
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiazolidinedione derivatives as novel GPR120 agonists for the treatment of type 2 diabetes†
Xuekun Wang *a,
Guoxia Jiab,
Xinyu Hana,
Huiran Haoa,
Wenjing Liua,
Qidi Xuea,
Qinghua Guoa,
Shiben Wanga,
Kang Leia and
Yadi Liu*c
*a,
Guoxia Jiab,
Xinyu Hana,
Huiran Haoa,
Wenjing Liua,
Qidi Xuea,
Qinghua Guoa,
Shiben Wanga,
Kang Leia and
Yadi Liu*c
aSchool of Pharmaceutical Sciences, Liaocheng University, 1 Hunan Street, Liaocheng 252059, China. E-mail: wangxuekun@lcu.edu.cn; xuekunwang0610@126.com; Tel: +86-0635-823-9087
bSchool of Chemistry and Chemical Engineering, Liaocheng University, 1 Hunan Street, Liaocheng 252059, China
cState Key Laboratory of Food Science and Technology, Jiangnan University, Wuxi 214122, China. E-mail: liuyadi@jiangnan.edu.cn
First published on 16th February 2022
Abstract
GPR120, also called FFAR4, is preferentially expressed in the intestines, and can be stimulated by long-chain free fatty acids to increase the secretion of glucagon-like peptide-1 (GLP-1) from intestinal endocrine cells. It is known that GLP-1, as an incretin, can promote the insulin secretion from pancreatic cells in a glucose-dependent manner. Therefore, GPR120 is a potential drug target to treat type 2 diabetes. In this study, thiazolidinedione derivatives were found to be novel potent GPR120 agonists. Compound 5g, with excellent agonistic activity, selectivity, and metabolic stability, improved oral glucose tolerance in normal C57BL/6 mice in a dose-dependent manner. Moreover, compound 5g exhibited anti-diabetic activity by promoting insulin secretion in diet-induced obese mice. In summary, compound 5g might be a promising drug candidate for the treatment of type 2 diabetes.
1. Introduction
Diabetes is a chronic condition that has become a public health crisis worldwide. According to the International Diabetes Federation, approximately 463 million people worldwide suffer from diabetes as of 2019.1 Its incidence is increasing rapidly, and the total number is predicted to rise to 578 million (10.2%) by 2030 and 700 million (10.9%) by 2045.2 The main categories of diabetes are type 1, type 2, and gestational diabetes. Type 2 diabetes accounts for approximately 90% of all diabetes cases and is characterized by reduced insulin sensitivity and insulin resistance. Drugs that enhance insulin secretion, such as sulfonylureas and glinides, are commonly used to treat of type 2 diabetes. However, these drugs enhance insulin secretion by directly closing ATP-sensitive potassium channels (KATP channels) independent of blood glucose levels, leading to the side effect of hypoglycemia.3G protein-coupled receptors (GPRs) are important signaling molecules involved in many functions. Recently, several studies have reported that GPRS receptors, such as GPR40 (ref. 4) and GPR120,5 play an important role in regulating blood glucose homeostasis. GPR120 (also called free fatty acid receptor 4, FFAR4), preferentially expressed in intestines, can be stimulated by long-chain FFAs to increase the secretion of glucagon-like peptide-1 (GLP-1) from intestinal endocrine cells,5 which leads to a glucose-dependent increase in insulin secretion from pancreatic cells, stimulation of insulin biosynthesis, and a decrease in glucagon secretion. Moreover, GLP-1 has been reported to play a significant role in appetite and feeding control.6 Thus, GPR120 has emerged as an attractive target for the treatment of type 2 diabetes and obesity.7 Some of its other outstanding pharmacology includes promoting the secretion of hormones, augmenting PPARγ activity and having anti-inflammatory, neuroprotective, antiproliferative properties.8–10
Recently, some GPR120 agonists have been reported,11–16 and most of their structures contain polar head, aromatic or aliphatic ring, aromatic or heteroaromatic ring and pendant tail (Fig. 1).17 Polar groups are generally carboxylic acids, which play an important role in agonistic activity. TUG-891, the first potent and selective GPR120 agonist, developed at the University of Southern Denmark has been widely used to explore the physiological function of GPR120.18–20 However, TUG-891 displayed high plasma clearance in rats, possibly because phenylpropanoic acid is vulnerable to β-oxidation, similar to the previously reported GPR40 agonists with phenylpropanoic acid moiety (Fig. 2).21 Therefore, we attempted to develop a metabolically stable candidate drug by the strategy of replacement carboxyl acid with bioisostere.
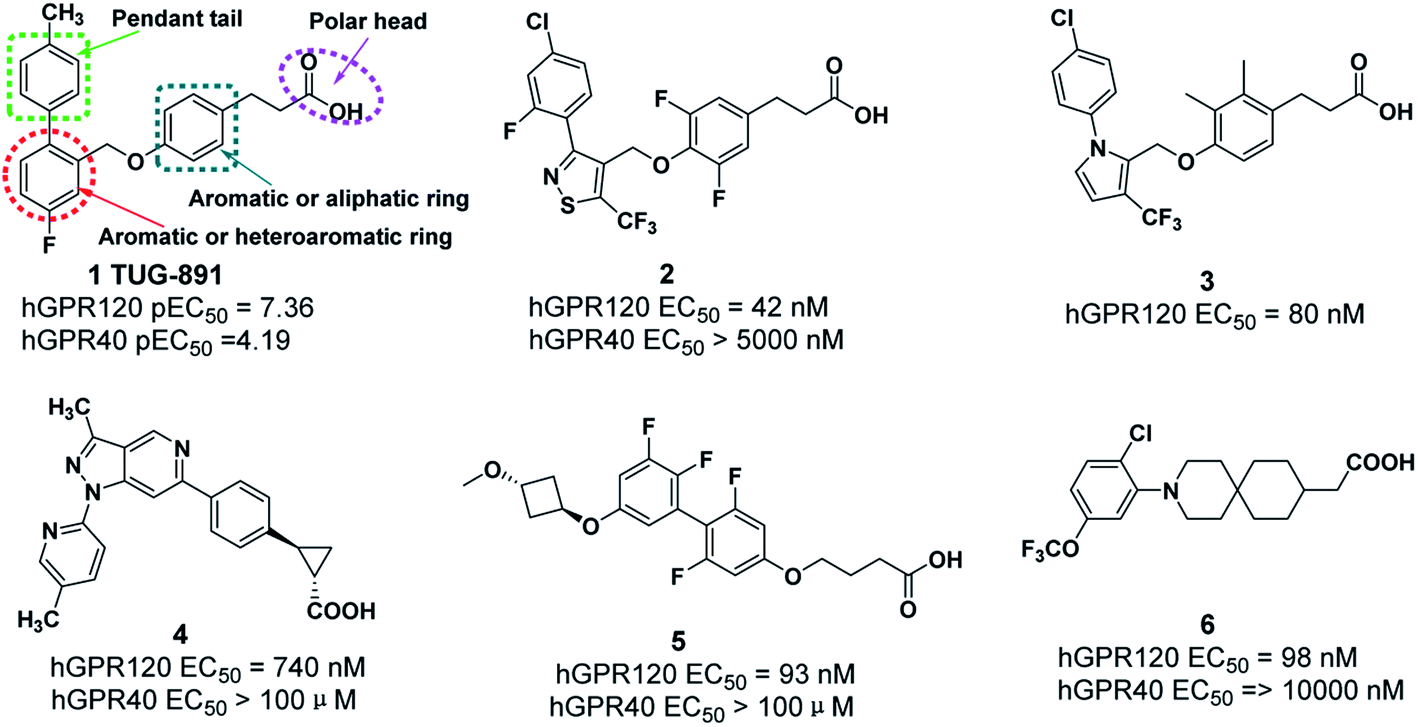 | ||
| Fig. 1 GPR120 agonists. | ||
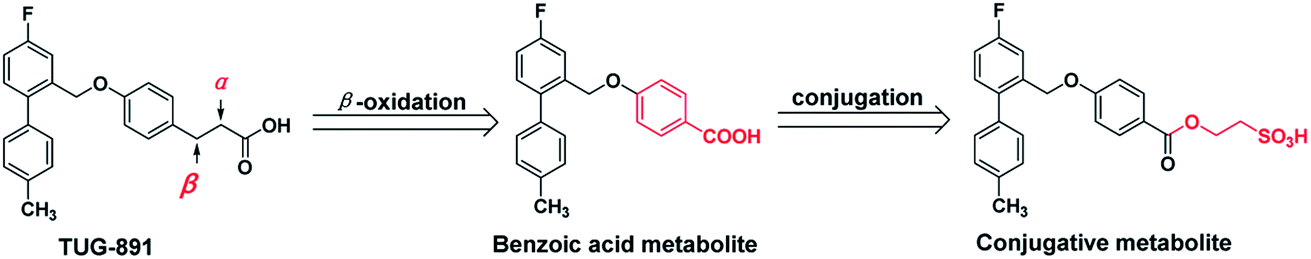 | ||
| Fig. 2 The speculative metabolic route of TUG-891. | ||
The thiazolidinedione heterocycle is a planar, moderately acidic (pKa = 6–7), and relatively lipophilic carboxylic acid bioisostere, employed as a carboxylic acid bioisostere in the design of PPARγ agonists22 and GPR40 agonists23,24 for the treatment of type 2 diabetes (Fig. 3). Moreover, the thiazolidinedione moiety is metabolically stable and the main metabolic routes of rosiglitazone are N-demethylation, hydroxylation, and subsequent conjugation with sulfate and glucuronic acid.25
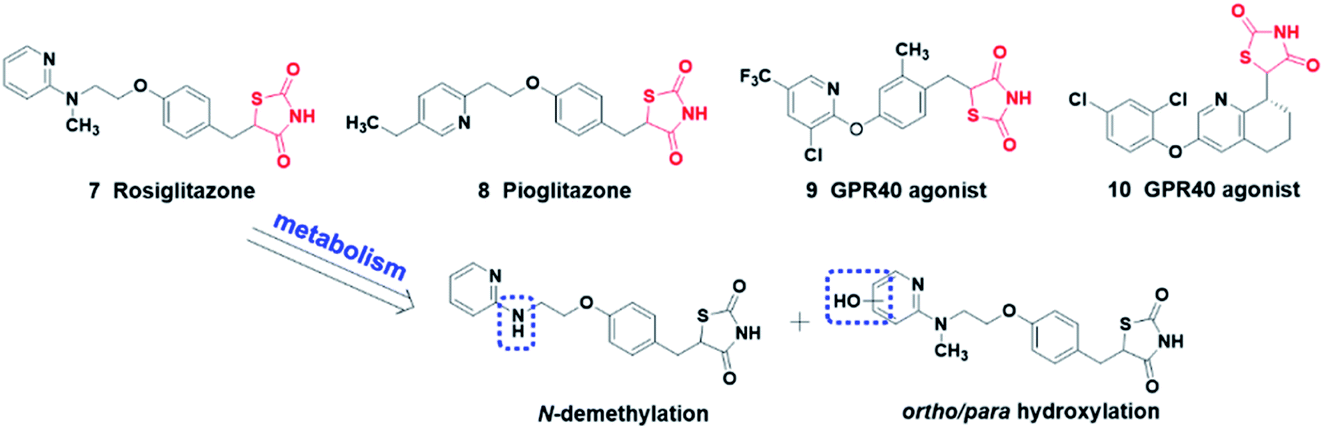 | ||
| Fig. 3 Drugs containing thiazolidinedione moiety and the metabolic route of rosiglitazone. | ||
Based on the previous study of the structure–activity relationship of TUG-891,14 a series of novel GPR120 agonists containing thiazolidinedione moiety were designed, and we expected that they are metabolically stable and have little effect on the physiological mechanism of TUG-891 (Fig. 4A). Molecular overlay studies were performed to simulate the difference in molecular conformation between compound 1g and TUG-891, and compound 1g with thiazolidinedione moiety exhibited an excellent degree of overlap with TUG-891 (Fig. 4B). Thereafter, homology modeling was performed with Modeler in Accelrys Discovery Studio 2020 using the crystal structure of turkey β1 adrenoceptor 20 (PDB code 6IBL),26 neurotensin receptor21 (PDB code 4XES)27 and β2-adrenoceptor (PDB code 3P0G) 28 as templates, and the structure with the lowest DOPE score was used for molecular docking. Molecular docking was completed using the CDOCKER in Accelrys Discovery Studio 2020. The molecular docking model showed that the thiazolidinedione moiety was engaged in double hydrogen bond interactions with Arg99, which plays a very important role in GPR120 activation,29 and a hydrogen bond interaction with Leu196 (Fig. 4C). All simulation results indicated that thiazolidinedione analogs were likely to exhibit the required GPR120 agonistic activity.
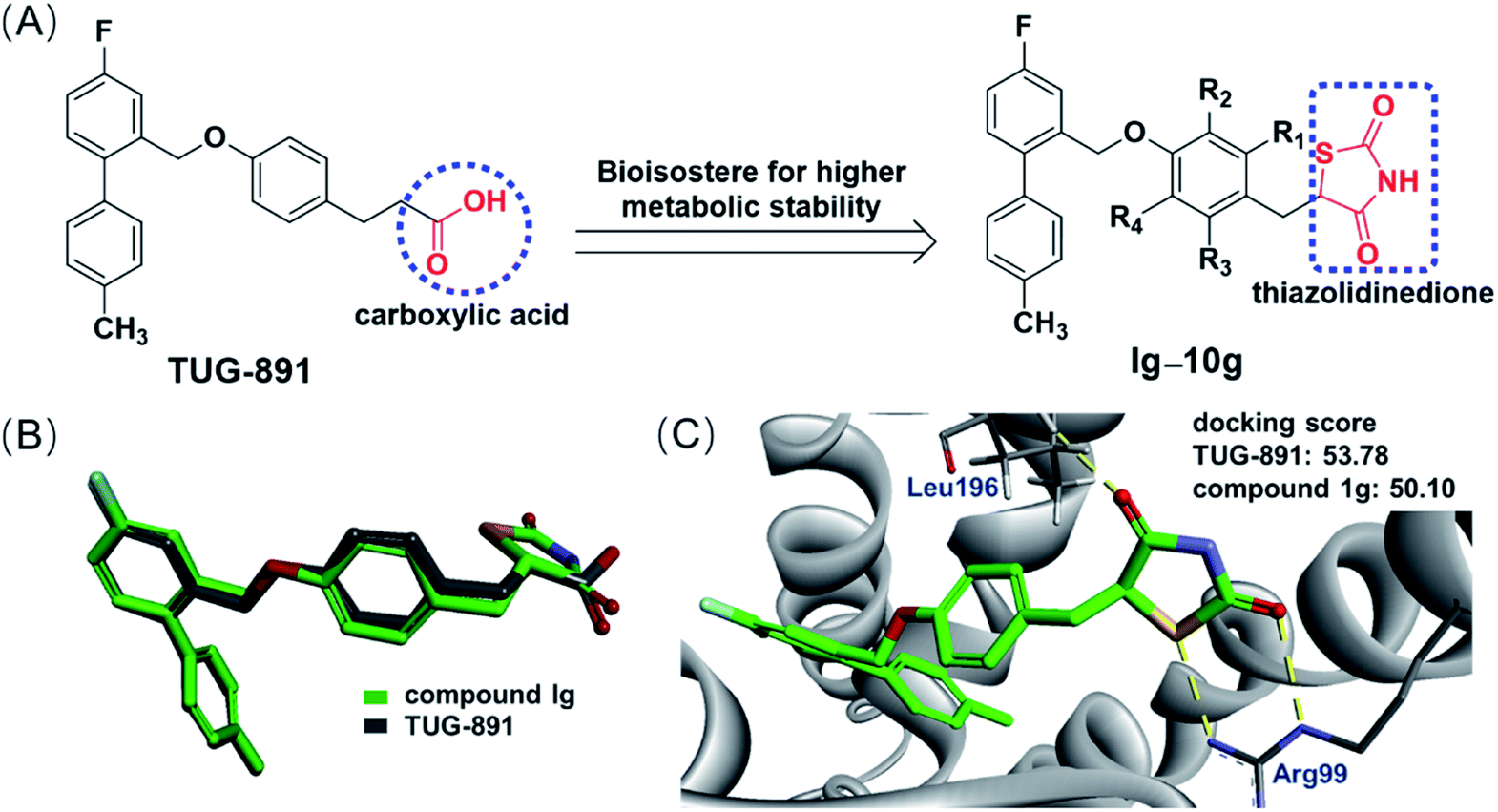 | ||
| Fig. 4 (A) The idea of substituting thiazolidinedione for the carboxylic acid of TUG-891. (B) The conformation overlap between TUG-891 and compound 1g. (C) Docking of compound 1g inside of a homology GPR120 model. | ||
Compound 5g showed ideal metabolic stability with higher maximum concentration, longer half-life than TUG-891, and excellent agonistic activity. In addition, compound 5g dose-dependently improved oral glucose tolerance in normal mice and reduced blood glucose by promoting the secretion of insulin diet-induced obese (DIO) mice.
2. Materials and methods
2.1 Chemistry
All commercially available materials and reagents were used without purification unless otherwise indicated. Purification by column chromatography was performed using silica gel (200–300 mesh). The NMR spectra (500 MHz for 1H NMR and 125 MHz for 13C NMR spectra) were recorded using a on Bruker AVANCE NEO 500 instrument. Chemical shifts are shown as values relative to the internal standard (tetramethylsilane), and coupling constants (J values) are given in hertz (Hz). High-resolution mass spectrometry was conducted using a UPLC G2-XS Qtof spectrometer (Waters) with the electrospray ionization Fourier transform ion cyclotron resonance (ESI-FTICR) technique. High-performance liquid chromatography (HPLC) data were obtained using a Shimadzu LC-20AT (Japan). The NMR and HRMS spectra of compounds 1g–10g and TUG-891 are presented in ESI Fig. S1–S44.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as eluent to afford the desired product 1b (4.29 g, 20.04 mmol, 81%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 9.83 (d, J = 3.2 Hz, 1H), 7.64–7.58 (m, 2H), 7.58–7.54 (m, 1H), 7.33 (s, 4H), 2.39 (s, 3H); HRMS (ES+) for C14H11FO (M + H)+: calcd 215.0872; found, 215.0872.:1, v/v) as eluent to afford a white solid (3.16 g, 11.37 mmol, 58%). 1H NMR (500 MHz, DMSO-d6) δ 7.34 (dd, J = 10.4, 2.9 Hz, 1H), 7.26–7.19 (m, 5H), 7.12 (td, J = 8.5, 2.9 Hz, 1H), 4.39 (s, 2H), 2.35 (s, 3H); HRMS (ES+) for C14H12BrF (M + H)+: calcd 279.0185; found, 279.0191.:1, v/v) as eluent to afford the desired product.
:1, v/v) as eluent to afford the desired product 1b (4.29 g, 20.04 mmol, 81%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 9.83 (d, J = 3.2 Hz, 1H), 7.64–7.58 (m, 2H), 7.58–7.54 (m, 1H), 7.33 (s, 4H), 2.39 (s, 3H); HRMS (ES+) for C14H11FO (M + H)+: calcd 215.0872; found, 215.0872.:1, v/v) as eluent to afford a white solid (3.16 g, 11.37 mmol, 58%). 1H NMR (500 MHz, DMSO-d6) δ 7.34 (dd, J = 10.4, 2.9 Hz, 1H), 7.26–7.19 (m, 5H), 7.12 (td, J = 8.5, 2.9 Hz, 1H), 4.39 (s, 2H), 2.35 (s, 3H); HRMS (ES+) for C14H12BrF (M + H)+: calcd 279.0185; found, 279.0191.:1, v/v) as eluent to afford the desired product.
2.1.4.1 4-((4-Fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzaldehyde (1e). White solid 0.18 g, yield 78%; 1H NMR (500 MHz, DMSO-d6) δ 9.86 (s, 1H), 7.87–7.82 (m, 2H), 7.46 (dd, J = 9.9, 2.8 Hz, 1H), 7.37 (dd, J = 8.5, 5.9 Hz, 1H), 7.32–7.26 (m, 3H), 7.23 (d, J = 7.9 Hz, 2H), 7.08 (d, J = 8.7 Hz, 2H), 5.07 (s, 2H), 2.33 (s, 3H); HRMS (ES+) for C21H17FO2 (M + H)+: calcd 321.1291; found, 321.1292.
2.1.4.2 4-((4-Fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)-2-methylbenzaldehyde (2e). White solid 0.17 g, yield 71%; 1H NMR (500 MHz, DMSO-d6) δ 9.86 (s, 1H), 7.87–7.82 (m, 2H), 7.46 (dd, J = 9.9, 2.8 Hz, 1H), 7.37 (dd, J = 8.5, 5.9 Hz, 1H), 7.32–7.26 (m, 3H), 7.23 (d, J = 7.9 Hz, 2H), 7.08 (d, J = 8.7 Hz, 2H), 5.07 (s, 2H), 2.33 (s, 3H); HRMS (ES+) for C22H19FO2 (M + H)+: calcd 335.1447; found, 335.1450.
2.1.4.3 4-((4-Fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)-2-methoxybenzaldehyde (3e). White solid 0.19 g, yield 75%; 1H NMR (500 MHz, DMSO-d6) δ 10.16 (s, 1H), 7.63 (d, J = 8.7 Hz, 1H), 7.46 (dd, J = 9.9, 2.8 Hz, 1H), 7.37 (dd, J = 8.5, 5.8 Hz, 1H), 7.33–7.27 (m, 3H), 7.24 (d, J = 7.9 Hz, 2H), 6.68 (d, J = 2.2 Hz, 1H), 6.59 (dd, J = 8.6, 2.2 Hz, 1H), 5.07 (s, 2H), 3.86 (s, 3H), 2.33 (s, 3H); HRMS (ES+) for C22H19FO3 (M + H)+: calcd 351.1396; found, 351.1398.
2.1.4.4 2-Fluoro-4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzaldehyde (4e). White solid 0.20 g, yield 82%; 1H NMR (500 MHz, DMSO-d6) δ 10.18 (s, 1H), 7.81 (d, J = 8.7 Hz, 1H), 7.47 (dd, J = 9.9, 2.8 Hz, 1H), 7.37 (dd, J = 8.6, 5.9 Hz, 1H), 7.31 (dd, J = 8.5, 2.8 Hz, 1H), 7.27 (d, J = 8.2 Hz, 2H), 7.23 (d, J = 7.9 Hz, 2H), 7.13 (d, J = 2.4 Hz, 1H), 7.04 (dd, J = 8.7, 2.4 Hz, 1H), 5.09 (s, 2H), 2.33 (s, 3H); HRMS (ES+) for C21H16F2O2 (M + H)+: calcd 339.1197; found, 339.1198.
2.1.4.5 2-Chloro-4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzaldehyde (5e). White solid 0.22 g, yield 86%; 1H NMR (500 MHz, DMSO-d6) δ 10.06 (s, 1H), 7.77 (t, J = 8.5 Hz, 1H), 7.47 (dd, J = 9.9, 2.7 Hz, 1H), 7.37 (dd, J = 8.5, 5.8 Hz, 1H), 7.33–7.28 (m, 1H), 7.27 (d, J = 8.1 Hz, 2H), 7.23 (d, J = 8.2 Hz, 2H), 6.97 (dd, J = 12.9, 2.4 Hz, 1H), 6.92 (dd, J = 8.7, 2.4 Hz, 1H), 5.07 (s, 2H), 2.33 (s, 3H).
2.1.4.6 4-((4-Fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)-3-methylbenzaldehyde (6e). White solid 0.17 g, yield 71%; 1H NMR (500 MHz, DMSO-d6) δ 9.82 (s, 1H), 7.72–7.68 (m, 2H), 7.47 (dd, J = 9.9, 2.8 Hz, 1H), 7.36 (dd, J = 8.5, 5.9 Hz, 1H), 7.32–7.27 (m, 3H), 7.24 (d, J = 7.8 Hz, 2H), 6.98 (d, J = 9.1 Hz, 1H), 5.11 (s, 2H), 2.33 (s, 3H), 2.19 (s, 3H); HRMS (ES+) for C22H19FO2 (M + H)+: calcd 335.1447; found, 335.1453.
2.1.4.7 3-Fluoro-4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzaldehyde (7e). White solid 0.18 g, yield 73%; 1H NMR (500 MHz, DMSO-d6) δ 9.86 (d, J = 2.0 Hz, 1H), 7.74–7.72 (m, 1H), 7.72–7.69 (m, 1H), 7.47 (dd, J = 9.8, 2.8 Hz, 1H), 7.38 (dd, J = 8.5, 5.9 Hz, 1H), 7.32 (dd, J = 8.5, 2.8 Hz, 1H), 7.30–7.26 (m, 3H), 7.23 (d, J = 7.8 Hz, 2H), 5.13 (s, 2H), 2.32 (s, 3H); HRMS (ES+) for C21H16F2O2 (M + H)+: calcd 339.1197; found, 339.1188.
2.1.4.8 2,3-Difluoro-4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzaldehyde (8e). White solid 0.19 g, yield 74%; 1H NMR (500 MHz, DMSO-d6) δ 10.03 (s, 1H), 7.65–7.59 (m, 1H), 7.48 (dd, J = 9.8, 2.8 Hz, 1H), 7.38 (dd, J = 8.5, 5.9 Hz, 1H), 7.32 (td, J = 8.5, 2.8 Hz, 1H), 7.27 (d, J = 8.2 Hz, 2H), 7.23 (d, J = 7.9 Hz, 2H), 7.10 (t, J = 8.1 Hz, 1H), 5.17 (s, 2H), 2.32 (s, 3H); HRMS (ES+) for C21H15F3O2 (M + H)+: calcd 357.1102; found, 357.1106.
2.1.4.9 2,6-Difluoro-4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzaldehyde (9e). White solid 0.20 g, yield 78%; 1H NMR (500 MHz, DMSO-d6) δ 10.06 (s, 1H), 7.48 (dd, J = 9.8, 2.7 Hz, 1H), 7.37 (dd, J = 8.6, 5.8 Hz, 1H), 7.31 (td, J = 8.5, 2.8 Hz, 1H), 7.26 (d, J = 8.2 Hz, 2H), 7.23 (d, J = 8.3 Hz, 2H), 6.87 (d, J = 11.1 Hz, 2H), 5.06 (s, 2H), 2.33 (s, 3H); HRMS (ES+) for C21H15F3O2 (M + H)+: calcd 357.1102; found, 357.1101.
2.1.4.10 4-((4-Fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)-2,5-dimethylbenzaldehyde (10e). White solid 0.18 g, yield 72%; 1H NMR (500 MHz, DMSO-d6) δ 10.05 (s, 1H), 7.59 (s, 1H), 7.46 (dd, J = 9.9, 2.8 Hz, 1H), 7.36 (dd, J = 8.5, 5.9 Hz, 1H), 7.30 (dd, J = 8.2, 2.3 Hz, 3H), 7.24 (d, J = 7.9 Hz, 2H), 6.71 (s, 1H), 5.08 (s, 2H), 2.51 (s, 4H), 2.33 (s, 3H), 2.13 (s, 3H); HRMS (ES+) for C23H21FO2 (M + H)+: calcd 349.1604; found, 349.1606.
:1, v/v) as eluent to afford the desired product.To an ice-cold solution of CoCl2·6H2O (0.03 mmol) and dimethylglyoxime (0.23 mmol) in water (6 mL) was added a drop of 1 N NaOH, followed by NaBH4 (0.45 mmol). To the resulting dark olive-amber solution was added, over a 10 min period, a solution of 1f–10f (0.45 mmol) in a mixture of THF (3 mL) and DMF (0.25 mL). The solution was allowed to gradually warm to room temperature and stirred overnight. The mixture was then acidified to pH 5 with HOAc, diluted with water (6 mL), extracted with red at room temperature for another two hours. The mixture was extracted with dichloromethane (3 × 15 mL), and the organic fractions were combined, washed with water (2 × 30 mL) and saturated brine (2 × 30 mL) prior to drying over anhydrous sodium sulfate. After filtration and concentrate under vacuum, the residue was crystallized from petroleum ether/ethyl acetate (4:1, v/v) to afford the desired product.
2.1.5.1 5-(4-((4-Fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzyl)thiazolidine-2,4-dione (1g). White solid 108 mg, yield 57% and purity 97.9%; mp 114–116 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.00 (s, 1H), 7.42 (dd, J = 9.9, 2.7 Hz, 1H), 7.34 (dd, J = 8.5, 5.9 Hz, 1H), 7.32–7.25 (m, 3H), 7.22 (d, J = 8.0 Hz, 2H), 7.13 (d, J = 8.6 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 4.91 (s, 2H), 4.86 (dd, J = 9.0, 4.3 Hz, 1H), 3.29 (dd, J = 14.2, 4.3 Hz, 1H), 3.05 (dd, J = 14.2, 9.0 Hz, 1H), 2.33 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.14, 172.10, 162.83, 157.51, 138.08, 137.27, 136.96, 136.46, 132.33, 130.87, 129.57, 129.45, 129.36, 116.10, 115.45, 115.05, 67.56, 53.43, 36.76, 21.15; HRMS (ES−) for C24H20FNO3S (M − H)−: calcd 420.1070; found, 420.1071.
2.1.5.2 5-(4-((4-Fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)-2-methylbenzyl)thiazolidine-2,4-dione (2g). White solid 0.108 mg, yield 55% and purity 98.6%; mp 101–103 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.08 (s, 1H), 7.40 (dd, J = 9.9, 2.8 Hz, 1H), 7.34 (dd, J = 8.5, 6.0 Hz, 1H), 7.30–7.25 (m, 3H), 7.23 (d, J = 7.8 Hz, 2H), 7.04 (d, J = 8.4 Hz, 1H), 6.72 (d, J = 2.7 Hz, 1H), 6.66 (dd, J = 8.4, 2.8 Hz, 1H), 4.90 (s, 2H), 4.83 (dd, J = 10.5, 4.2 Hz, 1H), 3.38 (dd, J = 14.5, 4.2 Hz, 1H), 2.99 (dd, J = 14.5, 10.5 Hz, 1H), 2.34 (s, 3H), 2.24 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.32, 172.17, 162.82, 160.86, 157.37, 138.30, 138.09, 137.27, 137.04, 136.48, 132.33, 130.69, 129.47, 129.37, 128.53, 67.43, 52.79, 34.60, 21.19, 19.78; HRMS (ES−) for C25H22FNO3S (M − H)−: calcd 434.1226; found, 434.1230.
2.1.5.3 5-(4-((4-Fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)-2-methoxybenzyl)thiazolidine-2,4-dione (3g). White solid 106 mg, yield 52% and purity 98.7%; mp 155–157 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.01 (s, 1H), 7.43 (dd, J = 9.9, 2.8 Hz, 1H), 7.35 (dd, J = 8.6, 5.8 Hz, 1H), 7.31–7.25 (m, 3H), 7.23 (d, J = 8.0 Hz, 2H), 7.00 (d, J = 8.3 Hz, 1H), 6.55 (d, J = 2.4 Hz, 1H), 6.38 (dd, J = 8.3, 2.4 Hz, 1H), 4.93 (s, 2H), 4.77 (dd, J = 9.9, 4.5 Hz, 1H), 3.74 (s, 3H), 3.39 (dd, J = 13.9, 4.6 Hz, 1H), 3.33 (s, 3H), 2.86 (dd, J = 13.9, 10.0 Hz, 1H), 2.33 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.35, 172.29, 162.82, 160.88, 159.14, 158.64, 138.10, 137.28, 136.94, 136.46, 132.35, 131.27, 129.46, 129.38, 118.23, 116.29, 115.49, 105.58, 99.83, 67.74, 55.91, 52.14, 32.71, 21.15; HRMS (ES−) for C25H22FNO4S (M − H)−: calcd 450.1175; found, 450.1180.
2.1.5.4 5-(2-Chloro-4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzyl)thiazolidine-2,4-dione (4g). White solid 119 mg, yield 60% and purity 100%; mp 107–109 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.14 (s, 1H), 7.43 (dd, J = 9.8, 2.8 Hz, 1H), 7.35 (dd, J = 8.5, 5.8 Hz, 1H), 7.30–7.25 (m, 3H), 7.25–7.21 (m, 3H), 7.00 (d, J = 2.6 Hz, 1H), 6.86 (dd, J = 8.5, 2.7 Hz, 1H), 4.95 (s, 2H), 4.84 (dd, J = 9.8, 4.7 Hz, 1H), 3.50 (dd, J = 14.3, 4.7 Hz, 1H), 3.13 (dd, J = 14.3, 9.8 Hz, 1H), 2.33 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 175.96, 171.96, 162.81, 160.87, 158.29, 138.28, 137.31, 136.38, 134.37, 133.15, 129.45, 129.35, 127.43, 116.34, 115.99, 115.66, 114.54, 68.05, 51.94, 34.89, 21.14; HRMS (ES−) for C24H19F2NO3S (M − H)−: calcd 438.0975; found, 438.0973.
2.1.5.5 5-(2-Fluoro-4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzyl)thiazolidine-2,4-dione (5g). White solid 131 mg, yield 64% and purity 98.5%; mp 116–118 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.08 (s, 1H), 7.43 (dd, J = 9.8, 2.8 Hz, 1H), 7.35 (dd, J = 8.6, 5.8 Hz, 1H), 7.27 (d, J = 7.7 Hz, 3H), 7.25–7.15 (m, 3H), 6.79 (dd, J = 11.7, 2.5 Hz, 1H), 6.72 (dd, J = 8.4, 2.5 Hz, 1H), 4.94 (s, 2H), 4.82 (dd, J = 8.9, 4.7 Hz, 1H), 3.41–3.34 (m, 1H), 3.08 (dd, J = 14.3, 8.9 Hz, 1H), 2.33 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 175.96, 171.99, 162.63, 160.68, 158.98, 138.27, 137.30, 136.37, 132.39, 132.20, 129.45, 129.35, 116.41, 116.26, 115.65, 111.35, 102.79, 68.08, 52.28, 30.62, 21.13; HRMS (ES−) for C24H19ClF2NO3S (M − H)−: calcd 454.0692; found, 454.0694.
2.1.5.6 5-(4-((4-Fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)-3-methylbenzyl)thiazolidine-2,4-dione (6g). White solid 108 mg, yield 55% and purity 98.2%; mp 172–173 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.01 (s, 1H), 7.43 (dd, J = 9.9, 2.8 Hz, 1H), 7.34 (dd, J = 8.5, 5.9 Hz, 1H), 7.31–7.25 (m, 3H), 7.23 (d, J = 7.8 Hz, 2H), 7.03 (d, J = 2.3 Hz, 1H), 6.96 (dd, J = 8.4, 2.3 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 4.94 (s, 2H), 4.83 (dd, J = 9.4, 4.3 Hz, 1H), 3.27 (dd, J = 14.2, 4.3 Hz, 1H), 2.99 (dd, J = 14.2, 9.3 Hz, 1H), 2.34 (s, 3H), 2.13 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.16, 172.13, 162.86, 160.92, 155.57, 137.89, 137.23, 136.53, 132.32, 131.90, 129.46, 129.26, 128.04, 126.36, 115.71, 115.32, 111.81, 67.56, 53.51, 36.90, 21.16, 16.56; HRMS (ES−) for C25H22FNO3S (M − H)−: calcd 434.1226; found, 434.1226.
2.1.5.7 5-(3-Fluoro-4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzyl)thiazolidine-2,4-dione (7g). White solid 115 mg, yield 58% and purity 99.9%; mp 101–103 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.05 (s, 1H), 7.44 (dd, J = 9.8, 2.8 Hz, 1H), 7.35 (dd, J = 8.5, 5.8 Hz, 1H), 7.31–7.25 (m, 3H), 7.22 (d, J = 7.8 Hz, 2H), 7.18–7.13 (m, 1H), 7.02–6.91 (m, 2H), 4.98 (s, 2H), 4.90 (dd, J = 8.9, 4.5 Hz, 1H), 3.32 (dd, J = 14.2, 4.5 Hz, 1H), 3.08 (dd, J = 14.2, 9.0 Hz, 1H), 2.33 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.05, 172.00, 162.80, 160.86, 152.81, 150.87, 145.27, 138.31, 137.33, 136.34, 132.40, 130.86, 129.44, 129.33, 125.98, 117.39, 116.30, 115.72, 68.75, 52.99, 36.59, 21.13; HRMS (ES−) for C24H19F2NO3S (M − H)−: calcd 438.0975; found, 438.0978.
2.1.5.8 5-(2,3-Difluoro-4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzyl)thiazolidine-2,4-dione (8g). White solid 121 mg, yield 59% and purity 100%; mp 207–209 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.10 (s, 1H), 7.45 (dd, J = 9.8, 2.8 Hz, 1H), 7.36 (dd, J = 8.5, 5.9 Hz, 1H), 7.30 (dd, J = 8.5, 2.8 Hz, 1H), 7.27 (d, J = 8.1 Hz, 2H), 7.22 (d, J = 7.9 Hz, 2H), 7.00 (t, J = 8.3 Hz, 1H), 6.87 (t, J = 7.4 Hz, 1H), 5.03 (s, 2H), 4.85 (dd, J = 8.7, 4.9 Hz, 1H), 3.39 (dd, J = 14.5, 5.0 Hz, 1H), 3.16 (dd, J = 14.4, 8.7 Hz, 1H), 2.33 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 175.83, 171.88, 162.79, 160.84, 150.52, 148.57, 146.86, 141.73, 139.77, 138.45, 137.35, 136.32, 135.93, 132.47, 129.44, 129.31, 125.45, 118.39, 116.46, 115.89, 110.80, 69.22, 51.94, 30.60, 21.13; HRMS (ES−) for C24H18F3NO3S (M − H)−: calcd 456.0881; found, 456.0890.
2.1.5.9 5-(2,6-Difluoro-4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzyl)thiazolidine-2,4-dione (9g). White solid 118 mg, yield 56% and purity 100%; mp 123–125 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.13 (s, 1H), 7.45 (dd, J = 9.8, 2.8 Hz, 1H), 7.36 (dd, J = 8.5, 5.8 Hz, 1H), 7.32–7.24 (m, 3H), 7.22 (d, J = 7.9 Hz, 2H), 6.72 (d, J = 9.6 Hz, 2H), 4.95 (s, 2H), 4.73 (dd, J = 8.6, 5.6 Hz, 1H), 3.30 (dd, J = 14.4, 5.6 Hz, 1H), 3.16 (dd, J = 14.4, 8.7 Hz, 1H), 2.33 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 175.60, 171.85, 163.30, 161.68, 159.17, 138.45, 137.33, 136.34, 135.87, 132.44, 129.45, 129.36, 116.63, 115.85, 105.27, 99.46, 99.23, 68.56, 51.33, 25.41, 21.12; HRMS (ES−) for C24H18F3NO3S (M − H)−: calcd 456.0881; found, 456.0884.
2.1.5.10 5-(2,6-Difluoro-4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)benzyl)thiazolidine-2,4-dione (10g). White solid 105 mg, yield 52% and purity 95.1%; mp 198–200 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.06 (s, 1H), 7.42 (dd, J = 9.9, 2.8 Hz, 1H), 7.34 (dd, J = 8.5, 5.9 Hz, 1H), 7.29 (d, J = 8.2 Hz, 2H), 7.28–7.23 (m, 3H), 6.93 (s, 1H), 6.55 (s, 1H), 4.93 (s, 2H), 4.79 (dd, J = 10.6, 4.1 Hz, 1H), 3.41–3.32 (m, 1H), 2.94 (dd, J = 14.4, 10.6 Hz, 1H), 2.34 (s, 3H), 2.18 (s, 3H), 2.08 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.49, 172.31, 162.85, 160.91, 155.32, 153.52, 138.03, 137.28, 136.58, 135.21, 132.37, 131.91, 129.40, 127.91, 123.68, 115.98, 115.39, 114.05, 67.53, 53.04, 34.65, 21.17, 19.58, 16.12; HRMS (ES−) for C26H24FNO3S (M − H)−: calcd 448.1383; found, 448.1382.
2.2 Biological methods
2.2.3.1 Pharmacokinetic analysis in C57BL/6 mice. The pharmacokinetic properties of TUG-891 and compound 5g were determined in C57BL/6 mice. Male C57BL/6 mice were fasted for 12 h, weighed, and randomized into two groups (four mice per group). Oral doses were prepared at a concentration of 10 mg mL−1 in 0.5% methylcellulose (0.5% MC) aqueous solution and administered in a volume of 1 mL kg−1. Blood samples were collected from the retro-orbital plexus into EDTA-containing microcentrifuge tubes at 5, 15 and 30 min and 1, 2, 4, 6, 8, 12, 24, and 36 h after oral administration. The plasma was separated by centrifugation at 5000 × g for 10 min and then stored at −80 °C until analysis. The proteins in the plasma were precipitated with two volumes of acetonitrile containing an internal standard, and centrifuged at 10000 × g for 10 min after vortexing. The supernatant was diluted 10 times with acetonitrile and centrifuged again, and 10 μL of supernatant was analyzed by Waters LC-PDA-MS/MS to determine plasma drug levels. Pharmacokinetic parameters were determined using the mean data from four mice at each time point. Statistical analysis of the data was performed using the DAS 2.1.1 statistical software program.
2.2.3.2 Oral glucose tolerance test of compound 5g in normal C57BL/6 mice. For glucose tolerance testing, male C57BL/6 mice aged 5 weeks were fasted overnight (12 h), weighed, bled via tail tip, and randomized into groups (n = 8). Mice were orally administered a single dose of vehicle (0.5% MC aqueous solution), TUG-891 (suspended in vehicle; 30 mg kg−1), or compound 5g (suspended in vehicle; 3 mg kg−1, 10 mg kg−1, 30 mg kg−1, 100 mg kg−1), 30 min before oral glucose loading (3 g kg−1). Blood samples were collected before drug administration (−30 min), before glucose loading (time 0), and 15, 30, 60, and 120 min after glucose loading. Blood glucose levels were measured using blood glucose test strips (Sannuo GA-3 type, Changsha, China).
2.2.3.3 Hypoglycemic effects of compound 5g explored in DIO mice. Male C57BL/6 mice, after one week adaptation, were fed a high-fat diet (45% calories from fat, from Mediscience Ltd, Yangzhou, China) ad libitum for an additional 12 weeks to induce insulin resistance, which was used as an acute model of type 2 diabetes. The diabetic C57BL/6 mice were fasted overnight (12 h), weighed, bled via the tail tip, and randomized into four groups (six mice per group) based on body weight and blood glucose levels (−30 min). Thereafter, the diabetic C57BL/6 mice were dosed orally with single doses of vehicle, TUG-891(suspended in vehicle; 20 mg kg−1), or 5g (suspended in vehicle; 20 mg kg−1), 30 min before oral glucose load (2 g kg−1). Blood samples were collected and measured in accordance with the oral glucose tolerance test in normal mice. In addition, plasma insulin levels were measured using mouse insulin RIA kit (Beijing North Institute of Biological Technology, China).
2.3 Molecular modeling
The amino sequence of GPR120 was obtained from the UniProtKB Database (identifier: Q5NUL3). Similar sequences were identified using the NCBI BLAST. Turkey β1 adrenoceptor20 (PDB code 6IBL), neurotensin receptor21 (PDB code 4XES) and β2-adrenoceptor (PDB code 3P0G) were selected as the templates to construct a homology model of the GPR120 receptor using Discovery Studio software. The CHARMm force field was added to the model with the lowest discrete optimized protein energy (DOPE) score. The model was used for molecular docking after energy optimization and evaluation. The structures of the ligands were etched using ChemBioDraw Ultra 14.0. Cdocker protocols were followed, and the top Cdocker Score was used for further analysis. The docking results were analyzed using Discovery Studio software. The complete molecular modeling protocol and parameters were given in the ESI.†3. Results discussion
3.1 Chemistry
The target compounds 1g–10g were obtained according to the synthetic route summarized in Scheme 1. Intermediate 1b was synthesized by Suzuki–Miyaura cross-coupling of 4-tolylboronic acid with 2-bromo-5-fluorobenzaldehyde in the catalysis of Pd(PPh3)4, which was further converted to brominated intermediate 1d by reduction with sodium borohydride and bromination with phosphorus tribromide. Condensation of 1d with appropriate 4-hydroxybenzaldehyde by Williamson ether synthesis afforded intermediate e–10e, followed by condensation with thiazolidine-2,4-dion and reduction with sodium borohydride to generate the target compounds 1g–10g. The structures of the target compounds were confirmed by 1H-NMR, 13C-NMR, and HRMS. | ||
| Scheme 1 Reagents and conditions: Synthesis of target compounds 1g–10g. (a) 4-Tolylboronic acid, Pd(PPh3)4, Na2CO3, toluene, ethanol, H2O, 80 °C, 12 h, 81%; (b) NaBH4, CH3OH, THF, 0 °C, 1 h; (c) PBr3, CH2Cl2, 0 °C, 1 h, 58%(two-steps); (d) K2CO3, DMF, rt., 12 h; 71–86% (e) NH4OAc, AcOH, 100 °C, 12 h; (f) biacetyl dioxime, NaBH4, CoCl2, NaOH, H2O, 0 °C, 52–64%(two-steps). | ||
3.2 GPR120 agonistic activity and selectivity
The agonistic activities of GPR120 of the target compounds 1g–10g were investigated early by monitoring the Ca2+ signal, and the GPR120 agonist TUG-891 was used as a positive control. The results showed that replacement of the carboxylic acid of TUG-891 with thiazolidinedione was well tolerated as the agonistic activity of compound 1g with unsubstituted was moderately reduced compared with TUG-891(1g: EC50 182.4 nM and TUG-981: EC50 51.2 nM). Compounds with a 2-position substituent, whether it was an electron-donating group or an electron-withdrawing substituent, could increase agonistic activity, while compounds with a 3-position substituent could significantly reduce agonistic activity compared with 1g (2g: EC50 = 133.7 nM and 6g, EC50 = 460.5 nM; 4g: EC50 = 90.5 nM, 7g, EC50 = 276.3 nM). Compounds substituted with electron withdrawing group at the 2-position (4g: EC50 = 90.5 nM) exhibited higher agonistic activity than compounds substituted with electron-donating group (2g: EC50 = 133.7 nM), and the same trend was found in the 3-position substitution (7g: EC50 = 276.3 nM and 6g: EC50 = 460.5 nM). These results indicate that electron-withdrawing substituents are beneficial for increasing the agonistic activity of GPR120. In addition, the size of the substituent also affects agonistic activity. Substituting a large-size 2-methoxy group for the 2-methyl group slightly increased the agonistic activity (3g: EC50 = 122.4 nM), and replacing 2-F with 2-Cl also improved the agonistic activity, which was similar to that of TUG-891 (5g: EC50 = 76.5 nM). Since 2-substitution was beneficial for increasing activity, some compounds with two substituents (2-position substitution and 3-or 5-position substitution) were synthesized and evaluated for agonistic activity. Consistent with the result of single substitution, the agonistic activity compounds with two electron-withdrawing groups were better than those with two electron-donating groups; however, their agonistic activity was lower than that of TUG-891 and 5g (8g: EC50 141.7 nM, 9g: EC50 137.3 nM, and 10g: EC50 = 400.2 nM) (Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | Docking scoreb | GPR120a (EC50, nM) |
| a EC50 values for GPR120 activities represent the mean of three determinations, values are expressed as the mean ± SD.b Docking score values for -CDOCKER interaction energy. | ||||||
| TUG-891 | — | — | — | — | 53.78 | 51.2 ± 7.2 |
| 1g | H | H | H | H | 50.10 | 182.4 ± 13.2 |
| 2g | CH3 | H | H | H | 54.55 | 133.7 ± 11.8 |
| 3g | OCH3 | H | H | H | 55.05 | 122.4 ± 13.1 |
| 4g | F | H | H | H | 52.38 | 90.5 ± 6.2 |
| 5g | Cl | H | H | H | 53.49 | 76.4 ± 8.9 |
| 6g | H | CH3 | H | H | 51.29 | 460.5 ± 36.1 |
| 7g | H | F | H | H | 50.73 | 276.3 ± 20.5 |
| 8g | F | F | H | H | 51.11 | 141.7 ± 12.5 |
| 9g | F | H | F | H | 51.27 | 137.3 ± 16.3 |
| 10g | CH3 | H | H | CH3 | 53.82 | 400.2 ± 34.6 |
Molecular docking of the designed compounds with GPR120 model was accomplished using CDOCKER-CHARMm-based technique in the interface of Accelrys Discovery Studio 2020 to provide insights of molecular binding modes of the tested molecules inside the pocket of GPR120 receptor built by the homology hypothesis. After that the docking scores (-CDOCKER interaction energy) of the best-fitted conformation of each of the docked molecules with the amino acids at the GPR120 binding pocket were recorded (Table 1). The results showed that although compound 5g and TUG-891 interacted with GPR120 in slightly different ways (Fig. 5A and B), compound 5g had similar docking score with TUG-891, and the agonistic activity of GPR120 was also comparable. When other electron-withdrawing groups were introduced, the docking scores were reduced, and the agonistic activities of GPR120 were also reduced. However, electron-donating groups were introduced into the benzene ring, the GPR120 agonistic activities were slightly lower than TUG-891, such as 3g and 4g, although the docking scores were higher than that of TUG-891. This might be due to the presence of electron releasing groups on benzene ring which could not significantly impart the biological activity.32
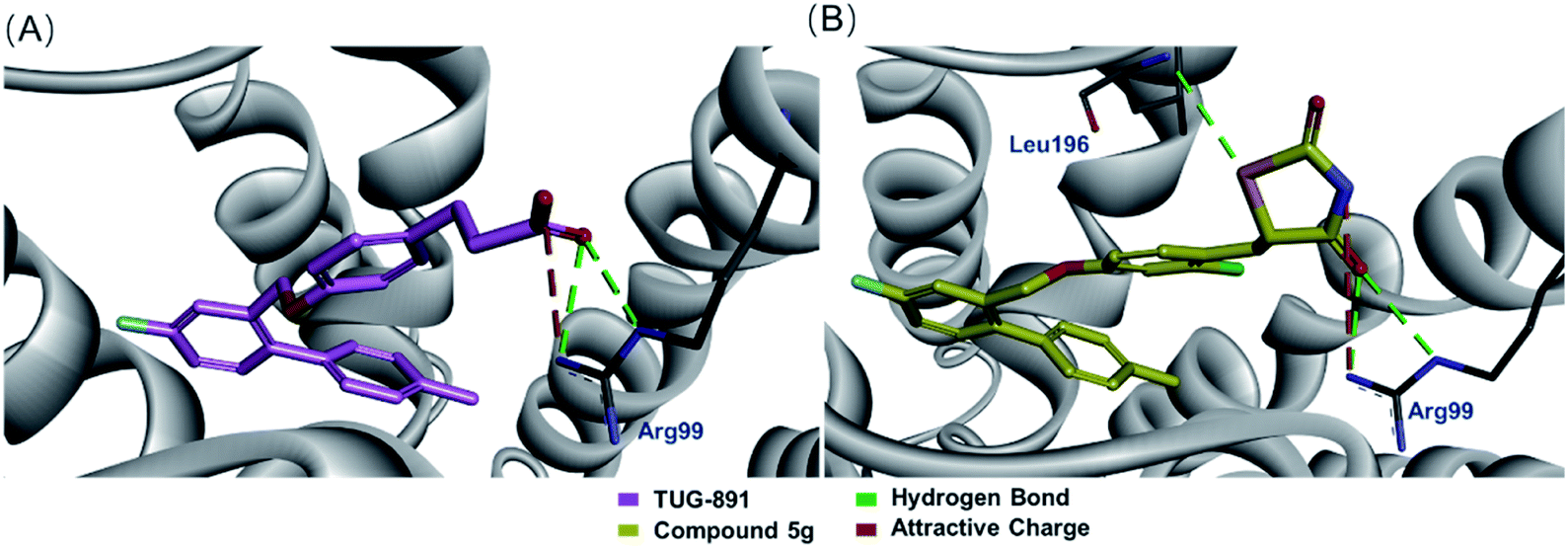 | ||
| Fig. 5 (A) Docking of TUG-891 inside of a homology GPR120 model; (B) docking of compound 5g inside of homology GPR120 model. | ||
The selectivity of compounds 4g and 5g with excellent GPR120 agonistic activity was evaluated, and the results revealed that the selectivity of compounds 4g and 5g was slightly improved compared with TUG-891, and the PPARγ agonistic activity of compounds 4g, 5g and TUG-891 could be ignored (EC50 > 100 μM) (Table 2). Therefore, the potent agonist 5g, with excellent selectivity and potency on GPR120 was selected for further investigation.
| Comp. | GPR120 (EC50) | GPR40 (EC50)a | PPARγ (EC50)b | Selectivity (GPR40/GPR120) | Selectivity (PPARγ/GPR120) |
|---|---|---|---|---|---|
| a EC50 values for GPR40 activities represent the mean of three determinations, values are expressed as the mean ± SD.b EC50 values for PPARγ activities represent the mean of three determinations, values are expressed as the mean ± SD. | |||||
| TUG-891 | 51.2 ± 7.2 nM | 42.8 ± 5.1 μM | >100 μM | 836 | >1953 |
| 4g | 90.5 ± 6.2 nM | 94.1 ± 10.2 μM | >100 μM | 1040 | >1105 |
| 5g | 76.4 ± 8.9 nM | 78.9 ± 6.7 μM | >100 μM | 1033 | >1309 |
3.3 Pharmacokinetic evaluation of compound 5g
The pharmacokinetics of compound 5g were obtained in normal C57BL/6 mice to determine if it had a suitable profile to investigate the in vivo effects. As a result, compound 5g revealed excellent PK profiles; in particular, a higher maximum plasma concentration (Cmax = 3872 ng mL−1) and longer half-life (t1/2 = 2.58 h), leading to a 8-fold higher exposure (AUC0–24h = 12805 ng mL−1 h−1) than TUG-891 (AUC0–24h = 1495 ng mL−1 h−1) (Table 3). These results indicated that it a successful strategy to improve stability by substituting thiazolidinedione for carboxylic acid, and compound 5g was sufficient for in vivo studies in rodents.| Comp. | Dose (mg kg−1) | Cmax (ng mL−1) | Tmax (min) | AUC (ng.h mL−1) | t1/2 (h) |
|---|---|---|---|---|---|
| a Data are average of four C57BL/6 mice, TUG-891 and compound 5g were suspended in 0.5% MC aqueous solution, values are expressed as the mean ± SD. **p ≤ 0.01 compared with TUG-891-treated by Student's t-test; ***p ≤ 0.001 compared with TUG-891-treated by Student's t-test. | |||||
| TUG-891 | 10 | 2298 ± 121 | 15 | 1495 ± 87 | 0.62 ± 0.14 |
| 5g | 10 | 3873 ± 163*** | 30 | 12805 ± 564*** | 2.58 ± 0.46** |
3.4 Oral glucose tolerance test of compound 5g in normal mice
Based on the experimental results, compound 5g with excellent activity, high selectivity and metabolic stability was selected for acute efficacy evaluation in normal C57BL/6 mice by oral glucose tolerance tests (oGTTs). Compound 5g was administered orally at doses of 3 mg kg−1, 10 mg kg−1, 30 mg kg−1, and 100 mg kg−1 30 min before the oral glucose challenge at a 3.0 g kg−1 dose. TUG-891 was used as a control, and glucose levels were measured from 30 min pre-glucose to 120 min post-glucose. The results illustrated that compound 5g lowered glucose in a dose-dependent manner, and the hypoglycemic effects were better than those of TUG-891 at 30 mg kg−1. In addition, no hypoglycemia was observed even at a dose of 100 mg kg−1 during the experiment (Fig. 6A and B).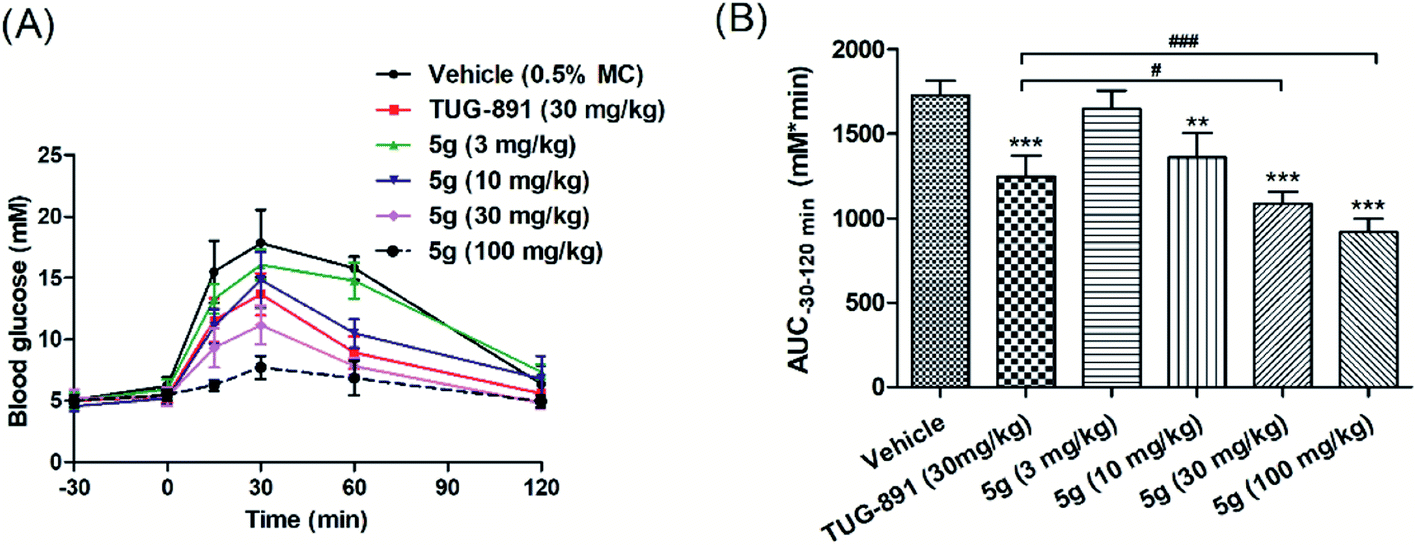 | ||
| Fig. 6 Effects of compound 5g and TUG-891 on plasma glucose levels during oGTT in fasting C57BL/6 mice. (A) Time-dependent changes of blood glucose levels after oral administration of compound 5g, followed by oral glucose load (3 g kg−1). (B) AUC−30–120min of plasma glucose levels. Values are expressed as the mean ± SD. (n = 6). **P ≤ 0.01 compared to vehicle-treated normal mice by Student's t-test; ***P ≤ 0.001 compared to vehicle-treated normal mice by Student's t-test; #P ≤ 0.05 compared to TUG-891-treated normal mice by Student's t-test; ###P ≤ 0.001 compared to TUG-891-treated normal mice by Student's t-test. | ||
3.5 Hypoglycemic effects of compound 5g explored in DIO mice
Thereafter, the anti-hyperglycemic and insulinotropic effects of compound 5g were evaluated in diet-induced obese (DIO) mice to assess the consequences of GPR120 agonist in an acute model of type 2 diabetes. The 20 mg kg−1 dose of 5g significantly reduced glucose excursion during oGTTs, and showed better antihyperglycemic effects than TUG-891(Fig. 7A and B). In addition, compound 5g significantly increased the secretion of insulin, which was more effective than TUG-891 (Fig. 7C and D). The results indicated that compound 5g increased insulin secretion and reduced blood sugar by activating GPTR120.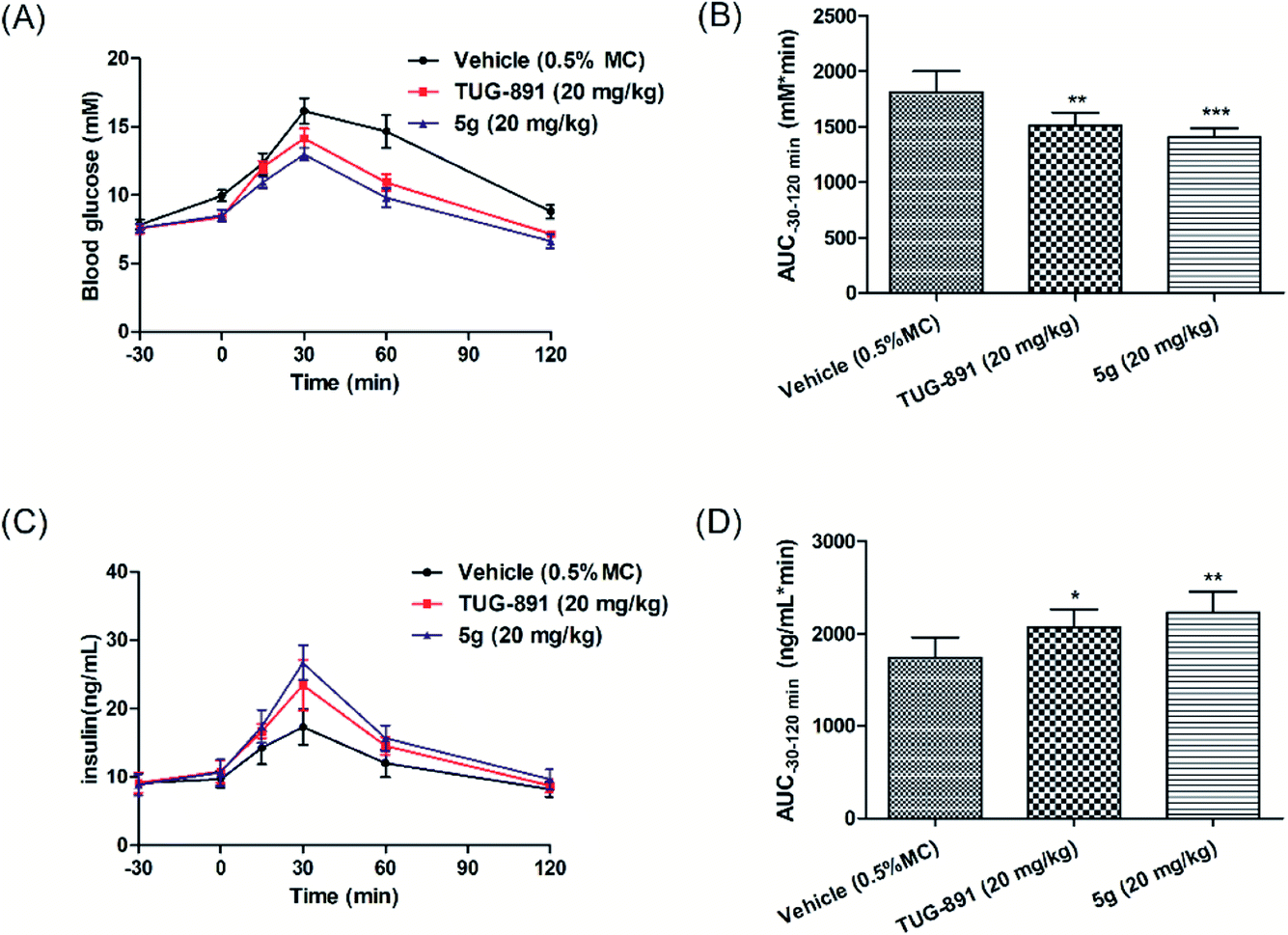 | ||
| Fig. 7 Effects of compound 5g and TUG-891 on plasma glucose levels and insulin levels during oGTT in fasting DIO mice. (A and C) The changes of blood glucose levels and insulin levels after oral administration of compound 5g, followed by oral glucose load (2 g kg−1), respectively. (B and D) AUC−30–120min of plasma glucose levels and insulin levels, respectively. Values are expressed as the mean ± SD. (n = 6). *P ≤ 0.05 compared to vehicle-treated DIO mice by Student's t-test; **P ≤ 0.01 compared to vehicle-treated DIO mice by Student's t-test; ***P ≤ 0.001 compared to vehicle-treated DIO mice by Student's t-test. | ||
4. Conclusions
GPR120 is a promising target for the treatment of type 2 diabetes. Although many GPR120 agonists have been reported in the literature, there is no clinical research on GPR120 agonists. For example, TUG-891, with good GPR120 agonistic activity, was easily metabolized in vivo and exhibited a short half-life. Based on the structure–activity relationship of TUG-891, this study substituted the carboxylic acid of TUG-891 with the bioisostere thiazolidinedione of carboxylic acid to synthesize a series of novel agonists with good GPR120 agonistic activity, high selectivity, and stable metabolism in vivo. The thiazolidinedione derivative 5g showed excellent GPR120 agonistic activity and selectivity. The metabolic stability of compound 5g was significantly improved, with a higher AUC and longer half-life compared with TUG-891. In summary, compound 5g, with excellent activity and suitable pharmacokinetic properties, is a promising candidate for treating type 2 diabetes.Author contributions
Xuekun Wang and Yadi Liu are roles for conceptualization, writing – original draft-preparation, and writing – review & editing-preparation. Xinyu Han, Huiran Hao, Wenjing Liu, Qidi Xue, Qinghua Guo, Shiben Wang, Kang Lei are roles for formal analysis, investigation and methodology.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This study was supported by the National Natural Science Foundation of China (81803360 and 32000037), Major Project of Research and Development of Shandong Province (2019GSF108226), the Provincial Natural Science Foundation of Jiangsu Province (BK20200614), Shandong Provincial Administration of Traditional Chinese Medicine (No. 2017-525) and the Doctoral Foundation of Liaocheng University (No. 318051745).References
- J. White, Diabetes Res. Clin. Pract., 2019, 157, 107932–107933 CrossRef PubMed.
- S. G. d. Lapertosa, A. F. d. Moura, C. Decroux, L. Duke, L. Hammond, E. Jacobs, A. Kaundal, J. Li, J. Liu, A. W. Ohlrogge, I. Petersohn, L. Piemonte, M. R. S. Prosser, E. Sung, M. Wilson, B. Y. Jiménez, W. Yang and M. Ysebaert, IDF Diabetes Atlas, International Diabetes Federation, Brussells, 9th edn, 2019, Available online: http://www.diabetesatlas.org Search PubMed.
- M. E. Doyle and J. M. Egan, Pharmacol. Rev., 2003, 55, 105–131 CrossRef CAS PubMed.
- Y. Itoh, Y. Kawamata and M. Harada, Nature, 2003, 1600, 2001–2004 Search PubMed.
- A. Hirasawa, K. Tsumaya, T. Awaji, S. Katsuma, T. Adachi, M. Yamada, Y. Sugimoto, S. Miyazaki and G. Tsujimoto, Nat. Med., 2005, 11, 90–94 CrossRef CAS PubMed.
- P. E. MacDonald, W. El-Kholy, M. J. Riedel, A. M. F. Salapatek, P. E. Light and M. B. Wheeler, Diabetes, 2002, 51, S434–S442 CrossRef CAS PubMed.
- G. V. Rayasam, V. K. Tulasi, J. A. Davis and V. S. Bansal, Expert Opin. Ther. Targets, 2007, 11, 661–671 CrossRef CAS PubMed.
- G. Milligan, B. Shimpukade, T. Ulven and B. D. Hudson, Chem. Rev., 2017, 117, 67–110 CrossRef CAS PubMed.
- I. Kimura, A. Ichimura, R. Ohue-Kitano and M. Igarashi, Physiol. Rev., 2019, 100, 171–210 CrossRef PubMed.
- V. A. Paschoal, E. Walenta, S. Talukdar, A. R. Pessentheiner, O. Osborn, N. Hah, T. J. Chi, G. L. Tye, A. M. Armando, R. M. Evans, N. W. Chi, O. Quehenberger, J. M. Olefsky and D. Y. Oh, Cell Metab., 2020, 31, 1173–1188 CrossRef CAS PubMed.
- M. P. Winters, Z. Sui, M. Wall, Y. Wang, J. Gunnet, J. Leonard, H. Hua, W. Yan, A. Suckow, A. Bell, W. Clapper, C. Jenkinson, P. Haug, T. Koudriakova, N. Huebert and W. V. Murray, Bioorg. Med. Chem., 2018, 28, 841–846 CrossRef CAS PubMed.
- W. McCoull, A. Bailey, P. Barton, A. M. Birch, A. J. Brown, H. S. Butler, S. Boyd, R. J. Butlin, B. Chappell, P. Clarkson, S. Collins, R. M. Davies, A. Ertan, C. D. Hammond, J. L. Holmes, C. Lenaghan, A. Midha, P. Morentin-Gutierrez, J. E. Moore, P. Raubo and G. J. Robb, Med. Chem., 2017, 60, 3187–3197 CrossRef CAS PubMed.
- X. Zhang, C. Cai, Z. Sui, M. Macielag, Y. Wang, W. Yan, A. Suckow, H. Hua, A. Bell, P. Haug, W. Clapper, C. Jenkinson, J. Gunnet, J. Leonard and W. V. Murray, ACS Med. Chem. Lett., 2017, 8, 947–952 CrossRef CAS PubMed.
- B. Shimpukade, B. D. Hudson, C. K. Hovgaard, G. Milligan and T. Ulven, J. Med. Chem., 2012, 55, 4511–4515 CrossRef CAS PubMed.
- R. Sheng, L. Yang, Y. Zhang, E. Xing, R. Shi, X. Wen, H. Wang and H. Sun, Bioorg. Med. Chem., 2018, 28, 2599–2604 CrossRef CAS PubMed.
- G. Ji, Q. Guo, Q. Xue, R. Kong, S. Wang, K. Lei, R. Liu and X. Wang, Molecules, 2021, 26, 6907–6921 CrossRef CAS PubMed.
- G. Carullo, S. Mazzotta, M. Vega-Holm, F. Iglesias-Guerra, J. M. Vega-Perez, F. Aiello and A. Brizzi, J. Med. Chem., 2021, 64, 4312–4332 CrossRef CAS PubMed.
- A. Kiepura, K. Stachyra, A. Wiśniewska, K. Kuś, K. Czepiel, M. Suski, M. Ulatowska-Białas, M. Surmiak and R. Olszanecki, Int. J. Mol. Sci., 2021, 22, 9772–9784 CrossRef CAS PubMed.
- T.-t. Wei, L.-t. Yang, F. Guo, S.-b. Tao, L. Cheng, R.-s. Huang, L. Ma and P. Fu, Acta Pharmacol. Sin., 2021, 42, 252–263 CrossRef CAS PubMed.
- B. D. Hudson, B. Shimpukade, A. E. Mackenzie, A. J. Butcher, J. D. Pediani, E. Christiansen, H. Heathcote, A. B. Tobin, T. Ulven and G. Milligan, Mol. Pharmacol., 2013, 84, 710–725 CrossRef CAS PubMed.
- Z. Li, X. Xu, G. Li, X. Fu, Y. Liu, Y. Feng, M. Wang, Y. Ouyang and J. Han, Bioorg. Med. Chem., 2017, 25, 6647–6652 CrossRef CAS PubMed.
- B. R. Henke, S. G. Blanchard, M. F. Brackeen, K. K. Brown, J. E. Cobb, J. L. Collins, W. W. Harrington, M. A. Hashim, E. A. Hull-Ryde and I. Kaldor, J. Med. Chem., 1998, 41, 5020–5036 CrossRef CAS PubMed.
- C. Zhou, C. Tang, E. Chang, M. Ge, S. Lin, E. Cline, C. P. Tan, Y. Feng, Y.-p. Zhou, G. J. Eiermann, A. Petrov, G. Salituro, P. Meinke, R. Mosley, T. E. Akiyama, M. Einstein, S. Kumar, J. Berger, A. D. Howard, N. Thornberry, S. G. Mills and L. Yang, Bioorg. Med. Chem. Lett., 2010, 20, 1298–1301 CrossRef CAS PubMed.
- J. C. Medina and J. B. Houze, Annu. Rep. Med. Chem., 2008, 43, 75–85 CAS.
- P. J. Cox, D. A. Ryan, F. J. Hollis, A.-M. Harris, A. K. Miller, M. Vousden and H. Cowley, Drug Metab. Dispos., 2000, 28, 772–780 CAS.
- Y. Lee, T. Warne, R. Nehmé, S. Pandey, H. Dwivedi-Agnihotri, M. Chaturvedi, P. C. Edwards, J. García-Nafría, A. G. Leslie and A. K. Shukla, Nature, 2020, 583, 862–866 CrossRef CAS PubMed.
- B. E. Krumm, J. F. White, P. Shah and R. Grisshammer, Nat. Commun., 2015, 6, 1–10 CAS.
- S. G. Rasmussen, H.-J. Choi, J. J. Fung, E. Pardon, P. Casarosa, P. S. Chae, B. T. DeVree, D. M. Rosenbaum, F. S. Thian and T. S. Kobilka, Nature, 2011, 469, 175–180 CrossRef CAS PubMed.
- B. D. Hudson, B. Shimpukade, G. Milligan and T. Ulven, J. Biol. Chem., 2014, 289, 20345–20358 CrossRef CAS PubMed.
- Z. Li, Y. Xu, Z. Cai, X. Wang, Q. Ren, Z. Zhou and R. Xie, Bioorg. Chem., 2020, 99, 103803 CrossRef CAS PubMed.
- Q. Ren, L. Deng, Z. Zhou, X. Wang, L. Hu, R. Xie and Z. Li, Bioorg. Chem., 2020, 101, 103963 CrossRef CAS PubMed.
- N. Siddiqui, M. S. Alam, M. Sahu, M. J. Naim, M. S. Yar and O. Alam, Bioorg. Chem., 2017, 71, 230–243 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08925k |
| This journal is © The Royal Society of Chemistry 2022 |