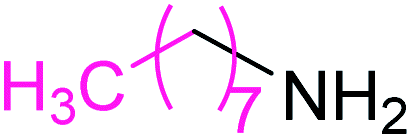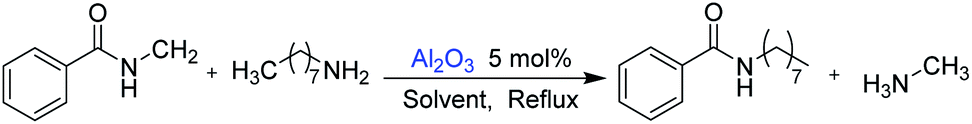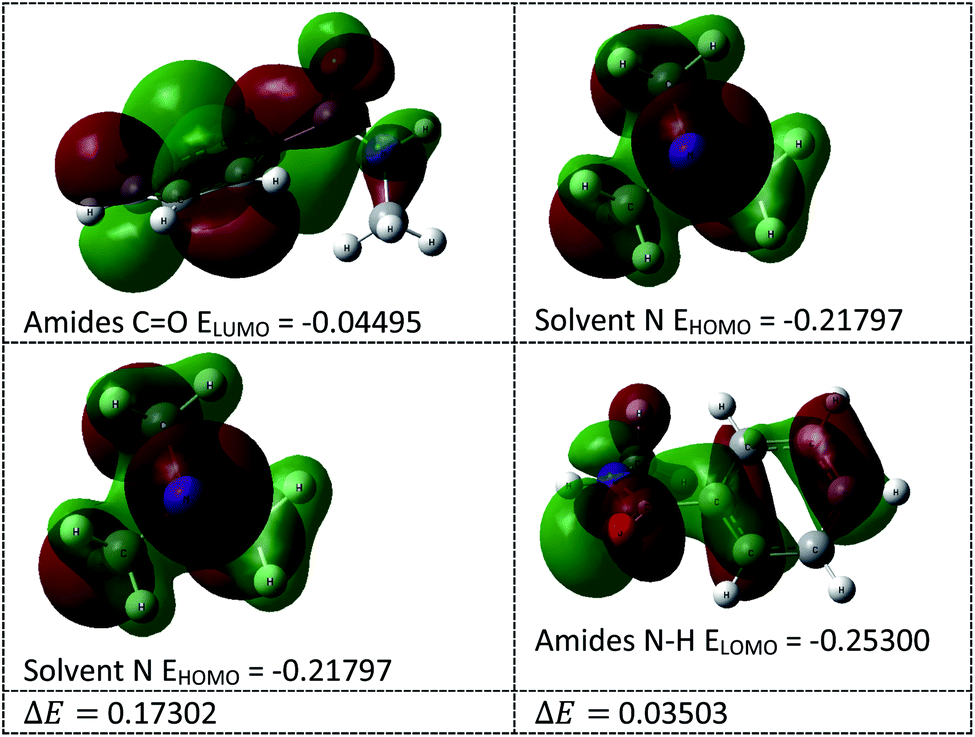DOI:
10.1039/D2RA00450J
(Paper)
RSC Adv., 2022,
12, 11255-11261
Combined experimental and computational study of Al2O3 catalyzed transamidation of secondary amides with amines†
Received
21st January 2022
, Accepted 21st March 2022
First published on 11th April 2022
Abstract
Amides are the most extensively used substances in both synthetic organic and bioorganic chemistry. Unfortunately, the traditional synthesis of amides suffers from some important drawbacks, including low atom efficiency, high catalyst loading, separation of products from the reaction mixture and production of byproducts. Al2O3 is an amphoteric catalyst that activates the carbonyl carbon of the secondary amide group and helps the C–N cleavage of the reactant amide group by attacking the N–H hydrogen. By using the concepts of amphoteric properties of Al2O3, amides were synthesized from secondary amides and amines in the presence of triethylamine solvent. Several aliphatic and aromatic amines were used for the transamidation of N-methylbenzamide in the presence of the Al2O3 catalyst. Moreover, using the Gaussian09 software at the DFT level, HUMO, LUMO and the intrinsic reaction coordinates (IRCs) have also been calculated to find out the transition state of the reaction and energy. In this study, five successful compounds were synthesized by the transamidation of secondary amides with amines using a reusable Al2O3 catalyst. The catalyst was reused several times with no significant loss in its catalytic activity. The products were purified by recrystallization and column chromatography techniques. This catalytic method is effective for the simultaneous activation of the carbonyl group and N–H bond by using the Al2O3 catalyst.
Introduction
The amide bond is an inevitable ingredient of synthetic and biological polymers1,2 and is a significant functional group in organic chemistry, particularly in synthetic organic chemistry. Amidation reactions are the most accomplished chemical processes in the pharmaceutical industry and drug discovery activities.3–6 The world's leading pharmaceutical companies GSK, AstraZeneca and Pfizer conducted a small survey of 128 drug candidate molecules, and from which, they found that around 10% of the reactions used in their synthesis involved an amide bond formation reaction.7–9
A consecutive scheme for the synthesis of amides has emerged on amides and the coupling of activated carboxylic acid derivatives.10,11 However, there are boundaries such as the lability of the activated acid derivatives and exhausting procedures.12 Also, a greener procedure for direct amide bond formation has been investigated.13,14 Therefore, the ACS Green Chemistry Institute has evaluated the amide bond formation with well atom economy as one of the biggest challenges for organic chemists.15,16 The most successful example of direct amide synthesis is the coupling of carboxylic acids with amines flourished by boric acids.17,18 Another example is metal-free transamidation by direct nucleophilic addition to the amide bond.19,20 Amide bond formation21 via unconventional methods has been reviewed, and some substitutes were obtainable to carry out acylation on nitrogen.22 For straight amidation,23 at present, most researchers are focusing on heterogeneous24 catalysis from carboxylic acids25 with amines. Most of them pass through from low activity, low selectivity, or narrow scope.26 Examples of transamidation reaction is occasional, and it's application is finite in organic synthesis, and mostly used in intermolecular reactions.27 However, the trouble with the presence of an acidic N–H bond in amides causes this reaction to grow from the intrinsic strength of the amide C–N bond together.28 Transamidation usually focuses on harsh conditions (>250 °C), long reaction times or stoichiometric reagents to cleave the chemically jolly amide bond.29
For more observations on transamidation, some reported methods used density functional theory (DFT)30,31 calculations. Moreover, the use of computational investigations gave more information on these routes of transamidation and insights into the reaction mechanism.
Enlightened by these prior studies, in this study, we have demonstrated a novel and sustainable method for the transamidation of secondary amides with amines using Al2O3 as a reusable, inexpensive, and commercially available heterogeneous catalyst with tolerance to the co-presence of base molecules and performed the computational study for probable transition states and reaction mechanism.
Result and discussions
Synthesis of N,N-octyl benzamide
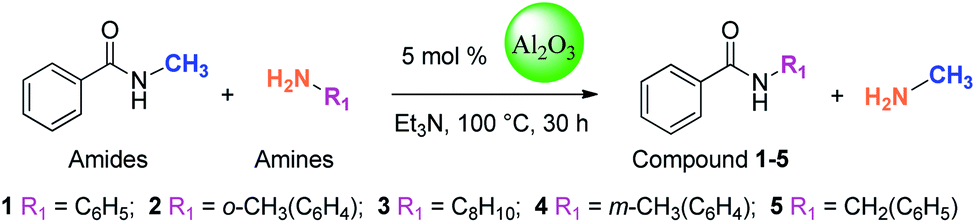
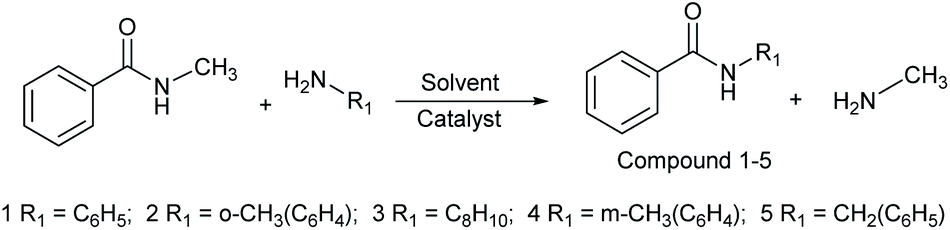
In this study, amides were synthesized via transamidation reaction with secondary amides and amines in the presence of a heterogeneous catalyst. Typically, in an amide to amine ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 4 mL triethylamine and 50 mg of Al2O3 were added to a reaction vessel (RB flask). The reaction mixtures were heated on a hot plate at 100 °C in a sand bath and stirred at 300 rpm. Upon the completion of the reaction, 2-propanol (4 mL) was added to the mixture, and then, the Al2O3 catalyst was separated from the reaction mixtures by centrifugation. The pure amide products compound 1–5 (Scheme 1, Table 1, and Schemes S1–S5†) were confirmed by TLC. The as-synthesized pure amide products were identified by FT-IR spectroscopy, 1H-NMR, and 13C-NMR.
:1, 4 mL triethylamine and 50 mg of Al2O3 were added to a reaction vessel (RB flask). The reaction mixtures were heated on a hot plate at 100 °C in a sand bath and stirred at 300 rpm. Upon the completion of the reaction, 2-propanol (4 mL) was added to the mixture, and then, the Al2O3 catalyst was separated from the reaction mixtures by centrifugation. The pure amide products compound 1–5 (Scheme 1, Table 1, and Schemes S1–S5†) were confirmed by TLC. The as-synthesized pure amide products were identified by FT-IR spectroscopy, 1H-NMR, and 13C-NMR.
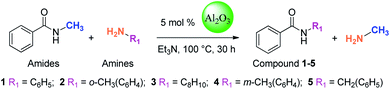 |
| | Scheme 1 Synthesis of amide derivatives 1–5. | |
Table 1 Optimize structure of synthesized amides compound 1–5
Synthesis of N-phenyl benzamide 1
Molecular weight: 197 g mol−1; molecular formula: C13NOH11; solubility: soluble in chloroform; FT-IR (υ KBr): 3347, 3055, 1659, 1536, 1439, 1659, 1075 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.02 (br s, 1H, –NH), 7.088 (m, 2H), 7.67 (m, 2H), 7.48 (t, 1H), 7.38 (t, 2H), 7.27 (m, 2H), 7.17 (t, 1H); 13C-NMR (100 MHz, CDCl3): δ 165.895 (1C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 137.969 (1C), 135.010 (1C), 131.844 (1C), 129.100–129.438 (2C), 128.783–129.074 (2C), 127.080 (2C), 124.550 (1C), 120.30 (2C). Calc. mass: 197; GC-152 MS (CDCl3): m/z 197, 105 (base peak), 77, 65, 51.
O), 137.969 (1C), 135.010 (1C), 131.844 (1C), 129.100–129.438 (2C), 128.783–129.074 (2C), 127.080 (2C), 124.550 (1C), 120.30 (2C). Calc. mass: 197; GC-152 MS (CDCl3): m/z 197, 105 (base peak), 77, 65, 51.
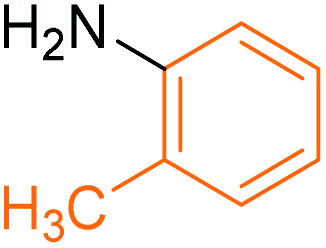
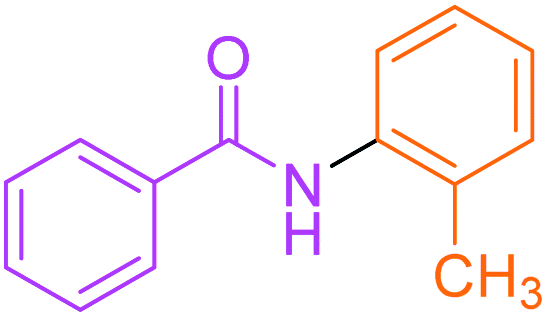
Synthesis of N-(o-methyl) phenyl benzamide 2
Molecular weight: 212 g mol−1; molecular formula: C14NOH14; solubility: soluble in chloroform; FT-IR (υ KBr): 3227, 3060, 1645, 1592, 1434, 1294, 1074 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.067 (br s, 1H, –NH), 7.86–7.88 (m, 2H), 7.53–7.56 (t, 2H), 7.44–7.47 (t, 3H), 7.23–7.28 (m, 2H), 6.69–6.99 (m, 1H), 2.36 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 165.924 (1C, CO), 138.989 (1C), 137.913 (1C), 131.764 (1C), 129.391 (1C), 128.86 (1C), 128.729 (2C), 125.401 (1C), 127.092 (2C), 121.031 (1C), 117.462 (1C), 21.51 (1C). Calc. mass: 211; GC-152 MS (CDCl3): m/z 211, 105 (base peak), 77, 51.
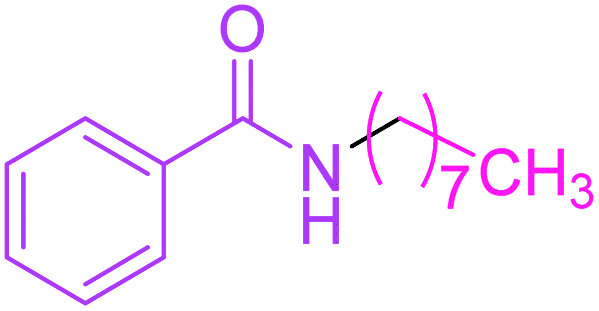
Synthesis of N-n-octyl benzamide 3
Molecular weight: 233 g mol−1; molecular formula: C15NOH23; solubility: soluble in chloroform; FT-IR (υ KBr): 3360, 3058, 1653, 1549, 1493, 1447, 1076 cm−1; 1H-NMR (400 MHz, CDCl3): δ 6.368 (br s, 1H, –NH), 7.77–7.78 (m, 2H), 7.47–7.51 (t, 1H), 7.40–7.44 (t, 2H), 3.42–3.47 (m, 2H), 2.362–2.398 (m, 2H), 1.58–1.65 (m, 2H), 1.28–1.34 (m, 8H), 0.87–0.91 (t, 3H); 13C-NMR (100 MHz, CDCl3): δ 167.626 (1C, CO), 134.811 (1C), 131.313 (1C), 128.525 (1C), 126.889 (1C), 40.181 (1C), 31.806 (1C), 29.675 (1C), 29.225 (1C), 29.308 (1C), 27.029 (1C), 14.097 (1C). Calc. mass: 233; GC-152 MS (CDCl3): m/z 233, 105 (base peak), 205, 190, 176, 162, 148, 124, 122, 77.
Synthesis of N-(m-methyl) phenyl benzamide 4
Molecular weight: 212 g mol−1; molecular formula: C14NOH14; solubility: soluble in chloroform; FT-IR (υ KBr): 3240, 3030, 2829, 1651, 1603, 1526, 1440 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.722 (br s, 1H, –NH), 7.962–7.982 (m, 1H), 7.907–7.925 (m, 1H), 7.576–7.612 (t, 1H), 7.50–7.54 (m, 3H), 7.24–7.27 (m, 1H), 7.13 (m, 1H), 2.36 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ 165.683 (1C, CO), 135.789 (1C), 135.039 (1C), 131.885 (1C), 130.591 (1C), 129.244 (1C), 128.873–128.918 (2C), 127.074 (1C), 126.947 (1C), 125.399 (1C), 123.139 (1C), 17.860 (1C). Calc. mass: 211; GC-152 MS (CDCl3): m/z 211, 105 (base peak), 77, 51.
Synthesis of N-benzyl benzamide 5
Molecular weight: 212 g mol−1; molecular formula: C14NOH13; solubility: soluble in chloroform; FT-IR (υ KBr): 3329, 3056, 2940, 1639, 1578, 1555, 1492 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.270 (br s, 1H, –NH), 5.150 (s, 1H), 4.460 (s, 1H), 6.825–6.834 (m, 2H), 7.396–7.404 (m, 2H), 7.185–7.283 (m, 1H), 8.001–8.183 (m, 2H), 7.597–7.641 (m, 2H), 7.826–7.844 (m, 1H). 13C-NMR (100 MHz, CDCl3): Calc. mass: 211; GC-152 MS (CDCl3): m/z 211.
Catalyst screening
The “model reaction” was checked if it was catalytic or not. For this purpose, we have set a reaction condition, where the catalyst was not used and all the parameters remained the same. Under such conditions, the reaction does not give any products. Therefore, it was clear that the reaction was catalytic. Catalysts for the model reaction of N-methyl benzamide with n-octylamine at 100 °C under reflux conditions for 30 h were screened. The model reaction (Scheme 2, Table 2, and Fig. S1†) summarizes the yield of the corresponding amide for various heterogeneous catalysts. We screened 6 types of metal oxides. Among these catalysts, Al2O3 shows the highest catalytic activity and obtained 76% yield. In the reaction, methylamine produced is volatile and leaves the reaction immediately, which explains why the reaction is not reversible.
 |
| | Scheme 2 Model reaction. | |
Table 2 Catalyst screening for the model reaction
| Entry |
Catalyst |
Yields (%) |
| 01 |
— |
0 |
| 02 |
SnO2 |
5 |
| 03 |
Cu2O |
3 |
| 04 |
Al2O3 |
76 |
| 05 |
Nb2O5 |
8 |
| 06 |
CeO2 |
10 |
| 07 |
TiO2 |
6 |
Solvent screening
In solvent screening, five solvents were used in the model reaction of N-methyl benzamide with n-octylamine. Among these, triethylamine was a suitable solvent for amide synthesis. Interestingly, it was found that aluminium oxide showed the highest catalytic activity when triethylamine was used as a solvent. It was also observed that without solvent, no reaction occurred. The model reaction (Scheme 2, Table 3, and Fig. S2†) shows the result of the solvent screening.
Table 3 Solvent screening for model reaction
| Entry |
Solvent |
Yields (%) |
| 01 |
No solvent |
0 |
| 02 |
O-Xylene |
9 |
| 03 |
Benzene |
5 |
| 04 |
Triethylamine |
76 |
| 05 |
Toluene |
7 |
Reusability of Al2O3
We checked the reusability of the Al2O3 catalyst for the reaction of N-methyl benzamide with n-octylamine after the completion of the reaction, whereby the catalyst was separated from the mixture by centrifugation, followed by washing with acetone and drying at 100 °C for 3 h. The recovered Al2O3 catalyst was reused for three cycles without a marked loss of its catalytic activity, but after three cycles, the product of this reaction decreased remarkably (Table S3 and Fig. S3†).
Al2O3-Catalyzed transamidation of secondary amides
Usually, amidation and transamidation proceed through the activation of the carbonyl group by using Lewis acid catalysts, where Lewis acid sites of the catalyst interact with carbonyl oxygen and increase the electrophilicity of the carbonyl carbon. However, in this case, secondary amides were more inactive due to higher electron density on the nitrogen in the amide functional group. Therefore, the simultaneous activation of the carbonyl oxygen and N–H proton were required to proceed with this reaction. Since Al2O3 is amphoteric in nature, it was used as a catalyst for the transamidation of secondary amides with amines. In this reaction, the Lewis acid site Al3+ activated the carbonyl oxygen, and the O2− site interacted with N–H proton simultaneously. Moreover, the basic solvent triethylamine enhanced the reaction by activating N–H proton (Table 4 and Fig. 3).
Table 4 Optimization energy of Al2O3
| Al2O3 (method: RB3LYP, basis set: 6-31G(d,p) |
| Electronic energy |
−710.599253 Hartree |
| Polarizability |
53.599723 a.u. |
| Dipole moment |
0.002013 Debye |
Characterization of Al2O3
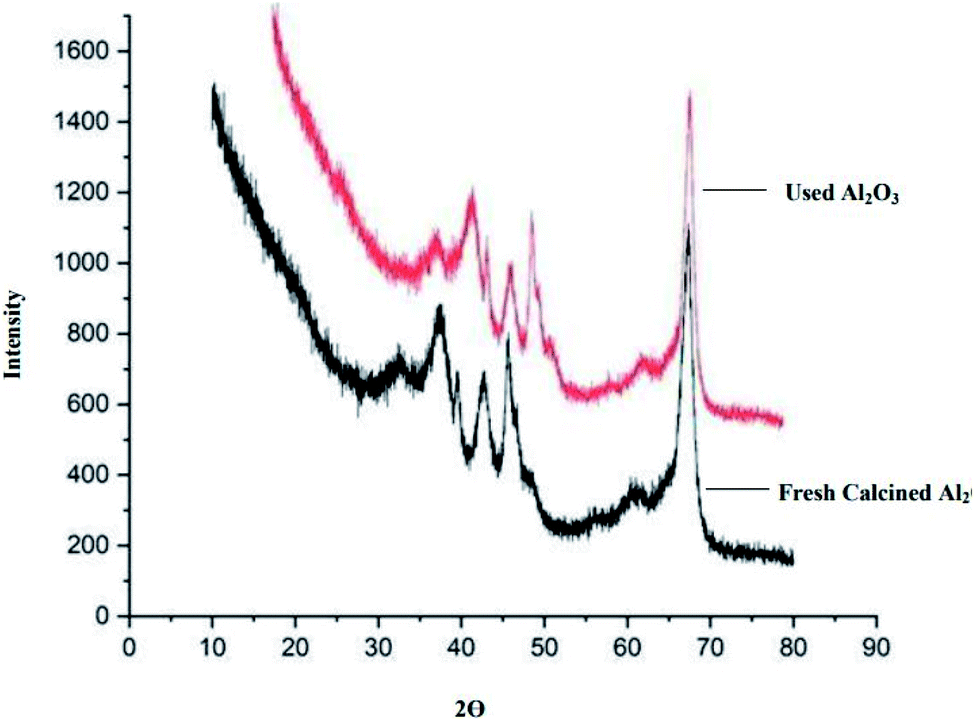
Powder X-ray diffraction (XRD) analysis. The X-ray powder diffraction (XRD) of freshly calcined Al2O3 and reused Al2O3 was performed at 0°–90° at an angle of 2θ (Fig. 1). The results of the XRD data reflect the γ-Al2O3 pattern. The pattern is similar for both the freshly calcined Al2O3 and reused Al2O3. The peaks observed in XRD can be indexed to the (111), (220), (311), (222), (400), (511) and (440) reflections of γ-Al2O3 according to the JCPDS card: 100425.32
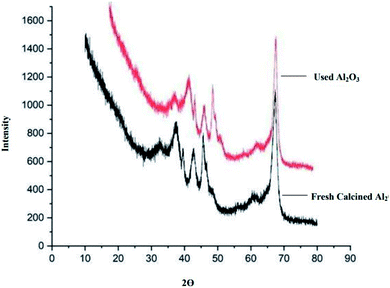 |
| | Fig. 1 XRD of freshly calcined and used Al2O3. | |
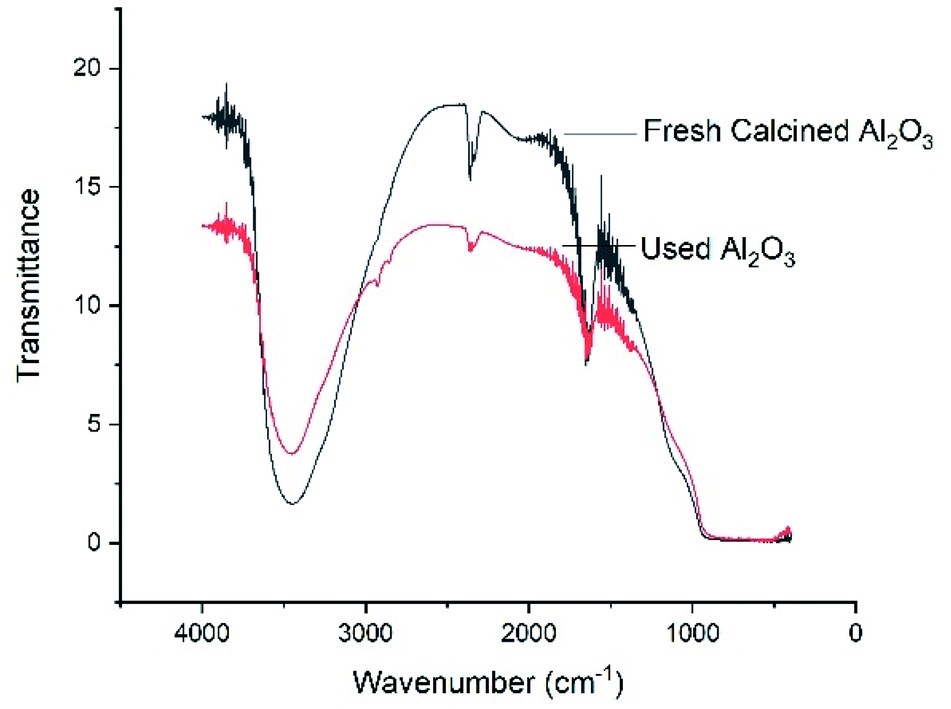
Fourier transform infrared spectroscopy (FT-IR) analysis. Fourier transform infrared (FTIR) spectroscopy was also performed for both of freshly calcined Al2O3 and reused Al2O3 (Fig. 2). The FTIR patterns showed a broad band at 3400–3600 cm−1, which is characteristic of the stretching vibrations of –OH that is bonded to Al3+, and another band at 1620–1650 cm−1 corresponding to physisorbed water are observed. A slightly wide peak (hub) at 1100 cm−1 is responsible for Al–O in γ-Al2O3. The bands appear near ∼2800 cm−1 are ascribed to organic compounds, which might have been deposited from the reaction mixture. Due to this reason, the percentage yield of the product slightly decreases during the re-usability test.
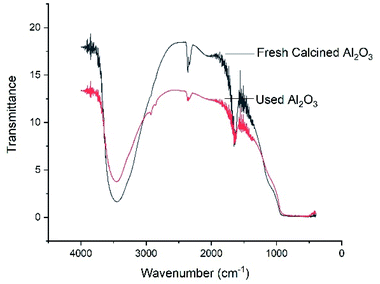 |
| | Fig. 2 FT-IR of freshly calcined Al2O3 and used Al2O3. | |
 |
| | Fig. 3 Al2O3 Mulliken charge distribution. | |
Computational investigations
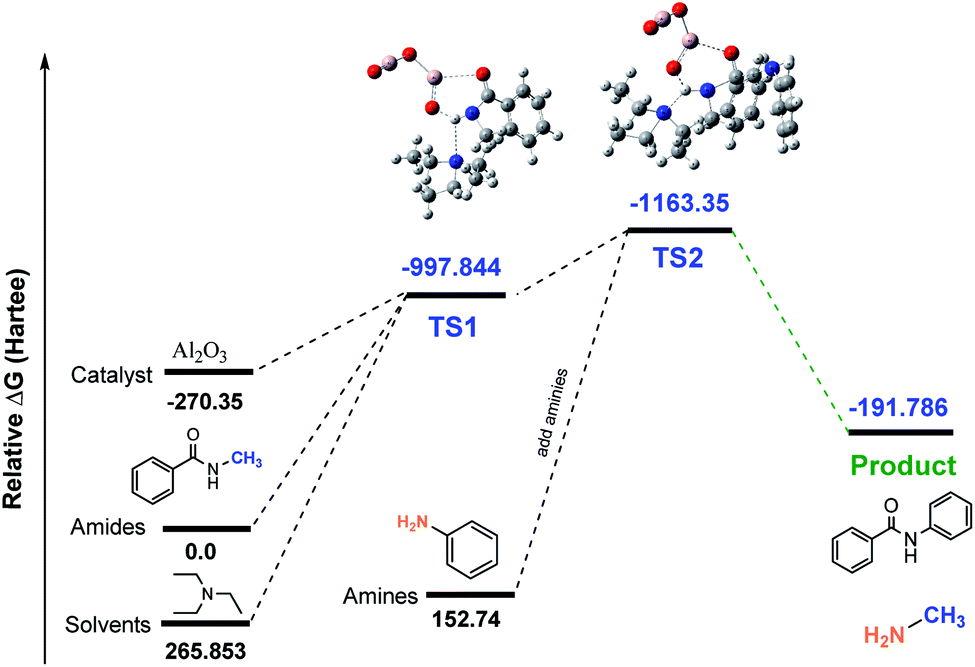
Studies of AI2O3-catalyzed transamidation. In order to evaluate the energies that raised in intermediates of the reaction, we used density functional theory (DFT) calculations as well as the reaction coordinates for the Al-catalyzed transamidation of secondary amides. The initial relative free energies of the reactants and solvents are 0, 265.853, and −270.35 for amides, solvents (Et3N) and catalyst (Al2O3), respectively. The calculated relative free energies of the Al-complex (TS1) was −997.844 (Hartee) with amides and solvent. After adding amines to the Al-complex (TS2), the energy of the relative transition state was found to be −1163.35 Hartee. Finally, the energy dropped to −191.786 Hartee after releasing the product (compound 2). A simplified transamidation model system, consisting of TS1 and TS2 as transition states was used to probe the reaction profile for the Al-catalyzed transamidation of tertiary amides, as shown in Fig. 4. The difference between relative energies of the reactant and product was reasonable in light of our observation, indicating that the substrate structure can have a significant influence on the rate of transamidation.
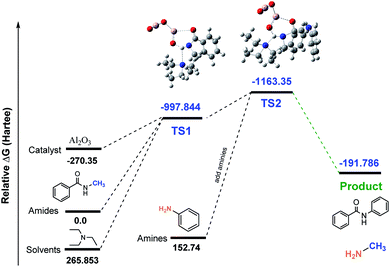 |
| | Fig. 4 Calculated free energy profile (Hartree) for the synthesis of N-phenyl benzamide 1 using Al2O3. | |
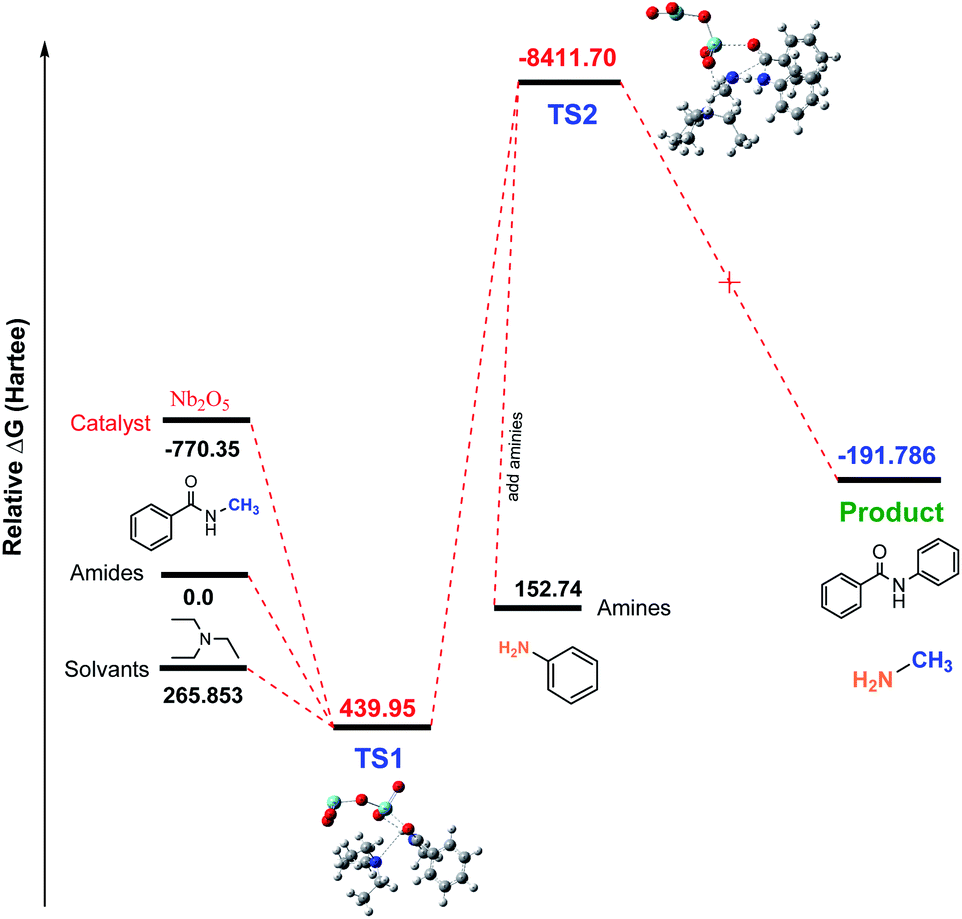
Studies of Nb2O5-catalyzed transamidation. In most of the cases, the CO activation occurred when Nb2O5 catalyst was used. However, in our research, we did not obtain any good results when using the Nb2O5 catalyst. To investigate why this base tolerant catalyst was not effective for this transamidation reaction, we used density functional theory (DFT) calculations as well as the reaction coordinates for the Nb2O5-catalyzed transamidation of secondary amides. Initial relative free energies of reactants and solvent were 0, 265.853, and −770.35 for amides, solvents (Et3N) and the catalyst (Nb2O5), respectively. The calculated relative free energies of Al-complex (TS1) was 439.95 (Hartree) with amide solvents. After adding amines to the Al-complex (TS2), the energy of the relative transition state was found at −8411.70 Hartree. Finally, there was a drop to −191.786 Hartree after releasing the product (compound 2) in Fig. 5. Therefore, if we consider the transition state with Nb2O5, the energy is less than the reactant molecules, which is not possible for a reaction. This possibly occurred due to the non-interaction of reactants with the catalyst. This evidence is also found in the intrinsic reaction coordinate (IRC) calculations.
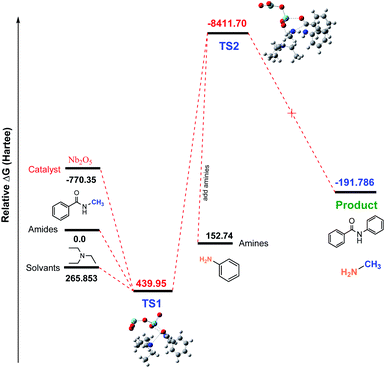 |
| | Fig. 5 Calculated free energy profile (Hartree) for the synthesis of N-phenyl benzamide 1 using Nb2O5. | |
Reaction mechanism
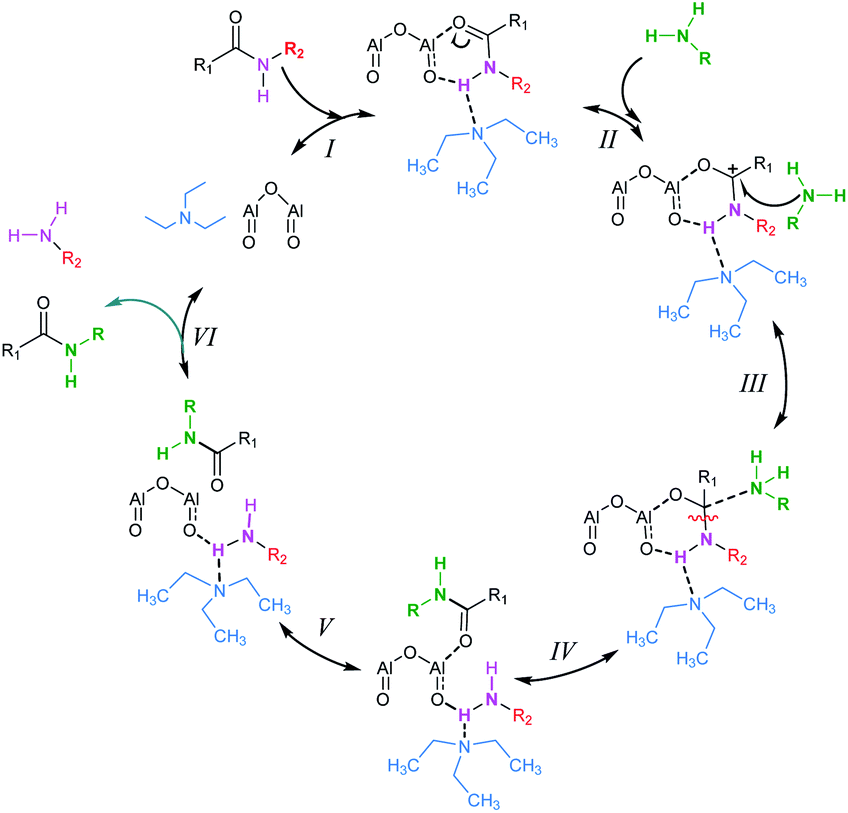
To investigate the reaction mechanism of the Al2O3-catalyzed transamidation, we studied DFT calculations. A simple catalytic phase correlative with its data is shown below in Scheme 3. At the first step (I), Al2O3, amides and solvent (Et3N) make a complex. The amides CO π electron participate in the Al–O bond formation. Thus, the amines N attack on the carbonyl carbon of amides. Following this complex, amides C–N bond cleavage and formation of a new N–C bond occur. Finally, amines are removed and new amides are formed.
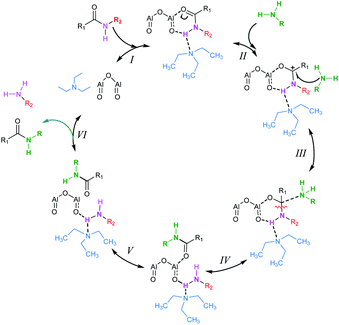 |
| | Scheme 3 Proposed mechanism for Al2O3 catalyzed transamidation. | |
Efficiency of the Al2O3/Et3N system
In this transamidation reaction, the solvent (Et3N) plays a significant role. From the DFT calculations, we found that the solvent's N HOMO and amide's N–H LUMO energy gap (ΔE = 0.035) was less than the amide's CO LUMO and solvent's N HOMO energy gap (ΔE = 0.173), as shown in Fig. 6. For this reason, the solvent's N makes an intermediate bond with the amide's N–H. Therefore, the use of Et3N was an important part of the research. Lone pair of electrons on the nitrogen of the solvent interacts with the proton of amide that helps to remove a proton from amide, and this helps to cleave to the C–N bond of amide.
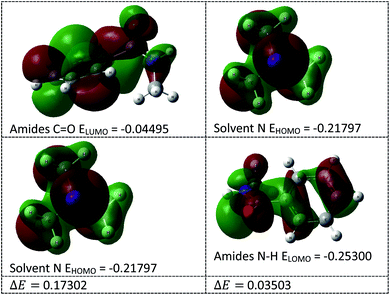 |
| | Fig. 6 HOMO–LUMO energy calculation of solvents and amides. | |
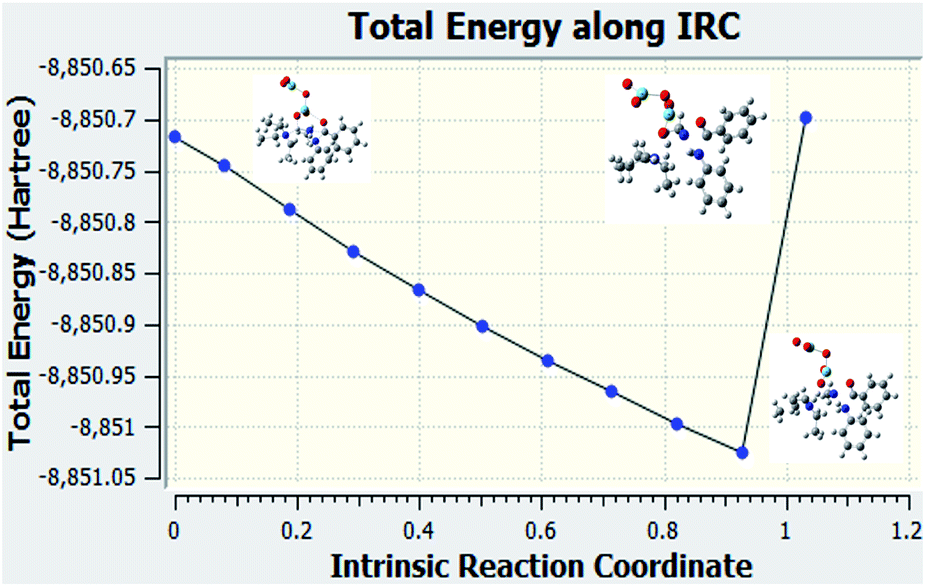
IRC calculation of reaction when Al2O3 is used as the catalyst
The IRC calculations were performed. This calculations helped us to verify that the correct transition state was used for the reaction when examining the structures that were downhill from the saddle point.
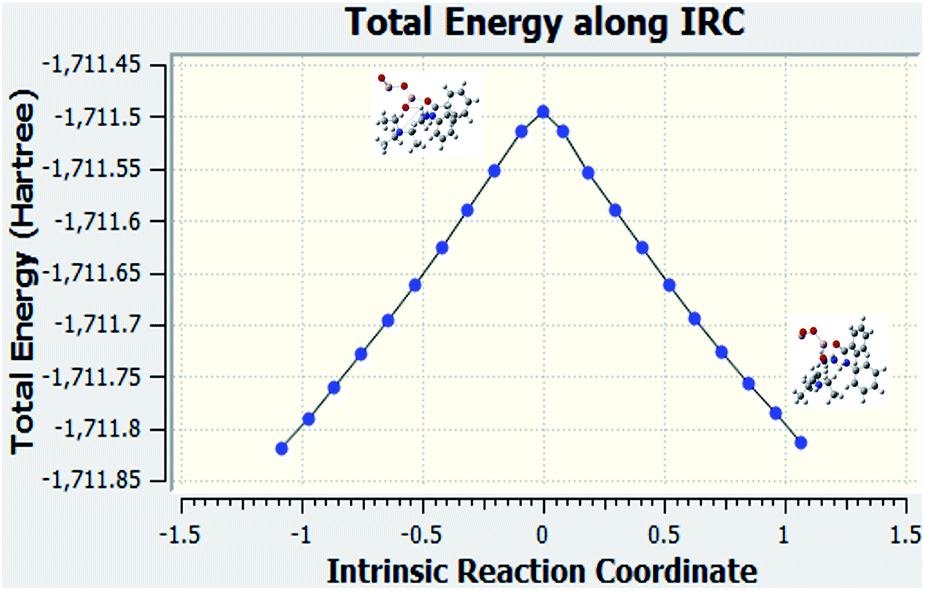
Fig. 7 is the graph of the total energy vs. intrinsic reaction coordinate of the reaction between N-methylbenzamide with aniline, where alumina is used as the catalyst. This graph is very similar to the reference graph of any reaction with a transition state where reactants have lower energy that gradually increases, and the transition state with higher energy gradually decreases to form the product. We also examined the structures that were downhill from the saddle point. These structures supported our proposed mechanism when alumina was used as the catalyst.
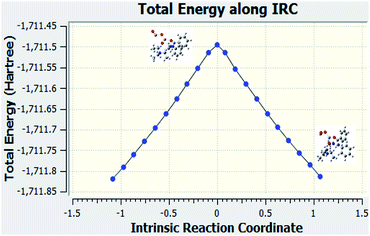 |
| | Fig. 7 IRC calculation for Al2O3 used as the catalyst for compound 1. | |
IRC calculation of the reaction when Nb2O5 used as the catalyst
Fig. 8 is the graph of the total energy vs. intrinsic reaction coordinate of the reaction between N-methyl benzamide with aniline, where niobium pentoxide was used as the catalyst. This graph is not similar to the reference graph of any reaction with a transition state. It can be concluded from this IRC calculation that this reaction is very difficult to proceed with the niobium pentoxide catalyst. We also examined the structures when Nb2O5 was used as the catalyst. All the structures are given below for every point.
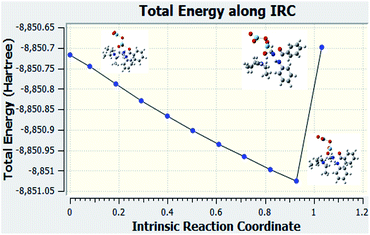 |
| | Fig. 8 IRC calculation: Nb2O5 used as the catalyst for compound 1. | |
Experimental
Computational methods
For the density functional theory (DFT) calculations, the Gaussian 16 program31 and Becke3LYP functional33 were performed. The final total energies were obtained with the 6-311+G(d,p) on all atoms. The nature of all stationary points was confirmed by performing frequency analyses. Subsequent intrinsic reaction coordinate (IRC) and transition states (TS) calculations were performed to demonstrate their connection to the adjacent ground states.
Synthesis methods
Melting points were determined using open capillary tubes on an Gallenkamp (England) melting point apparatus and were uncorrected. IR spectra were recorded on a Shimadzu FTIR spectrophotometer. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX-400 spectrophotometer (400 MHz) using tetramethylsilane as the internal reference. Analytical thin-layer chromatography (TLC) was performed on precoated silica gel 60 F-254 (E. Merck), and column chromatography was performed on silica gel (60–120 mesh). Bis(triphenylphosphine) palladium(II) chloride and other reagents were purchased from E. Merck (Germany) and Fluka (Switzerland).
Calcination of catalyst
The methodology was arranged by connecting two parts: first, calcination34 of the catalyst, and second, use of the calcinated catalyst for the synthesis of amides via transamidation reaction. All purchased catalysts were stored at room temperature. For the catalyst preparation, we used the calcination method. The catalyst was burned at a high temperature to make it suitable for use in the reaction and this burning process is called calcination. The catalyst shows different properties at different temperatures. The activity of the catalyst depends on various characteristics such as phase, surface area, porosity, and particle size. All these properties of the catalyst are dependent on the calcination temperature. Alumina shows a high surface area in the gamma phase35 when calcined at 500 °C. Commercially available Al2O3 was calcined to remove water and other possible organic impurities. In this method, 5 g aluminium oxide was placed in a crucible that was kept in a furnace and calcined at 500 °C for 3 h. After completing calcination, Al2O3 was used in the transamidation for amide synthesis in Fig. 9. The other catalysts, such as TiO2, Nb2O5, SnO2, and Cu2O, were also prepared via the same method by calcination at 500 °C for 3 h.
 |
| | Fig. 9 Calcination of Al2O3. | |
Conclusions
In conclusion, the present study showed the new heterogeneous catalytic method for the transamidation of secondary amides with amines using commercially available, inexpensive and non-pollutant Al2O3. This method provides a general and practical route for the synthesis of desired amide products by the transamidation of secondary amides. DFT calculations reveal that the Al2O3 catalyst forms a complex with reactant molecules in the presence of a solvent. The data for the intrinsic reaction coordinate showed the actual transition state with Al2O3 rather than Nb2O5. A comparison study with more expensive Nb2O5 was performed because of its high applicability for amidation reaction and base tolerant properties. Our developed method is also tolerant to base because it shows higher activity in the presence of trimethylamine solvents, which are highly basic. This general catalytic method might replace the existing method for the synthesis of targeted drugs with amide functionality.
Author contributions
Md Ayub Ali: supervision, conceptualization, methodology, writing –review & editing, investigation, validation. Ashutosh Nath: conceptualization, software, visualization, validation, writing – original manuscript. Md Midul Islam: methodology, synthesis, investigating product synthesis. Sharmin Binte Shahid: methodology and spectroscopic analysis. Ifat Nur Dibbo: methodology and spectroscopic analysis. All authors reviewed the paper.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We are grateful to the Bangladesh University of Engineering and Technology (BUET), Dhaka, Dhaka-1000, and University Grants Commission of Bangladesh for all kinds of experimental and financial supports.
Notes and references
- L. I. Anderson, D. J. O'Shannessy and K. Mosbach, J. Chromatogr. A, 1990, 513, 167–179 CrossRef.
- A. Greenberg, C. M. Breneman and J. F. Liebman, The amide linkage: Structural significance in chemistry, biochemistry, and materials science, John Wiley & Sons, 2002 Search PubMed.
- M. A. Ali, A. Nath, M. Jannat and M. M. Islam, ACS Omega, 2021, 6, 25002–25009 CrossRef CAS PubMed.
- A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC.
- H. M. Geysen, F. Schoenen, D. Wagner and R. Wagner, Nat. Rev. Drug Discovery, 2003, 2, 222–230 CrossRef PubMed.
- M. D. Eastgate, M. A. Schmidt and K. R. Fandrick, Nat. Rev. Chem., 2017, 1, 1–16 CrossRef.
- J. Liu, J. Han, K. Izawa, T. Sato, S. White, N. A. Meanwell and V. A. Soloshonok, Eur. J. Med. Chem., 2020, 112736 CrossRef CAS PubMed.
- F. Roberts and K. Hellgardt, Chemical Processes for a Sustainable Future, 2014, p. 49 Search PubMed.
- S. M. Mennen, C. Alhambra, C. L. Allen, M. Barberis, S. Berritt, T. A. Brandt, A. D. Campbell, J. Castañón, A. H. Cherney and M. Christensen, Org. Process Res. Dev., 2019, 23, 1213–1242 CrossRef CAS.
- R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed.
- T. Dalidovich, K. A. Mishra, T. Shalima, M. Kudrjašova, D. G. Kananovich and R. Aav, ACS Sustainable Chem. Eng., 2020, 8, 15703–15715 CrossRef CAS.
- V. V. Dunina, O. N. Gorunova, P. Zykov and K. A. Kochetkov, Russ. Chem. Rev., 2011, 80, 51 CrossRef CAS.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
- M. Tamura, T. Tonomura, K.-i. Shimizu and A. Satsuma, Green Chem., 2012, 14, 717–724 RSC.
- J. Andraos, Org. Process Res. Dev., 2005, 9, 404–431 CrossRef CAS.
- J. L. Young and R. Peoples, J. Chem. Educ., 2013, 90, 513–514 CrossRef CAS.
- D. Matheau-Raven, P. Gabriel, J. A. Leitch, Y. A. Almehmadi, K. Yamazaki and D. J. Dixon, ACS Catal., 2020, 10, 8880–8897 CrossRef CAS.
- C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed.
- M. M. Rahman, G. Li and M. Szostak, J. Org. Chem., 2019, 84, 12091–12100 CrossRef CAS PubMed.
- G. Li and M. Szostak, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- M. A. Ali, S. H. Siddiki, K. Kon and K.-i. Shimizu, Tetrahedron Lett., 2014, 55, 1316–1319 CrossRef.
- A. Pettignano, A. Charlot and E. Fleury, Polym. Rev., 2019, 59, 510–560 CrossRef CAS.
- M. A. Ali, S. Siddiki, W. Onodera, K. Kon and K.-i. Shimizu, ChemCatChem, 2015, 7, 3555–3561 CrossRef CAS.
- M. A. Ali, S. H. Siddiki, K. Kon, J. Hasegawa and K. i. Shimizu, Chem.–Eur. J., 2014, 20, 14256–14260 CrossRef CAS PubMed.
- S. H. Siddiki, M. N. Rashed, M. A. Ali, T. Toyao, P. Hirunsit, M. Ehara and K. i. Shimizu, ChemCatChem, 2019, 11, 383–396 CrossRef CAS.
- N. Nakajima and Y. Ikada, Bioconjugate Chem., 1995, 6, 123–130 CrossRef CAS PubMed.
- E. De Canck, D. Esquivel, F. J. Romero-Salguero and P. Van Der Voort, Comprehensive Guide For Mesoporous Materials, 2015, p. 197 Search PubMed.
- J. P. Agrawal and R. Hodgson, Organic chemistry of explosives, John Wiley & Sons, 2007 Search PubMed.
- A. Chardon, E. Morisset, J. Rouden and J. Blanchet, Synthesis, 2018, 50, 984–997 CrossRef CAS.
- C. Zhu, J. Dong, X. Liu, L. Gao, Y. Zhao, J. Xie, S. Li and C. Zhu, Angew. Chem., 2020, 132, 12917–12921 CrossRef.
- A. Nath, A. Kumer and M. W. Khan, J. Mol. Struct., 2021, 1224, 129225 CrossRef CAS.
- R. Romero Toledo, V. Ruíz Santoyo, D. Moncada Sánchez and M. Martínez Rosales, Nova Scientia, 2018, 10, 83–99 CrossRef.
- J. Kong and E. Proynov, J. Chem. Theory Comput., 2016, 12, 133–143 CrossRef CAS PubMed.
- H.-S. Roh, I.-H. Eum, D.-W. Jeong, B. E. Yi, J.-G. Na and C. H. Ko, Catal. Today, 2011, 164, 457–460 CrossRef CAS.
- T. Itoh, T. Uchida, I. Matsubara, N. Izu, W. Shin, H. Miyazaki, H. Tanjo and K. Kanda, Ceram. Int., 2015, 41, 3631–3638 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Ashutosh Nath
*a,
Ashutosh Nath