
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy of the Pauson–Khand reaction in flow over alkynylphenyl vinyl ethers: towards the synthesis of tricyclic multisubstituted benzofurans†
Jorge García-Lacuna ,
Maialen Alonso,
Gema Domínguez and
Javier Pérez Castells*
,
Maialen Alonso,
Gema Domínguez and
Javier Pérez Castells*
Department of Chemistry and Biochemistry, Facultad de Farmacia, Universidad San Pablo-CEU, CEU Universities, Urbanización Montepríncipe, 28660 Boadilla del Monte, Madrid, Spain. E-mail: jpercas@ceu.es
First published on 4th March 2022
Abstract
The use of flow methodology allows the use of alkynylphenyl vinyl ethers (benzo-fused 1,7 enynes) as substrates for the intramolecular Pauson–Khand reaction (PKr). Forced temperature and pressure conditions during a short reaction time minimize the substrate decomposition allowing the formation of the PK adduct. Substrates substituted at the internal position of the double bond and with internal triple bonds give better yields. The resulting products are cyclopentabenzofuranones present in diverse natural products and drugs that can be further functionalised.
Introduction
The Pauson–Khand reaction (PKr) is a formal [2 + 2 + 1] cycloaddition of an alkyne, an alkene and a carbonyl unit to give a cyclopentenone. Until the late 1990s it was mediated by a stoichiometric amount of metal, generally a cobalt carbonyl complex. The industrial applicability of this reaction was limited due to the use of either stoichiometric or harsh catalytic conditions (high carbon monoxide pressures and elevated temperatures).1 In 2017, our group developed a flow protocol for both intra- and intermolecular Pauson–Khand reactions, using catalytic amounts of Co2(CO)8 in a plug flow reactor.2 The applicability, robustness and safety of the process were proved in several gram scale syntheses and it was also applied in the key step of the synthesis of the commercial drug treprostinil.3 A photochemically triggered stoichiometric PKr in flow was described using preformed alkyne–cobalt complexes.4The field of flow chemistry has gained considerable attention along the last decade. Flow methods are generally faster, safer and greener. They enable safer handling of hazardous reagents and better mixing of biphasic regimes, in some cases reactions that are difficult or impossible to do in the batch mode are made possible.5 In particular, flow technologies are highly advantageous when using toxic gases like CO, particularly if harsh conditions are needed.6 All these advantages explain the high number of continuous flow protocols being used for the synthesis of pharmaceuticals or important building blocks, with special attention to industrial applicability.7
Despite the great number of substrates used to date, vinyl ethers continue to be challenging substrates for the PKr.8 Only three reports, where these motifs were used as PK substrates have been reported. In 1981, Croudace and Schore attempted to use several vinyl ethers in an intramolecular PKr. They observed the expected reaction with cobalt complexes, but the subsequent isolation of the cyclopentenone was unsuccessful.9 Later, examples of the intramolecular version provided only moderate yields and the cleavage of the vinyl ether structure in the final product. The vinyl ether acted, thus, as ethylene surrogate (Scheme 1).10
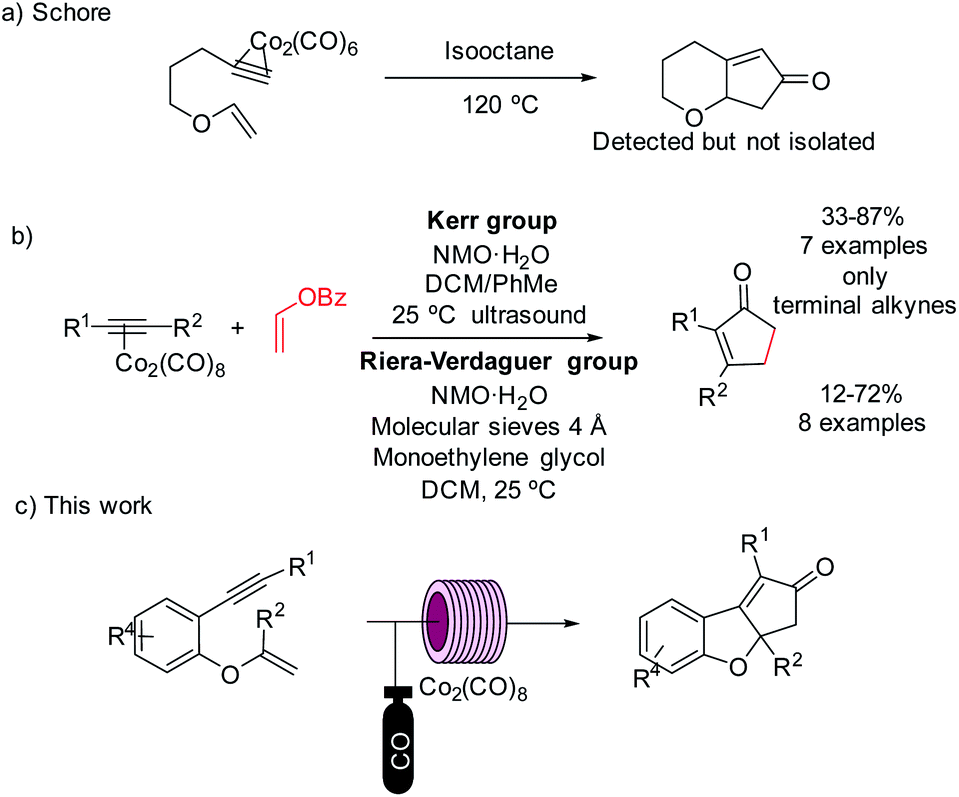 | ||
| Scheme 1 Previous works using vinyl ethers in the PKr and our strategy. | ||
The use of 1,7-enynes tethered through an aromatic ring where the olefinic part is a vinyl ether has no precedents in PKr chemistry. These substrates are very reactive, but we envisioned that using flow technology we could find specific conditions to enable the PKr. This would provide a new efficient, scalable and versatile synthesis of tricyclic multi-substituted benzofurans.
The benzofuran structure is ubiquitous in bioactive molecules, natural products, pharmaceuticals and functional materials. The broad spectrum of pharmacological activity in individual benzofurans indicates that these compounds are of an undoubted interest.11 In addition, this motif is a useful scaffold in organic synthesis. Ohno and Arisawa recently reviewed the use of these, and other kinds of benzo-fused substrates to form heterocycles.12 Therefore, extensive attention has been paid to develop versatile methods for the synthesis of these structurally diverse heterocycles.13 Among tricyclic benzofurans that would be similar to our Pauson–Khand adducts, we highlight silvestrol,14 a natural product which is a potent inhibitor of Ebola virus replication; beraprost,15 a PGI analog; and natural sesquiterpenes, isolated from the red alga Laurencia and the sea hare Aplysia specie (Fig. 1).16
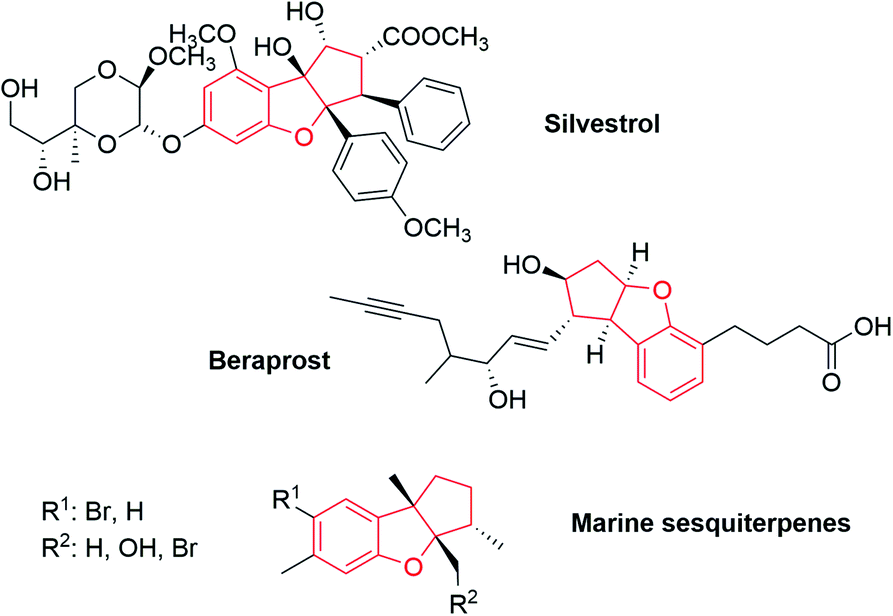 | ||
| Fig. 1 Representative cycopenta benzofurans. | ||
Results and discussion
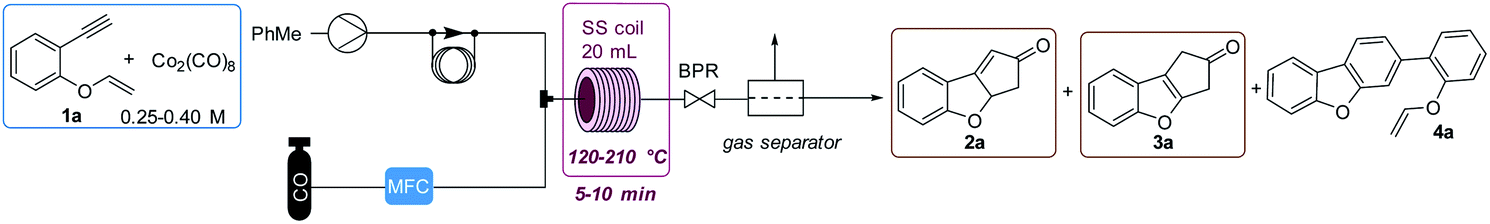
We used substrate 1a as the model for the reaction optimization. This reaction produced variable amounts of the expected PKr product 2a, the isomerized product 3a and the resulting product of a [2 + 2 + 2] cycloaddition, 4a. This latter cycloaddition reaction has been already described with this catalyst at high temperatures.17 Preliminary experiments indicated that, in the presence of the catalyst and at high temperatures, there is an important decomposition of the starting material. Therefore, we introduced an internal standard (4,5-dibromo-o-xylene, 15 mol%) to calculate the amount of decomposition/polymerization in each reaction. We compared similar conditions at temperatures from 120 to 210 °C, both in batch and flow. Two batch conditions were studied for comparison under low (5 bar, batch 1) or high (20 bar, batch 2) CO pressure. We measured the amount of detected products and the selectivity towards the Pauson–Khand reaction (Fig. 2). With both methodologies, at 120 °C, the conversion was poor and starting material was still detected in the crude mixture. At 150 °C in flow we observed the highest conversion (70%) but with poor selectivity towards 3a, and at 180 °C there was a slightly better selectivity, not detecting 2a, but with some more decomposition. This latter temperature was selected to optimize the rest of the flow parameters. We could confirm our initial hypothesis i.e. that we could benefit from the flow conditions to get fast reactions that avoid partially the decomposition of the starting material. In fact, yields in batch measured by NMR were very low and starting material was never recovered. Possibly, in the flow reactor we have achieved a chemical intensification due to the finding of a novel process window.18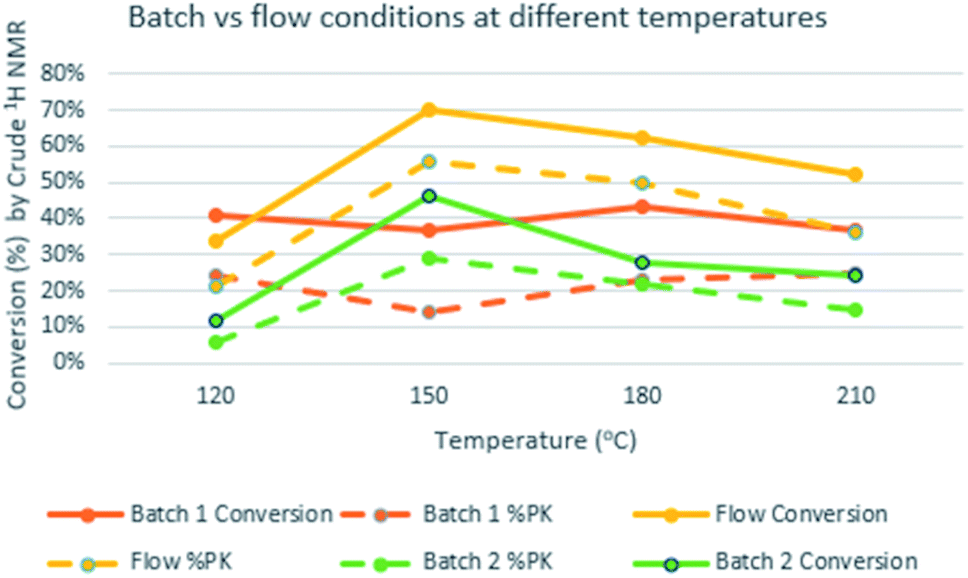 | ||
Fig. 2 Flow/batch comparison at different temperatures with 5 mol% of Co2(CO)8. Flow: 5 equiv. of CO, 7–15 min of residence time, 28 bar of system pressure. Batch 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 bar of CO, 45 min of reaction time. Batch 2:20 bar of CO, 45 min of reaction time. Conversion refers to the NMR yield of 2a + 3a + 4a, while dashed lines only 2a + 3a. :5 bar of CO, 45 min of reaction time. Batch 2:20 bar of CO, 45 min of reaction time. Conversion refers to the NMR yield of 2a + 3a + 4a, while dashed lines only 2a + 3a. | ||
Then, as shown in Table 1, flow parameters were optimized. Best results were achieved fixing the residence time to 13 min (entries 2–4) and using high pressures at 180 °C with 5 equiv. of CO (entry 4). The concentration was an important fact as high concentrations (entry 5) favored the [2 + 2 + 2] side product, while low concentrations, (entry 6), decreased the speed of the reaction and increased decomposition. Reducing the amount of gas (entry 8) or the catalyst loading (entry 7) decreased the selectivity by forming more of the [2 + 2 + 2] cycloadduct. With the conditions of entry 4, a long run was performed with 720 mg and an isolated yield of 50% was achieved. This result shows a fall in the yield compared to the IS yield (64%), that can be explained because of the instability of the final products. To check this point, we replicated three times the optimized reaction obtaining similar results.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entrya | Temp. (°C) | Res. time (min) | Conc. (M) | CO equiv. | P (bar) | Cat. (mol%) | % of conversionb | % of 3a |
| a All reactions in PFR (20 mL). See ESI for system pump and MFC flows.b % of conversion is measured in the 1H NMR spectrum of the crude mixture using 4,5-dibromo-o-xylene as internal standard.c 0.12 equiv. of DME were added as additive.d A long run with 720 mg (20 mL of volume) was performed to check the reliability of the IS, reaching an isolated yield of 50%. In this experiment 4a was isolated in 6% yield. | ||||||||
| 1 | 150 | 11 | 0.25 | 5 | 28 | 5 | 70 | 56 |
| 2 | 180 | 8 | 0.25 | 5 | 28 | 5 | 62 | 50 |
| 3 | 180 | 8 | 0.25c | 5 | 28 | 5 | 60 | 52 |
| 4d | 180 | 13 | 0.25 | 5 | 35 | 5 | 72 | 64 |
| 5 | 180 | 10 | 0.40 | 5 | 28 | 5 | 35 | 5 |
| 6 | 180 | 15 | 0.15 | 5 | 28 | 5 | 53 | 46 |
| 7 | 180 | 8 | 0.25 | 5 | 28 | 2.5 | 49 | 42 |
| 8 | 180 | 16 | 0.25 | 3 | 28 | 5 | 50 | 35 |
Regarding the scope of the protocol, all the compounds with a terminal alkyne showed important decomposition and consequently poor to moderate yields (products 3b–e, 24–40% yield, Fig. 3). An example using a vinyl thioether was also included (3f: 36% yield). In the same way, structures with two quaternary carbons surrounding the triple bond (1g–h) did not show any reaction. The substrates with substitution at the external carbon of the vinyl ether (1i–j), only produced [2 + 2 + 2] cycloadducts, 1i formed the dimer 4i (21%), while 1j formed the cyclotrimerization product 4j (18%). With substrates 1k–l bearing internal alkyne units, no isomerization and low decomposition of the PK product was observed (2k–l). However, the reaction produced several different products, giving only moderate yields (31 and 29% respectively).
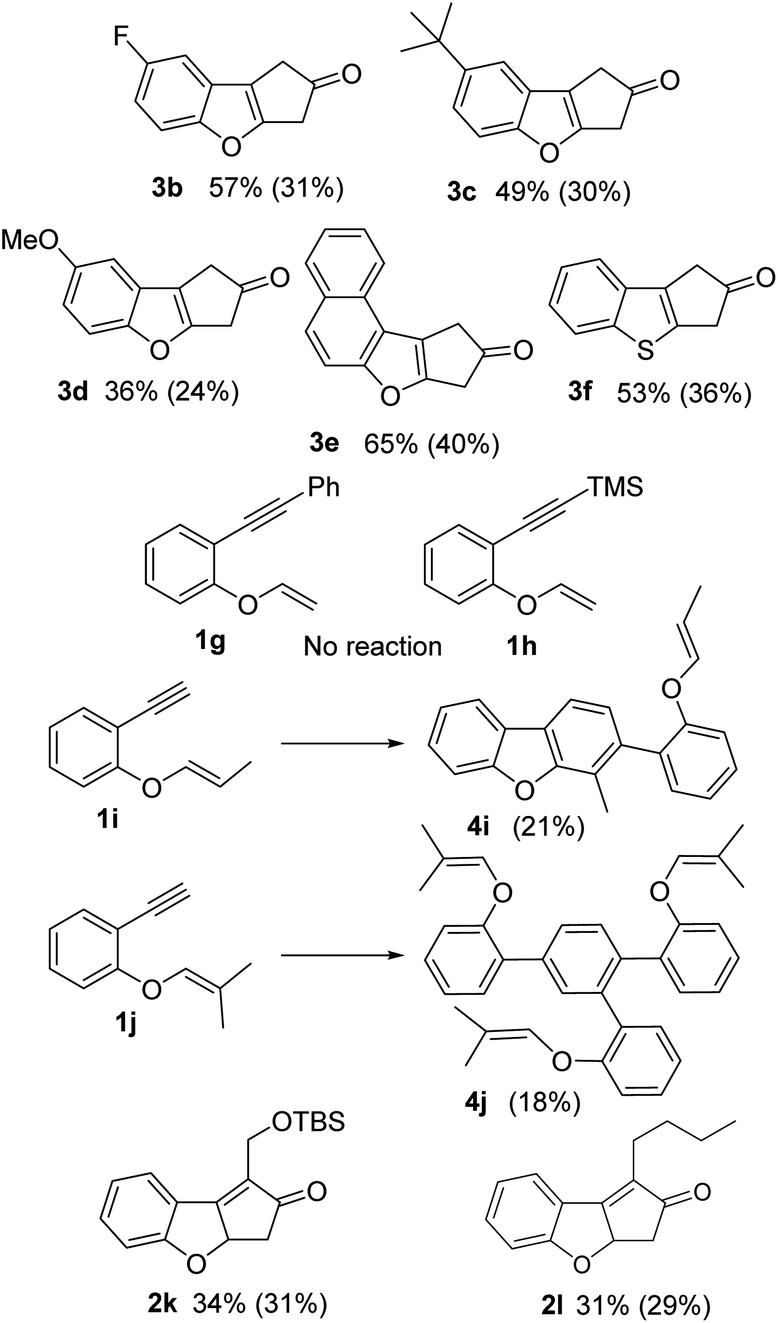 | ||
| Fig. 3 Scope of the reaction. NMR yield (isolated yield). | ||
Up to this point, the results were interesting as the first and only efficient example achieved to date using vinyl ethers as substrates in PKr, albeit yields were not satisfactory. Interestingly, with substrate 1m, substituted at the internal carbon of the double bond, and under the conditions used above, neither decomposition nor [2 + 2 + 2] side product formation was observed. However, the PK product 2m was detected in only 30% in the NMR spectrum of the crude, mainly because of a low conversion and the appearance of another side product. This latter product was identified as 5 and is the result of a migratory cycloisomerization: which has been described using other catalytic systems and conditions, but not using cobalt complexes.19 In view of this result we carried out an optimization study for the reaction of 1m (Table 2). First experiments showed relatively low conversion (entries 1–4). Increasing the temperature improved this parameter but the selectivity towards 2m decreased. Using 10 mol% of catalyst with high system pressures and 5 equiv. of gas gave 2m as the only detectable product (entry 7). Finally, a relatively long residence time (67 min, entry 9) provided almost full conversion with excellent selectivity. The isolated yield was 74%. Furthermore, a long run with 1.21 g, operating for 102 minutes afforded a 79% of isolated yield.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entrya | Temp. (°C) | Res. time (min) | Conc. (M) | CO equiv. | P (bar) | Cat. (mol%) | % of conv.b | 2m:5 ratio (%) |
| a All reactions in PFR (20 mL). See ESI for system pump and MFC flows.b % of conversion is measured in the 1H NMR spectrum of the crude mixture.c A 60 mL reactor was used in these entries.d A long run with 1.21 g was performed, an isolated yield of 79% was achieved, total time: 102 minutes. | ||||||||
| 1 | 150 | 11 | 0.25 | 5 | 20 | 5 | 7 | 99 |
| 2 | 150 | 26 | 0.40 | 5 | 35 | 5 | 14 | 99 |
| 3 | 180 | 14 | 0.40 | 3 | 28 | 5 | 66 | 79 |
| 4 | 180 | 19 | 0.40 | 5 | 35 | 5 | 48 | 81 |
| 5 | 180 | 16 | 0.25 | 5 | 28 | 5 | 64 | 80 |
| 6 | 210 | 7 | 0.25 | 5 | 28 | 5 | 86 | 67 |
| 7 | 150 | 26 | 0.40 | 5 | 35 | 10 | 62 | >99 |
| 8c | 150 | 91 | 0.40 | 5 | 35 | 10 | 82 | 73 |
| 9bc | 170 | 67 | 0.40 | 5 | 35 | 10 | 97 | 88d |
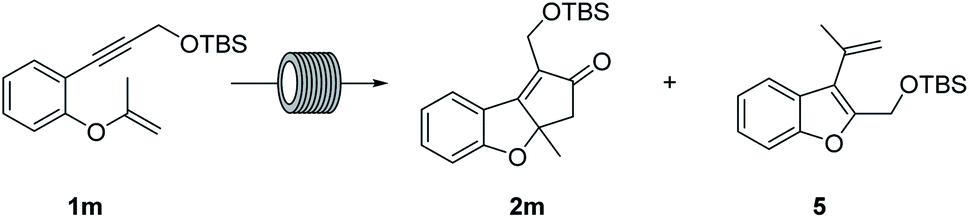
These good results show that using substrates with internal alkynes and double bonds with substitution at the internal carbon precludes decomposition and avoids the possibility of isomerization giving very clean and efficient reactions. Thus, we prepared a bunch of substrates with these requirements with which we used the optimized conditions (entry 9, Table 2). Yields in these cases were good (66–76%, Fig. 4). These examples include different alkynes that can be easily functionalized and various substituents at the aromatic ring. The productivity obtained with these substrates is only possible due the advantages derived from the use of continuous flow processes and the possibility of handling high temperatures, pressures and a hazardous gas. These conditions are difficult to reproduce in batch.
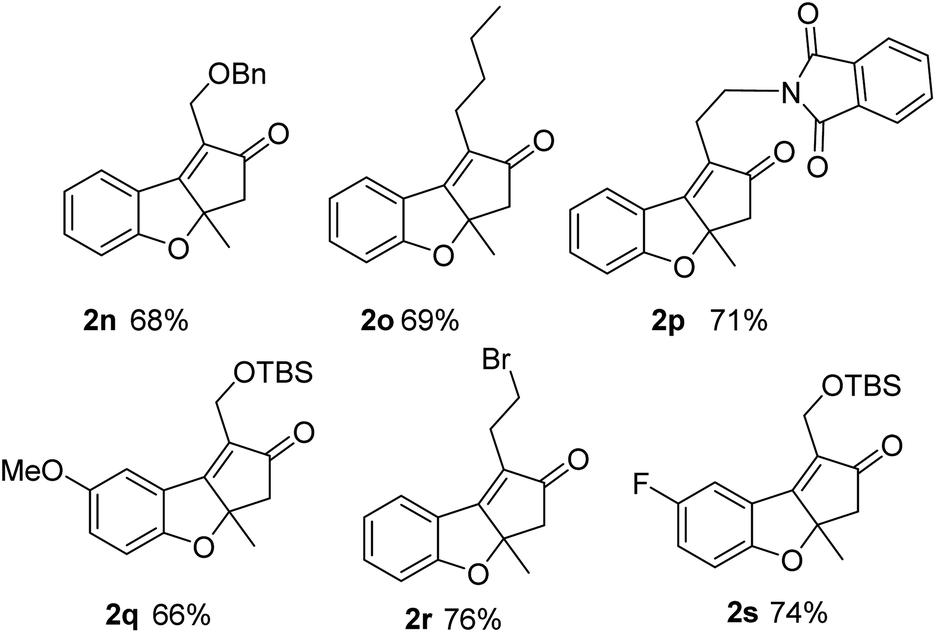 | ||
| Fig. 4 Scope of the reaction using substrates with internal substituted double bonds (isolated yields). | ||
Conclusions
We have described for the first time an efficient Pauson–Khand reaction where the double bond is a vinyl ether. Good yields are achieved if the double bond is substituted at the internal position, whereas with other substrates the results are moderate. We believe that this reaction has been possible because of the use of continuous flow methodology, where the conditions have been intensified in relatively short time. This minimises the decomposition and allows the reaction. The process leads to cyclopentabenzofuranes which are very interesting scaffold's present in natural products and other interesting molecules.Author contributions
Conceptualization of the work, J. P.-C. and G. D.; methodology, all authors; synthetic work, J. G.-L. and M. A.; writing—original draft preparation, J. G.-L.; writing—review and editing, J. P.-C. and G. D.; and funding acquisition, J. P.-C. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding of this project by the Spanish MINECO, grant No. RTI2018-095588-B-I00 (Co-funded by European Regional Development Fund/European Social Fund, “Investing in your future”) and FUSP-CEU (PC17/17) is acknowledged. J. G. thanks the Fundación San Pablo-CEU for a pre-doctoral fellowship.Notes and references
- (a) G. Domínguez and J. Pérez-Castells, in Science Of Synthesis: Metal-Catalyzed Cyclization Reactions, ed. S. Ma, Thieme Chem, 2017, vol. 2, p. 99 Search PubMed; (b) The Pauson-Khand Reaction: Scope, Variations and Applications, ed. R. R. Torres, Wiley, Hoboken NJ, 2012 Search PubMed; (c) H. Lee and F. Kwong, Eur. J. Org. Chem., 2010, 2010, 789 CrossRef.
- J. Garcia-Lacuna, G. Dominguez, J. Blanco-Urgoiti and J. Perez-Castells, Chem. Commun., 2017, 53, 4014 RSC.
- J. García-Lacuna, G. Domínguez, J. Blanco-Urgoiti and J. Pérez-Castells, Org. Biomol. Chem., 2019, 17, 9489 RSC.
- K. Asano, Y. Uesugi and J. Yoshida, Org. Lett., 2013, 15, 2398 CrossRef CAS PubMed.
- For recent reviews on flow chemistry see: (a) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688 CrossRef CAS PubMed; (b) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796 CrossRef CAS PubMed; (c) L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481 RSC; (d) J. García-Lacuna, G. Domínguez and J. Pérez-Castells, ChemSusChem, 2020, 13, 5138 CrossRef PubMed; (e) M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802 CrossRef CAS.
- C. J. Mallia and I. R. Baxendale, Org. Process Res. Dev., 2016, 20, 327 CrossRef CAS.
- (a) V. R. L. J. Bloemendal, M. A. C. H. Janssen, J. C. M. van Hest and F. P. J. T. Rutjes, React. Chem. Eng., 2020, 5, 1186 RSC; (b) R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2 CrossRef CAS.
- See for recent PK reviews with uncommon substrates: (a) J. D. Ricker and L. M. Geary, Top. Catal., 2017, 60, 609 CrossRef CAS PubMed; (b) J. Escorihuela, D. M. Sedgwick, A. Llobat, M. Medio-Simón, P. Barrio and S. Fustero, Beilstein J. Org. Chem., 2020, 16, 1662 CrossRef CAS PubMed.
- M. C. Croudace and N. E. Schore, J. Org. Chem., 1981, 46, 5357 CrossRef CAS.
- (a) A. Cabré, X. Verdaguer and A. Riera, Synthesis, 2017, 49, 3945 CrossRef; (b) W. Kerr, M. McLaughlin, P. Pauson and S. Robertson, J. Organomet. Chem., 2001, 630, 104 CrossRef CAS.
- H. Khanam and Shamsuzzaman, Eur. J. Med. Chem., 2015, 97, 483 CrossRef CAS PubMed.
- S. Ohno and M. Arisawa, J. Org. Chem., 2020, 85, 6831 CrossRef CAS PubMed.
- L. Chiummiento, R. D'Orsi, M. Funicello and P. Lupattelli, Molecules, 2020, 25, 2327 CrossRef CAS PubMed.
- N. Biedenkopf, K. Lange-Grünweller, F. W. Schulte, A. Weißer, C. Müller, D. Becker, S. Becker, R. K. Hartmann and A. Grünweller, Antiviral Res., 2017, 137, 76 CrossRef CAS PubMed.
- S. Umemiya, D. Sakamoto, G. Kawauchi and Y. Hayashi, Org. Lett., 2017, 19, 1112 CrossRef CAS PubMed.
- S. Biswas, A. Ghosh and R. V. Venkateswaran, J. Org. Chem., 1990, 55, 3498 CrossRef CAS.
- J. García-Lacuna, G. Domínguez, J. Blanco-Urgoiti and J. Pérez-Castells, Org. Lett., 2018, 20, 5219 CrossRef PubMed.
- (a) V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, ChemSusChem, 2013, 6, 746 CrossRef CAS PubMed; (b) S. Borukhova and V. Hessel, in Process Intensif. Green Chem., ed. K. Boodhoo and A. Harvey, Wiley, Chichester, UK, 2013, pp. 91–156 Search PubMed; (c) T. Razzaq, T. N. Glasnov and C. O. Kappe, Chem. Eng. Technol., 2009, 32, 1702 CrossRef CAS.
- (a) S. Ohno, J. Qiu, R. Miyazaki, H. Aoyama, K. Murai, J. Hasegawa and M. Arisawa, Org. Lett., 2019, 21, 8400 CrossRef CAS PubMed; (b) N. Sakiyama, K. Noguchi and K. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 5976 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, spectroscopical data and copies of NMR spectra of all new compounds. See DOI: 10.1039/d2ra01062c |
| This journal is © The Royal Society of Chemistry 2022 |