
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclodextrin and its derivatives as effective excipients for amorphous ulipristal acetate systems†
Peng Wanga,
Yan Wangb,
Zili Suoa,
Yuanming Zhai c and
Hui Li*a
c and
Hui Li*a
aCollege of Chemical Engineering, Sichuan University, Chengdu, Sichuan, China. E-mail: lihuilab@sina.com; Fax: +86 028 85401207; Tel: +86 028 85405149
bSichuan Center for Disease Control and Prevention, Chengdu, Sichuan, China
cAnalytical & Testing Center, Sichuan University, P. R. China
First published on 23rd March 2022
Abstract
Many efforts have been devoted to screening new solid-state forms of poorly soluble drugs in the pharmaceutical industry, thus modulating the drug properties without changing the pharmacological nature. It is a wise strategy to prepare amorphous series with cyclodextrin (CD) and its derivatives as excipients to enhance the aqueous solubility, dissolution, and bioavailability of water-insoluble drugs. In this study, four binary amorphous mixtures of ulipristal acetate (UPA) with CDs (β-CD, γ-CD, dimethyl-β-CD, hydroxypropyl-β-CD) were prepared by the co-milling method and characterized in the solid-state. According to powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC), UPA existed in the noncrystalline form in the four binary amorphous mixtures. Fourier transform infrared spectroscopy (FT-IR) and nuclear magnetic resonance (NMR) indicated that UPA interacted with the four CDs, which was also verified by molecular docking. Compared with the crystalline and amorphous UPA, the solubility, dissolution, and stability of the drug in the four amorphous UPA systems were significantly improved, so they were considered potentially advantageous solid forms. Our research shows that CDs can be used as new effective excipients in amorphous systems for active pharmaceutical ingredients (API).
1. Introduction
Approximately 90% of current candidates exhibit limited water solubility, which often causes oral absorption problems and increases the side effects of poorly water-soluble drugs.1 Converting crystalline drugs into an amorphous form is an effective solubilization technology.2 In the amorphous state, the absence of long-range molecular order and the higher Gibbs free energy in amorphous drugs are the main reason for their higher solubility, and hence dissolution rate and bioavailability.3 However, due to the high thermodynamic energy, most amorphous drugs alone are not sufficiently physically stable during the process, storage, and dissolution.4In some cases, polymers with a high glass transition temperature are often added to prepare amorphous solid dispersions (ASDs) in order to prevent the crystallization of an amorphous drug.5 However, this technique has some disadvantages that cannot be ignored.6 For example, the limited miscibility of the drug with the polymer sometimes requires the addition of a large amount of polymer, which is disadvantageous for the usability, manufacturing, and cost.7 Besides, ASDs also have problems of poor long-term stability as well as difficulty in manufacturing and processing.8 Co-amorphous systems have been extensively studied in the pharmaceutical industry because of their excellent properties, such as promoting drug solubilization, enhancing drug stability, and improving drug delivery and absorption.9,10 They are combinations of a drug and other small molecules, such as organic acids, amino acids, and other drugs.11,12 A drug and small molecule may interact via hydrogen bonds or other noncovalent bonds, which can reduce the mobility of long-range disordered molecules in amorphous drugs, thereby improving their physical properties.13 Whilst there are clear advantages of co-amorphous systems, there are also some challenges, such as the high hygroscopicity of small molecules that sometimes affect their stability, thus limiting their practical applications.14
Cyclodextrins (CDs) have been recognized as pharmaceutical excipients, which are cyclic oligosaccharides composed of D-glucopyranose units with a slightly conical hollow cylinder stereoscopic ring structure.15,16 In terms of the number of D-glucose units, the common natural CD can be classified as alpha-CD (α-CD), beta-CD (β-CD), and gamma-CD (γ-CD).17 With the development of pharmaceutics, more CD derivatives have been developed and applied. Among those derivatives, 2,6-dimethyl-β-CD (DM-β-CD) and 2-hydroxypropyl-β-CD (HP-β-CD) with good water solubility are the most commonly used.18 They all have satisfactory inclusion ability, surface activity, and cavity depth suitable for drug loading.19 Due to the special molecular structure with hydrophilic exteriors and hydrophobic internal cavity,20 CDs exhibit vast application potential in the pharmaceuticals, drug delivery systems, and chemical industries.21,22 The high density of hydroxyl groups makes it possible to interact with drugs groups, which also makes it a versatile molecule for modifying drug properties.23 CDs can effectively increase the solubility and dissolution rate in an aqueous medium of compounds with low solubility and pharmacodynamic properties of drug molecules.24 More generally, the importance of substitution patterns may also affect the structure–activity relationships on poorly soluble drugs (interactions, complexations, toxicity, stability, and solubilizing effects…).25 However, most of the current research focuses on the applications of CDs, and there are relatively few studies on the screening and properties of binary amorphous materials prepared by CDs.
Ulipristal acetate (UPA) is the first progestin receptor modulator after unprotected sexual intercourse within 5 days. It has a high inhibitory effect on progesterone receptors.26 UPA belongs to BCS II class compound and most of the commercially available raw materials are crystalline, whose low solubility limits its clinical application.27 As a contraceptive, it is important to improve the dissolution and release of UPA. Due to the small cavity structure, α-CD is not suitable for the system of larger molecules like UPA. Therefore, β-CD, γ-CD, DM-β-CD, and HP-β-CD were used as excipients in this paper, we have prepared four UPA–CD binary amorphous materials by the co-milling method. Powder X-ray diffraction (PXRD) was used to monitor the preparation process while differential scanning calorimetry (DSC), infrared spectroscopy (FT-IR), and nuclear magnetic resonance (NMR) were used to characterize. Besides, the interaction between UPA and CDs was investigated by molecular docking. The properties of the different solid drug forms were investigated from three aspects of solubility, dissolution rate, and stability to evaluate their potential as a drug product with high bioavailability.
2. Experimental
2.1. Materials
The main drug crystalline UPA (purity ≥99%) was obtained from Chengdu D-innovation Pharmaceutical Technology Co., Ltd. β-CD, γ-CD, DM-β-CD, and HP-β-CD was purchased from Kelong Company, Ltd (Chengdu, China) (Fig. 1). Phosphoric acid and potassium dihydrogen phosphate (chromatographic pure) were purchased from Ourchem (Chengdu, China) and Fisher Scientific (Fair Lawn, NJ, USA), respectively. Water used in the experiment was purified using a Milli Q water purification system (Millipore Corp., MA, USA).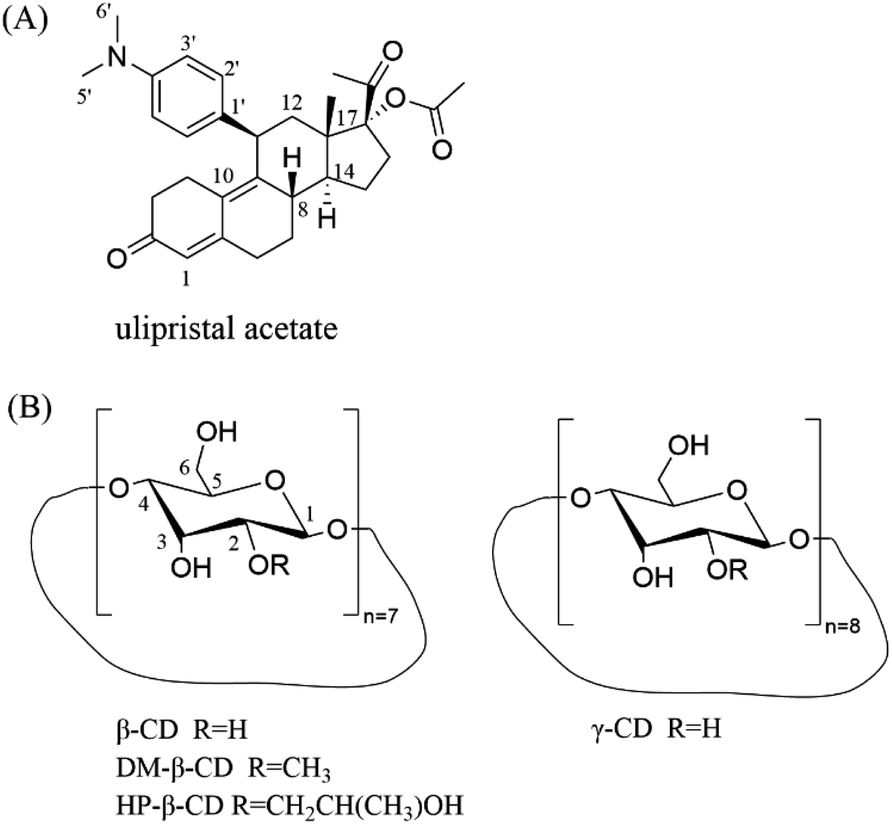 | ||
| Fig. 1 Chemical structures of (A) UPA and (B) CDs. | ||
2.2. Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were placed into agate ball jars and co-milling without solvent added. All the solid powders were withdrawn and analyzed by PXRD. The grinding stopped when all diffraction peaks disappeared. Physical mixtures (PM) were prepared by geometric mixing of crystalline UPA with each CD at equal mass ratios using a motor and pestle for a few minutes.200000 lux h) were investigated. The stability of different solid forms was monitored by PXRD at 5, 10, and 30 days under different conditions. At the same time, the impurities in UPA during the different storage states were quantitatively analyzed, and the relevant substances of different UPA solid forms were also detected by the high-performance liquid chromatography (HPLC).
:1 were placed into agate ball jars and co-milling without solvent added. All the solid powders were withdrawn and analyzed by PXRD. The grinding stopped when all diffraction peaks disappeared. Physical mixtures (PM) were prepared by geometric mixing of crystalline UPA with each CD at equal mass ratios using a motor and pestle for a few minutes.200000 lux h) were investigated. The stability of different solid forms was monitored by PXRD at 5, 10, and 30 days under different conditions. At the same time, the impurities in UPA during the different storage states were quantitatively analyzed, and the relevant substances of different UPA solid forms were also detected by the high-performance liquid chromatography (HPLC).
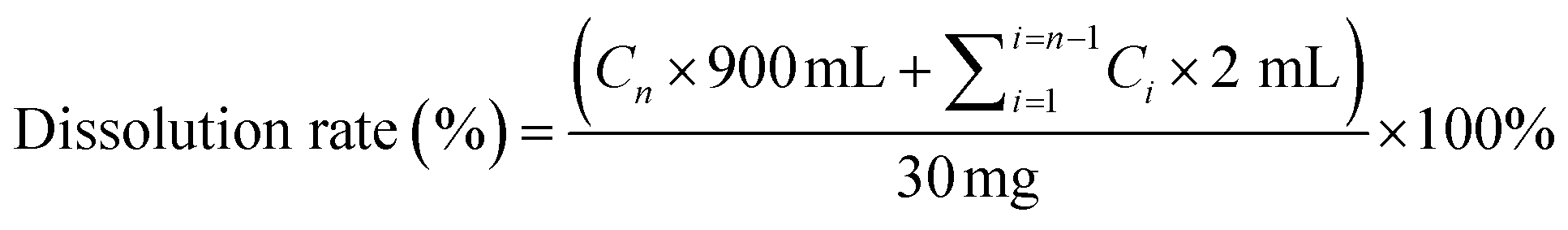 | (1) |
2.3 Analytical techniques
:10) mixture as mobile phase 1 mL min−1. The retention time is 7.59 min.The related substances of different UPA solid form samples during storage were quantified by external calibration at 40 °C using an Xtimate® C18 column (250 mm × 4.6 mm, 5 μm). The mobile phase was a mixture of acetonitrile and water (70:30 v/v) at a flow rate of 1.0 mL min−1 and the retention time is 5.25 min (Fig. S1†).
3. Results and discussion
3.1. Characterization of solid dispersion
Four CDs and their derivatives (β-CD, γ-CD, DM-β-CD, HP-β-CD) were selected to evaluate whether they could successfully convert into the binary amorphous systems. The co-milled solid powders were monitored by PXRD to confirm the successful preparation of the amorphous mixtures. As illustrated in Fig. 2, the four UPA–CD systems were gradually transformed into amorphous counterparts as the milling time increased. The formation time of UPA-γ-CD, UPA-DM-β-CD, and UPA-HP-β-CD amorphous mixed systems was greatly shortened. After grinding UPA-β-CD for 2 hours, the diffraction peaks also disappeared, which is attributed to dilution and surface coverage of the UPA particles by CDs.28 Moreover, UPA is presented in the amorphous form in the four UPA–CD binary amorphous systems. The phenomenon initially indicated the formations of amorphous UPA and UPA-CD amorphous mixtures (CM). UPA interacts with CDs to a certain extent, which promotes the formation of amorphous systems.
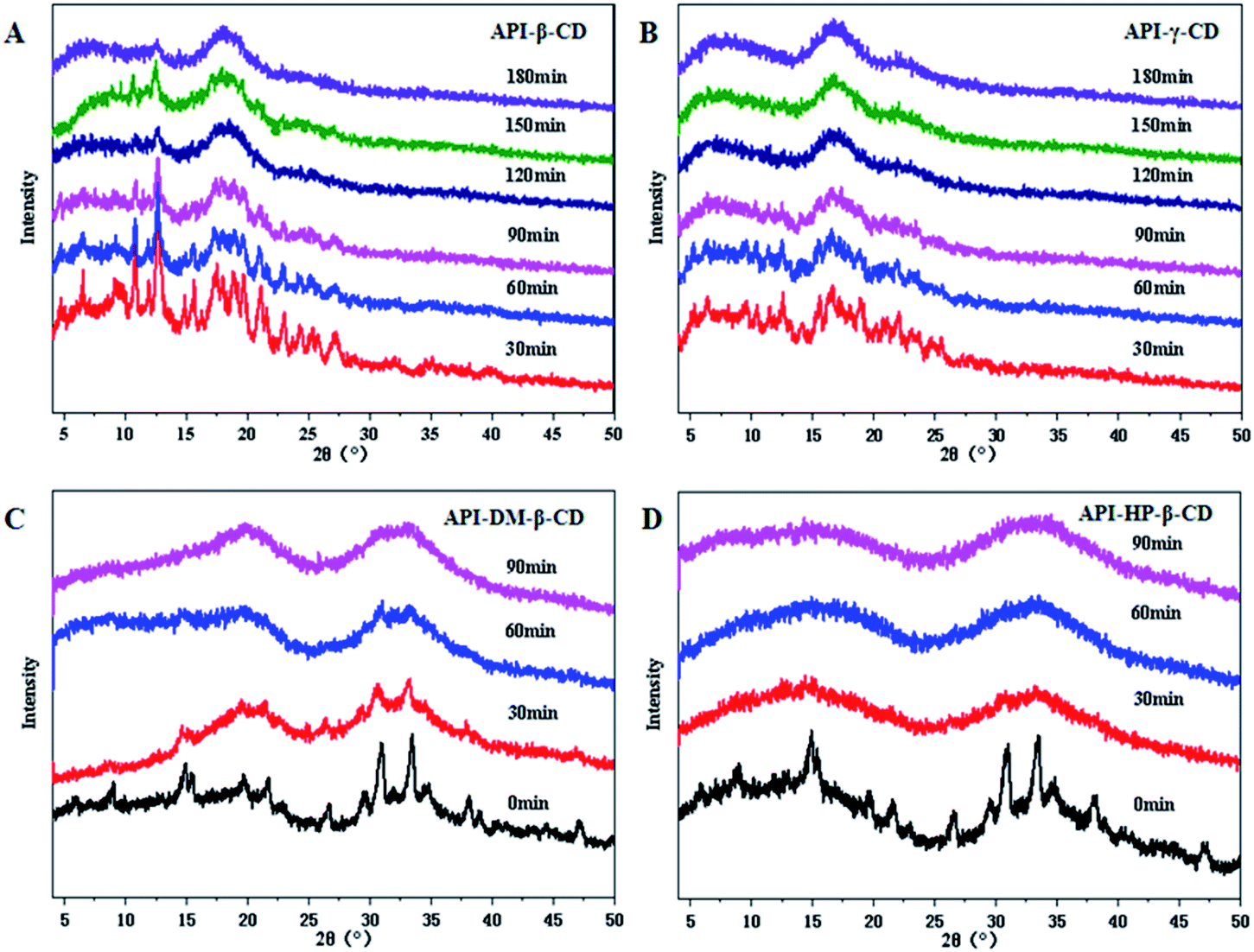 | ||
| Fig. 2 Preparation of amorphous systems of PXRD patterns of UPA-β-CD (A), UPA-γ-CD (B), UPA-DM-β-CD (C), and UPA-HP-β-CD (D) obtained at different time points during milling. | ||
 | ||
| Fig. 3 SEM photographs of UPA (A), amorphous UPA (B), UPA-β-CD amorphous (C), UPA-γ-CD amorphous (D), UPA-DM-β-CD amorphous (E), and UPA-HP-β-CD amorphous (F). | ||
There are significant differences in the structures of several single components. In our previous study, it was observed that β-CD was an irregular massive crystal, while HP-β-CD was a granular sphere with the cavity, DM-β-CD was spherical and accompanied by a large number of spherical ruptures, and HP-β-CD was a hollow spherical structure.29 However, the amorphous samples co-milled with UPA and CDs showed irregular block structures with different sizes and uneven surfaces, which were completely different from the morphology of single UPA and four CDs. Amorphous UPA was agglomerated together due to the static electricity produced during the grinding process. The amorphous mixture of UPA and four CDs have a uniform size, which is dispersed evenly. This finding may be due to the introduction of CD increasing the hydrophilicity of the samples, thus reducing static electricity and making the grinding more uniform.30
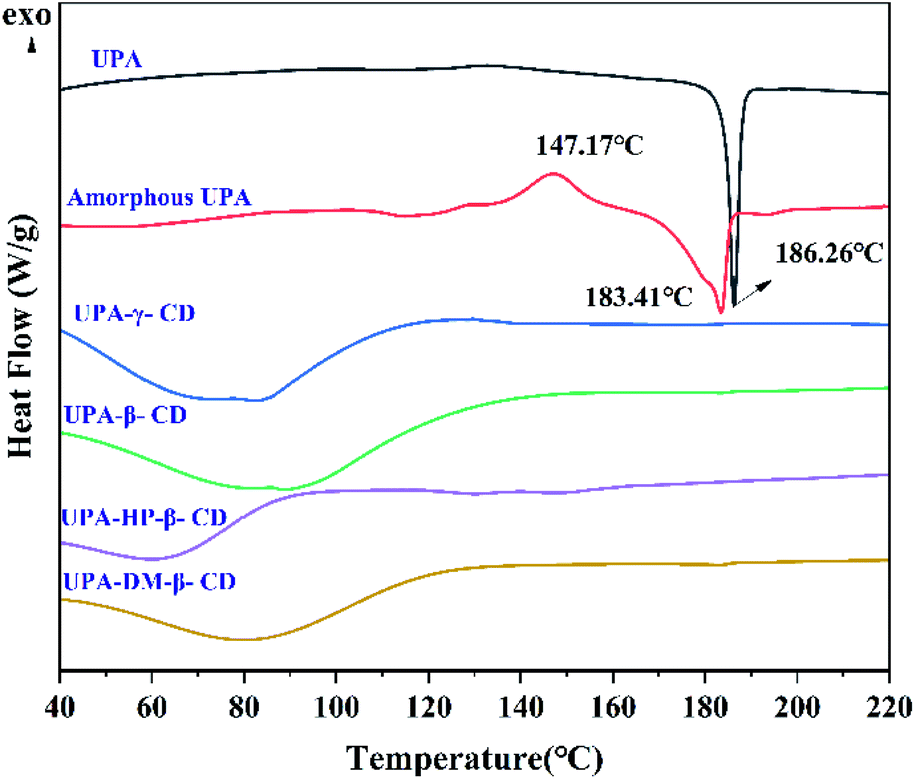 | ||
| Fig. 4 DSC curves of pure UPA, amorphous UPA, the UPA–CD amorphous. | ||
3.2. Interactions between UPA and CDs
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in UPA respectively. The CO peak intensity of amorphous UPA decreased significantly. The FT-IR spectra of the physical mixtures corresponded to the superposition of the FT-IR spectra of pure UPA and CDs. The FT-IR spectra of UPA-β-CD, UPA-γ-CD, UPA-DM-β-CD, and UPA-HP-β-CD showed some characteristic absorption peaks of UPA and CD, but the intensity of the UPA peak decreased significantly. Compared to the corresponding physical mixtures, after forming the UPA-β-CD amorphous, the absorption peaks of UPA at 1716 cm−1 for strong CO stretching vibration band, at 1598 cm−1 for C–O bending vibration, and at 1351 cm−1 for N–C covalent stretching band were masked by β-CD. The absorption peaks of UPA at 1438 cm−1 for C–H asymmetrical bending vibration and at 1365 cm−1 for C–H symmetric bending vibration showed reduced intensity and merged into a peak at 1456 cm−1. The same results were obtained in the FT-IR spectra of UPA-γ-CD, UPA-DM-β-CD, and UPA-HP-β-CD amorphous systems. The results showed that the interaction between UPA and CDs had a certain influence on the stretching vibration of the group, and then changed the peak shape or peak position. These results indicated the formation of binary amorphous systems.
O in UPA respectively. The CO peak intensity of amorphous UPA decreased significantly. The FT-IR spectra of the physical mixtures corresponded to the superposition of the FT-IR spectra of pure UPA and CDs. The FT-IR spectra of UPA-β-CD, UPA-γ-CD, UPA-DM-β-CD, and UPA-HP-β-CD showed some characteristic absorption peaks of UPA and CD, but the intensity of the UPA peak decreased significantly. Compared to the corresponding physical mixtures, after forming the UPA-β-CD amorphous, the absorption peaks of UPA at 1716 cm−1 for strong CO stretching vibration band, at 1598 cm−1 for C–O bending vibration, and at 1351 cm−1 for N–C covalent stretching band were masked by β-CD. The absorption peaks of UPA at 1438 cm−1 for C–H asymmetrical bending vibration and at 1365 cm−1 for C–H symmetric bending vibration showed reduced intensity and merged into a peak at 1456 cm−1. The same results were obtained in the FT-IR spectra of UPA-γ-CD, UPA-DM-β-CD, and UPA-HP-β-CD amorphous systems. The results showed that the interaction between UPA and CDs had a certain influence on the stretching vibration of the group, and then changed the peak shape or peak position. These results indicated the formation of binary amorphous systems.
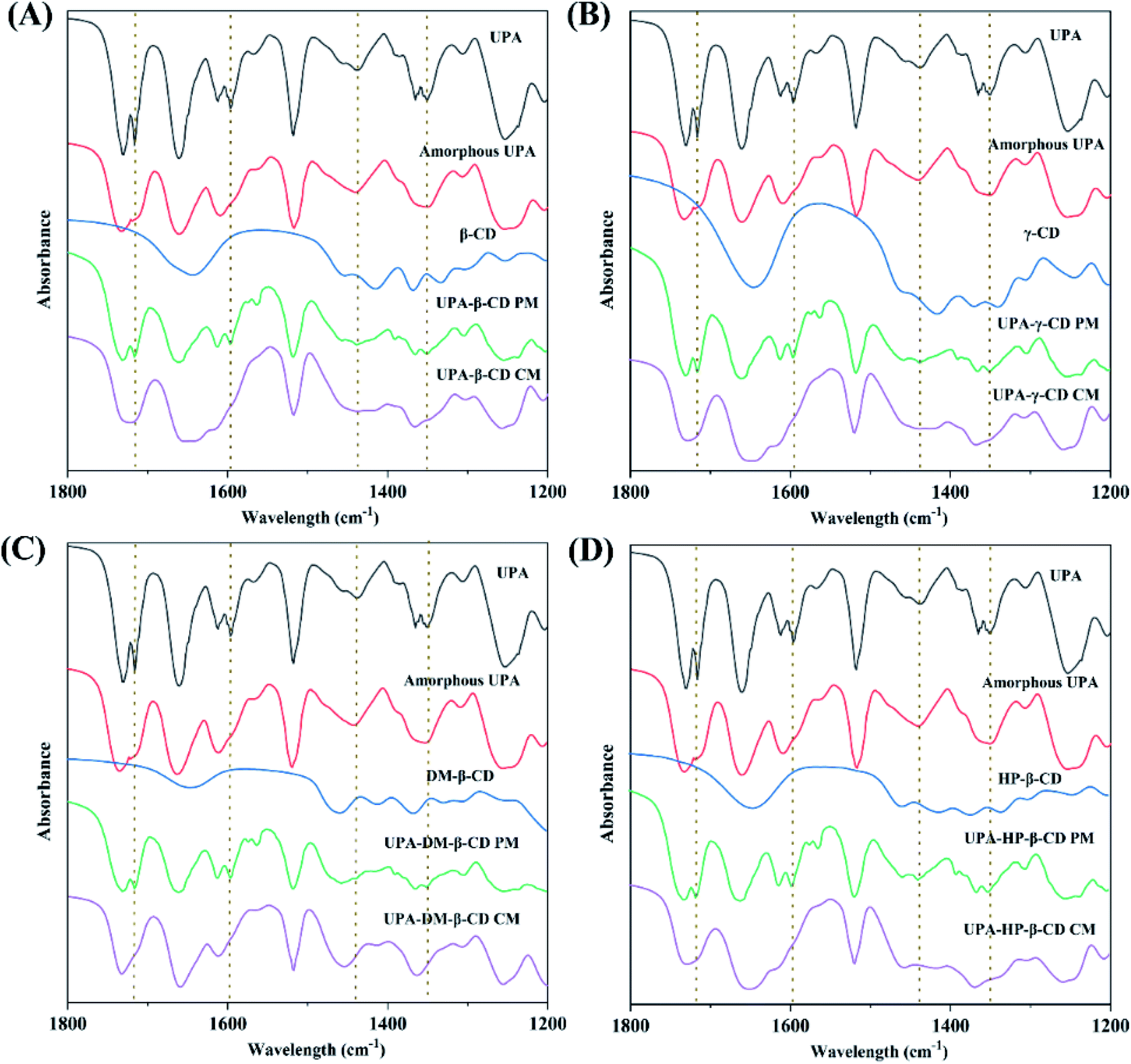 | ||
| Fig. 5 FT-IR spectra of UPA-β-CD (A), UPA-γ-CD (B), UPA-DM-β-CD (C), and UPA-HP-β-CD (D). Each picture from top to bottom: crystalline UPA, amorphous UPA, CD, UPA-CD PM, and UPA-CD CM. | ||
| Samples | UPA | CD | |||||
|---|---|---|---|---|---|---|---|
| H-2′ | H-3′ | H-1 | H-5′, H-6′ | H-3 | H-5 | ||
| UPA-β-CD | Free | 6.765 | 6.419 | 5.452 | 3.122 | 3.629 | 3.544 |
| Complex | 6.777 | 6.412 | 5.474 | 3.143 | 3.408 | 3.367 | |
| Δδ | 0.012 | −0.007 | 0.022 | 0.021 | −0.221 | −0.177 | |
| UPA-γ-CD | Free | 6.765 | 6.419 | 5.453 | 3.122 | 3.607 | 3.564 |
| Complex | 6.766 | 6.417 | 5.450 | 3.133 | 3.521 | 3.385 | |
| Δδ | 0.001 | −0.002 | −0.003 | 0.011 | −0.086 | −0.179 | |
| UPA-HP-β-CD | Free | 6.765 | 6.419 | 5.452 | 3.122 | 3.557 | 3.493 |
| Complex | 6.768 | 6.418 | 5.455 | 3.117 | 3.485 | 3.334 | |
| Δδ | 0.003 | −0.001 | 0.003 | −0.005 | −0.072 | −0.159 | |
| UPA-DM-β-CD | Free | 6.765 | 6.419 | 5.452 | 3.122 | 3.622 | 3.519 |
| Complex | 6.767 | 6.418 | 5.454 | 3.136 | 3.399 | 3.319 | |
| Δδ | 0.002 | −0.001 | 0.002 | 0.014 | −0.223 | −0.200 | |
As shown in Fig. 6, host–guest complexation can be formed but the depth of inclusion of the guest molecule into the CD cavity varies. The results showed that β-CD and γ-CD entered the cavity more deeply than DM-β-CD and HP-β-CD, which may be related to the molecular size of CD. Besides, the UPA molecules and CD molecules (β-CD, γ-CD, DM-β-CD, and HP-β-CD) adjust the conformation each other and combine to form a stable complex under the action of hydrophobic force. These results theoretically confirmed that there was a certain degree of interaction between UPA and the four CDs.
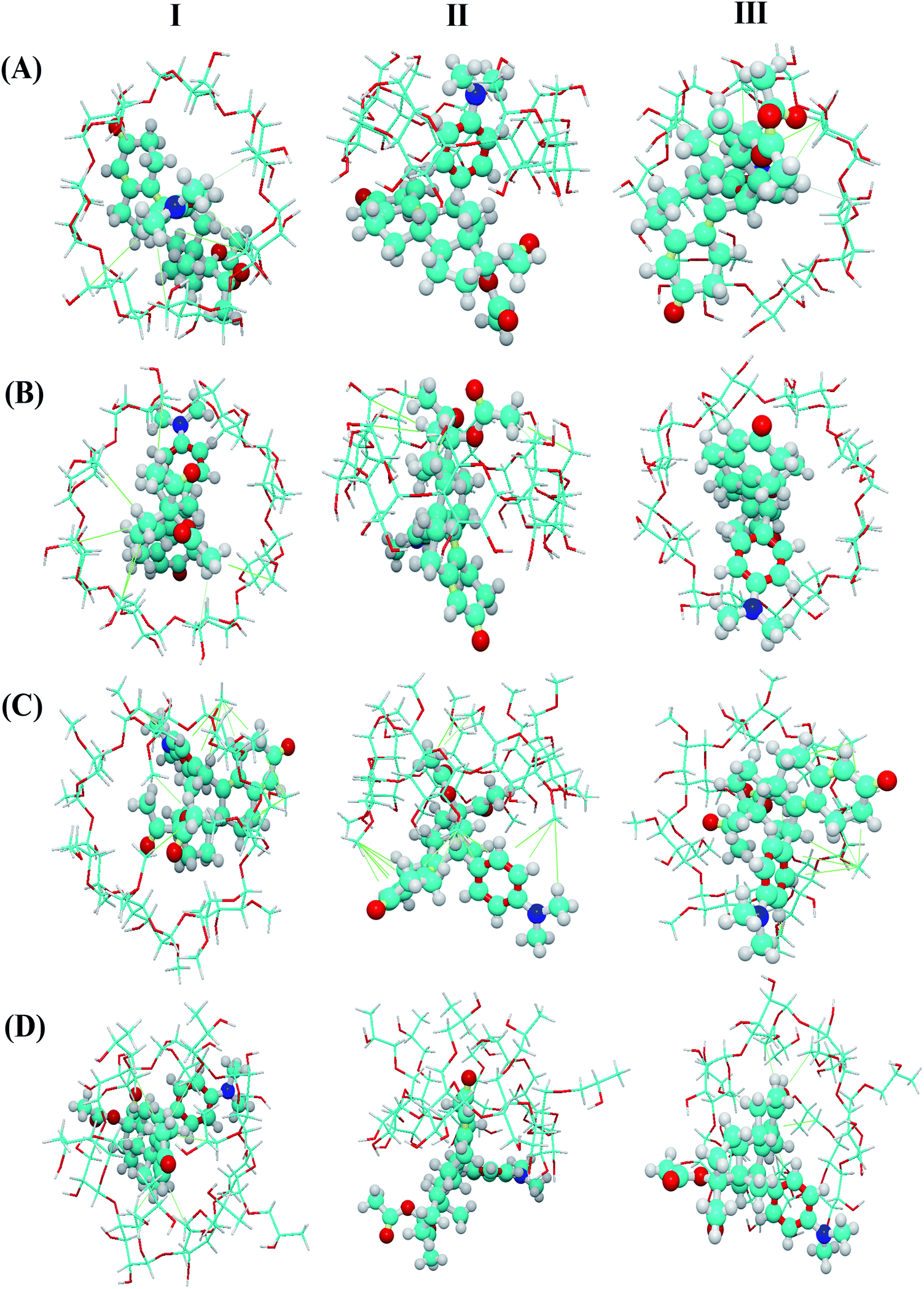 | ||
| Fig. 6 Molecular models of the inclusion complexes of UPA with β-CD, γ-CD, DM-β-CD, and UPA-HP-β-CD (I, II and III were corresponding to the top, side and bottom views of the models). | ||
3.3. Physical stability
This study explored the effects of temperature, humidity, and photolysis on the different UPA amorphous solid forms. The stabilities of the different amorphous samples were monitored and the results were shown in Fig. S6–S10.† After 10 days of storage at temperature and humidity, the pure amorphous UPA showed a tendency of crystallization, which is confirmed by the change of PXRD diffraction peaks. Amorphous UPA maintained an amorphous halo under light storage conditions, indicating that light had little effect on the stability of amorphous UPA, and temperature and humidity were the main reasons for their stability. The four UPA–CD amorphous mixture systems showed excellent stability at different stress conditions. Under the conditions of temperature and humidity for 30 days, four UPA–CD systems maintained an amorphous halo and no obvious diffraction peak appeared. The improved stability is due to the hollow structure of CDs, which is the result of wrapping UPA in the process of forming an amorphous mixture, thus having a certain protective effect.Table 2 showed the contents of related substances in different UPA amorphous systems under temperature, humidity, and photolysis. According to the data, the total impurity contents of amorphous UPA are 1.00%, 0.96%, and 2.76%, respectively, after being stored under temperature, humidity, and photolysis for 10 days, which is slightly higher than that of crystalline UPA. The single impurity of UPA was N-desmethylulipristal acetate with a run time of 5.6 min (Fig. S1†). The total impurity and single impurity content of the four amorphous UPA–CD systems were lower than that of the crystalline UPA under photolysis and close to that of the crystalline UPA under temperature and humidity conditions, indicating that the addition of CDs did not change the properties of the UPA, and the growth rate of related substances was not higher than that of the UPA. The stabilizing effect of CDs on amorphous drugs may due to the interaction of CDs with UPA, thereby interfering with the interaction between drug molecules and enhancing the stability of systems.
| Sample | Initial | Temperature 60 °C | Humidity 75% RH | Photolysis 1200000 lux h |
|
|---|---|---|---|---|---|
| UPA | Single impurity% | 0.53 | 0.70 | 0.69 | 1.62 |
| Total impurity% | 0.67 | 0.94 | 0.94 | 2.76 | |
| Amorphous UPA | Single impurity% | 0.55 | 0.74 | 0.70 | 1.70 |
| Total impurity% | 0.69 | 1.00 | 0.96 | 2.94 | |
| UPA-β-CD | Single impurity% | 0.54 | 0.74 | 0.68 | 1.46 |
| Total impurity% | 0.68 | 1.00 | 0.93 | 2.45 | |
| UPA-γ-CD | Single impurity% | 0.55 | 0.72 | 0.71 | 1.40 |
| Total impurity% | 0.69 | 1.03 | 0.96 | 2.34 | |
| UPA-DM-β-CD | Single impurity% | 0.54 | 0.73 | 0.68 | 1.40 |
| Total impurity% | 0.68 | 1.00 | 1.13 | 2.33 | |
| UPA-HP-β-CD | Single impurity% | 0.56 | 0.74 | 0.68 | 1.32 |
| Total impurity% | 0.72 | 1.00 | 0.94 | 2.21 | |
3.4 Solubility and dissolution tests
Improving the water solubility of drugs is one of the main reasons for preparing new solid forms of UPA. The apparent solubility results are shown in Fig. 7. Compared with crystalline UPA, amorphous UPA and UPA–CD binary systems displayed different degrees of advantage in solubility. The solubility of amorphous UPA was significantly improved compared with 1.48 mg mL−1 of crystalline UPA in simulated gastric juice, which was 1.62 times higher than those of the UPA.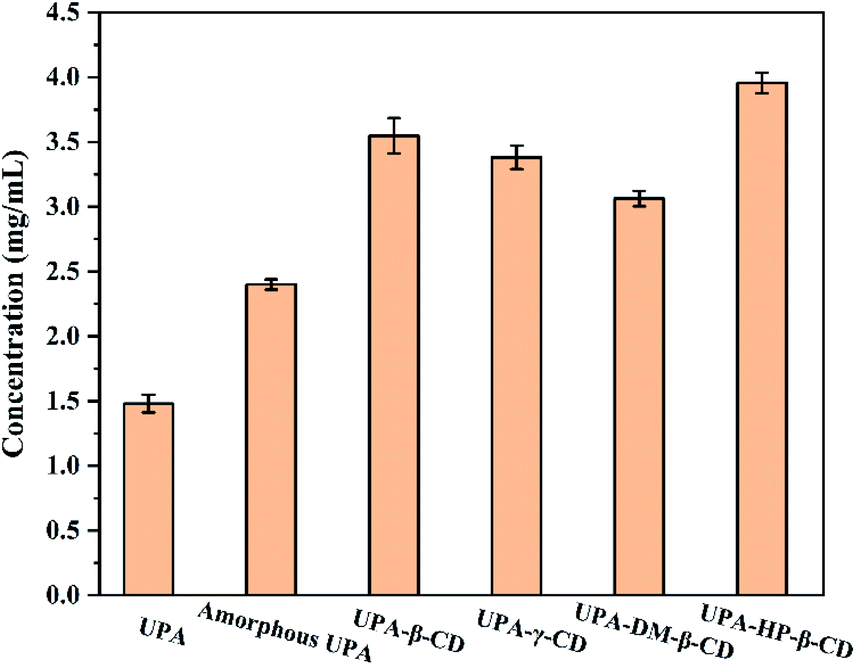 | ||
| Fig. 7 Solubility histograms of crystalline UPA, amorphous UPA, and UPA-CD in artificial gastric juice at pH = 2 (mean ± SD, n = 3). | ||
DM-β-CD was the fastest among them, 40% of the drug was released within 5 min, and the cumulative dissolution rate of UPA-DM-β-CD was 62% after 150 min, which represented a 4.43-fold and 1.63-fold increase compared with crystalline and amorphous UPA, respectively. The cumulative dissolution rate of the UPA-HP-β-CD binary amorphous system was similar to that of UPA-β-CD, and UPA-γ-CD amorphous, both about 75%, which was 5 times that of UPA, indicating that the interaction between UPA and CD molecules greatly promoted the dissolution and release of UPA. Among the four UPA–CD amorphous mixtures, UPA-HP-β-CD showed the best solubility advantages and higher cumulative dissolutions over pure crystalline drugs.
The UPA-HP-β-CD amorphous preparation showed a significant advantage, which was 2.8 times (3.95 mg mL−1) higher than the crystalline UPA. The interaction between UPA and CDs promoted the dissolution of UPA and improved the solubility of API. The solubility order of the samples was UPA-HP-β-CD > UPA-β-CD > UPA-γ-CD > UPA-DM-β-CD > amorphous UPA > crystalline UPA. These results indicated that the solubility of the binary amorphous system was not only attributed to the amorphous structure of UPA, but also to the addition of CDs.
Fig. 8 shows the dissolution rates of the different UPA amorphous samples. Crystalline UPA only exhibited a cumulative release of 14%. Compared with crystalline UPA, the cumulative dissolution of amorphous UPA is improved to a certain extent, which is about 38%. The dissolution rate of UPA-DM-β-CD was the fastest among them, 40% of the drug was released within 5 min, and the cumulative dissolution rate of UPA-DM-β-CD was 62% after 150 min, which represented a 4.43-fold and 1.63-fold increase compared with crystalline and amorphous UPA, respectively. The cumulative dissolution rate of the UPA-HP-β-CD binary amorphous system was similar to that of UPA-β-CD, and UPA-γ-CD amorphous, both about 75%, which was 5 times that of UPA, indicating that the interaction between UPA and CD molecules greatly promoted the dissolution and release of UPA. Among the four UPA–CD amorphous mixtures, UPA-HP-β-CD showed the best solubility advantages and higher cumulative dissolutions over pure crystalline drugs.
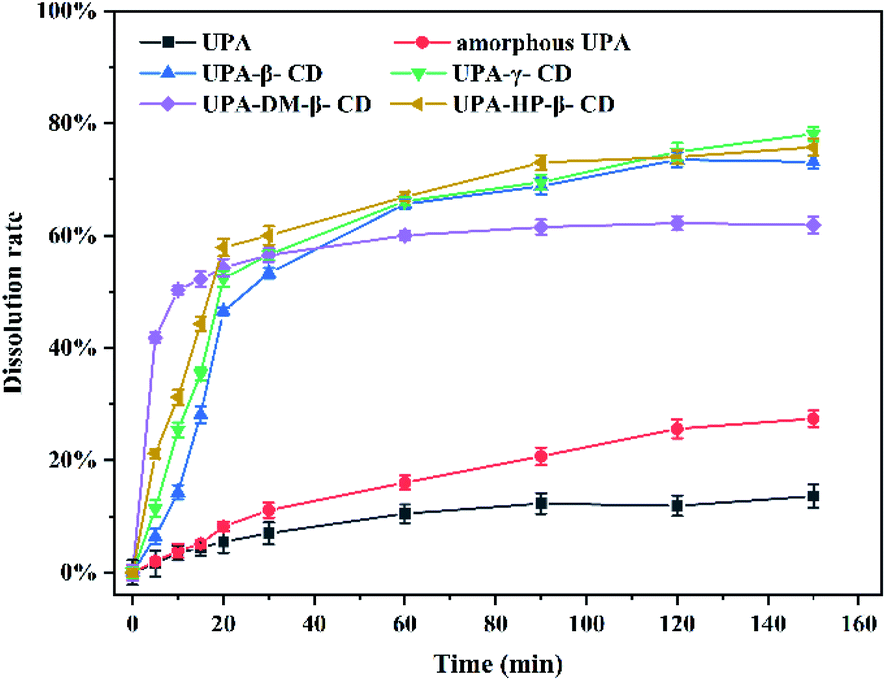 | ||
| Fig. 8 Dissolution rate curves of crystalline UPA, amorphous UPA, and UPA–CD amorphous in an artificial gastric juice at pH = 2. | ||
4. Conclusions
In this study, CDs and their derivatives were selected as effective excipients in poorly soluble drug UPA amorphous system. Four UPA–CD amorphous systems were successfully prepared by co-milling and characterized through PXRD, SEM, DSC, TGA, FT-IR, and 1H NMR. Our results revealed that the novel binary UPA–CD formulations were showed good performance compared with the crystalline and amorphous UPA. The low solubility of crystalline UPA and poor physical stability of amorphous UPA were effectively improved in the four UPA–CD binary amorphous mixture systems. The stability of UPA-β-CD, UPA-γ-CD, UPA-DM-β-CD, and UPA-HP-β-CD was better than that of amorphous UPA under temperature, humidity, and photolysis. The addition of CDs did not change the properties of the UPA, and the growth rate of related substances was not higher than that of UPA. Among the four amorphous mixtures, the UPA-HP-β-CD amorphous showed solubility advantages and it might be in favour of its oral bioavailability. The interaction between UPA and CDs greatly promoted the dissolution and release of UPA. These researches on the UPA–CDs amorphous systems presented a safe and effective formulation technology for the development of new drug solid forms, which is a promising formulation for improving oral absorption.Author contributions
Peng Wang: investigation, visualization, writing – original draft. Yan Wang: investigation, supervision. Zili Suo: investigation, supervision. Yuanming Zhai: writing – review and editing. Hui Li: writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Analytical Testing center of Sichuan University for providing scanning electron microscopy.References
- H. Ueda, W. Wu, K. Löbmann, H. Grohganz, A. Müllertz and T. Rades, Mol. Pharm., 2018, 15, 2036–2044 CrossRef CAS PubMed.
- M. Jara, Z. Warnken and O. Williams, Pharmaceutics, 2021, 13, 1–15 CrossRef PubMed.
- P. Pandi, R. Bulusu, N. Kommineni, W. Khan and M. Singh, Int. J. Pharm., 2020, 586, 115960 CrossRef PubMed.
- W. Wu, Y. Wang, K. Löbmann, H. Grohganz and T. Rades, Mol. Pharm., 2019, 16, 1294–1304 CrossRef CAS PubMed.
- A. Schittny, J. Huwyler and M. Puchkov, Drug Delivery, 2020, 27, 110–127 CrossRef CAS PubMed.
- Y. Song, D. Zemlyanov, X. Chen, H. Nie, Z. Su, K. Fang, X. Yang, D. Smith, S. Byrn and W. Lubach, Mol. Pharm., 2016, 13, 483–492 CrossRef CAS PubMed.
- M. Zhang, N. Xiong, L. Suo, Q. Hou, N. Gan, X. Tang, H. Ding and H. Li, RSC Adv., 2019, 9, 3946–3955 RSC.
- X. Lin, Y. Hu, L. Liu, L. Su, N. Li, J. Yu, B. Tang and Z. Yang, Pharm. Res., 2018, 35, 125 CrossRef PubMed.
- L. Chambers, H. Grohganz, H. Palmelund, K. Löbmann, T. Rades, M. Musa and W. Steed, Eur. J. Pharm. Sci., 2021, 157, 105636 CrossRef CAS PubMed.
- A. Santos, C. Bastos, F. Pinto and I. Fernandes, Ann. Med., 2021, 53, S111–S112 CrossRef.
- J. Dengale, H. Grohganz, T. Rades and K. Löbmann, Adv. Drug Delivery Rev., 2016, 100, 116–125 CrossRef PubMed.
- S. Wairkar and R. Gaud, AAPS PharmSciTech, 2016, 17, 673–681 CrossRef CAS PubMed.
- W. Wu, K. Löbmann, J. Schnitzkewitz, A. Knuhtsen, S. Pedersen, T. Rades and H. Grohganz, Eur. J. Pharm. Biopharm., 2019, 134, 68–76 CrossRef CAS PubMed.
- M. Zhang, L. Suo, X. Peng, N. Gan, D. Zhao, X. Tang, X. Wei and H. Li, Carbohydr. Polym., 2020, 229, 115476 CrossRef CAS PubMed.
- T. Loftsson and E. Brewster, J. Pharm. Sci., 2012, 101, 3019–3032 CrossRef CAS PubMed.
- T. Loftsson and M. Masson, Int. J. Pharm., 2001, 225, 15–30 CrossRef CAS PubMed.
- M. Zhang, H. Liu and Y. Liu, Adv. Mater., 2020, 32, 1806158 CrossRef PubMed.
- T. Loftsson and E. Brewster, J. Pharm. Pharmacol., 2010, 62, 1607–1621 CrossRef CAS PubMed.
- S. Gould and C. Scott, Food Chem. Toxicol., 2005, 43, 1451–1459 CrossRef CAS PubMed.
- T. Loftsson and E. Brewster, J. Pharm. Sci., 1996, 85, 1017–1025 CrossRef CAS PubMed.
- A. Celebioglu and T. Uyar, Food Chem., 2020, 317, 126397 CrossRef CAS PubMed.
- H. Lin, C. Chen, C. Lin and Y. Fang, J. Food Drug Anal., 2017, 25, 219–234 CrossRef PubMed.
- Z. Jiang, C. Liu, K. Xu, L. Pan, H. Hou, P. Hu and K. Yang, Carbohydr. Polym., 2018, 182, 106–114 CrossRef PubMed.
- B. Carvalho, K. Burusco, C. Jaime, T. Venâncio, A. Carvalho, L. Murgas and L. Pinto, Carbohydr. Polym., 2018, 179, 386–393 CrossRef PubMed.
- M. Malanga, J. Szemán, É. Fenyvesi, I. Puskás, K. Csabai, G. Gyémánt, F. Fenyvesi and L. Szente, J. Pharm. Sci., 2016, 105, 2921–2931 CrossRef CAS PubMed.
- P. Fine, H. Mathé, S. Ginde, V. Cullins, J. Morfesis and E. Gainer, Obstet. Gynecol., 2010, 115, 257–263 CrossRef PubMed.
- O. Pohl, I. Osterloh, V. Lecomte and P. Gotteland, Int. J. Clin. Pharmacol. Ther., 2013, 51, 26–33 CrossRef CAS PubMed.
- M. Li, N. Ioannidis, C. Gogos and E. Bilgili, Eur. J. Pharm. Biopharm., 2017, 119, 68–80 CrossRef CAS PubMed.
- P. Tang, S. Li, L. Wang, H. Yang, J. Yan and H. Li, Carbohydr. Polym., 2015, 131, 297–305 CrossRef CAS PubMed.
- W. Kaialy, Int. J. Pharm., 2016, 503, 262–276 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra09420c |
| This journal is © The Royal Society of Chemistry 2022 |