
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light photocatalytic one pot synthesis of Z-arylvinyl halides from E-arylvinyl acids with N-halosuccinimide†
Qiong Yua,
Kun Yi Yua,
Cai Feng Xua and
Man-Kin Wong *ab
*ab
aThe Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen, 518057, China
bState Key Laboratory of Chemical Biology and Drug Discovery, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hum, Hong Kong, China. E-mail: mankin.wong@polyu.edu.hk
First published on 31st January 2022
Abstract
An efficient visible light photocatalytic strategy to synthesize thermodynamically less stable Z-arylvinyl halides (with up to >99/1 Z/E ratio and 86% yield) was developed. The reaction combined base-mediated halodecarboxylation of E-arylvinyl acids with N-halosuccinimide and visible light Ir-photocatalyzed isomerization of E-arylvinyl halides in a one pot sequential catalytic process.
Visible light photocatalysis has received considerable attention in recent years owing to the mild reaction conditions, green and sustainable chemistry features, and high atom-economy.1 As reported in the literature, two different activation modes are commonly used. Most of the photochemical reactions proceed through a single-electron transfer (SET) process from the excited photosensitizers to the organic substrates or reagents. The other activation mode is an energy transfer (EnT) process in which no charge separation is involved in the whole process. This EnT activation pathway mainly depends on the triplet-state energies of the photosensitizers and the organic substrates. Synthetically useful visible light-induced organic transformation reactions through the EnT process have been successfully developed in the past decades.1n,2
The synthesis of multisubstituted alkenes is an important reaction because of their versatile utility as synthetic building blocks for organic synthesis and as structural elements contributing to the significant biological properties of natural products and pharmaceuticals.3 Unlike E-alkenes, the strategies for synthesis of thermodynamically less stable Z-alkenes are not readily accessible.4 Hammond and Arai developed pioneering photochemical E → Z isomerisations of stilbenes and styrenes and delineated the reaction mechanisms.5 Inspired by these mechanistic studies, visible light induced E → Z isomerization has attracted great interest. In 2014, Weaver and co-workers reported Ir(ppy)3 catalyzed E to Z isomerization of allylamines proceeding via an EnT mechanism.6 In 2015, the Gilmour group developed photoisomerization of activated alkenes using (−)-riboflavin as an EnT photocatalyst.7 Furthermore, Gilmour and co-workers have reported photoisomerization of styrenyl boron species and selective isomerization of β-borylacrylates.8 In 2020, the same group reported a synthetic procedure for E → Z isomerization of β-borylacrylates via EnT using thioxanthone as the sensitizer eliminating the need of an aryl unit for alkene isomerization, and the inert aryl rings were replaced by a traceless BPin handle.9
Arylvinyl halides are versatile synthetic intermediates for organic synthesis. In particular, transition metal-catalyzed cross couplings of vinyl halides with organometallics, such as, organo boronic and organozinc reagents, are efficient methods for synthesis of multisubstituted alkenes.10 However, synthesis of the thermodynamically less stable Z-isomer still poses a great challenge. In 2019, Yu's group demonstrated a synthetically useful E → Z photocatalytic isomerization of styrenyl halides (Scheme 1).11 On the other hand, decarboxylation of α,β-unsaturated arylvinyl acids accompanied by simultaneous replacement by halogen is a useful reaction for the synthesis of styrenyl halides.12 Considering the importance of Z-vinyl halides in the synthesis of multisubstituted alkenes, we hypothesize that it would be interesting to combine the halodecarboxylation of α,β-unsaturated arylvinyl acids and photoisomerization of E-arylvinylhalides in a one pot sequential catalytic process. Herein, we report a novel method to synthesize Z-arylvinyl halides by visible light Ir-photocatalyzed reaction of E-arylvinyl acids with N-halosuccinimide.
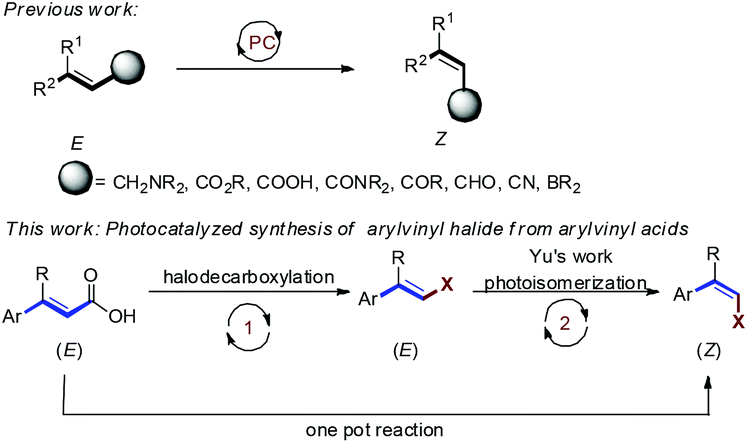 | ||
| Scheme 1 Photocatalytic synthesis of multisubstituted alkenes. | ||
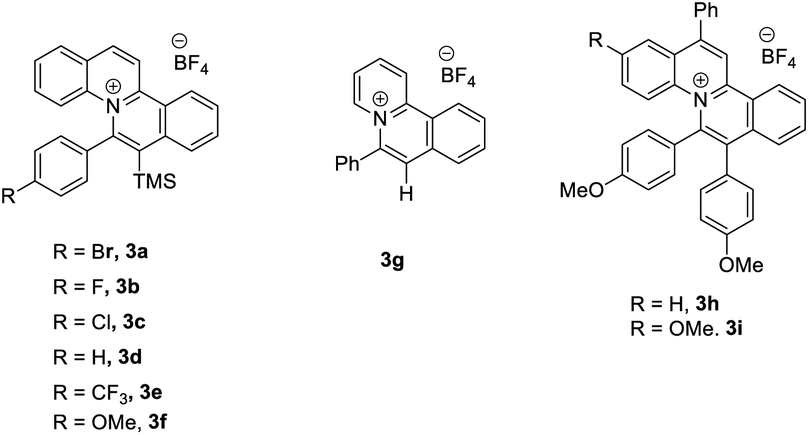
Our group has synthesized a series of new fluorescent quinolizinium compounds from quinolines and alkyne substrates.13 Due to the high tunability and high excited state reduction potentials of the fluorescent quinolizinium compounds, we proposed that the quinolizinium compounds could be used as photocatalysts for the synthesis of Z-arylvinyl halides from α,β-unsaturated arylvinyl acids.
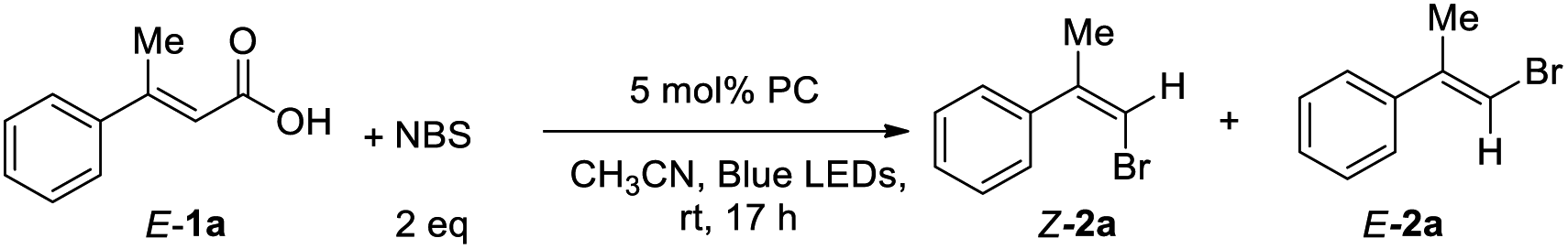
With this idea in mind, we started the initial investigation by treatment of (2E)-3-phenyl-2-butenoic acid (1a) with 2 equiv. of N-bromosuccinimide (NBS) in the presence of 5 mol% of photocatalyst 3a in CH3CN at room temperature under 30 W blue LEDs irradiation. 36% NMR yield of the desired product (Z/E)-(1-bromoprop-1-en-2-yl)benzene (Z/E-2a) was obtained with modest selectivity (Z/E = 44/56). Next, we optimized the reaction conditions by varying the additives. Adding K2CO3 and TBAI, E-2a was obtained only (entries 2–3, Table 1). Interestingly, adding 1.0 equiv. of acetic acid as the additive, improvement in the Z/E ratio of 2a (56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :44) was observed (entry 4, Table 1). Then, various acids were examined in this reaction. 0.3 equiv. of benzoic acid was found to be the best additive (entry 8, Table 1). Next, studies were conducted for the photocatalysts. It was found that the reaction could proceed to afford the product 2a with modest to excellent Z/E ratios using most of the quinolizinium compounds synthesized by our group (entries 9–16, Table 1). In particular, quinolizinium 3f was found to be the best photocatalyst giving product (Z)-2a (Z/E = 94:6) in 33% yield (entry 13, Table 1). With these reaction conditions, we tried to expand the scope of this reaction with different E-arylvinylcarboxylic acids. However, no desired product was observed regardless of the substituent on the aryl ring, such as, methyl, electron-donating group (OMe) or electron-withdrawing group (CF3).
:44) was observed (entry 4, Table 1). Then, various acids were examined in this reaction. 0.3 equiv. of benzoic acid was found to be the best additive (entry 8, Table 1). Next, studies were conducted for the photocatalysts. It was found that the reaction could proceed to afford the product 2a with modest to excellent Z/E ratios using most of the quinolizinium compounds synthesized by our group (entries 9–16, Table 1). In particular, quinolizinium 3f was found to be the best photocatalyst giving product (Z)-2a (Z/E = 94:6) in 33% yield (entry 13, Table 1). With these reaction conditions, we tried to expand the scope of this reaction with different E-arylvinylcarboxylic acids. However, no desired product was observed regardless of the substituent on the aryl ring, such as, methyl, electron-donating group (OMe) or electron-withdrawing group (CF3).
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst (PC) | Additive | Yieldb | Z/Ec |
| a Reaction conditions: treatment of E-1a (0.2 mmol), NBS (0.4 mmol) and photocatalyst (5 mol%) in 2 mL of CH3CN under N2 and blue LEDs light for 17 hours at room temperature.b Yield was determined by 1H NMR using dibromomethane as internal standard.c The Z/E ratio was determined by 1H NMR spectroscopy. | ||||
| 1 | 3a | — | 45 | 44/56 |
| 2 | 3a | 1 eq. K2CO3 | 88 | 0/100 |
| 3 | 3a | 1 eq. TBAI | 75 | 0/100 |
| 4 | 3a | 1 eq. CH3CO2H | 43 | 56/44 |
| 5 | 3a | 2 eq. CH3CO2H | 35 | 77/23 |
| 6 | 3a | 3 eq. CH3CO2H | 58 | 66/34 |
| 7 | 3a | 2 eq. PhCO2H | 21 | 99/1 |
| 8 | 3a | 0.3 eq. PhCO2H | 32 | 94/6 |
| 9 | 3b | 0.3 eq. PhCO2H | 29 | 86/14 |
| 10 | 3c | 0.3 eq. PhCO2H | 22 | 91/9 |
| 11 | 3d | 0.3 eq. PhCO2H | 38 | 84/16 |
| 12 | 3e | 0.3 eq. PhCO2H | 24 | 88/12 |
| 13 | 3f | 0.3 eq. PhCO2H | 33 | 94/6 |
| 14 | 3g | 0.3 eq. PhCO2H | 35 | 86/14 |
| 15 | 3h | 0.3 eq. PhCO2H | 49 | 69/31 |
| 16 | 3i | 0.3 eq. PhCO2H | 63 | 60/40 |
 |
||||
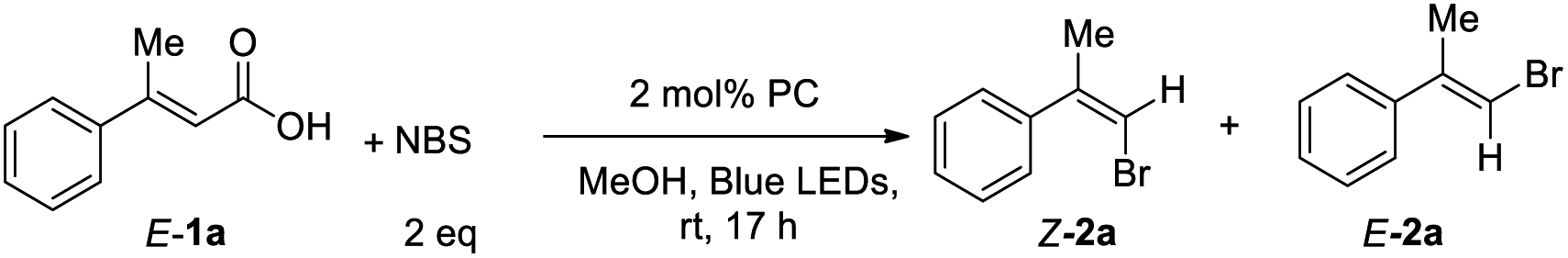
Then, we screened the reaction conditions with metal photocatalyst Ir(ppy)3. Styrenyl hailde 2a was obtained in 48% NMR yield in a Z/E ratio (52:48) (entry 1, Table 2). When the reaction conditions of 2 mol% Ir(ppy)3 with 0.3 equiv. of K2CO3 were used, the reaction proceeded to give the desired product 2a with 61:39 Z/E ratio. Increasing the base loading from 0.3 equiv. to 2 equiv. improved the NMR yield of 2a from 31% to 60% and gave a 97:3 Z/E ratio (entry 5, Table 2). When we further increased the loadings of the base, the yield dropped slightly (entries 6 and 7, Table 2). Next, various inorganic and organic bases were examined in this reaction, and α,β-unsaturated arylvinyl acid 1a was found to be successfully converted to 2a in moderate to good Z/E ratios (entries 8–13, Table 2). Several other photocatalysts were also screened, such as, Ir(Fppy)3, Ir(diFppy)3, Mes-Acr-BF4, Ru(bpy)3(PF6)2 and Ru(bpy)3Cl2, but none of them gave better yields or Z/E ratios (entries 14–18, Table 2). Only E-2a was obtained in the absence of light (entry 19, Table 2), and a trace amount of Z-2a was observed without photocatalyst (entry 20, Table 2).
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst (PC) | Additive | Yieldb | Z/Ec |
| a Reaction conditions: treatment of E-1a (0.2 mmol), NBS (0.4 mmol) and photocatalyst (2 mol%) in 2 mL of MeOH under N2 and blue LEDs light for 17 hours at room temperature.b Yield was determined by 1H NMR using dibromomethane as internal standard.c The Z/E ratio was determined by 1H NMR spectroscopy.d The reaction solvent was CH3CN.e No light irradiation. | ||||
| 1d | Ir(ppy)3 | 0.3 eq PhCO2H | 48 | 52/48 |
| 2 | Ir(ppy)3 | 0.3 eq PhCO2H | 0 | — |
| 3 | Ir(ppy)3 | — | 0 | — |
| 4 | Ir(ppy)3 | 0.3 eq. K2CO3 | 31 | 61/39 |
| 5 | Ir(ppy)3 | 2 eq. K2CO3 | 60 | 97/3 |
| 6 | Ir(ppy)3 | 2.5 eq. K2CO3 | 56 | 95/5 |
| 7 | Ir(ppy)3 | 3 eq. K2CO3 | 49 | 96/4 |
| 8 | Ir(ppy)3 | 2 eq. Na2CO3 | 57 | 95/5 |
| 9 | Ir(ppy)3 | 2 eq. Cs2CO3 | 54 | 94/6 |
| 10 | Ir(ppy)3 | 2 eq. K3PO4 | 64 | 89/11 |
| 11 | Ir(ppy)3 | 2 eq. tBuOK | 38 | 92/8 |
| 12 | Ir(ppy)3 | 2 eq. DBU | 47 | 94/6 |
| 13 | Ir(ppy)3 | 2 eq. Et3N | 9 | 67/33 |
| 14 | Ir(Fppy)3 | 2 eq. K2CO3 | 56 | 89/11 |
| 15 | Ir(diFppy)3 | 2 eq. K2CO3 | 73 | 79/21 |
| 16 | Mes-Acr-BF4 | 2 eq. K2CO3 | 30 | 10/90 |
| 17 | Ru(bpy)3(PF6)2 | 2 eq. K2CO3 | 64 | 5/95 |
| 18 | Ru(bpy)3Cl2 | 2 eq. K2CO3 | 48 | 0/100 |
| 19e | Ir(ppy)3 | 2 eq. K2CO3 | 74 | 0/100 |
| 20 | — | 2 eq. K2CO3 | 54 | 4/96 |
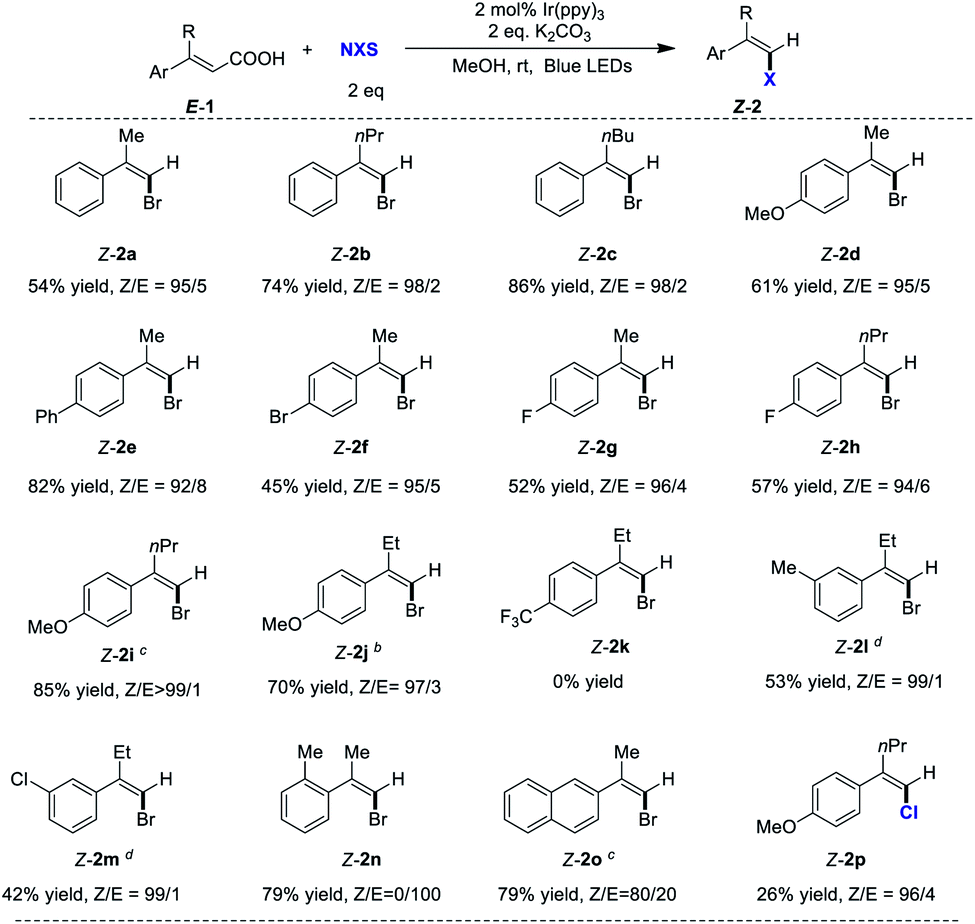
After optimizing the reaction conditions, we next sought to explore the scope of the reaction. The results summarized in Table 3 show that this reaction provides a straightforward route to a variety of Z-arylvinyl halides 2 directly from E-arylvinyl acids 1. Different alkyl substitutions at the α position (R), such as methyl, ethyl, propyl and butyl were compatible in the reaction, affording the corresponding Z-arylvinyl bromides in high stereoselectivities (up to 99/1 Z/E ratios). The reaction was affected by the electronic effect of the substituents on the aryl ring. Substrates bearing electron-donating group (OMe), phenyl, and halogens gave the desired Z-2 in high Z/E ratios (up to >99/1) (2d–2j, Table 3). However, E-arylvinyl acid 1k with electron-withdrawing group (CF3) did not afford the corresponding bromide 2k. The efficiency of the reaction was not impeded by meta-substituents on the aryl ring (2l and 2m, 53% and 42% yield, respectively, 99/1 Z/E ratios). In contrast, the ortho-substituent hindered the reaction, only providing the halodecarboxylation product E-2n. In addition, naphthyl ring was also compatible under these mild reaction conditions giving Z-2o (Z/E = 80/20) in 79% yield. N-Chlorosuccinimide (NCS) can also be used in this halodecarboxylation/isomerization smoothly to afford Z-2p in 96:4 Z/E ratios. When 1.03 g of 1j was used, the corresponding styrenyl bromide Z-2j was obtained without compromising on yield and stereoselectivity, representing the robustness of this reaction.
To gain mechanistic insight on this reaction, the reaction progress was monitored under the optimized reaction conditions (Scheme 2). Complete halodecarboxylation of E-1c was observed in 1 hour to give E-2c, which suggests that the isomerisation is the rate-determining step. Some product Z-2c was obtained after 2 hours. Almost complete E → Z isomerization was observed over the course of 16 hours.
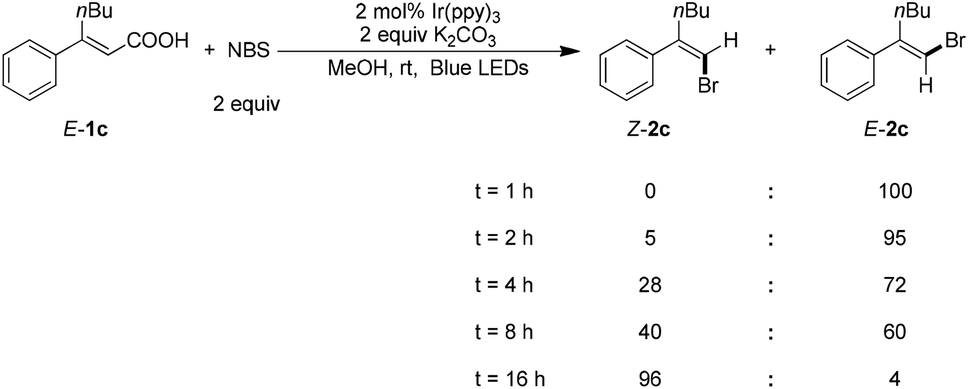 | ||
| Scheme 2 Reaction progress monitoring of E-1c. | ||
At last, we performed two Pd-catalyzed coupling reactions with styrenyl bromide Z-2j to further illustrate the synthetic utility of this cascade reaction (Scheme 3). When 4-methoxyphenylboronic acid was treated with bromide Z-2j using Pd(PtBu3)2 as catalyst, Suzuki–Miyaura cross coupling reaction14 successfully afforded trisubstituted alkene Z-4 (Z/E = 98/2) in 78% yield. Moreover, Sonogashira coupling reaction15 with 1-ethynyl-4-methylbenzene gave enyne Z-5 (Z/E = 98/2) in 87% yield.
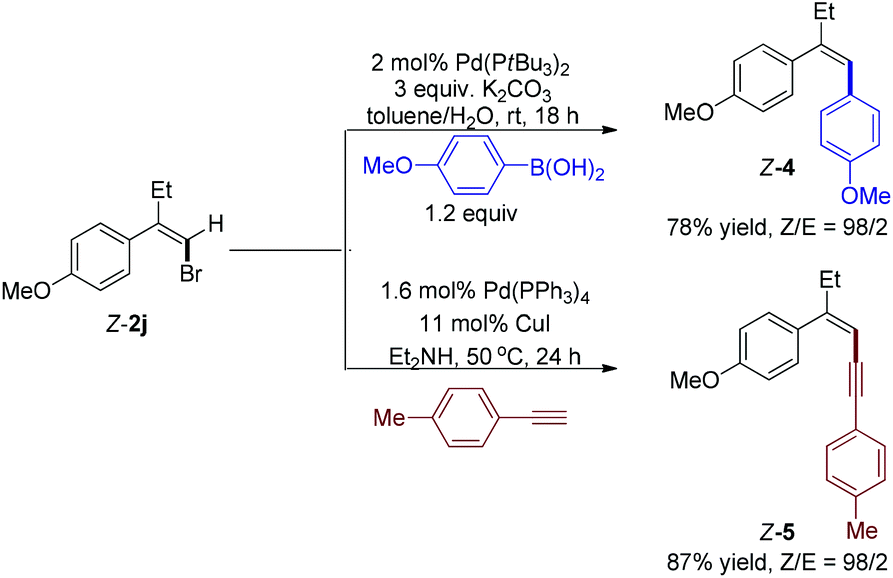 | ||
| Scheme 3 Pd-catalyzed coupling reactions of styrenyl bromide Z-2j. | ||
Conclusions
In conclusion, we have developed an efficient visible light photocatalytic strategy to synthesize Z-arylvinyl halides directly from E-arylvinyl acid with N-halosuccinimide through a sequential halodecarboxylation/photoisomerization sequence with up to >99/1 Z/E ratio and 86% yield. A series of E-arylvinyl acids were converted to Z-arylvinyl halides with high stereoselectivity under mild reaction conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the Shenzhen Science and Technology Innovation Commission (JCYJ20170818104257975), the State Key Laboratory of Chemical Biology and Drug Discovery, and The Hong Kong Polytechnic University (G-ZVQZ).Notes and references
- For selected recent reviews: (a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (b) J. D. Nguyen, E. M. D'Amato, J. M. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed; (c) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (e) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734 CrossRef CAS PubMed; (f) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872 CrossRef CAS PubMed; (g) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (h) M. D. Kärkäs, J. A. Porco Jr. and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683 CrossRef PubMed; (i) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef CAS PubMed; (j) T. Hering, A. U. Meyer and B. König, J. Org. Chem., 2016, 81, 6927 CrossRef CAS PubMed; (k) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (l) Q. Liu and L.-Z. Wu, Natl. Sci. Rev., 2017, 4, 359 CrossRef CAS; (m) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034 CrossRef CAS PubMed; (n) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586 CrossRef CAS PubMed; (o) S. K. Pagire, T. Föll and O. Reiser, Acc. Chem. Res., 2020, 53, 782 CrossRef CAS PubMed; (p) R. Cannalire, S. Pelliccia, L. Sancineto, E. Novellino, G. C. Tron and M. Giustiniano, Chem. Soc. Rev., 2021, 50, 766 RSC.
- F. S. Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190 RSC.
- For selected reviews, see: (a) E.-i. Negishi, Z. Huang, G. Wang, S. Mohan, C. Wang and H. Hattori, Acc. Chem. Res., 2008, 41, 1474 CrossRef CAS PubMed; (b) J.-L. Li, T.-Y. Liu and Y.-C. Chen, Acc. Chem. Res., 2012, 45, 1491 CrossRef CAS PubMed; (c) C. Oger, L. Balas, T. Durand and J. M. Galano, Chem. Rev., 2013, 113, 1313 CrossRef CAS PubMed; (d) J. L. Bras and J. Muzart, Chem. Soc. Rev., 2014, 43, 3003 RSC; (e) E. McNeill and T. Ritter, Acc. Chem. Res., 2015, 48, 2330 CrossRef CAS PubMed; (f) S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592 RSC; (g) B. L Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060 CrossRef PubMed.
- W.-Y. Siau, Y. Zhang and Y. Zhao, Top. Curr. Chem., 2012, 327, 33 CrossRef CAS PubMed.
- (a) G. S. Hammond, N. J. Turro and P. A. Leermakers, J. Phys. Chem., 1962, 66, 1144 CrossRef CAS; (b) G. M. Wyman, Chem. Rev., 1955, 55, 625 CrossRef CAS; (c) G. S. Hammond, J. Saltiel, A. A. Lamola, N. J. Turro, J. S. Bradshaw, D. O. Cowan, R. C. Counsell, V. Vogt and C. Dalton, J. Am. Chem. Soc., 1964, 86, 3197 CrossRef CAS; (d) F. D. Lewis, D. K. Howard, J. D. Oxman, A. L. Upthagrove and S. L. Quillen, J. Am. Chem. Soc., 1986, 108, 5964 CrossRef CAS PubMed; (e) T. Arai, H. Sakuragi and K. Tokumaru, Chem. Lett., 1980, 9, 261 CrossRef.
- (a) K. Singh, S. J. Staig and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 5275 CrossRef CAS PubMed; (b) A. Singh, C. J. Fennell and J. D. Weaver, Chem. Sci., 2016, 7, 6796 RSC.
- (a) J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2015, 137, 11254 CrossRef CAS PubMed; (b) J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 1040 CrossRef CAS PubMed; (c) J. B. Metternich, D. G. Artiukhin, M. C. Holland, M. V. Bremen-Kühne, J. Neugebauer and R. Gilmour, J. Org. Chem., 2017, 82, 9955 CrossRef CAS PubMed; (d) J. B. Metternich, S. Sagebiel, A. Lückener, S. Lamping, B. J. Ravoo and R. Gilmour, Chem. -Eur. J., 2018, 24, 4228 CrossRef CAS PubMed.
- J. J. Molloy, J. B. Metternich, C. G. Daniliuc, A. J. B. Watson and R. Gilmour, Angew. Chem., Int. Ed., 2018, 57, 3168 CrossRef CAS PubMed.
- J. J. Molloy, M. Schäfer, M. Wienhold, T. Morack, C. G. Daniliuc and R. Gilmour, Science, 2020, 369, 302 CrossRef CAS PubMed.
- For selected reviews, see: (a) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS; (b) J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177 CrossRef CAS; (c) A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412 RSC; (d) L. Xu, S. Zhang and P. Li, Chem. Soc. Rev., 2015, 44, 8848 RSC.
- H. Zhang, Q. Xu, L. Yu and S. Yu, Eur. J. Org. Chem., 2020, 1472 CrossRef CAS.
- (a) C. Kuang, H. Senboku and M. Tokuda, Synlett, 2000, 10, 1439 Search PubMed; (b) K. C. Rajanna, N. Maasi Reddy, M. Rajender Reddy and P. K. Saiprakash, J. Dispers. Sci. Technol., 2007, 28, 613 CrossRef CAS; (c) V. N. Telvekar and S. N. Chettiar, Tetrahedron Lett., 2007, 48, 4529 CrossRef CAS; (d) D. Hazarika and P. Phukan, Tetrahedron Lett., 2018, 59, 4593 CrossRef CAS; (e) D. Crich and K. Sasaki, Comprehensive Organic Synthesis II, 2014, 7, 818 CAS; (f) A. Varenikov, E. Shapiro and M. Gandelman, Chem. Rev., 2021, 121, 412 CrossRef CAS PubMed.
- (a) J.-R. Deng, W.-C. Chan, N. C.-H. Lai, B. Yang, C.-S. Tsang, B. C.-B. Ko, S. L.-F. Chan and M.-K. Wong, Chem. Sci., 2017, 8, 7537 RSC; (b) W.-M. Yip, Q. Yu, A. Tantipanjaporn, W.-C. Chan, J.-R. Deng, B. C.-B. Ko and M.-K. Wong, Org. Biomol. Chem., 2021, 19, 8507 RSC.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) M. Wakioka, Y. Mutoh, R. Takita and F. Ozawa, Bull. Chem. Soc. Jpn., 2009, 82, 1292 CrossRef CAS.
- (a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef; (b) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed; (c) H. Zhou and C. Moberg, J. Am. Chem. Soc., 2012, 134, 15992 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08753c |
| This journal is © The Royal Society of Chemistry 2022 |