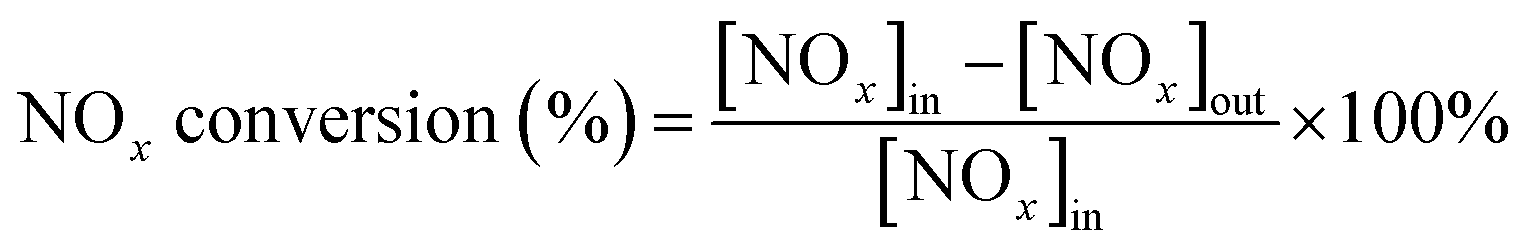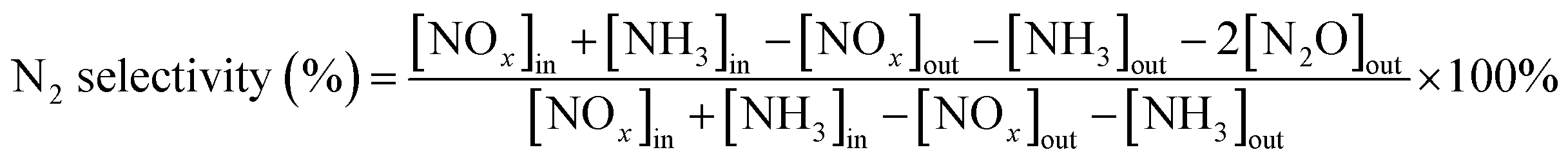DOI:
10.1039/D2RA00316C
(Paper)
RSC Adv., 2022,
12, 10142-10153
DeNOx performance enhancement of Cu-based oxides via employing a TiO2 phase to modify LDH precursors†
Received
16th January 2022
, Accepted 24th March 2022
First published on 31st March 2022
Abstract
CuAl-LDO, CuAl-LDO/TiO2 and CuAl-LDO/TiO2NTs catalysts were obtained from TiO2 modified LDHs precursor which were prepared by in situ assembly method. Then catalysts were evaluated in the selective catalytic reduction of NOx with NH3(NH3-SCR), and the results showed that the CuAl-LDO/TiO2NTs catalyst exhibited preferable deNOx performance (more than 80% NOx conversion and higher than 90% N2 selectivity at a temperature range of 210–330 °C) as well as good SO2 resistance. With the aid of series of characterizations such as XRD, N2 adsorption/desorption, XPS, NH3-TPD, H2-TPR, and in situ DRIFTS, it could be concluded that, doping TiO2NTs afforded the catalyst larger specific surface area, more abundant surface chemisorption oxygen species and more excellent redox performance. Meanwhile, In situ DRIFTS evidenced that CuAl-LDO/TiO2NTs catalyst has a strong adsorption capacity for the reaction gas, which is more conducive to the progress of the SCR reaction.
1 Introduction
Nitrogen oxides (NOx), released from stationary sources (such as coal-fired power plants), are major atmospheric pollutants which seriously threaten the ecological environment and human health.1,2 Selective catalytic reduction with NH3 (NH3-SCR) is considered as one of the most efficient technologies for removing NOx.3–5 Nowadays, the commercial V2O5-WO3/TiO2 catalysts are widely applied in the NH3-SCR field and exhibit excellent catalytic activity in the temperature range of 300–400 °C.3,6,7 However, there are still some practical problems of the catalysts, such as the toxicity of vanadium, low N2 selectivity due to the formation of N2O and high conversion of SO2 to SO3 at high temperature.8,9 Generally, the poison of SO2 and dust can be avoided by placing an SCR reactor behind the dust removal and desulfurization equipment, resulting in the debasement of exit gas temperature, which cannot meet the optimum activity temperature window of V2O5-WO3/TiO2 catalysts.10–12 Thus, it would be necessary to develop more efficient and eco-friendlier deNOx catalysts with non-vanadium-based at low temperatures.
At the present stage, many researchers consider the copper oxides as a potential candidate for the low-temperature SCR catalysts, including pure copper oxides, copper-based composite oxides and supported copper-based catalysts.13–16 Among them, copper-based composite oxides catalysts with excellent catalytic performance have recently attracted much attention. In recent years, it is well known that layered double oxides (LDO) from layered-double hydroxides (LDHs) demonstrated fine reaction activity in NH3-SCR, especially the typical CuAl-LDO catalyst.17,18 For instance, Yan et al. reported a high-performance Cu-based oxides catalyst prepared from CuAl-LDHs precursor.19 In our previous work, it is also found that the CuAl-LDO catalysts derived from LDHs demonstrated better reaction activity in NH3-SCR.18 However, due to its insufficient acidity, the CuAl-LDO catalyst afforded poor reaction activity at medium and high temperature in NH3-SCR, which limited its further application.
As well known, titanium dioxide (TiO2) has been found to be a valuable and environmentally friendly material with stable chemical properties.20 At the same time, for transition metal oxide catalyst, the introduction of TiO2 could improve the acid sites and chemical adsorption oxygen over the catalyst surface, enhance the adsorption and activation of NH3 as well as resistance of SO2.21–24 Based on the characteristic advantages of LDHs and TiO2, it is promising to prepare well-behaved Cu–Ti composite oxide catalysts derived by LDHs. However, due to the Jahn–Teller distortion of Cu2+ ions, it is difficult to prepare CuTi-based LDHs by one step preparation. Here, desirable SCR catalyst of CuAl-LDO/TiO2 was tentatively fabricated by modifying CuAl-LDHs with TiO2 phase.
Herein, the CuAl-LDHs with titanium-based materials were assembled and calcined at high temperature. Subsequently, the catalytic performance of the obtained composite oxide catalysts was evaluated in NH3-SCR reaction. At the same time, the structural characteristics and the SCR performance of the catalysts were explored through a series of characterizations, such as XRD, N2 adsorption/desorption, TEM, XPS, H2-TPR, NH3-TPD. The intrinsic difference between CuAl-LDO and CuAl-LDO/TiO2NTs catalysts derived by LDHs over the deNOx activity was studied by In situ DRIFTS. This research might provide significant reference value for LDHs precursor in SCR application.
2 Materials and methods
2.1. Materials
TiO2(P25), Cu(NO3)2·3H2O(99%), Al(NO3)3·9H2O(99%), NaOH(96%) and Na2CO3(99%) were obtained from the Sinopharm Chemical Reagent Company. All chemicals were of analytical grade and were employed without further purification.
2.2. Sample preparation
2.2.1. Synthesis of TiO2NTs. The titanium-based materials used in this study were TiO2(P25) and TiO2 nanotubes (TiO2NTs) where the TiO2NTs were synthesized by hydrothermal method via TiO2(P25) as the precursor. The specific procedure is as follows: the P25 was added to the NaOH solution (10 M) to form a homogeneous mixture. After that, the mixture was transferred into a Teflon autoclave and reacted at 150 °C for 30 h. The slurry was filtered and washed initially with deionized water until the pH reached about 7 and mixed with 0.1 M H+ solution to reach a pH value about 1. Finally, the obtained precipitate was washed until the filtrate was neutral and dried at 70 °C for 30 h. Then, the obtained powder was calcined at 400 °C for 2 h (the heating rate being 2 °C min−1) to obtain titanium dioxide nanotubes (marked as TiO2NTs).
2.2.2. Preparation of catalyst. The CuAl-LDHs/TiO2NTs and CuAl-LDHs/TiO2 precursors were prepared by in situ assembly method. In detail, a certain amount of TiO2NTs or TiO2 was dispersed in 100 mL deionized water to form dispersion liquid (solution A). The mixed nitrate solution (0.12 M Cu(NO3)2·3H2O and 0.04 M Al(NO3)3·9H2O) and mixed base solution (0.3 M NaOH and 0.1 M Na2CO3) were slowly dripped into a round-bottomed flask within solution A. After that, the mixed solution was hydrothermally treated at 65 °C for 10 h. The obtained LDHs slurry was filtered and washed, dried overnight at 70 °C to obtain LDHs powder which was denoted as CuAl-LDHs/TiO2NTs or CuAl-LDHs/TiO2. The CuAl-LDHs were also fabricated via the identical method without any addition of titanium dioxide in solution A. Then CuAl-LDO, CuAl-LDO/TiO2NTs and CuAl-LDO/TiO2 catalysts were prepared via calcining LDHs powders at 500 °C for 5 h under nitrogen (80 mL min−1).
2.3. Catalyst characterization
The phase structure of the LDHs and catalyst samples was analyzed by the Rigaku DX-2700 X-ray diffractometer instrument, with Cu/Kα radiation (λ = 1.54184 Å) at the 2θ of 5° to 85°. The morphology of the precursors was scanned and analyzed by the scanning electron microscope (SEM, Hitachi's SU8010 JEOL). The morphology and surface atomic concentration of catalyst was scanned and analyzed by SEM and EDS-mapping (Carl Zeiss GeminiSEM300, Germany). The surface textural structures of catalysts were operated on the ASAP-2460 physical adsorption instrument (Micromeritics, USA) at −196 °C. Brunauer–Emmett–Teller (BET) method and Barrett–Joyner–Halenda (BJH) model were used to calculated the specific surface area, pore volume and pore diameter of catalysts. The transmission electron microscope (TEM) analysis was used to research the morphology images of the catalysts by JEM-2100F, JEOL microscope with an accelerating voltage of 200 kV. The surface property of the catalysts was analyzed by X-ray photoelectron spectroscopy (XPS, Thermo Fisher ESCALAB 250xi, England) with Al Kα radiation (hν = 1486.6 eV). Both NH3-temperature programmed desorption (NH3-TPD) and H2 temperature-programmed reduction (H2-TPR) tests were performed on the FINESORB-3010 temperature programmed adsorption instrument (Zhejiang Pantech) with using 80 mg catalysts. For NH3-TPD test, the catalysts were pretreated in He atmosphere at 150 °C for 1 h, and then cooled to 50 °C in the same stream. Then the input gas was shifted a 3% NH3 + He (25 mL min−1) mixed atmosphere at 50 °C, and the adsorption in this atmosphere lasted for 30 minutes. Before starting the TPD experiment, the catalyst was flushed with He at a flow rate of 30 mL min−1 to remove physically adsorbed NH3 on its surface. The desorption process was performed by heating the samples in He (30 mL min−1) from 50 °C to 600 °C at a heating rate of 10 °C min−1, the data was detected and recorded online by a thermal conductivity detector (TCD), then analyzed by the Origin software. Each desorption peak was integrated to obtain the corresponding peak area, as well as the total acid content and the ratio of strong and weak acid sites were calculated. For H2-TPR experiment, 80 mg catalyst (particle size: 40–60 mesh) was putted into a laboratory-made quartz tube, and pre-treated in pure Ar atmosphere (flow rate: 20 mL min−1) for 1 h at 300 °C. Then cooled down the temperature to 30 °C and stabilize for a period of time, followed by changing the gas to 10% H2/Ar, and the reduction process occurred from 30 °C to 600 °C at a heating rate of 10 °C min−1. The corresponding data was detected and recorded by TCD. Origin software was employed to fit the H2-TPR curve through splitting the peaks, and calculate the hydrogen consumption based on the peak area. In situ diffuse reflectance infrared Fourier transform spectroscopy (In situ DRIFTS) experiments were performed using In situ infrared spectrometer (Bruker TENSOR 27) from Germany, with a Pike DRIFTS sample cell and a high-precision MCT detector cooled by liquid nitrogen. At first, the sample was pretreated at 300 °C under N2 about 10 min, and the background spectrum was collected in the same condition at the test temperature (240 °C). Then, each spectrum was recorded by subtracting the background spectrum in the range of 2000–1000 cm−1. The gas flow during reaction process included: 600 ppm NO, 600 ppm NH3, 5 vol% O2, 100 ppm SO2, with N2 as the balance gas. The acid sites over the samples were investigated by infrared spectroscopy of adsorbed pyridine (Py-IR) using Bruker INVENIO S from Germany. The spectra were recorded with 2 cm−1 spectral resolution on a Frontier FT-IR Spectrometer at 240 °C.
2.4. SCR activity test
The NH3-SCR performance of the catalysts (40–60 mesh) was tested over the fixed-bed quartz tube reactor. The reaction gas was set as follows: 600 ppm NO, 600 ppm NH3, 5 vol% O2, 100 ppm SO2 (when used), with N2 as the balance gas and the space velocity (GHSV) was 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. Experimental configurations for evaluating catalytic performance were presented in Fig. S1.†
000 h−1. Experimental configurations for evaluating catalytic performance were presented in Fig. S1.†
The IR flue gas analyser (MKS) was hired to detect online gas concentration and recorded the reaction data in a steady state at each temperature point (from 150–360 °C with 30 °C as an interval). NOx conversion and N2 selectivity were calculated by eqn (1) and (2):
| |
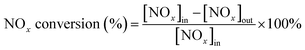 | (1) |
| |
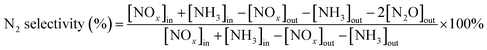 | (2) |
In the above formula, the subscript “[X]in” and “[X]out” indicate the inlet and outlet concentration of gas in a steady state flow mode, respectively.
3 Results and discussion
3.1. Catalytic performance of the catalysts
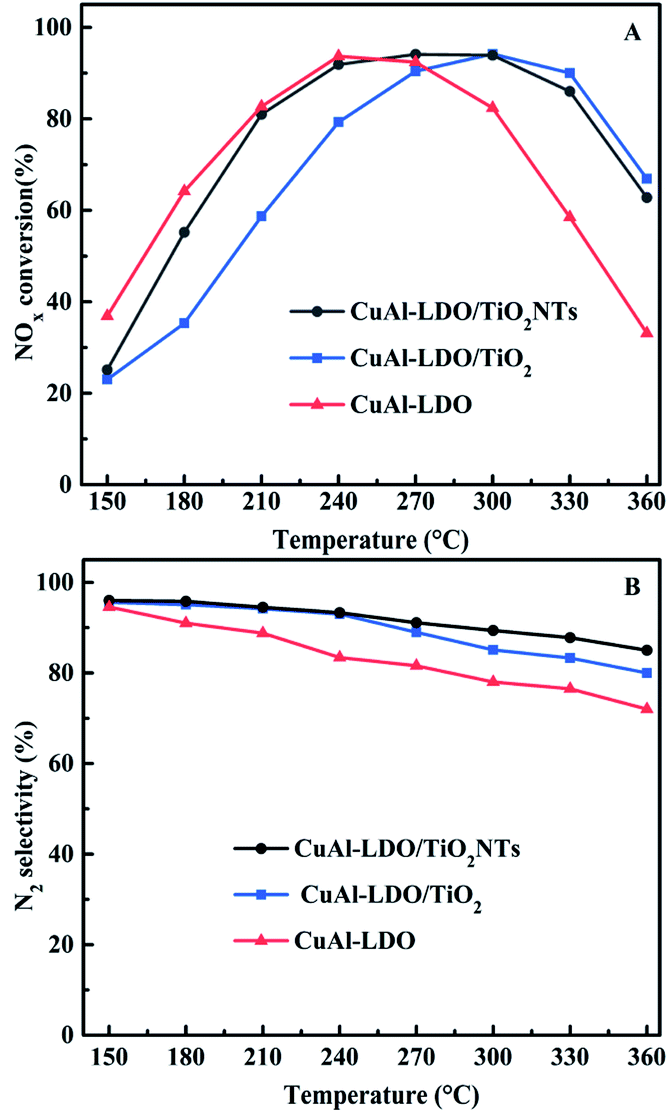
3.1.1. Catalytic activity and N2 selectivity. The deNOx performance of CuAl composite oxide catalysts (CuAl-LDO, CuAl-LDO/TiO2 and CuAl–TiO2NTs) were tested from 150 °C to 360 °C, and the results are shown in Fig. 1. It can be seen from Fig. 1 that modifying CuAl-LDO catalyst by different Ti-based materials significantly improved the NH3-SCR performance. As displayed in Fig. 1(A), the catalytic activity of CuAl-LDO catalyst was better than the CuAl-LDO/TiO2 and CuAl-LDO/TiO2NTs catalysts below 240 °C, the catalytic activity of CuAl-LDO/TiO2 catalyst was better than that of the CuAl-LDO and CuAl-LDO/TiO2NTs catalysts above 300 °C. However, at the test temperature ranges of 210–330 °C, the CuAl-LDO/TiO2NTs catalyst showed better catalytic activity with above 80% NOx conversion than other catalysts. As demonstrated in Fig. 1(B), the N2 selectivity of all catalysts showed clear downward trend with increasing temperature, suggesting that the N2 selectivity was susceptible to ammonia oxidation at high temperature. During the whole test temperature range, CuAl-LDO catalyst afforded lowest selectivity while the CuAl-LDO/TiO2NTs presented best N2 selectivity of more than 90% in 150–360 °C, as well as higher than 80% for CuAl-LDO/TiO2. The results indicated that the NH3-SCR performance of CuAl-LDO catalyst was observably affected by introducing different Ti-based materials. In addition, the stability of CuAl-LDO/TiO2NTs catalyst was tested and results are shown in Fig. S2.† As presented, the better catalytic activity of CuAl-LDO/TiO2NTs catalyst was kept even after seven cycle tests, indicating the superior stability.
 |
| | Fig. 1 The (A) NOx conversion, (B) N2 selectivity of catalysts. | |
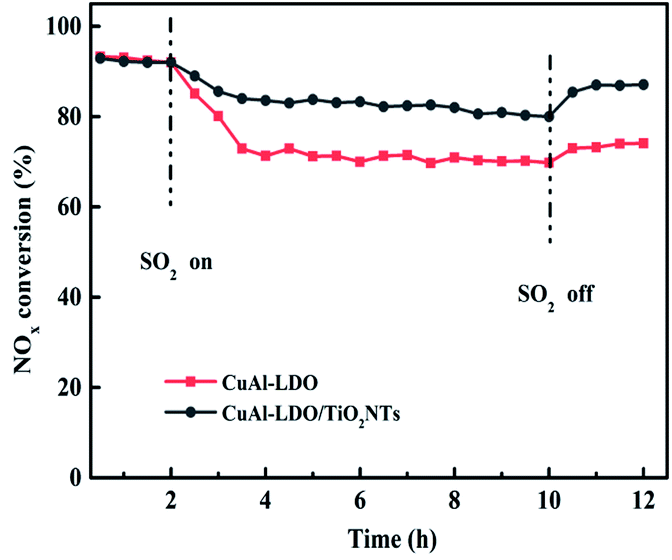
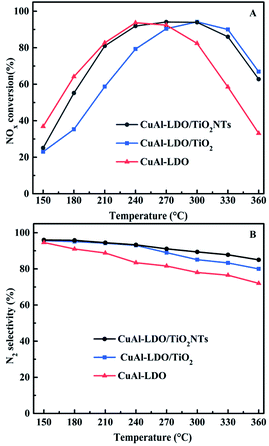
3.1.2. The SO2 resistance test. In NH3-SCR reaction, the catalyst is usually deactivated by the SO2 in the flue gas. So, the catalysts with excellent SO2 durability are vital in practical process. Therefore, the SO2 resistance of CuAl-LDO and CuAl-LDO/TiO2NTs catalysts at 240 °C were tested and the corresponding results are displayed in Fig. 2. As shown in Fig. 2, after adding SO2 into flue gas, the NOx conversion decreased from 92% to 80% for CuAl-LDO/TiO2NTs catalyst and 93% to 70% for CuAl-LDO catalyst, respectively. After the SO2 were purged off, the activity recovered to 86% for the CuAl-LDO/TiO2NTs catalyst, but only 73% for CuAl-LDO catalyst. The results indicated that the CuAl-LDO/TiO2NTs catalysts possessed better SO2 resistance than CuAl-LDO catalyst. The capacity of SO2 adsorption over both catalysts were researched by In situ DRIFTS measurement as shown in the Fig. S3.† The results displayed that the intensity of SO2 adsorption peaks and the species of sulfate over CuAl-LDO were higher than CuAl-LDO/TiO2NTs. As summary, it is difficult to form sulfate on the surface of CuAl-LDO/TiO2NTs catalyst after the introduction of SO2, which was consistent with the result of resistance SO2 performance test.
 |
| | Fig. 2 SO2 resistance test over CuAl-LDO/TiO2NTs and CuAl-LDO catalysts at 240 °C. Reaction conditions: [NO] = [NH3] = 600 ppm, [O2] = 5 vol%, [SO2] = 100 ppm, N2 as balance gas, under a GHSV of 45000 h−1. | |
3.2. XRD patterns and SEM images of LDHs precursors
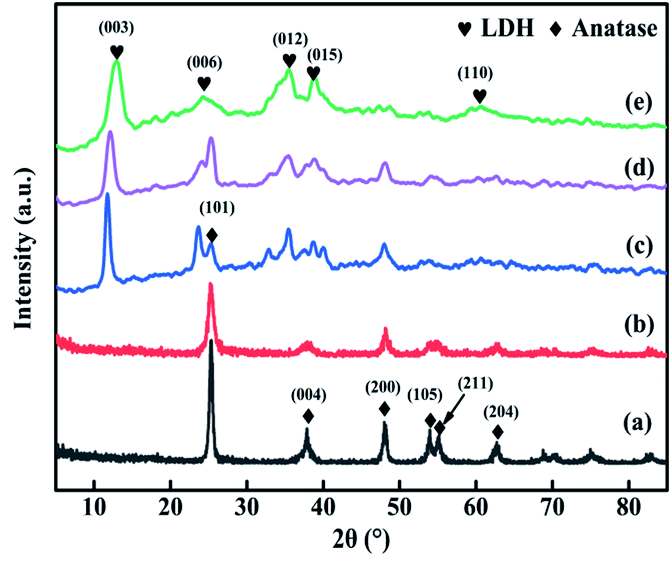
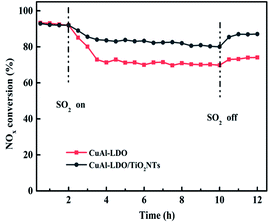
The XRD patterns of TiO2, TiO2NTs (prepared by the hydrothermal method), CuAl-LDHs, CuAl-LDHs/TiO2 and CuAl-LDHs/TiO2NTs are presented in Fig. 3. For TiO2 and TiO2NTs, it could be confirmed that the diffraction peaks at 25.3°, 37.8°, 48.1°, 53.9°, 55.1°, 62.7°, 68.8°, 70.3° and 75.1° might be assigned to the crystal planes of anatase TiO2 phase (JCPDS # 21-1272).20,23 The peaks located at 11.9°, 23.8°, 34.8°, 39.1° and 60.9° in CuAl-LDHs, CuAl-LDHs/TiO2 and CuAl-LDHs/TiO2NTs were corresponded to the (003), (006), (012), (015) and (110) crystal planes of LDHs (JCPDS # 35-0964), respectively, indicating the successful preparation of LDHs precursors.18,19 For CuAl-LDHs/TiO2NTs and CuAl-LDHs/TiO2, the peaks around 25.28°, 36.95°, 37.80° and 48.05° could be ascribed to the (101), (103), (004) and (200) of anatase. The TEM images of TiO2NTs are presented in Fig. S4(A) and (B).† The TiO2NTs has a regular morphology and a complete hollow tubular structure.
 |
| | Fig. 3 The XRD patterns of (a) TiO2(P25), (b) TiO2NTs, (c) CuAl-LDHs/TiO2NTs, (d) CuAl-LDHs/TiO2 and (e) CuAl-LDHs. | |
The SEM images of CuAl-LDHs, CuAl-LDHs/TiO2 and CuAl-LDHs/TiO2NTs are displayed in Fig. 4. It could be seen from SEM images that all samples displayed morphology of assembled nanosheets, which was the typical structure of LDHs, thus further witnessing the successful preparation of LDHs. As shown Fig. 4(C), the nanosheets of CuAl-LDHs/TiO2NTs were effectively separated by TiO2NTs, which effectively alleviates the stacking and aggregation of LDHs laminates.
 |
| | Fig. 4 The SEM images of the precursors: (A) CuAl-LDHs, (B) CuAl-LDHs/TiO2 and (C) CuAl-LDHs/TiO2NTs. | |
3.3. Characterization of the catalysts
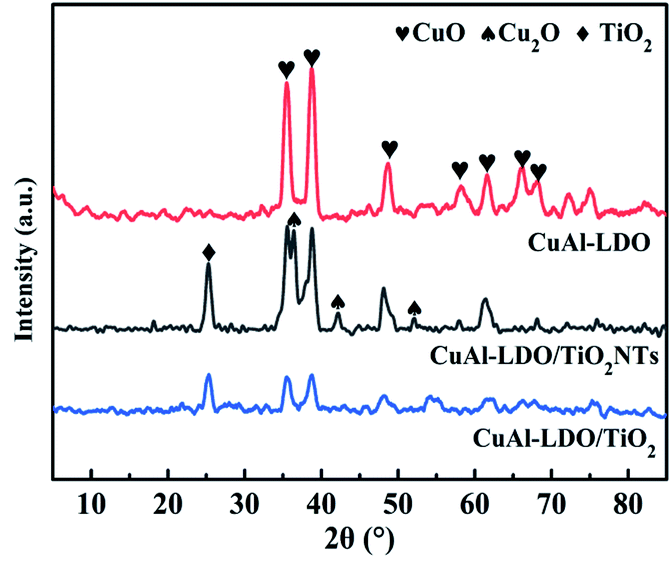
3.3.1. XRD analysis. The XRD spectra of CuAl-LDO, CuAl-LDO/TiO2 and CuAl-LDO/TiO2NTs catalysts are shown in Fig. 5. As can be seen in Fig. 5, all catalysts presented the diffraction peaks of CuO (JCPDS #02-1040), while for the CuAl-LDO/TiO2NTs catalyst, the peaks at 36.4° and 42.3° corresponding to Cu2O crystalline phase (JCPDS # 05-0667). In addition, the diffraction peak located at 25.35° for CuAl-LDO/TiO2 and CuAl-LDO/TiO2NTs catalysts can be attributed to TiO2 (JCPDS # 21-1272).
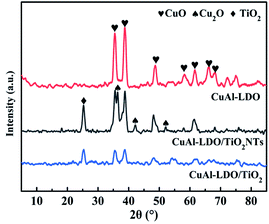 |
| | Fig. 5 The XRD patterns of catalysts. | |
3.3.2. N2 adsorption/desorption analysis. The N2 adsorption–desorption isotherms and pore diameter distribution curves of CuAl-LDO, CuAl-LDO/TiO2 and CuAl-LDO/TiO2NTs catalysts are showed in Fig. S5.† According to Fig. S5(A),† all samples displayed isotherms of type III which exhibited hysteresis loops mostly of type H3, indicating mesoporous structure of catalysts. The pore diameter distribution curves of catalysts were calculated by the Barrett–Joyner–Halenda (BJH) model as shown in Fig. S5(B).† It can be found that the CuAl-LDO/TiO2NTs catalyst had more abundant pore structures than others. The BET specific surface area, pore volume and pore size of different catalysts are listed in Table 1. As shown in Table 1, the specific surface area, pore volume and pore diameter of CuAl-LDO/TiO2 and CuAl-LDO/TiO2NTs catalysts increased in comparison to that of CuAl-LDO catalysts. Meanwhile, the specific surface area sequence of the three catalysts was CuAl-LDO/TiO2NTs (80.5 m2 g−1) > CuAl-LDO/TiO2 (51.7 m2 g−1) > CuAl-LDO (49.7 m2 g−1), hinting that the surface area of CuAl-LDO/TiO2NTs catalyst increased more significantly. The abundant pore structures and larger surface area of CuAl-LDO/TiO2NTs catalyst was not only conducive to the explosion of active centers but also beneficial to the adsorption and activation reaction gases, thus leading to the better SCR activity, being consistent with the previous literature.25
Table 1 The specific surface area, pore volume and pore size of catalysts
| Catalysts |
SBET (m2 g−1) |
Pore volume (cm³ g−1) |
Pore diameter (nm) |
| CuAl-LDO |
49.7 |
0.35 |
27.3 |
| CuAl-LDO/TiO2 |
51.7 |
0.48 |
34.6 |
| CuAl-LDO/TiO2NTs |
80.5 |
0.67 |
28.7 |
| TiO2 |
55.8 |
0.19 |
14.8 |
| TiO2NTs |
192.8 |
0.8 |
15.2 |
3.3.3. TEM analysis. The microstructure of all catalysts was investigated by TEM tests and the results are shown in Fig. 6. It can be seen form Fig. 6(A)–(C), the thin layers still existed in CuAl-LDO, CuAl-LDO/TiO2 and CuAl-LDO/TiO2NTs catalyst during the annealing process. For CuAl-LDO/TiO2NTs catalyst, it could be observed that the hollow tubular structure of TiO2NTs was maintained after roasting. During the LDHs annealing process, it could effectively slow down the stacking phenomenon of LDHs laminates, thereby facilitating the adsorption of reaction gas and accelerating the SCR reaction. Besides, the HR-TEM image (Fig. 6(D)) of the CuAl-LDO/TiO2NTs presented two different lattice fringes, corresponding to the (111) crystal plane of CuO and (101) crystal plane of anatase (TiO2), respectively. This conclusion was consistent with the results of XRD.
 |
| | Fig. 6 The (A–C) TEM and (D) HR-TEM images of catalysts: (A) CuAl-LDO, (B) CuAl-LDO/TiO2 and (C and D) CuAl-LDO/TiO2NTs. | |
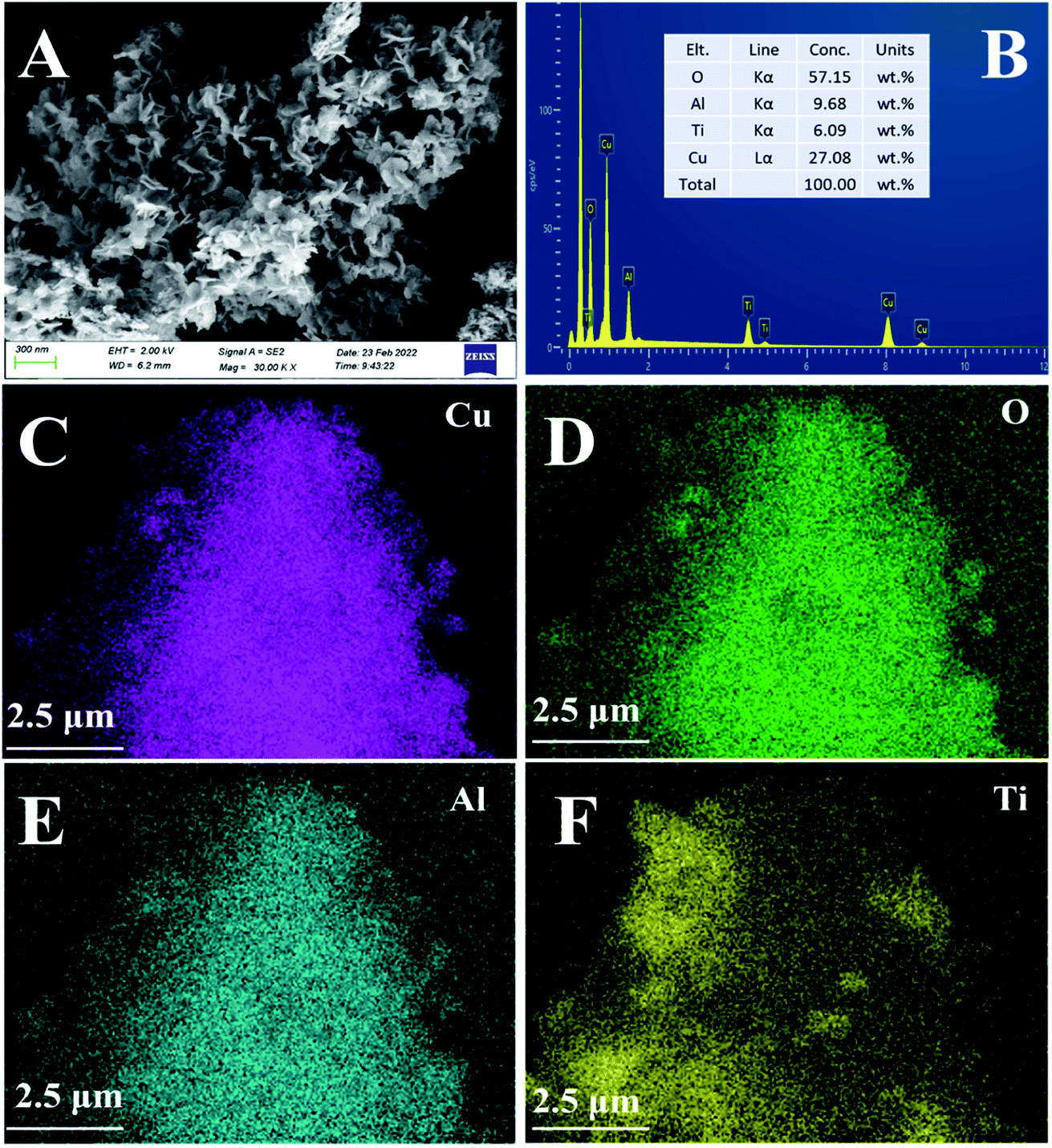
3.3.4. SEM & EDS-mapping analysis. The morphology and surface atomic concentration of the CuAl-LDO/TiO2NTs catalyst were investigated by SEM and EDS-mapping analysis and the results are presented in Fig. 7. It was observed in Fig. 7(A) that the CuAl-LDO/TiO2NTs catalyst could maintain the ‘‘flower-like” morphology after calcination. In Fig. 7(B)–(F), the EDS-mapping was applied to carry out the surface atomic concentration analysis of CuAl-LDO/TiO2NTs catalyst. It was presented that the molar ratio of Cu/Al was about 2.8. The EDS mapping images of Cu, O, Al and Ti elements are shown in Fig. 7(C) and (D). The overlapping of all elements of Cu, O, Al and Ti verified a uniform distribution of all elements of CuAl-LDO/TiO2NTs catalyst.
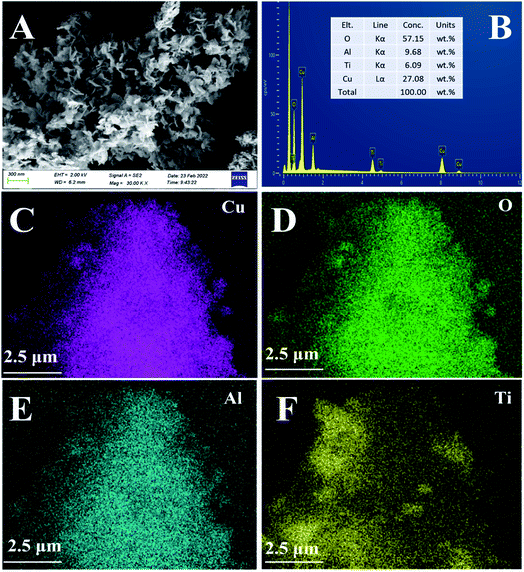 |
| | Fig. 7 The (A) SEM images and (B–F) EDS-mapping of CuAl-LDO/TiO2NTs catalyst: (C) Cu, (D) O, (E) Al and (F) Ti. | |
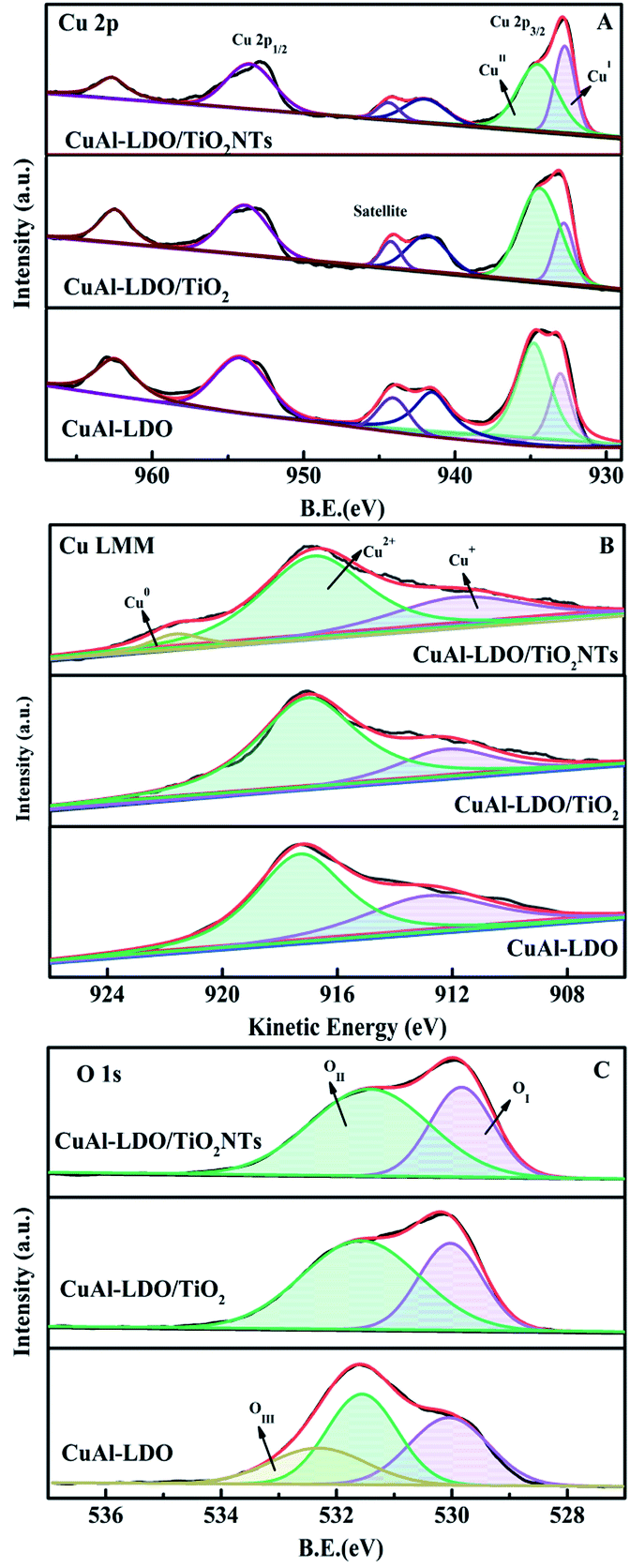
3.3.5. XPS analysis. The XPS tests were employed to study the surface element composition and electronic interaction of catalysts. The Cu 2p3/2, Cu LMM XAES and O1s spectra are presented in Fig. 8, coupled with the relative calculated parameters from the fitting curves as listed in Table 2. In Fig. 8(A), the Cu 2p3/2 spectra at 930–937 eV of all catalysts could be divided into two peaks, corresponding to Cu2+ (∼934 eV) and Cu+ (∼932 eV) or Cu0 (∼932 eV), respectively.19 The peaks at 940–945 eV of all these catalysts were attributed to the satellite peak of Cu2+. Usually, the binding energy of Cu0 was similar to that of Cu+, thus the Cu LMM XAES spectrums were tested to distinguish them as displayed in Fig. 8(B).26 It can be seen from Fig. 8(B) that all catalysts presented the peaks located at 912–914 eV and 917 eV, which could be assigned to Cu+ (912–914 eV) and Cu2+ (917 eV). In addition, the CuAl-LDO/TiO2NTs catalyst showed the peak at ∼921 eV, contributing to the characteristics of Cu0.18 Meanwhile, the relative intensity of Cu+/(Cu2+ + Cu+) was calculated as demonstrated in Table 2, with the following order: CuAl-LDO (38.4%) > CuAl-LDO/TiO2NTs (33.4%) > CuAl-LDO/TiO2 (24.1%). Therefore, the synergistic effect between Cu+ and Cu2+ seems to be the main reason for the good deNOx performance at low temperature range.
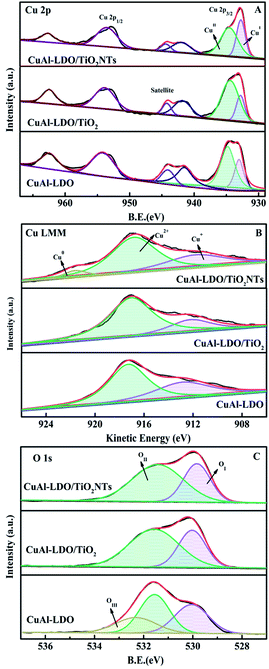 |
| | Fig. 8 The XPS spectra of the CuAl-LDO, CuAl-LDO/TiO2 and CuAl-LDO/TiO2NTs catalysts: (A) Cu 2p, (B) Cu LMM XAES, and (C) O 1s. | |
Table 2 The surface components of catalysts obtained by XPS analysis
| Catalysts |
Cu LMM XAES kinetic energy (eV) |
Relative concentration ratio/% |
| Cu+ |
Cu2+ |
Cu0 |
Cu+/(Cu2+ + Cu+)a |
OII/(OI + OII) |
| Intensity ratio between Cu+ and (Cu+ + Cu2+) by deconvolution of Cu LMM XAES spectra. |
| CuAl-LDO |
913.0 |
917.4 |
— |
38.4 |
54.4 |
| CuAl-LDO/TiO2 |
914.0 |
917.2 |
920.6 |
24.1 |
62.3 |
| CuAl-LDO/TiO2NTs |
913.1 |
917.0 |
921.4 |
33.7 |
65.2 |
The O 1s spectra of the CuAl-LDO, CuAl-LDO/TiO2 and CuAl-LDO/TiO2NTs catalysts are showed in Fig. 8(C). As displayed in Fig. 8(C), the peaks at 530 eV could be assigned to the lattice oxygen species (denoted as OI), accompanied by the chemisorbed oxygen species (denoted as OII) located at 532 eV, which was closely related with the number of surface oxygen vacancies and defects of catalyst. Moreover, the peak at 533 eV for CuAl-LDO catalyst was corresponded to the adsorbed molecular water (denoted as OIII).27,28 Besides, due to the high mobility ratio, OII species were beneficial to the NO → NO2 process and favored to the “fast SCR” reaction, thus resulting in the enhancement of low temperature SCR activity.8 As listed in Table 2, the relative concentrations of OII/(OI + OII) for CuAl-LDO/TiO2NTs (65.2%) was higher than CuAl-LDO (54.4%) and CuAl-LDO/TiO2 (62.3%). It could be concluded that the CuAl-LDO/TiO2NTs owned more abundant surface oxygen than CuAl-LDO and CuAl-LDO/TiO2, which might be helpful to the improvement of catalytic activity.
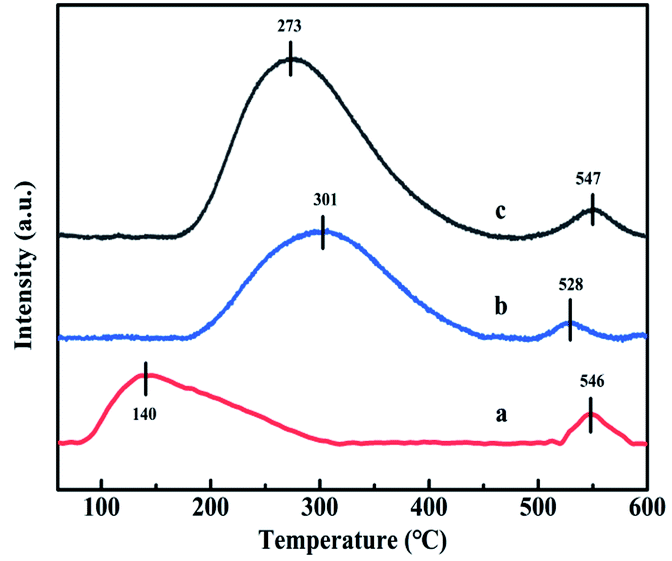
3.3.6. H2-TPR analysis. The redox ability of catalysts is closely related to the NH3-SCR performance, thus the H2-TPR technique has been widely used and the results are showed in Fig. 9. Meanwhile, the reduction peak temperature and H2 consumption of each catalyst are listed in Table 3. Two reduction peaks could be observed over TPR profiles for all catalysts. The first peak at 190 °C (CuAl-LDO), 224 °C (CuAl-LDO/TiO2NTs) and 285 °C (CuAl-LDO/TiO2) might be assigned to the reduction of highly dispersed CuO particles, being related to the reduction of Cu2+ →Cu+ → Cu.29,30 While the second peak located at 240 °C (CuAl-LDO), 271 °C (CuAl-LDO/TiO2NTs) and 327 °C (CuAl-LDO/TiO2) could be attributed to the reduction of the small crystalline CuO, the results above are consistent with the XRD results in Fig. 5.30 As presented in Fig. 9(A), both peaks for CuAl-LDO/TiO2NTs catalyst shifted to lower temperature comparing with CuAl-LDO/TiO2, indicating the better reduction ability of the former one, which might be profited from the property of TiO2NTs further resulting in the higher NOx conversion of CuAl-LDO/TiO2NTs catalyst at low temperature. It was noteworthy that the reduction temperature of CuAl-LDO/TiO2NTs catalyst was slightly higher than CuAl-LDO, which was consistent with the results of low temperature NH3-SCR activity. Moreover, the calculated results shown in Table 3 further illuminating that the CuAl-LDO/TiO2NTs catalyst with higher H2 consumption was more favorable for the redox reaction.
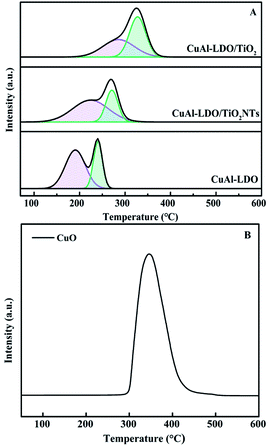 |
| | Fig. 9 The H2-TPR profiles of (A) catalysts and (B) CuO standard sample. | |
Table 3 The reduction peak attribution, amount of hydrogen consumption and normalized NH3 desorption amount of catalysts
| Catalysts |
Reduction temperature (°C) |
H2 consumptiona (mmol g−1) |
Total amount of NH3 desorptionb |
| T1 |
T2 |
| The H2 consumption of catalysts was calculated with CuO standard sample. Normalized by the NH3 total desorption peak area of CuAl-LDO. |
| CuAl-LDO |
190 |
240 |
1.3 |
1 |
| CuAl-LDO/TiO2 |
285 |
327 |
1.4 |
1.7 |
| CuAl-LDO/TiO2NTs |
224 |
271 |
1.5 |
2.7 |
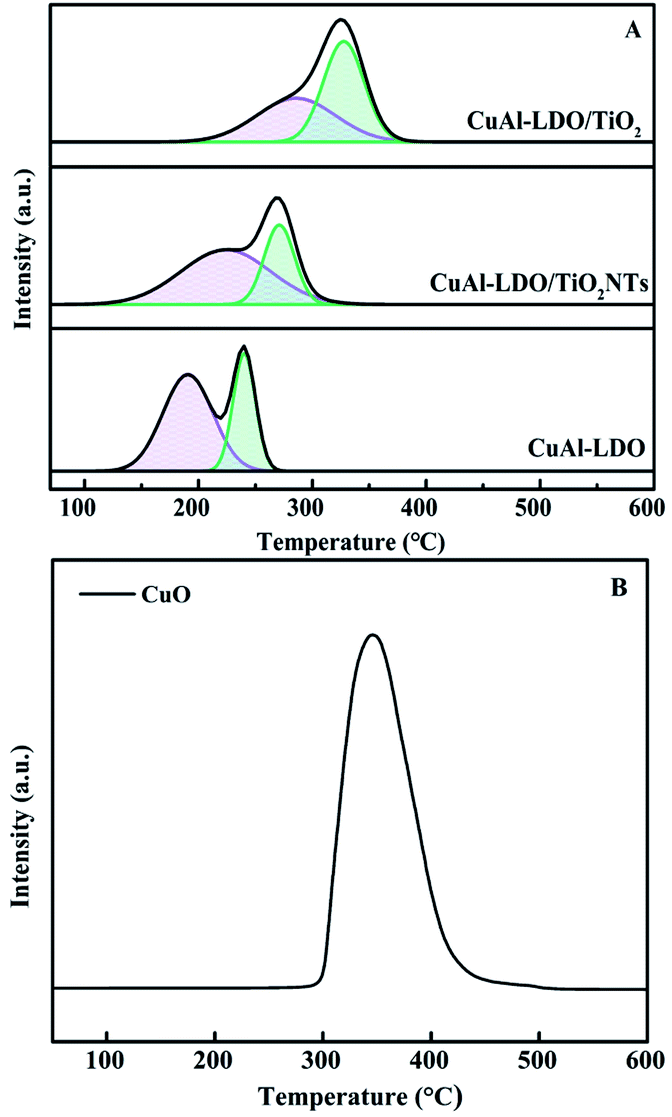
3.3.7. NH3-TPD analysis. In addition to redox ability, the acidity of catalyst was also the main factor for SCR activity. The NH3-TPD profiles of CuAl-LDO, CuAl-LDO/TiO2 and CuAl-LDO/TiO2NTs catalysts are shown in Fig. 10, which researched the quantity and intensity of acidity over the catalyst surface. The relative acid contents of the catalysts calculated by normalizing the area of the NH3 desorption peak of CuAl-LDO catalyst are listed in Table 3. It can be seen from the Fig. 10 that the CuAl-LDO catalyst showed a larger NH3 desorption peak at 140 °C, indicating the existence of more weak acid sites. Compared with CuAl-LDO catalyst, the CuAl-LDO/TiO2 and CuAl-LDO/TiO2NTs catalysts presented significantly desorption peaks near 300 °C and 270 °C, respectively, which contributed to the medium-strong acid sites. While the small desorption peak located at around 530 °C for three catalysts could be assigned to the ammonia desorption from strong acid sites. As summarized in Table 3, the total amount of acid sites over CuAl-LDO/TiO2NTs catalyst was richer than CuAl-LDO and CuAl-LDO/TiO2 catalysts, which could be attributed to the outstanding catalytic performance of the CuAl-LDO/TiO2NTs catalyst in the total test temperature range.31
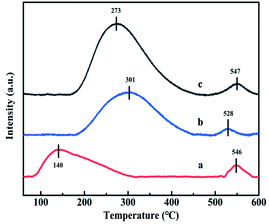 |
| | Fig. 10 The NH3-TPD profiles of catalysts: (a) CuAl-LDO, (b) CuAl-LDO/TiO2 and (c) CuAl-LDO/TiO2NTs. | |
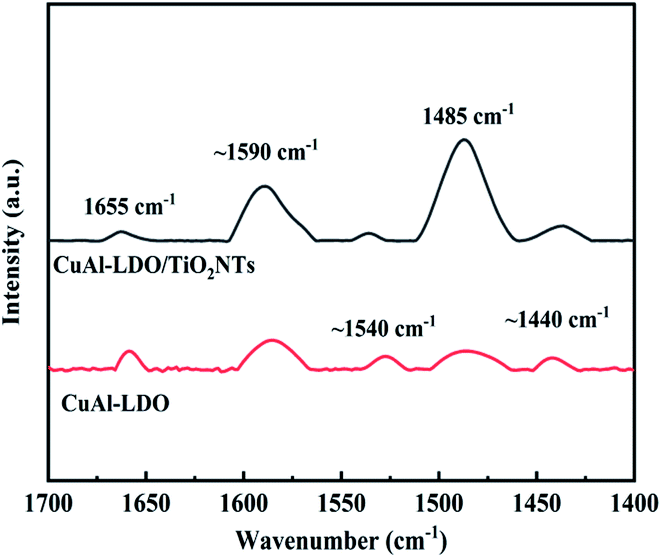
3.3.8. Py-IR analysis. The properties of acid species on the surface of CuAl-LDO and CuAl-LDO/TiO2NTs catalysts were investigated by Pyridine Adsorption Fourier-transform Infrared (Py-IR) and the results are presented in Fig. 11. For CuAl-LDO and CuAl-LDO/TiO2NTs catalysts, both the Brønsted and Lewis acid sites were detected simultaneously over the surface. The bands located at ∼1440 cm−1, ∼1540 cm−1, 1590 cm−1 and 1655 cm−1 were corresponded to the Lewis acid sites (∼1440 cm−1 and 1590 cm−1) and Brønsted acid sites (∼1540 cm−1 and 1655 cm−1), respectively.32,33 The bands around 1485 cm−1 were assigned to both Brønsted and Lewis acid sites over catalysts.34 The literature was reported that there was only Lewis acid sites over the anatase (TiO2).32,33,35 Therefore, the intensity of the band of the Lewis acid sites over CuAl-LDO/TiO2NTs catalyst was stronger than CuAl-LDO catalyst. In addition, compared with CuAl-LDO, CuAl-LDO/TiO2NTs catalyst have the most abundant and strongest acid sites over the surface, which was in good agreement with their NH3-TPD results.
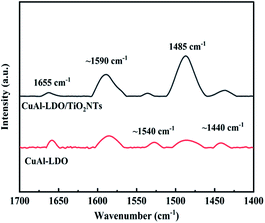 |
| | Fig. 11 Py-IR spectra of CuAl-LDO catalyst and CuAl-LDO/TiO2NTs catalyst. | |
3.4. In situ DRIFTS analysis
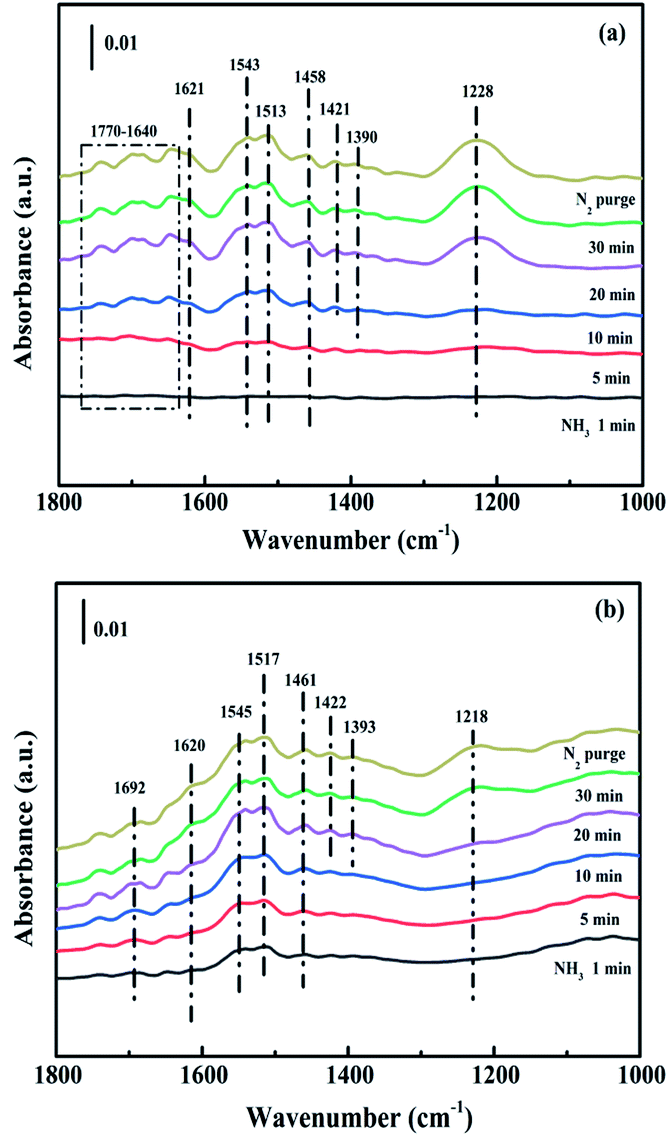
3.4.1. Adsorption of NH3. The capacity of NH3 adsorption over CuAl-LDO and CuAl-LDO/TiO2NTs catalysts were studied by In situ DRIFTS measurement. As shown in Fig. 12(a), for CuAl-LDO catalyst, the bands appeared at 1770–1640 cm−1, 1458 cm−1 and 1421 cm−1 could be assigned to NH4+ species on Brønsted acid sites.36,37 The peaks of coordinated NH3 linked to Lewis acid sites were observed at 1621 cm−1, 1543 cm−1 and 1228 cm−1.38 And the bands located at 1513 cm−1 and 1390 cm−1 were attributed to the –NH2 scissoring and wagging of N2H4 species, separately.16,39 The peaks for CuAl-LDO/TiO2NTs catalyst (in Fig. 12(b)) at 1692 cm−1, 1620 cm−1, 1545 cm−1, 1517 cm−1, 1461 cm−1, 1422 cm−1, 1393 cm−1 and 1218 cm−1 could be correspondingly assigned to the symmetric and asymmetric vibration of NH3 species adsorbed on Lewis acid sites (1620 cm−1, 1545 cm−1 and 1218 cm−1), the –NH2 scissoring and wagging of N2H4 species (1517 cm−1 and 1393 cm−1) and NH4+ ions bounded on Brønsted acid site (1692 cm−1, 1461 cm−1 and 1422 cm−1).16,38,40 It could be concluded that the Lewis acid sites and Brønsted acid sites coexisted on the surface of both catalysts. In Fig. 12, it could be found that the peak intensity of the bands assigned to Brønsted acid sites and Lewis acid sites of CuAl-LDO/TiO2NTs catalyst was higher than that of the CuAl-LDO catalyst, indicating the better NH3 adsorption capacity of CuAl-LDO/TiO2NTs catalyst.13
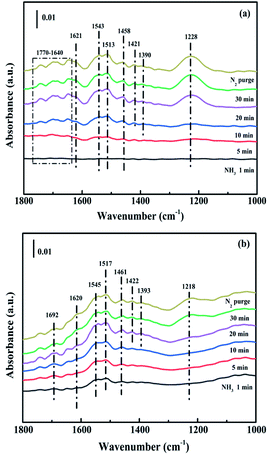 |
| | Fig. 12 In situ DRIFTS spectra of NH3 adsorption at 240 °C over (a) CuAl-LDO and (b) CuAl-LDO/TiO2NTs catalysts. | |
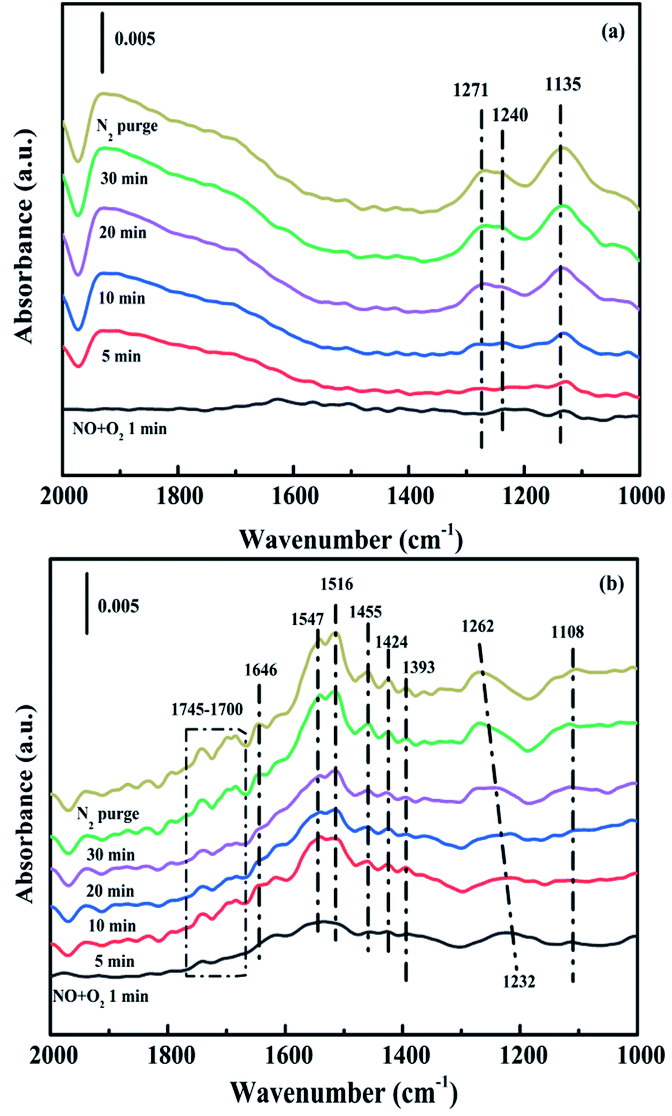
3.4.2. Co-adsorption of NO and O2. In situ DRIFTS experiments of NO + O2 adsorption were applied to research the formation and transformation of NOx species over the CuAl-LDO and CuAl-LDO/TiO2NTs catalysts. In Fig. 13(a), the bands at 1271 cm−1, 1240 cm−1 and 1135 cm−1 were detected for CuAl-LDO catalyst, which could be ascribed to linear nitrite (1454 cm−1), monodentate nitrite (1271 cm−1 and 1135 cm−1) and bridged nitrate (1240 cm−1), respectively.36,41 For CuAl-LDO/TiO2NTs catalyst, the bands located at 1745–1700 cm−1, 1646 cm−1, 1547 cm−1, 1516 cm−1, 1455 cm−1, 1424 cm−1, 1393 cm−1, 1232 cm−1 and 1108 cm−1 could be assigned to the absorbed N2O4 (1745–1700 cm−1), bridged nitrate (1646 cm−1 and 1232 cm−1), bidentate nitrate (1547 cm−1 and 1516 cm−1), linear nitrite (1455 cm−1), monodentate nitrite (1424 cm−1, 1393 cm−1 and 1108 cm−1).16,36,41,42 The intensity of peak located at 1232 cm−1 was gradually decreased along with the appearance of the band of monodentate nitrate (1262 cm−1). The results displayed that the intensity of NOx adsorbed peaks over CuAl-LDO/TiO2NTs were higher than CuAl-LDO. It could be concluded that there was larger amount of NO absorbed on the CuAl-LDO/TiO2NTs catalyst surface and oxidized to a variety of nitrate species, which resulting in the excellent SCR performance.
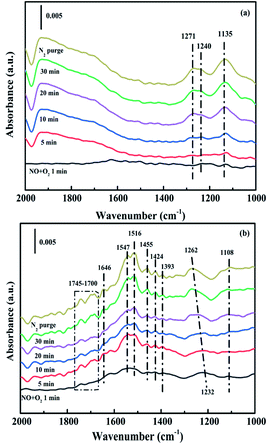 |
| | Fig. 13 In situ DRIFTS spectra of NO + O2 adsorption at 240 °C over (a) CuAl-LDO and (b) CuAl-LDO/TiO2NTs catalyst. | |
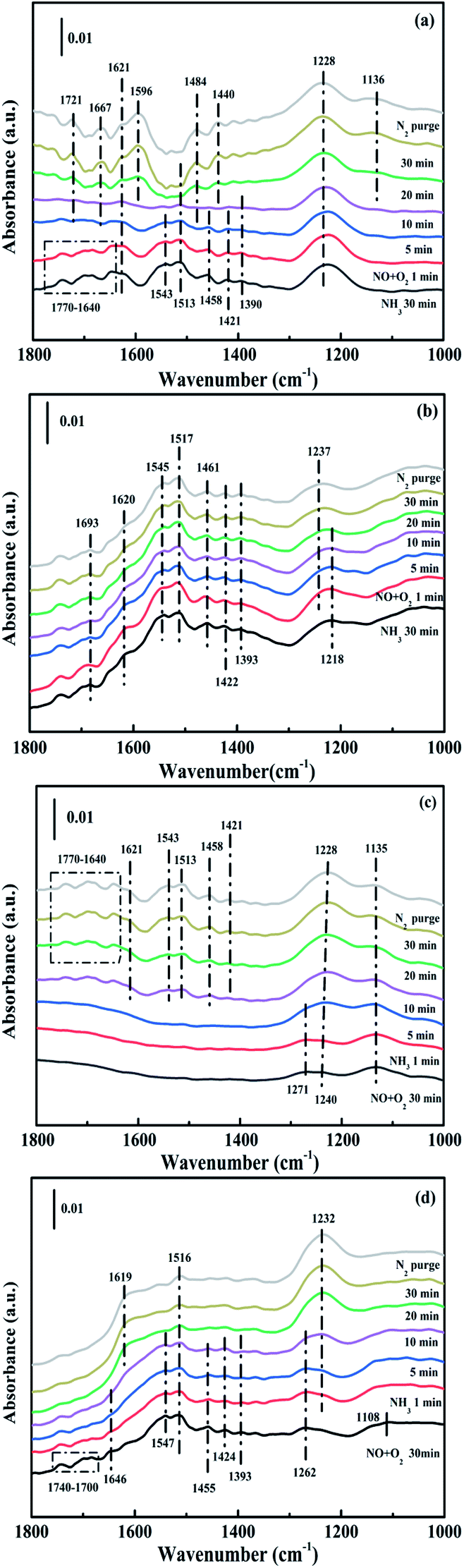
3.4.3. Reaction between nitrogen oxides and ammonia ad-species. In order to explore the potential reaction species and possible pathway, the transient reactions between NH3 and NO + O2 over CuAl-LDO and CuAl-LDO/TiO2NTs catalysts were further carried out at 240 °C. As shown in Fig. 14(a), the CuAl-LDO catalyst was firstly absorbed with NH3 for 30 min. The peaks located at 1770–1640 cm−1, 1458 cm−1 and 1421 cm−1 (NH4+ species on Brønsted acid sites), 1621 cm−1, 1543 cm−1 and 1228 cm−1 (NH3 linked to Lewis acid sites) and 1513 cm−1 and 1390 cm−1 (–NH2 scissoring and wagging of N2H4 species) were detected.16,36,38,39,43 After inletting NO + O2 for 10 min, except for the peak located at 1228 cm−1, other peaks almost disappeared, might propose the reaction between adsorbed NH3 species and NOx species. After imported NO + O2 for 20 min, the news bands located at 1721 cm−1, 1667 cm−1, 1621 cm−1, 1596 cm−1, 1484 cm−1, 1440 cm−1 and 1136 cm−1 appeared which could be assigned to the absorbed N2O4 (1721 cm−1), bridged nitrate (1667 cm−1), gaseous NO2 (1621 cm−1), bidentate nitrate (1596 cm−1), linear nitrite (1440 cm−1), monodentate nitrate (1484 cm−1 and 1136 cm−1), respectively.16,25,36,42 When the reactant gases were introduced as reverse order, the corresponding results are displayed in Fig. 14(c). As can be seen, the pre-adsorption of NO + O2 lead to the formation of linear nitrite (1454 cm−1), monodentate nitrite (1271 cm−1 and 1135 cm−1) and bridged nitrate (1240 cm−1), respectively. The further introduction of NH3 caused the generation of peaks located at 1770–1640 cm−1, 1458 cm−1 and 1421 cm−1 (NH4+ species on Brønsted acid sites), 1621 cm−1, 1543 cm−1 and 1228 cm−1 (coordinated NH3 linked to Lewis acid sites) and 1513 cm−1 (–NH2 scissoring of N2H4 species) were detected.41,44,45 It was worth noting that the intensity of peak located at 1240 cm−1 ascribed to bridged nitrite further increased after introducing NH3, which might be due to the coverage of coordinated NH3 linked to Lewis acid sites (1228 cm−1). The results indicated that the Eley–Rideal (E–R) mechanisms and Langmuir–Hinshelwood (L–H) mechanisms were occurred simultaneously over the surface of CuAl-LDO catalysts at 240 °C, and the E–R mechanisms were the main reaction way.
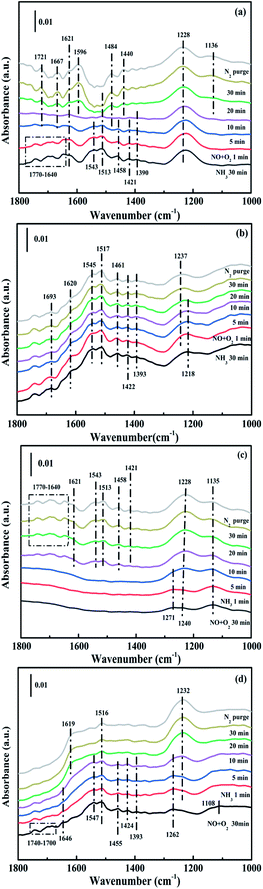 |
| | Fig. 14 In situ DRIFTS spectra of (a and b) NO + O2 adsorption with pre-absorbed NH3 and (c and d) NH3 adsorption with pre-absorbed NO + O2 at 240 °C over (a and c) CuAl-LDO and (b and d) CuAl-LDO/TiO2NTs catalysts. | |
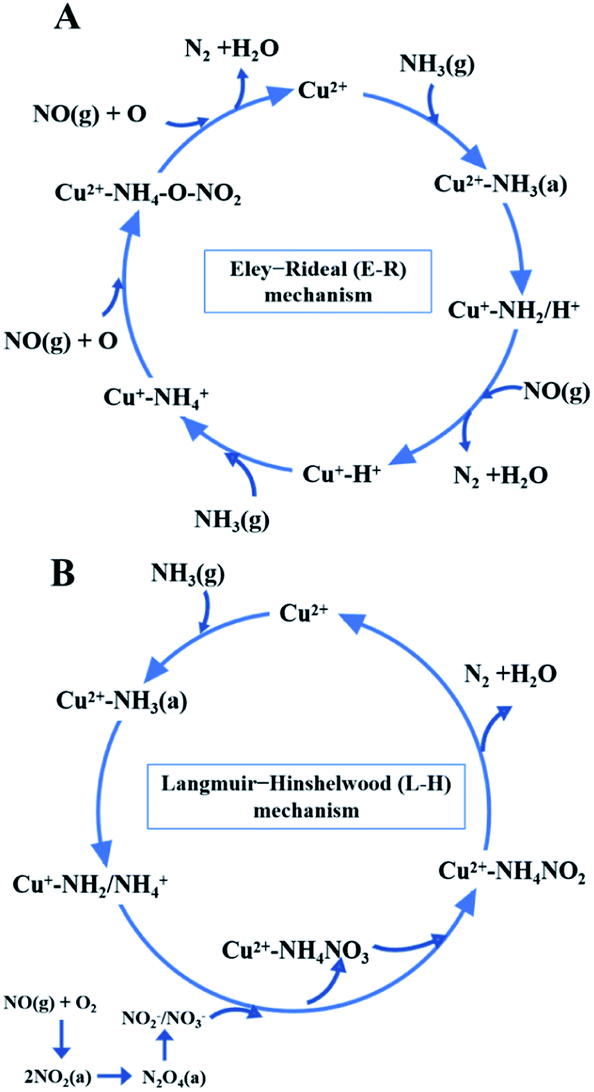
While for CuAl-LDO/TiO2NTs catalysts, the transient reaction spectra are displayed in Fig. 14(b) and (d). After pre-adsorption of NH3, the following bands emerged, NH4+ ions bounded on Brønsted acid site (1692 cm−1, 1461 cm−1 and 1422 cm−1), the symmetric and asymmetric vibration of NH3 species adsorbed on Lewis acid sites (1620 cm−1, 1545 cm−1 and 1218 cm−1), the –NH2 scissoring and wagging of N2H4 species (1517 cm−1 and 1393 cm−1).16,39,46 Further importing NO + O2, the intensity of all peaks gradually impaired, replaced by the arising of peak located at 1237 cm−1 corresponding to monodentate nitrate species. In comparison with the Fig. 13(b), there was no significant accumulation of nitrate species participating in the NH3-SCR reaction. When the reactant gases were introduced as the inverse order, the results are presented in Fig. 14(d). While NO + O2 were imported for 30 min, the corresponding species appeared as follows: the absorbed N2O4 (1745–1700 cm−1), bridged nitrate (1646 cm−1 and 1232 cm−1), bidentate nitrate (1547 cm−1 and 1516 cm−1), linear nitrite (1455 cm−1), monodentate nitrite (1424 cm−1, 1393 cm−1 and 1108 cm−1), monodentate nitrate (1262 cm−1).16,25,42,47 With an increase in the adsorption time of NH3, the bands of NOx species gradually reduced (1745–1700 cm−1, 1646 cm−1, 1547 cm−1, 1455 cm−1, 1424 cm−1, 1393 cm−1 1262 cm−1 and 1108 cm−1), accompanied by the emergence of bands located at 1619 cm−1 and 1232 cm−1, which were consistent with the asymmetric and symmetric bending vibrations of NH3 adsorbed on Lewis acid sites (1619 cm−1 and 1232 cm−1).13,48 Consequently, the NH3 species absorbed on the Lewis acid sites could react with nitrate and nitrite species on the surface of CuAl-LDO/TiO2NTs catalyst in the SCR reaction. The results indicated that CuAl-LDO/TiO2NTs catalysed the SCR reaction in both E–R mechanism and L–H mechanism. Based on In situ DRIFTS, the reaction mechanisms for CuAl-LDO/TiO2NTs catalyst were proposed, which was displayed in Fig. 15.
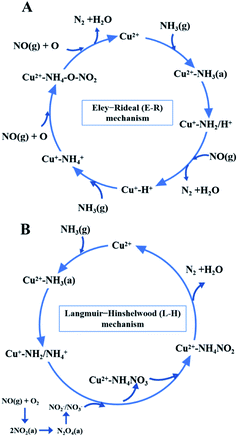 |
| | Fig. 15 The NH3-SCR reaction mechanisms over CuAl-LDO/TiO2NTs catalyst. | |
4 Conclusions
In summary, CuAl-LDO, CuAl-LDO/TiO2 and CuAl-LDO/TiO2NTs catalysts were obtained by calcining the LDHs precursor, and then applied in NH3-SCR reaction. The results demonstrated that the introduction of TiO2NTs with hollow tubular structure effectively avoided the stacking and accumulation of laminates during calcination process and enhanced the number of acidic sites on the catalyst. Consequently, the CuAl-LDO/TiO2NTs catalyst showed excellent NOx conversion, N2 selectivity as well as the good SO2 resistance. XRD results illustrated that a new phase of Cu2O was formed in the CuAl-LDO/TiO2NTs catalyst. Other characterization results indicated that the excellent deNOx performance of the CuAl-LDO/TiO2NTs catalyst depended on the synergistic effect between CuO and Cu2O, which could contribute to the larger surface area, more acidic sites, better redox performance, more abundant surface chemisorption oxygen species and stronger adsorption capacity of reactant gases. This work presents the preparation of Cu-based catalysts with wide temperature window via modifying LDHs template, which might serve as an important reference for LDHs precursor to be tailored in NH3-SCR filed.
Author contributions
Yali Du: validation, formal analysis, writing – review & editing, funding acquisition. Xuezhen Liu: methodology, formal analysis, investigation, resources, data curation, writing – original draft. Jiangning Liu: writing – review & editing, formal analysis. Rongting Du: writing – review & editing. Xu Wu: conceptualization, funding acquisition, project administration, supervision.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51978436), the National Natural Science Foundation of China (52000092), the Natural Science Foundation of Shanxi Province, China (201901D211445) and China Postdoctoral Science Foundation (2020M670706).
References
- D. Damma, P. Ettireddy, B. Reddy and P. Smirniotis, Catalysts, 2019, 9, 349 CrossRef CAS.
- V. Praveena and M. L. J. Martin, J. Energy Inst., 2018, 91, 704–720 CrossRef CAS.
- M. Fu, C. Li, P. Lu, L. Qu, M. Zhang, Y. Zhou, M. Yu and Y. Fang, Catal. Sci. Technol., 2014, 4, 14–25 RSC.
- Q. Yan, S. Chen, L. Qiu, Y. Gao, D. O'Hare and Q. Wang, Dalton Trans., 2018, 47, 2992–3004 RSC.
- W. Wang, Z. Xiong, W. He, W. Lu and H. Shi, J. Energy Inst., 2021, 98, 322–333 CrossRef CAS.
- C. Chen, Y. Cao, S. Liu, J. Chen and W. Jia, Chin. J. Catal., 2018, 39, 1347–1365 CrossRef CAS.
- Z. Song, Y. Xing, X. Zhang, H. Zhao, M. Zhao, J. Zhao, Z. Ma and Q. Zhang, Appl. Organomet. Chem., 2019, 33, e5160 Search PubMed.
- C. Li, Z. Xiong, Y. Du, X. Ning, Z. Li, J. He, X. Qu, W. Lu, S. Wu and L. Tan, J. Energy Inst., 2020, 93, 1809–1818 CrossRef CAS.
- S. Zang, G. Zhang, W. Qiu, L. Song, R. Zhang and H. He, Chin. J. Catal., 2016, 37, 888–897 CrossRef CAS.
- Z. Fan, J.-W. Shi, C. Gao, G. Gao, B. Wang, Y. Wang, C. He and C. Niu, Chem. Eng. J., 2018, 348, 820–830 CrossRef CAS.
- K. Cheng, W. Song, Y. Cheng, H. Zheng, L. Wang, J. Liu, Z. Zhao and Y. Wei, RSC Adv., 2018, 8, 19301–19309 RSC.
- B. Hou, Y. Du, X. Liu, C. Ci, X. Wu and X. Xie, RSC Adv., 2019, 9, 24377–24385 RSC.
- S. Ali, L. Chen, Z. Li, T. Zhang, R. Li, S. H. Bakhtiar, X. Leng, F. Yuan, X. Niu and Y. Zhu, Appl. Catal. B Environ., 2018, 236, 25–35 CrossRef CAS.
- X. Gao, X. Du, L. Cui, Y. Fu, Z. Luo and K. Cen, Catal. Commun., 2010, 12, 255–258 CrossRef CAS.
- S. Suárez, J. A. Martín, M. Yates, P. Avila and J. Blanco, J. Catal., 2005, 229, 227–236 CrossRef.
- X. Yao, L. Zhang, L. Li, L. Liu, Y. Cao, X. Dong, F. Gao, Y. Deng, C. Tang, Z. Chen, L. Dong and Y. Chen, Appl. Catal. B Environ., 2014, 150–151, 315–329 CrossRef CAS.
- X. Wu, R. Wang, Y. Du, C. Zou, H. Meng and X. Xie, Mol. Catal., 2019, 467, 150–160 CrossRef CAS.
- X. Wu, H. Meng, Y. Du, J. Liu, B. Hou and X. Xie, J. Catal., 2020, 384, 72–87 CrossRef CAS.
- Q. Yan, Y. Nie, R. Yang, Y. Cui, D. O'Hare and Q. Wang, Appl. Catal. Gen., 2017, 538, 37–50 CrossRef CAS.
- R. Fan, Z. Li, Y. Wang, C. Zhang, Y. Wang, Z. Ding, X. Guo and R. Wang, RSC Adv., 2020, 10, 5845–5852 RSC.
- D. K. Pappas, T. Boningari, P. Boolchand and P. G. Smirniotis, J. Catal., 2016, 334, 1–13 CrossRef CAS.
- S.-A. Chen, J.-N. Nian, C.-C. Tsai and H. Teng, J. Air Waste Manage. Assoc., 2007, 57, 600–605 CrossRef CAS PubMed.
- Z. Cai, G. Zhang, Z. Tang and J. Zhang, ACS Appl. Nano Mater., 2021, 4, 6201–6211 CrossRef CAS.
- C. Liu, L. Chen, J. Li, L. Ma, H. Arandiyan, Y. Du, J. Xu and J. Hao, Environ. Sci. Technol., 2012, 46, 6182–6189 CrossRef CAS PubMed.
- D. Meng, W. Zhan, Y. Guo, Y. Guo, L. Wang and G. Lu, ACS Catal., 2015, 5, 5973–5983 CrossRef CAS.
- Z. He, H. Lin, P. He and Y. Yuan, J. Catal., 2011, 277, 54–63 CrossRef CAS.
- L. Dou, T. Fan and H. Zhang, Catal. Sci. Technol., 2015, 5, 5153–5167 RSC.
- Y. Geng, W. Shan, F. Liu and S. Yang, J. Hazard. Mater., 2021, 405, 124223 CrossRef CAS PubMed.
- M. Ma, R. Yang, Z. Jiang, C. Chen, Q. Liu, R. Albilali and C. He, Fuel, 2021, 303, 121244 CrossRef CAS.
- Q. Yan, Y. Gao, Y. Li, M. A. Vasiliades, S. Chen, C. Zhang, R. Gui, Q. Wang, T. Zhu and A. M. Efstathiou, Appl. Catal. B Environ., 2019, 255, 117749 CrossRef CAS.
- J. Zhang, H. Tian, Y. Yu, Z. Jiang, M. Ma and C. He, Catal. Lett., 2021, 151, 2502–2512 CrossRef CAS.
- Y. Liu, Y. Guan, C. Li, J. Lian, G. Gan, E. Lim and F. Kooli, J. Catal., 2006, 244, 17–23 CrossRef CAS.
- L. Sun, S. Cao, Y. Huang, Y. Zhang, Y. Xiao, G. Dong and Y. Su, RSC Adv., 2019, 9, 30340–30349 RSC.
- W. Yao, Y. Liu, X. Wang, X. Weng, H. Wang and Z. Wu, J. Phys. Chem. C, 2016, 120, 221–229 CrossRef CAS.
- L. Li, H. Yue, T. Ji, W. Li, X. Zhao, L. Wang, J. She, X. Gu and X. Li, Appl. Catal. Gen., 2019, 574, 25–32 CrossRef CAS.
- L. Chen, Z. Si, X. Wu and D. Weng, ACS Appl. Mater. Interfaces, 2014, 6, 8134–8145 CrossRef CAS PubMed.
- Q. Zhang, T. Zhang, F. Xia, Y. Zhang, H. Wang and P. Ning, Appl. Surf. Sci., 2020, 500, 144044 CrossRef CAS.
- J. Ma, Y. Li, J. Liu, Z. Zhao, C. Xu, Y. Wei, W. Song, Y. Sun and X. Zhang, Ind. Eng. Chem. Res., 2019, 58, 2389–2395 CrossRef CAS.
- C. Yu, B. Huang, L. Dong, F. Chen and X. Liu, Catal. Today, 2017, 281, 610–620 CrossRef CAS.
- Y. Wei, P. Zhang, J. Xiong, Q. Yu, Q. Wu, Z. Zhao and J. Liu, Environ. Sci. Technol., 2020, 54, 6947–6956 CrossRef CAS PubMed.
- H. Hu, S. Cai, H. Li, L. Huang, L. Shi and D. Zhang, J. Phys. Chem. C, 2015, 119, 22924–22933 CrossRef CAS.
- G. Zhou, B. Zhong, W. Wang, X. Guan, B. Huang, D. Ye and H. Wu, Catal. Today, 2011, 175, 157–163 CrossRef CAS.
- T. Zhang, R. Qu, W. Su and J. Li, Appl. Catal. B Environ., 2015, 176–177, 338–346 CrossRef CAS.
- L. Wei, S. Cui, H. Guo, X. Ma and L. Zhang, J. Mol. Catal. Chem., 2016, 421, 102–108 CrossRef CAS.
- Z. Zhang, L. Chen, Z. Li, P. Li, F. Yuan, X. Niu and Y. Zhu, Catal. Sci. Technol., 2016, 6, 7151–7162 RSC.
- R. Gao, D. Zhang, X. Liu, L. Shi, P. Maitarad, H. Li, J. Zhang and W. Cao, Catal. Sci. Technol., 2013, 3, 191–199 RSC.
- Y. Wen, S. Cao, X. Fei, H. Wang and Z. Wu, Chin. J. Catal., 2018, 39, 771–778 CrossRef CAS.
- Z. Liu, H. Liu, X. Feng, L. Ma, X. Cao and B. Wang, Mol. Catal., 2018, 445, 179–186 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00316c |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc
*bc