluene, MeOH, dichloromethane (DCM) and dichloroethane (DCE) gave lower yields (Table 1, entries 2–7). When 1.5 equiv. of 2a was added in this transformation, 36% isolated yield of 3a was observed (Table 1, entry 8). Interestingly, the product yield was increased to 42% when the reaction time was shortened to 6 h (Table 1, entry 9). Besides, raising or lowering the reaction temperature cannot further promote the conversion of the reaction (Table S3,† entries 8 and 9). Generally, nitrogen-containing ligands play a vital role in copper-catalyzed coupling reactions, which could stabilize the active species of the catalyst in the catalytic cycle. However, bipyridine and o-phenanthroline ligands were also screened (Table S2, see ESI† for details), and the yield of the desired product 3a was not further improved. Further exploration of cupric dibromide revealed that the reaction worked efficiently in the presence of 1.0 equiv. of copper(II) bromide, producing the corresponding product 3a in 78% yield (Table 1, entry 11). Therefore, the optimized reaction conditions were as follows: 0.2 mmol of 1a and 1.5 equiv. of 2a were dissolved in MeCN under air atmosphere at 90 °C in the presence of CuBr2 (1 equiv.).
|
||||
|---|---|---|---|---|
| Entry | CuBr2 (equiv.) | Solvent | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (1.0 equiv.) in 2 mL solvent at 90 °C under air atmosphere.b Isolated yield.c 1a (0.2 mmol), 2a (1.5 equiv.). | ||||
| 1 | 0.2 | MeCN | 24 | 24 |
| 2 | 0.2 | DMSO | 24 | NR |
| 3 | 0.2 | Dioxane | 24 | Trace |
| 4 | 0.2 | Toluene | 24 | Trace |
| 5 | 0.2 | DCE | 24 | 12 |
| 6 | 0.2 | DCM | 24 | 12 |
| 7 | 0.2 | MeOH | 24 | Trace |
| 8c | 0.2 | MeCN | 24 | 36 |
| 9c | 0.2 | MeCN | 6 | 42 |
| 10c | 0.5 | MeCN | 6 | 48 |
| 11c | 1.0 | MeCN | 6 | 78 |
With the optimized reaction conditions in hand, the copper-assisted preparation of pyridinyl sulfonate esters from hydroxypyridines and sodium sulfinates were then explored, and the results are summarized in Tables 2 and 3. Various sodium sulfinates were tolerated; both electron-donating (–Me, –OMe and –tBu) and electron-withdrawing groups (–CF3, –F, –Cl and –Br) on the phenyl ring afforded the corresponding products in medium to good yields (Table 2). The position of the substituent also affected the reaction. Sodium sulfinates that bear electron-donating groups (as –Me) in the para-position of the phenyl ring were superior to their ortho- and meta-substituted counterparts in isolated yields (Table 2, 3a–3c). Remarkably, sodium naphthalene-2-sulfinate and sodium naphthalene-1-sulfinate also proved to be suitable substrates, providing products 3k and 3l in moderate yields. Apart from aromatic sodium sulfinates, less-reactive aliphatic sodium sulfinates, such as sodium methanesulfinate, were tolerated in this transformation, affording products 3m–3o with moderate to good yields.
| a Reaction conditions: 1a (0.2 mmol), 2a (1.5 equiv.), CuBr2 (1.0 equiv.) in 2 mL solvent at 90 °C under air atmosphere. |
|---|
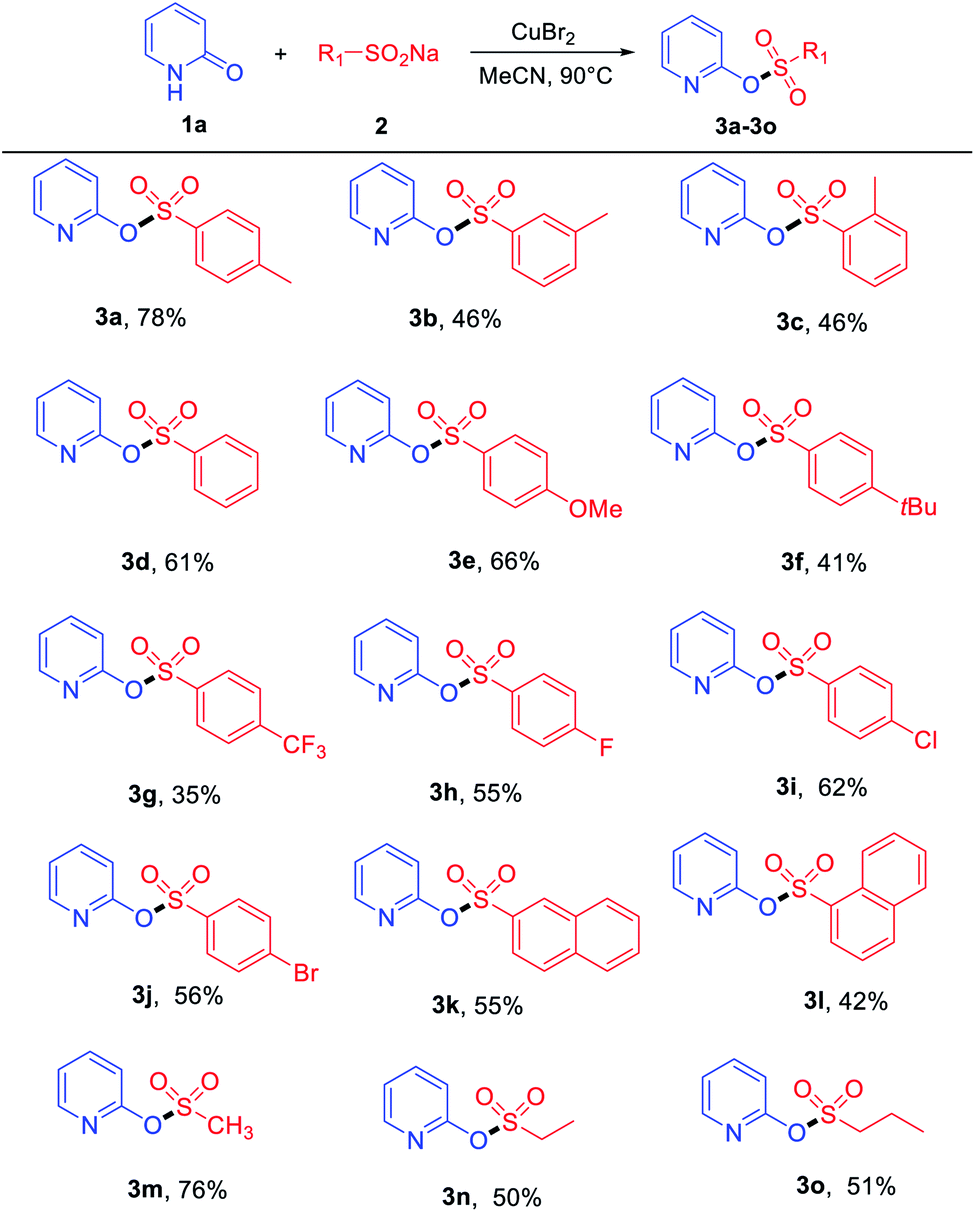 |
| a Reaction conditions: 1a (0.2 mmol), 2a (1.5 equiv.), CuBr2 (1.0 equiv.) in 2 mL solvent at 90 °C under air atmosphere. |
|---|
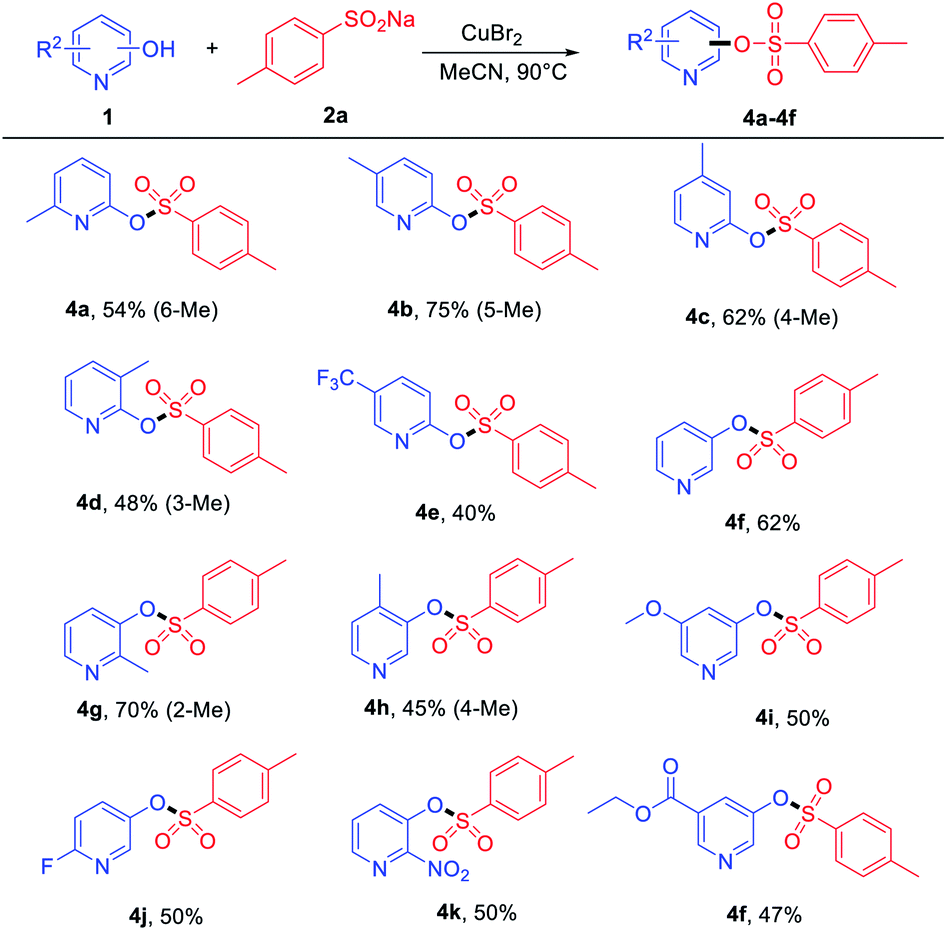 |
On the other hand, the reactions of some pyridinols with 2a and pyridin-3-ol were examined, and the results are summarized in Table 3. A series of ortho- and meta-substituted pyridinols by electron-donating (–Me and –OMe) and electron-withdrawing groups (–F, –CF3, and –NO2) all proceeded smoothly to afford the corresponding products (Table 3, 4a–4k) in medium to good yields. When some electron-donating group-substituted (as –Me) pyridinols were used, the 4-methyl and 5-methyl of the phenyl ring were better than their 3-methyl and 6-methyl substituted counterparts in isolated yields (Table 3, 4a–4d). This result may be attributed to the fact that the influence of steric hindrance is not conducive to the progress of the reaction. To our delight, the electron-withdrawing group (–NO2) performed with good yields (4k). In addition, ethyl 5-(tosyloxy) nicotinate (4f) was also obtained in a medium yield.
To further prove the practicability of this new synthetic method, a gram-scale reaction was performed. A 45% yield of 4j (1.02 g) was formed with 8 mmol 2-fluoro-5-hydroxypyridine and 12 mmol of 2a under the optimal conditions, which further demonstrate the practical synthetic value of the current methodology (Scheme 2a). In addition, further transformations of 4j were performed to demonstrate the potential application of the products in the construction of C–C and C–N bonds. Room-temperature Suzuki–Miyaura coupling of heteroaryl tosylates 4j with phenylboronic acid could afford 5b with 85% yield (Scheme 2b).42 Amination of 4j furnished heterocyclic amine 5a in 90% yield in the presence of robust acenaphthoimidazolylidene palladium complex (Scheme 2c).43
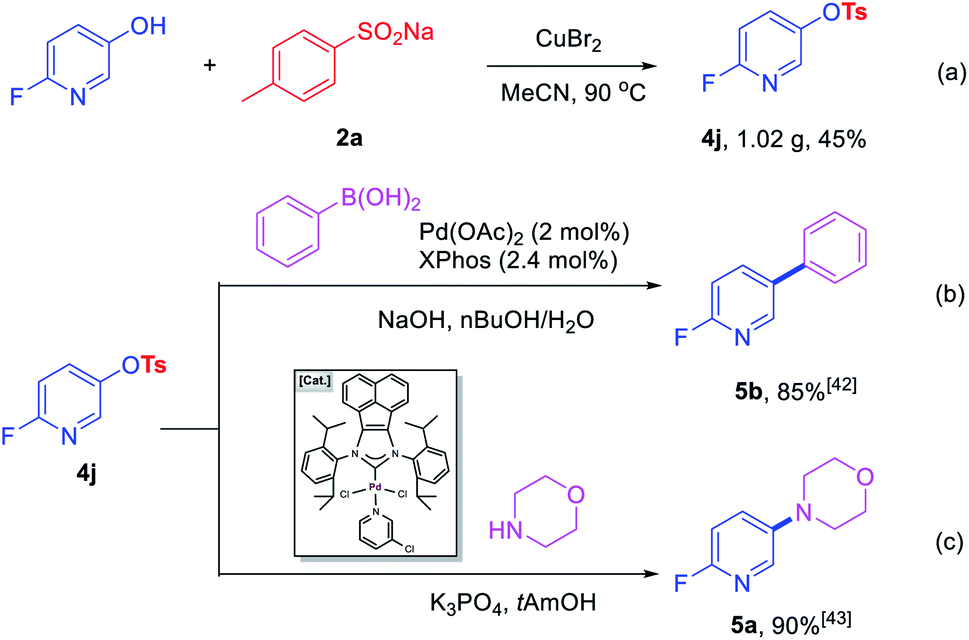 | ||
| Scheme 2 Gram-scale synthesis and further conversion of 4j. | ||
To investigate the possible reaction mechanism, radical trapping experiments were performed. No sulfonate ester product was detected in the presence of the radical scavengers TEMPO or BHT, which indicated that a radical pathway should be involved.44,45 According to the above results, a possible mechanism is proposed in Scheme 3. Initially, a sulfonyl intermediate A is generated by the coordination of copper to the sodium sulfinate,46,47 which decompose into the sulfonyl free radical intermediate and CuBr. The coordination of hydroxypyridine with copper occurs to generate intermediate B, which combines with the sulfonyl free radical to give the sulfonate ester with release of CuBr. Finally, CuBr can be oxidized by O2 to generate Cu(II) species.48
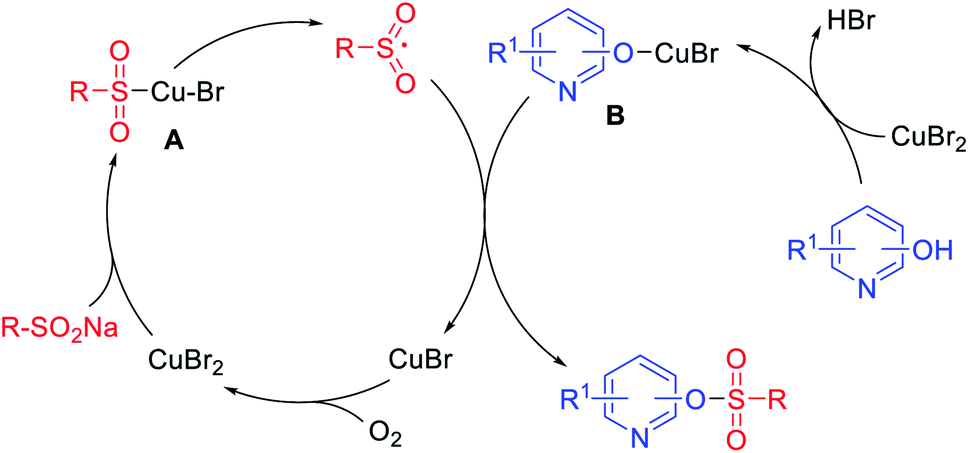 | ||
| Scheme 3 Possible mechanism. | ||
Conclusions
In conclusion, CuBr2 has been demonstrated as a highly efficient promoter in the direct hydroxylsulfonylation of readily available pyridinols with functional (hetero)aryl- or alkyl-sodium sulfinates. Remarkably, this protocol presented a direct, mild and efficient approach with good functional group tolerance to access diverse pyridinyl sulfonate esters. Further study on this reaction's application is currently ongoing in our laboratory.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21801040, 22102022), the Natural Science Foundation of Jiangxi Province (20192BAB213006, 20181BBH80007), the Opening Project of Jiangxi Province Key Laboratory of Polymer Micro/Nano Manufacturing and Devices (PMND202001), the Open fund of Jiangxi Key Laboratory for Mass Spectrometry and Instrumentation (No. JXMS202106), East China University of Technology Research Foundation for Advanced Talents (No. DHBK2017168) and School of Chemistry, Biology and Material Science at East China University of Technology is gratefully acknowledged.Notes and references
- L. M. Betts, N. C. Tam, S. M. H. Kabir, R. F. Langler and I. Crandall, Aust. J. Chem., 2006, 59, 277–282 CrossRef CAS.
- S. Fortin, L. Wei, E. Moreau, J. Lacroix, M. F. Cote, E. Petitclerc, L. P. Kotra and R. C.-Gaudreault, J. Med. Chem., 2011, 54, 4559–4580 CrossRef CAS PubMed.
- N. Jalalian, T. B. Petersen and B. Olofsson, Chem, 2012, 18, 14140–14149 CrossRef CAS PubMed.
- J. H. Park, G. E. Lee, S. D. Lee, T. T. Hien, S. Kim, J. W. Yang, J. H. Cho, H. Ko, S. C. Lim, Y. G. Kim, K. W. Kang and Y. C. Kim, J. Med. Chem., 2015, 58, 2114–2134 CrossRef CAS PubMed.
- D. Tondi, A. Venturelli, S. Ferrari, S. Ghelli and M. P. Costi, J. Med. Chem., 2005, 48, 913–916 CrossRef CAS PubMed.
- H.-B. Zhou, J. S. Comninos, F. Stossi, B. S. Katzenellenbogen and J. A. Katzenellenbogen, J. Med. Chem., 2005, 48, 7261–7274 CrossRef CAS PubMed.
- C.-H. Cho, H.-S. Yun and K. Park, J. Org. Chem., 2003, 68, 3017–3025 CrossRef CAS PubMed.
- Y.-Y. Gui, L.-L. Liao, L. Sun, Z. Zhang, J.-H. Ye, G. Shen, Z.-P. Lu, W.-J. Zhou and D.-G. Yu, Chem. Commun., 2017, 53, 1192–1195 RSC.
- Y. Zhang, G. Lavigne and V. César, J. Org. Chem., 2015, 80, 7666–7673 CrossRef CAS PubMed.
- T. Ogata and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 13848–13849 CrossRef CAS PubMed.
- H. N. Nguyen, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 11818–11819 CrossRef CAS PubMed.
- G. Song, Y. Cai and Y. Peng, J. Comb. Chem., 2005, 7, 561–566 CrossRef CAS PubMed.
- L. J. Gooßen, N. Rodríguez, P. P. Lange and C. Linder, Angew. Chem., Int. Ed., 2010, 49, 1111–1114 CrossRef PubMed.
- D. Joseph, M. A. Idris, J. Chen and S. Lee, ACS Catal., 2021, 11, 4169–4204 CrossRef CAS.
- V. K. K. Pampana, V. P. Charpe, A. Sagadevan, D. K. Das, C.-C. Lin, J. R. Hwu and K. C. Hwang, Green Chem., 2021, 23, 3569–3574 RSC.
- J. Wen, W. Shi, F. Zhang, D. Liu, S. Tang, H. Wang, X.-M. Lin and A. Lei, Org. Lett., 2017, 19, 3131–3134 CrossRef CAS PubMed.
- G. Zhang, L. Zhang, H. Yi, Y. Luo, X. Qi, C.-H. Tung, L.-Z. Wu and A. Lei, Chem. Commun., 2016, 52, 10407–10410 RSC.
- Y. Liu, H. Zhu, L. Yang, Z. Xie, G. Jiang, Z.-G. Le and T. Tu, Asian J. Org. Chem., 2020, 9, 247–250 CrossRef CAS.
- H. Zhu, Y. Shen, Q. Deng and T. Tu, Chem. Commun., 2015, 51, 16573–16576 RSC.
- G. Zhang, L. Zhang, H. Yi, Y. Luo, X. Qi, C.-H. Tung, L.-Z. Wu and A. Lei, Chem. Commun., 2016, 52, 10407–10410 RSC.
- H. Zhu, Y. Zhang, Y. Liu, L. Yang, Z. Xie, G. Jiang and Z.-G. Le, Tetrahedron Lett., 2020, 61, 151975 CrossRef CAS.
- B. Gong, H. Zhu, Y. Liu, Q. Li, L. Yang, G. Wu, Q. Fan, Z. Xie and Z. Le, Green Synth. Catal., 2021 DOI:10.1016/j.gresc.2021.10.002.
- W. Dong, Z.-Y. Fang, T.-Y. Cao, J.-H. Cao, Z.-Q. Zhao, L. Zhang, W. Li, L. Qi and L.-J. Wang, Org. Lett., 2021, 23, 5809–5814 CrossRef CAS PubMed.
- Z. Guo, Y. Zhao, Y. Wang, M. Xie and J. Zhang, J. Org. Chem., 2021, 86, 6247–6258 CrossRef CAS PubMed.
- Y. Wu, J. Chen, L. Li, K. Wen, X. Yao, J. Pang, T. Wu and X. Tang, Org. Lett., 2020, 22, 7164–7168 CrossRef CAS PubMed.
- Y.-S. Xiong, B. Zhang, Y. Yu, J. Weng and G. Lu, J. Org. Chem., 2019, 84, 13465–13472 CrossRef CAS PubMed.
- C. Dhonthulachitty, S. R. Kothakapu and C. K. Neella, Tetrahedron Lett., 2016, 57, 4620–4623 CrossRef CAS.
- J. Gao, X. Pan, J. Liu, J. Lai, L. Chang and G. Yuan, RSC Adv., 2015, 5, 27439–27442 RSC.
- H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed.
- Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed.
- Y. Wang, L. Deng, Y. Deng and J. Han, J. Org. Chem., 2018, 83, 4674–4680 CrossRef CAS PubMed.
- A. L. Tribby, I. Rodríguez, S. Shariffudin and N. D. Ball, J. Org. Chem., 2017, 82, 2294–2299 CrossRef CAS PubMed.
- T. S.-B. Lou, S. W. Bagley and M. C. Willis, Angew. Chem., Int. Ed., 2019, 58, 18859–18863 CrossRef CAS PubMed.
- M. Wang and X. Jiang, Chem. Rec., 2021, 21, 1–19 CrossRef.
- H. Zhang, M. Wang and X. Jiang, Green Chem., 2020, 22, 8238–8242 RSC.
- D. Zeng, M. Wang, W.-P. Deng and X. Jiang, Org. Chem. Front., 2020, 7, 3956–3966 RSC.
- Z. Tian, Q. Gong, T. Huang, L. Liu and T. Chen, J. Org. Chem., 2021, 86, 15914–15926 CrossRef CAS PubMed.
- H. Zhu, Y. Liu, Y. Zhang, L. Yang, J. Meng, Q. Li, B. Gong, Z. Xie and Z.-G. Le, Mol. Catal., 2021, 505, 111500 CrossRef CAS.
- H. Zhu, Y. Shen, Q. Deng, J. Chen and T. Tu, ACS Catal., 2017, 7, 4655–4659 CrossRef CAS.
- H. Zhu, Y. Shen, Q. Deng, J. Chen and T. Tu, Chem. Commun., 2017, 53, 12473–12476 RSC.
- H. Zhu, Y. Shen, D. Wen, Z.-G. Le and T. Tu, Org. Lett., 2019, 21, 974–979 CrossRef CAS PubMed.
- J. Yang, S. Liu, J.-F. Zheng and J. Zhou, Eur. J. Org. Chem., 2012, 2012, 6248–6259 CrossRef CAS.
- T. Tu, Z. Sun, W. Fang, M. Xu and Y. Zhou, Org. Lett., 2012, 14, 4250–4253 CrossRef CAS PubMed.
- G. O. Jones, P. Liu, K. N. Houk and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 6205–6213 CrossRef CAS PubMed.
- C. Tang and N. Jiao, J. Am. Chem. Soc., 2012, 134, 18924 CrossRef CAS PubMed.
- X. Tang, L. Huang, C. Qi, X. Wu, W. Wu and H. Jiang, Chem. Commun., 2013, 49, 6102–6104 RSC.
- X. Yu, X. Li and B. Wan, Org. Biomol. Chem., 2012, 10, 7479–7482 RSC.
- J. Chen, Y. Sun, B. Liu, D. Liu and J. Cheng, Chem. Commun., 2012, 48, 449–451 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08568a |
| This journal is © The Royal Society of Chemistry 2022 |