
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePO(CH2CH2CF3)3: an organic ultraviolet nonlinear optical material without any anionic group†
Liting Lin‡
a,
Xiaoqian Li‡a,
Kun Qian‡ *a,
Lin Cheng‡a,
Youquan Zhong‡a and
Hua Xu‡b
*a,
Lin Cheng‡a,
Youquan Zhong‡a and
Hua Xu‡b
aCollege of Pharmacy, Jiangxi University of Traditional Chinese Medicine, Nanchang, 330004, P. R. China. E-mail: qk0876@hotmail.com
bInternational Education College, Jiangxi University of Traditional Chinese Medicine, Nanchang, 330004, P. R. China
First published on 9th February 2022
Abstract
Researchers have focused on inorganic compounds to design deep ultraviolet (UV) nonlinear optical (NLO) materials according to anionic group theory, such as CO32−, NO3−, SO42− and PO43− anions. Here we provide a new route to design an UV NLO material using a pure organic compound without any anions. Compound PO(CH2CH2CF3)3 1, an UV NLO material, has light transmittance up to 83% in the UV spectral region which is larger than most inorganic UV NLO materials. It also displays a wide transparent band, a SHG response of 0.30 × KH2PO4, and a cut-off edge below 200 nm. It displays ladder-like nonlinear optical properties weakened by 1.4 times at around Tc, making compound 1 a potential temperature-controlled UV NLO material. Theoretical analyses reveal that the nonlinear optical properties are primarily due to O-2p, P-2p and F-2p.
UV NLO crystals have been a hot spot of scientific research because of their key role in modern laser technology.1–3 UV NLO materials should meet the following requirements, such as a large nonlinear optical coefficient, phase matching (PM) capability, a wide transparency band and a high light transmittance, a strong resistance to light damage, large size crystals, uniform refractive index and small scattering, being stable in physical and chemical properties, being hard to be hygroscopic and easy to be processed. In the past few decades, many researchers have made great efforts to study and design nonlinear optical materials according to anionic group theory. Many researchers focused on inorganic compounds, and they selected oxo anions, such as CO32−, NO3−, SO42− and PO43− as anions, because crystals containing these anions and ammonium are not fragile, have high solubility in water, and they are easy to grow to be bulk crystals. And monovalent cations = Li+, Na+, K+, Rb+, Cs+, NH4+ or divalent cations = Be2+, Mg2+, Ca2+, Sr2+, Ba2+ were selected as cations, because these cations do not have d–d and f–f electron transitions in the ultraviolet region, which is helpful for the ultraviolet ray to pass through.
KBBF is currently the only practically useable deep-UV nonlinear optical crystal. However, KBBF material suffers from its layered growth habits. Therefore, the study of high-performance ultraviolet optical materials has been focused on inorganic π conjugate systems, including borates, carbonates and nitrates. Many UV NLO materials with good optical properties were found, such as β-Rb2Al2B2O7,4 NH4B4O6F,5 Ca2B10O14F6,6 ABCO3F (A = K, Rb, Cs; B = Ca, Sr)7 and Re(NO3)2OH (Re = La, Y, Gd).8 In recent years, researchers have turned their enthusiasm to non-π conjugate NLO materials, which are considered as rich exploration systems.9–14 A case in point is the phosphate which was found to have thermal, chemical stability and strong rigid.15 Thus, many UV NLO phosphates with short UV-cutoff edges are also found, such as Ba3P3O10X (X = Cl, Br),16 LiPbPO4,17 K4Mg4(P2O7)3 (ref. 18) and Rb4Mg4(P2O7)3.18 For compound Rb4Mg4(P2O7)3, it has a deep-UV absorption edge at 185 nm, 3.0 × KH2PO4, and its light transmittance is up to 40%, undergoing a thermo-induced reversible phase transition and a SHG enhancement up to 1.5 times. Furthermore, a SHG enhancement strategies have been developed, such as constructing strong distortion and the introduction of stereo chemical active d0 or ns2 single pair cations.19–21 In addition, sulfate is one of non-π conjugate systems, anhydrous sulfate composed of non-π conjugate elements SO42− with UV penetration and phase matching were found, such as (NH4)2Na3Li9(SO4)7 and NH4NaLi2(SO4)2, of which SHG response is 1.1 × KH2PO4, and it is transparent down to the deep-UV region below 186 nm and its light transmittance is up to only 30%.22
However, organic nonlinear optical materials have high nonlinear optical coefficients with various structures. This kind of crystal is easy to grow to be larger one and is particularly suitable for nonlinear optical materials in the ultraviolet band. However, this kind of report is scare. When organic molecules absorb ultraviolet or visible radiation, their outer electrons will transition from the ground state to the excited state. There are four main transition modes, such as σ → σ*, n → σ*, π → π*, n → π*. For organic UV NLO crystals, the following chemical bonds are not included as far as possible, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C
C, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C and CO, which will produce π → π* electron transition. The following chemical bonds are not included as far as possible, such as CO, CS and NN, will undergo n → π* electron transition. The following groups are not included as far as possible, such as –NH2, –OH and –SH, which will produce n → σ* electron transition. Hence, we select a pure organic compound PO(CH2CH2CF3)3, as an UV NLO material. Fortunately, it displays a SHG response of 0.30 × KH2PO4, the light transmittance is up to 83% in the UV spectral region and its cut-off edge is below 200 nm, an above room temperature reversible phase transition, ladder-like nonlinear optical properties, obtaining a large size of high-quality single crystal easily.
C and CO, which will produce π → π* electron transition. The following chemical bonds are not included as far as possible, such as CO, CS and NN, will undergo n → π* electron transition. The following groups are not included as far as possible, such as –NH2, –OH and –SH, which will produce n → σ* electron transition. Hence, we select a pure organic compound PO(CH2CH2CF3)3, as an UV NLO material. Fortunately, it displays a SHG response of 0.30 × KH2PO4, the light transmittance is up to 83% in the UV spectral region and its cut-off edge is below 200 nm, an above room temperature reversible phase transition, ladder-like nonlinear optical properties, obtaining a large size of high-quality single crystal easily.
All starting reagents and solvents employed for synthesis were purchased from Energy Company (Shanghai, China) and without further purified before using. Compound 1 recrystallizes from a hot acetonitrile according to the ref. 23, formed as the fine needle-shaped crystals, melt point 193–195 °C. The infrared spectrum data were collected in the range of 4000–400 cm−1 on a Tensor 27 OPUS FT-IR spectrometer (Bruker) with solid KBr pellets (Fig. S1†).
Compound 1 was measured at temperature ranging from 285 to 380 K by differential scanning calorimetry (DSC) measurements. According to Fig. 1, a pair of abnormal peaks in DSC curves at 338/327 K upon heating/cooling are observed. It shows that a reversible phase transition occurs. Room temperature phase (RTP) is marked below 327 K. Correspondingly, high temperature phase (HTP) is marked above 338 K for convenience. It is very simple to calculate the entropy change ΔS of this system according to second law of thermodynamics ΔS = δQ/T, which is to be about 8.338 J mol−1 K−1. As we know, the Boltzmann formula ΔS = R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(N) illustrates the relationship between the possible number of disturbing ways of the atoms or molecular of a thermodynamic system and the entropy change ΔS. The N value is calculated to be 2.73 by the equation of Boltzmann, illustrating there is a structural phase transition. And it will be confirmed by the X-ray single crystal diffraction analysis.
ln(N) illustrates the relationship between the possible number of disturbing ways of the atoms or molecular of a thermodynamic system and the entropy change ΔS. The N value is calculated to be 2.73 by the equation of Boltzmann, illustrating there is a structural phase transition. And it will be confirmed by the X-ray single crystal diffraction analysis.
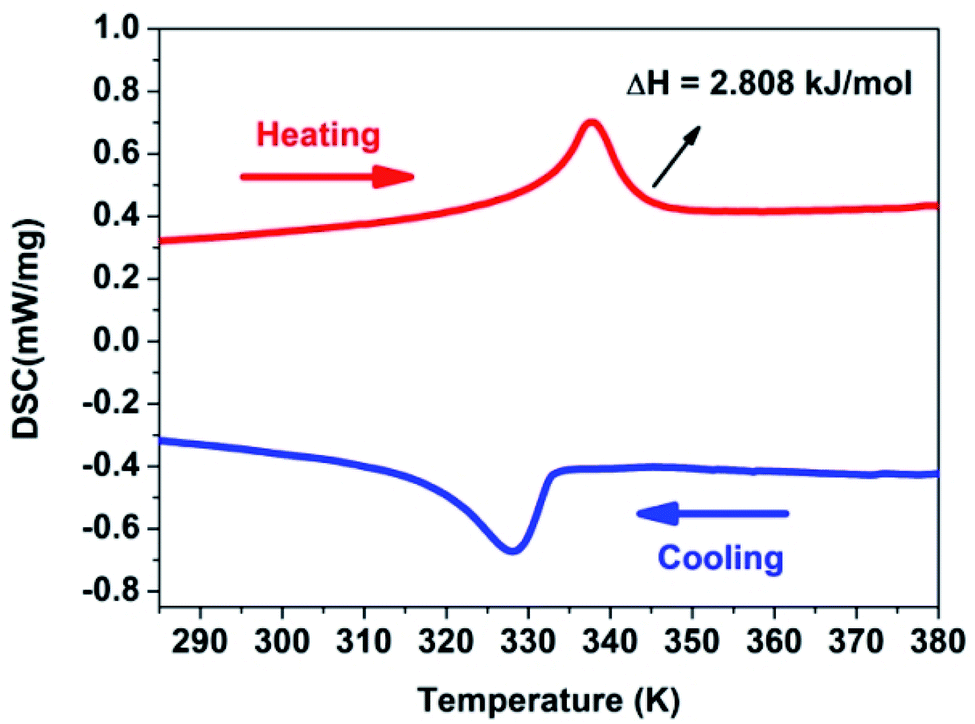 | ||
| Fig. 1 DSC curves of compound 1 shown in the temperature range from 285 to 380 K. | ||
In the same phase, all physical properties are essentially uniform. And phase transition is a physical process of transition. For revealing structural phase transition in detail, crystal structural data of compound 1 was gathered at two different temperatures 296(2) and 363.15 K by using variable-temperature X-ray single-crystal diffraction analysis.24–26 The crystal structure of compound 1 was determined at room temperature (RTP), which belongs to trigonal system with space group R3c at 296(2) K, with cell parameters of a = 15.35(5) Å, b = 15.35(5), c = 10.08(5) Å, V = 2057.46(17) Å3, γ = 120° and Z = 6 (Table S1†). Its asymmetric unit contains one phosphorus center, three [CH2CH2CF3] groups and one oxygen atom, forming a triangular cone geometry, as shown in Fig. 2a and S2.† The six symmetry elements (E, 3+, 3−, c, c, c) can be observed according to the International Tables for Crystallography A. The central phosphorus atom is seated in a 31 screw axis, being connected with three [CH2CH2CF3] groups and an oxygen atom of PO double bond. The phosphorus atom has a distorted tetrahedral coordination, where C–P–C bond angles of adjacent C atoms is 105.82(12)° and P–C bond distances is 1.803(3) Å and PO bond distances is 1.496(4) Å. The F–C–F angles of adjacent F atoms vary from 103.10(4)° to 107.30(4)° and bond distances between carbon atoms and fluorine atoms vary from 1.322(5) to 1.336(5) Å (Table S2,† Fig. 2b and c).
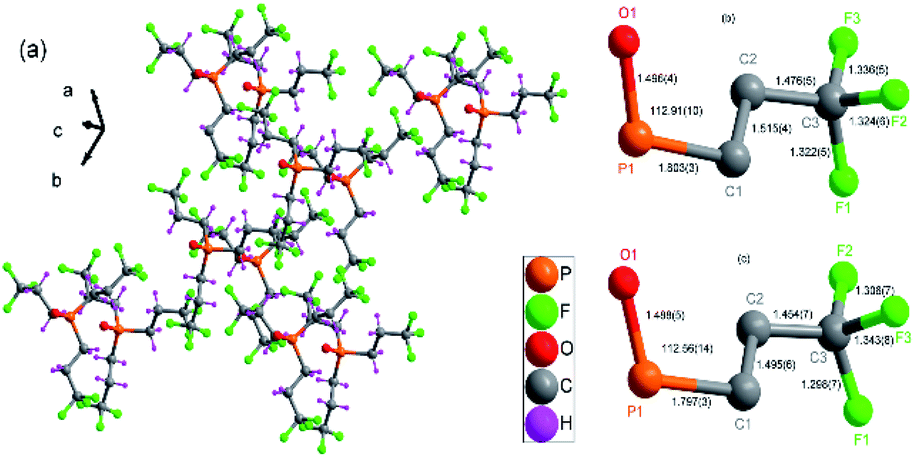 | ||
| Fig. 2 (a) Packing diagram of compound 1 at 296 K. (b) Selected bond distance and angle diagram of compound 1 (296(2) K). (c) Selected bond distance and angle diagram of compound 1 (363.15 K). | ||
When the same single crystal was heated to be at 363.15 K, cell parameters of compound 1 change a little with a = 15.54(7) Å, b = 15.54(7)Å, c = 10.13(7) Å, γ = 120°, V = 2118.40(2) Å3 and Z = 6 (Table S1†), adopts the same space group R3c. Each phosphorus atom, still lying in the 31 screw axis, is connected with three [CH2CH2CF3] groups and one oxygen atom, forming a triangular cone geometry. The phosphorus atoms still have a distorted tetrahedral coordination, where C–P–C angles of adjacent C atoms is 106.21(16)° and P–C bond distance is 1.797(3) Å and PO bond distances is 1.488(5) Å. The F–C–F angles of adjacent F atoms vary from 100.10(6)° to 106.00(5)° and bond distances between carbon atoms and fluorine atoms vary from 1.298(7) to 1.343(8) Å (Table S3†).
With temperature rising, cell volume increases by 1.03 times as much as obtained at 363.15 K. The length of the three edges of the cell becomes longer, and the three bond angles of the cell remain unchanged. And bond distances and bond angles change a little. The P–C bond distances is from 1.803(3) to 1.797(3) Å and PO bond distances is from 1.496(4) to 1.488(5) Å when phase change happens. Although space group keeps the same in different phases, it still reveals that an isomorphic phase transition occurs in the crystal (Table S1,† Fig. 1). The same six symmetry elements (E, 3+, 3−, c, c, c) can be observed in the HTP. The motion of atoms or the whole molecule under the heat stimuli is the mainly reason of the phase transition.
Enhanced/weakened nonlinear optical effect can be applied in the laser field to adjust the frequency of light. The nonlinearity is typically observed only at very high light intensity lasers, such as I = 1064 nm output from Nd:YAG lasers. Nonlinear optics measurements can effectively detect the inversion symmetry in the crystal, so it is used to verify space group of compound 1.27 The response of SHG signal of compound 1 over time were measured. According to Fig. 3 inserted, compound 1 has a SHG activity. Compared to the KDP, the SHG value of compound 1 is 0.30 times as much as the SHG value of KDP. The SHG value of compound 1 changes over time, it means compound 1 possesses polar space group at room temperature. At this stage, we measured nonlinear optics signal of compound 1 as a function of temperature. As shown in Fig. 3, SHG value keeps stable with a value of about 0.30 at temperature ranging from 285 to 325 K. But SHG intensity value changes abruptly at around 338 K. Above 345 K, SHG value decreases to 0.20. SHG value keeps stable with a value of about 0.20 at temperature ranging from 345 to 380 K. SHG value is not zero, which is consistent with the non-centrosymmetric space group R3c. The SHG anomalies are in conformity with the thermal anomalies of DSC measurement and X-ray single-crystal diffraction measurement (Fig. 3). Because SHG intensity of compound 1 can be adjusted by changing temperature even in the solid state, compound 1 has a potential application in temperature-controlled NLO materials.
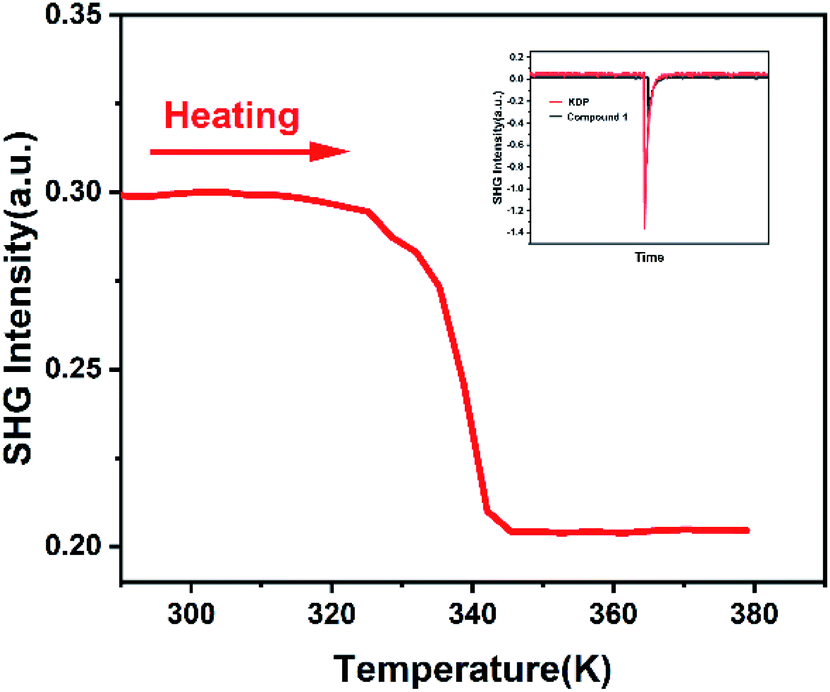 | ||
| Fig. 3 Temperature-dependent SHG intensity of compound 1 upon heating. And the inserted figure is oscilloscope traces of SHG signals of compound 1. | ||
The powder SHG signals of the different size crystals of compound 1 measured with a laser at 1064 nm are shown in Fig. 4 according to the rule proposed by Kurtz and Perry method. KDP samples were used as the reference for SHG measurement. The SHG intensity of compound 1 increases steadily with the increase of the particle size, which reveals that compound 1 is phase-matchable in UV region. The relative SHG intensities in the UV region for compound 1 is approximately 0.1 times that of KDP when the particle size is 250 μm. The powder SHG measurement indicates that compound 1 has a remarkable SHG conversion efficiency in UV region. The value is lower than some inorganic–organic NLO materials, such as [C6H14N2]2SbCl5 (0.30 × KH2PO4),28 [Et3P(CH2)2Cl][Cd(dca)3] (0.16 × KH2PO4),29 [R-3-hydroxy-pyrrolodinium]2SbBr5 (0.12 × KH2PO4).30
 | ||
| Fig. 4 Measured SHG intensity of compound 1 and KDP versus particle size. | ||
The optical transmittance spectrum of PO(CH2CH2CF3)3 was directly measured on its single-crystal sample without polishing, which was collected on a Lambda 900 UV/vis/NIR Spectrometer from 200 to 800 nm at room temperature (Fig. S3†). As shown in Fig. 5a, compound 1 is transparent down to the UV region, the light transmittance is up to 83% which is more than most of inorganic UV NLO materials. And its cutoff edge of compound 1 is below 200 nm, which is comparable to other inorganic UV NLO materials, such as KSrCO3F (˂ 200 nm),4 β-Rb2Al2B2O7 (<190 nm),7 and non-π-conjugated Ba3P3O10Cl (180 nm),16 RbNaMgP2O7 (185 nm).31 The light transmittance of compound 1 in the UV region is better than most of inorganic nonlinear optical crystals, such as NH4B4O6F (<13%),1 KLi(HC3N3O3)·2H2O (<60%),2 AZn2BO3X2 (A = K, Rb, NH4; X = Cl, Br) (<60%),3 Ca2Na3(CO3)3F (<40%)4 RbNaMgP2O7 (<40%),24 NH4NaLi2(SO4)2 (<30%)6 and NaZnCO3(OH) (<40%)7 (Table S4†). The light transmittance of compound 1 is slightly lower than NH4Be2BO3F2 (90%), Ba3P3O10Cl (85%) and Ba3P3O10Cl (90%).16 The ultraviolet-visible-near-infrared (UV-vis-NIR) diffuse reflectance spectrum of compound was collected on a UV-3600 UV/vis/NIR spectrophotometer from 200 to 700 nm at room temperature, as shown in Fig. 5b. It shows that the reflectance is 9.46% at 200 nm and it exhibits a wide transparency window in the UV region.
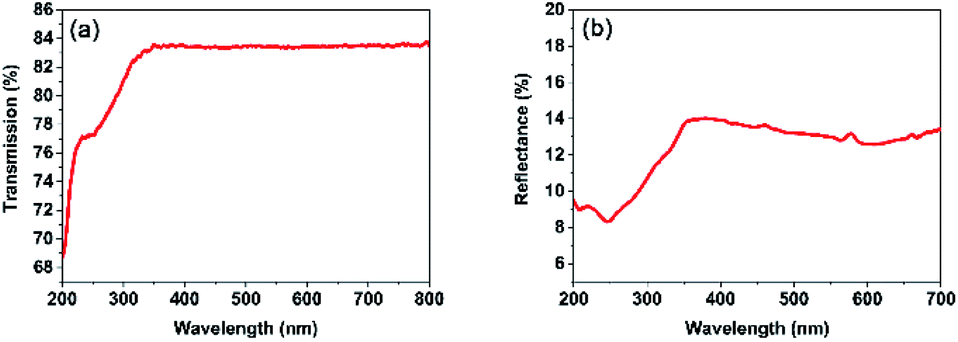 | ||
| Fig. 5 UV transmittance spectrum and UV-vis-NIR diffuse reflectance spectrum of compound 1. | ||
To disclose the microscopic mechanism for the thermo-induced SHG changes of compound 1, the first-principles calculations by the plane-wave pseudopotential method implemented in the CASTEP package were carried out.32,33 As shown in Fig. S4,† the electronic band structures are both direct-gap, and their bandgap values are 5.982 eV (corresponding to 200 nm), confirming the wide UV transparent range of compound 1. These results indicated that the crystal could be applied in the UV region. Meanwhile, compound 1 have similar energy band structural characteristics in the RTP and HTP. It also suggests that the difference of SHG effect between HTP and RTP is not determined by the band gap.
Since the optical properties mainly depend on the states close to the Fermi level, the upper region of the valence band (VB) and the bottom of the conduction band (CB) need to be analyzed. The total and partial densities of states (DOS and PDOS) for compound 1 in RTP and HTP are shown in Fig. 6. For compound 1 in RTP and HTP, the VB maximum is occupied by O-2p, C-2p and H-1s orbitals and the CB minimum is predominantly contributed from P-2p, C-2p and H-1s orbitals with slight hybridization with O-2p and F-2p orbitals. From the PDOS calculations, it could be concluded that the nonlinear optical properties of compound 1 in RTP and HTP are primarily due to the atom of O-2p, P-2p and F-2p. Compared to other high-performance ultraviolet optical materials, their oxo anions made a major contribution to the SHG effect, while O-2p, P-2p and F-2p in compound 1 made a major contribution to SHG effect. Similarly, for inorganic compound Rb4Mg4(P2O7)3,18 it undergoes a thermo-induced reversible phase transition and a SHG enhancement up to 1.5 times. However, its dominant NLO-active groups are due to its anion part according to theoretical analyses.
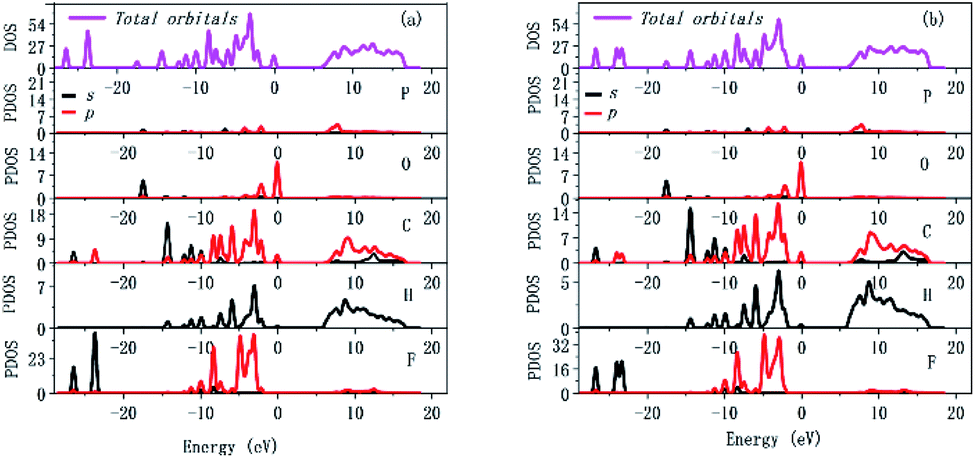 | ||
| Fig. 6 Total and partial density of states of compound 1 in (a) RTP and (b) HTP. | ||
In order to better understand the structural mechanism of SHG change, we further calculated the dipole moments in the unit cell of RTP and HTP with a simple bond-valence approach proposed by Poeppelmeier.34 According to the total and partial densities of states calculation results, O-2p, P-2p and F-2p are the dominant NLO-active groups for compound 1, so the whole molecular dipole moments are quantified. Detailed calculation results are given in the ESI (Tables S5 and S6†). As a result, vector summation of the whole unit gave a dipole moment of 300.35 D for RTP, but it gave a smaller value of 273.17 D for HTP. It can be rationalized that the weakened SHG from RTP to HTP is ascribed to the less collective alignment of the whole molecular along the polar axis. It is worth noting that the changes of dipole moment in compound 1 are caused by heating, which is different from traditional SHG weakened materials.
In summary, we put forward a new route to design UV NLO material using a pure organic compound without any anions, which is different from the traditional inorganic UV NLO materials according to anionic group theory. The organic compound PO(CH2CH2CF3)3, its light transmittance is up to 83% which is larger than most of inorganic UV NLO materials. It also displays a wide transparent band, a SHG response of 0.30 × KH2PO4, cut-off edge below 200 nm. The change of SHG responses specificity makes compound 1 as a potential temperature-controlled UV NLO material. Theoretical analyses reveal that the nonlinear optical properties are primarily due to the whole molecular, different from inorganic UV NLO materials dominated by their oxo anions parts.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China Program Project (Grants number 21761014, 22165014). Special thanks to Professor Ren Gen Xiong in Nanchang University for helping test second harmonic generation.References
- P. Becker, Borate Materials in Nonlinear Optics, Adv. Mater., 1998, 10, 979 CrossRef CAS.
- C. Chen, Z. Lin and Z. Wang, The development of new borate-based UV nonlinear optical crystals, Appl. Phys. B, 2005, 80, 1 CrossRef CAS.
- D. Cyranoski, Materials science: China's crystal cache, Nature, 2009, 457, 953 CrossRef CAS PubMed.
- T. T. Tran, N. Z. Koocher, J. M. Rondinelli and P. S. Halasyamani, Beryllium-Free β-Rb2Al2B2O7 as a possible deep-ultraviolet nonlinear optical material replacement for KBe2BO3F2, Angew. Chem., 2017, 56, 2969 CrossRef CAS PubMed.
- G. Q. Shi, Y. Wang, F. F. Zhang, B. B. Zhang, Z. H. Yang, X. L. Hou, S. L. Pan and K. R. Poeppelmeier, Finding the next deep-ultraviolet nonlinear optical material: NH4B4O6F, J. Am. Chem. Soc., 2017, 139, 10645 CrossRef CAS PubMed.
- M. Luo, F. Liang, Y. X. Song, D. Zhao, F. Xu, N. Ye and Z. S. Lin, Two noncentrosymmetric alkaline earth fluorooxoborates as promising next-generation deep-ultraviolet nonlinear optical materials, J. Am. Chem. Soc., 2018, 140, 3884 CrossRef CAS PubMed.
- G. H. Zou, N. Ye, L. Huang and X. S. Lin, Alkaline-alkaline earth fluoride carbonate crystals ABCO3F (A = K, Rb, Cs; B = Ca, Sr, Ba) as nonlinear optical materials, J. Am. Chem. Soc., 2011, 133, 20001 CrossRef CAS PubMed.
- Y. X. Song, M. Luo, C. S. Lin and N. Ye, Structural modulation of nitrate group with cations to affect SHG responses in RE(OH)2NO3 (RE = La, Y, and Gd): new polar materials with large NLO effect after adjusting pH values of reaction systems, Chem. Mater., 2017, 29, 896 CrossRef CAS.
- S. G. Zhao, P. F. Gong, S. Y. Luo, L. Bai, Z. S. Lin, C. M. Ji, T. L. Chen, M. C. Hong and J. H. Luo, Deep-Ultraviolet Transparent Phosphates RbBa2(PO3)5 and Rb2Ba3(P2O7)2 Show Nonlinear Optical Activity from Condensation of [PO4]3– Units, J. Am. Chem. Soc., 2014, 136, 8560 CrossRef CAS PubMed.
- K. C. Chen, Y. Yang, G. Peng, S. D. Yang, T. Yan, H. X. Fan, Z. S. Lin and N. Ye, A2Bi2(SO4)2Cl4 (A = NH4, K, Rb): achieving a subtle balance of the large second harmonic generation effect and sufficient birefringence in sulfate nonlinear optical materials, J. Mater. Chem. C, 2019, 32, 9900 RSC.
- H. W. Yu, W. G. Zhang, J. S. Young, J. M. Rondinelli and P. S. Halasyamani, Bidenticity-enhanced second harmonic generation from Pb chelation in Pb3Mg3TeP2O14, J. Am. Chem. Soc., 2016, 138, 88 CrossRef CAS.
- X. H. Dong, L. Huang, C. F. Hu, H. M. Zeng, Z. E. Lin, X. Wang, K. M. Ok and G. H. Zou, CsSbF2SO4: An excellent ultraviolet nonlinear optical sulfate with a KTiOPO4 (KTP)-type structure, Angew. Chem., Int. Ed., 2019, 58, 6528 CrossRef CAS PubMed.
- H. Lee and K. M. Ok, Na2Mg1−xZnxSiO4 (0≤x≤1): Noncentrosymmetric sodium metal silicate solid solutions with ultraviolet nonlinear optical properties, Bull. Korean Chem. Soc., 2020, 41, 139 CrossRef CAS.
- H. P. Wu, S. W. Liu, S. C. Cheng, H. W. Yu, Z. G. Hu, J. Y. Wang and Y. C. Wu, Syntheses, characterization, and theoretical calculation of Rb2Mg3(P2O7)2 polymorphs with deep-ultraviolet cutoff edges, Sci. China. Mater., 2020, 63, 593 CrossRef CAS.
- X. F. Lu, Z. H. Chen, X. R. Shi, Q. Jing and M. H. Lee, Two pyrophosphates with large birefringences and second-harmonic responses as ultraviolet nonlinear optical materials, Angew. Chem., Int. Ed., 2020, 59, 17648 CrossRef CAS.
- P. Yu, L. M. Wu, L. J. Zhou and L. Chen, Deep-ultraviolet nonlinear optical crystals: Ba3P3O10X (X = Cl, Br), J. Am. Chem. Soc., 2013, 136, 480 CrossRef PubMed.
- G. P. Han, Q. Liu, Y. Wang, X. Su, Z. H. Yang and S. L. Pan, Experimental and theoretical studies on the linear and nonlinear optical properties of lead phosphate crystals LiPbPO4, Phys. Chem. Chem. Phys., 2016, 28, 19123 RSC.
- H. W. Yu, J. S. Young, H. P. Wu, W. G. Zhang, J. M. Rondinelli and P. S. Halasyamani, (M4Mg4(P2O7)3 (M = K, Rb): structural engineering of pyrophosphates for nonlinear optical applications, Chem. Mater., 2017, 29, 1845 CrossRef CAS.
- L. Li, Y. Wang, B. H. Lei, S. J. Han, Z. H. Yang, R. Kenneth and P. S. L. Poeppelmeier, A new deep-ultraviolet transparent orthophosphate LiCs2PO4 with large second harmonic generation response, J. Am. Chem. Soc., 2016, 138, 9101 CrossRef CAS PubMed.
- O. A. Alekseeva, A. P. Dudka, N. E. Novikova, N. I. Sorokina, E. I. Agapova and V. I. Voronkova, Structure of the RbTi0.98Zr0.02OPO4 single crystal at temperatures of 293 and 105 K, Crystallogr. Rep., 2008, 53, 557 CrossRef CAS.
- A. A. Babaryk, I. V. Zatovsky, V. N. Baumer, N. S. Slobodyanik and K. V. Domasevitch, The complex phosphate K0.92In0.46Nb0.54OPO4: a new representative of the KTiOPO4 family, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, 1105 CrossRef PubMed.
- Y. Q. Li, F. Liang, S. G. Zhao, L. N. Li, Z. Y. Wu, Q. R. Ding, S. Liu, Z. S. Lin, M. C. Hong and J. H. Luo, Two non-π-Conjugated deep-UV nonlinear optical sulfates, J. Am. Chem. Soc., 2019, 141, 3833 CrossRef CAS PubMed.
- V. V. Semenov, N. F. Cherepennikova, L. G. Klapshina, M. A. Lopatin, T. G. Mushtina, N. P. Makarenko, S. Y. Khorshev, G. A. Domrachev, W. E. Douglas and B. A. Bushuk, The Er and Yb Complexes with silicon-containing phosphates and fluorine-containing phosphine oxide: synthesis, spectra, and films, Russ. J. Coord. Chem., 2004, 30, 435 CrossRef CAS.
- G. M. Sheldrick, Sadabs, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Göttingen (Germany), 1996 Search PubMed.
- G. M. Sheldrick, Shelxtl (version 6.1), Bruker AXS Inc, Madison, Wisconsin (USA), 2000 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr. A, 1990, 46, 467 CrossRef.
- O. Sato, J. Tao and Y. Z. Zhang, Control of magnetic properties through external stimuli, Angew. Chem., Int. Ed., 2007, 46, 2152 CrossRef CAS PubMed.
- J. Zhang, S. Han, X. Liu, Z. Wu, C. Ji, Z. Sun and J. Luo, A lead-free perovskite-like hybrid with above-room-temperature switching of quadratic nonlinear optical properties, Chem. Commun., 2018, 54, 5614 RSC.
- M. Szafrański, A. Katrusiak and K. Ståhl, Time-dependent transformation routes of perovskites CsPbBr3 and CsPbCl3 under high pressure, J. Mater. Chem. A., 2021, 9, 10769 RSC.
- T. Schmitt, S. Bourelle, N. Tye, G. Soavi, A. D. Bond, S. Feldmann, B. Traore, C. Katan, J. Even, S. E. Dutton and F. Deschler, Control of Crystal Symmetry Breaking with Halogen-Substituted Benzylammonium in Layered Hybrid Metal-Halide Perovskites, J. Am. Chem. Soc., 2020, 142, 5060 CrossRef CAS.
- S. G. Zhao, P. F. Gong, L. Bai, X. Xu, S. Q. Zhang, Z. H. Sun, Z. S. Lin, M. C. Hong, C. T. Chen and J. H. Luo, Beryllium-free Li4Sr(BO3)2 for deep-ultraviolet nonlinear optical applications, Nat. Commun., 2014, 5, 4019 CrossRef CAS.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, First principles methods using CASTEP, J. Phys.: Condens. Matter, 2005, 14, 2717 Search PubMed.
- P. A. Maggard, T. S. Nault, C. L. Stern and K. R. Poeppelmeier, Alignment of acentric MoO3F33- anions in a polar material: (Ag3MoO3F3)(Ag3MoO4)Cl, J. Solid State Chem., 2003, 175, 27 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, IR Spectrum, crystallographic data, powder X-ray diffraction, anisotropic displacement parameters, and selected bond length and bond angles. CCDC 2013852 and 2013829. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra08768a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |