
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous g-C3N4 layer-coated porous TiO2 fibers with enhanced photocatalytic activity toward H2 evolution and dye degradation†
Jing Liua,
Jinxiao Zhengb,
Guichu Yuea,
Huaike Lia,
Zhaoyue Liu a,
Yong Zhaoa,
Nü Wang*a,
Chenghua Sun*b and
Zhimin Cui*a
a,
Yong Zhaoa,
Nü Wang*a,
Chenghua Sun*b and
Zhimin Cui*a
aKey Laboratory of Bioinspired Smart Interfacial Science and Technology of Ministry of Education, Beijing Key Laboratory of Bioinspired Energy Materials and Devices, School of Chemistry, Beijing Advanced Innovation Center for Biomedical Engineering, Beihang University, Beijing, 100191, P. R. China. E-mail: wangn@buaa.edu.cn; sunchenghua@mail.ipc.ac.cn; cuizhm@buaa.edu.cn
bKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100029, P. R. China
First published on 1st April 2022
Abstract
TiO2/g-C3N4 composite photocatalysts with various merits, including low-cost, non-toxic, and environment friendliness, have potential application for producing clean energy and removing organic pollutants to deal with the global energy shortage and environmental contamination. Coating a continuous g-C3N4 layer on TiO2 fibers to form a core/shell structure that could improve the separation and transit efficiency of photo-induced carriers in photocatalytic reactions is still a challenge. In this work, porous TiO2 (P-TiO2)@g-C3N4 fibers were prepared by a hard template-assisted electrospinning method together with the g-C3N4 precursor in an immersing and calcination process. The continuous g-C3N4 layer was fully packed around the P-TiO2 fibers tightly to form a TiO2@g-C3N4 core/shell composite with a strong TiO2/g-C3N4 heterojunction, which greatly enhanced the separation efficiency of photo-induced electrons and holes. Moreover, the great length–diameter ratio configuration of the fiber catalyst was favorable for the recycling of the catalyst. The P-TiO2@g-C3N4 core/shell composite exhibited a significantly enhanced photocatalytic performance both in H2 generation and dye degradation reactions under visible light irradiation, owing to the specific P-TiO2@g-C3N4 core/shell structure and the high-quality TiO2/g-C3N4 heterojunction in the photocatalyst. This work offers a promising strategy to produce photocatalysts with high efficiency in visible light through a rational structure design.
1 Introduction
With rapid global industrialization and rapid population explosion, the global energy demand is ever growing. Traditional energy sources are non-renewable and thus exhaustible and also have hazardous combustion products, which put human beings under increasing threat from the energy dilemma and environmental contamination.1–3 Therefore, it is highly desirable to explore new sources of energy that are sustainable and pollution-free. Among the many candidates, hydrogen energy derived from photocatalytic water decomposition is regarded as the most green and has great potential to substitute conventional fuels in terms of sustainability and cleanliness.4–8 Besides, photocatalysts also show outstanding performance for the degradation of contaminants in water and air.9–13Among the numerous photocatalysts, TiO2 has been extensively investigated due to its many outstanding advantages, including non-toxic nature, low-cost, pollution-free, chemical and thermal stability. However, the fast recombination of photo-induced carriers and its limited response to visible light greatly hinder its photocatalysis application. Hence, numerous efforts have been dedicated to the modification of TiO2 for expanding its light absorption range and for separating the photogenerated holes and electrons rapidly, including surface sensitization, noble metal deposition, element doping, and heterojunction formation.14–17 Graphitic carbon nitride (g-C3N4), a metalloid and polymeric photocatalyst, has shown great potential application in hydrogen production under visible light since 2009.18 It has raised wide interested thanks to its excellent characteristics, for instance, chemical stability, cost-effectiveness, visible response, and environmental-friendly nature. The combination of TiO2 and g-C3N4 is a valid way to increase the separation efficiency of photon-induced carriers and expand the light absorption range.19,20 Many avenues have been conducted to fabricate different morphologies of g-C3N4/TiO2 nanocomposites. g-C3N4/TiO2 nanosheet composites were fabricated by Keller et al. via a sol–gel and thermal polycondensation synthesis. The nanocomposites exhibited efficient H2 production using very low amounts of sacrificial agents under visible light irradiation.21 Song et al. mixed urea with TiO2 nanoparticles to fabricate coupled photocatalysts by calcination. The TiO2/g-C3N4 nanocomposite presented better photocatalytic phenol degradation performance.22 TiO2/g-C3N4 nanospheres were fabricated by Wang et al. via a melt-infiltration of dicyandiamide into mesoporous TiO2 spheres followed by calcination. The melt-infiltration process made g-C3N4 undergo better fusion into mesoporous TiO2 and formed a firm interfacial contact, which showed a higher performance for the visible light degradation of organic pollutants.23 However, all these structures, such as nanosheets, nanoparticles, and nanospheres, are microscopic materials, which makes it difficult to recover and recycle the catalyst in practical application. Notably, fibers with a very large aspect ratio have structural advantages of a microscopic diameter as well as macroscopic length that make them very promising in catalyst recycling.
Among the various methods to fabricate fiber catalysts, electrospinning is a direct and effective approach to produce fibers with various fine structures.24–29 Wang and Lei et al. synthesized TiO2/g-C3N4 nanocomposites by a simple electrospinning process. Few g-C3N4 nanosheets were embedded and interspersed in the TiO2 nanofibers. The TiO2/g-C3N4 nanofibers showed improved photocatalytic activity for H2 evolution and rhodamine B (RhB) degradation under simulated solar irradiation.30 Park and Kim et al. prepared TiO2/g-C3N4 nanofibers via a two-nozzle electrospinning process combined with a calcination method. The TiO2/g-C3N4 nanofibers showed significantly improved performance for the photocatalytic degradation of RhB and reactive black 5 under solar light irradiation.31 However, the g-C3N4 content among the composite is typically restricted, which means it is unable to form a large contacted interface with TiO2 in these methods. Li and Yan et al. synthesized a TiO2/g-C3N4 heterojunction nanofibers photocatalyst via electrospinning and an in situ evaporation calcination method to control the ratio of g-C3N4 and TiO2 in the composite. The photocatalyst showed high activity in RhB degradation under simulated sunlight.32 Wang et al. fabricated TiO2/g-C3N4 nanocomposites by combining electrospun TiO2 nanofibers and calcinated g-C3N4 nanosheets with a hydrothermal reaction. The TiO2/g-C3N4 nanocomposites showed improved photocatalytic degradation performance for RhB solution.33 However, the massive g-C3N4 tended to aggregate on the TiO2 nanofibers. However, neither the blocks nor the adhesions of g-C3N4 were negligible on account of the intrinsic agglomerating properties of g-C3N4 on the TiO2 nanofibers during the calcination, which led to a limited surface area that is detrimental for forming homogeneous heterojunctions between TiO2 and g-C3N4. Heterojunction semiconductors achieved by infiltration approach method showed a greatly enhanced interfacial contact that surpassed that obtained by sol–gel chemistry or standard ball-milling methods.34 Porous materials possesses abundant active sites for mass transfer to build heterojunction semiconductors through the high pore volumes and large surface areas. Here, the precursor could facilely penetrate into the porous substrate material and nucleate, and then grow from inside to outside. As a result, substrate materials with a well-developed porosity could promote the growth of a secondary structure on the substrate to form strong contacted heterojunctions and could even enable a high structural stability.35,36 In our previous work, hollow porous TiO2/g-C3N4 nanofiber photocatalysts were prepared via a coaxial electrospinning combined with infiltration method.37 The hollow and porous TiO2/g-C3N4 nanofibers showed high photocatalytic performance for H2 evolution and RhB degradation, ascribed to the intact porous hollow structure and the TiO2/g-C3N4 heterojunctions. However, the g-C3N4 layers grown on the TiO2 nanofiber were cocked, leading to an incomplete contact with the TiO2 substrate. In addition, the photocatalytic performance of the composite was confined due to the blocking of the hollow structure through the high g-C3N4 content. Therefore, a continuous and uniform thin layer structure of g-C3N4 on TiO2 fibers would be more favorable for the construction of intimate contacted TiO2/g-C3N4 heterojunctions, which would be highly beneficial for photocatalysis reactions because of the fast photocarrier separation superiority and large specific surface area.38–41
Herein, we developed a template-assisted electrospinning approach together with a solution infiltration process to coat a continuous g-C3N4 layer on to porous TiO2 (P-TiO2) fibers to produce a porous TiO2@g-C3N4 core/shell composite. Abundant mesopores in the TiO2 fibers provided accommodating venues for the precursor of g-C3N4 in order that a continuous tightly wrapped g-C3N4 shell could be grown on the P-TiO2 fibers. Moreover, with the high content of g-C3N4, the porous TiO2@g-C3N4 composites still maintained the fibers' structure well with no aggregation or adhesion of g-C3N4. The intimate heterojunctions between the g-C3N4 shell and P-TiO2 fibers core inhibited the recombination of photo-induced carriers during the photocatalytic process. The H2 production and dye (RhB) degradation activities were both improved under visible light illumination, ascribed to the synergistic action of the continuous core/shell structure and the high-quality heterojunctions between the g-C3N4 layers and TiO2 fibers.
2 Experimental
2.1 Materials
Polyvinylpyrrolidone (PVP, Mw = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000) was provided by Aladdin (Shanghai, China). Titanium butoxide (TBOT), cyanamide (CY), triethanolamine (TEOA), acetylacetone, isopropanol (IPA), p-benzoquinone (p-BQ), RhB, methylene blue (MB), phenol and absolute ethanol were obtained from Macklin (Shanghai, China). The above reagents were all directly used without further treatment.
300000) was provided by Aladdin (Shanghai, China). Titanium butoxide (TBOT), cyanamide (CY), triethanolamine (TEOA), acetylacetone, isopropanol (IPA), p-benzoquinone (p-BQ), RhB, methylene blue (MB), phenol and absolute ethanol were obtained from Macklin (Shanghai, China). The above reagents were all directly used without further treatment.
2.2 Fabrication of porous TiO2 (P-TiO2) fibers
P-TiO2 fibers were produced via a template-assisted electrospinning technique. Hand-made candle soot carbon nanoparticles (CNPs) were used as the template to create nanopores in the TiO2 fibers. Generally, 1.2 g CNPs was added to 25.2 mL ethanol and then ultrasonically processed for 2 h to disperse the CNPs uniformly. Next, 4.8 g PVP was mixed with the above solution with magnetic stirring for 6 h. Afterward, 1.0 g TBOT and 2.4 g acetylacetone were added to the above homogeneous PVP/CS ethanol solution and then stirred for 12 h to obtain the TiO2 precursor sol. The ejection rate of the spinning fluid was 40 μL min−1. The distance from the syringe needle to collector was 20 cm, and the voltage was 9.0 kV. The as-prepared fibers were calcined at 500 °C for 3 h to prepare the P-TiO2 fibers. Solid TiO2 (S-TiO2) fibers with no nanopores were also fabricated by the same electrospinning condition without CNPs in the TiO2 precursor sol as a control to explore the role of nanopores in the formation of the TiO2@g-C3N4 core/shell structure.2.3 Preparation of porous TiO2@g-C3N4 (PTCN) core/shell heterojunction fiber photocatalysts
First, 0.5 mL cyanamide (CY) solution was added to the P-TiO2 fibers dropwise to make CY solution infiltrate into the P-TiO2 fibers fully and uniformly. The as-prepared mixture was transferred to a semi-enclosed silica crucible to calcinate at 550 °C for 4 h. PTCN photocatalysts with different CY concentrations were tagged as PTCN-x (x = 30%, 70%, 90%), where x represents the volume fraction of CY aqueous solution. As a control, the S-TiO2 fibers were also treated by the same process to synthesize solid TiO2/g-C3N4 (STCN). Besides, pure g-C3N4 was achieved via directly calcinating pristine CY under the same calcination conditions.2.4 Characterization
The morphologies of the samples were inspected by scanning electron microscopy (SEM) (FEI Quanta FEG 250) with an acceleration voltage of 10 kV and by transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) (JEM-F200). The Brunauer–Emmett–Teller (BET) surface areas (SBET) of the samples were determined using an ASAP 2420 (Micrometrics Instruments) nitrogen adsorption apparatus. The pore-size distributions were measured by the Barrett–Joyner–Halenda (BJH) model. X-ray diffraction (XRD) patterns were obtained on a Shimadzu XRD-6000, using a Cu Kα source. Fourier transform-infrared (FT-IR) spectra were obtained on a Thermo Scientific Nicolet 6700. X-ray photoelectron spectroscopy (XPS) analysis was performed on a Thermo Scientific ESCA Lab 250Xi. UV-Visible diffuse reflectance spectroscopy (DRS) was performed on a Shimadzu UV-3600. Photoluminescence (PL) spectra were obtained on a Cary Eclipse MY14110001. The UV-visible absorption spectra were obtained on an Agilent/Varian Cary 50.2.5 Photocatalytic hydrogen evolution
The H2-generation activity of the photocatalysts was characterized by a gas tracing and analysis system (Labsolar-6A, Beijing Perfectlight). Generally, the photocatalyst (50 mg) was dispersed in a mixture of distilled water (85 mL) and TEOA (15 mL) with constant stirring in a transparent quartz reactor. Next, 1 wt% Pt was loaded on to the photocatalyst by a photodeposition process. Previous to irradiation, the dissolved air in the reaction system was removed by a vacuum pump. Subsequently, a xenon lamp (300 W, λ ≥ 420 nm) (Microsolar 300, Beijing Perfectlight) was equipped to irradiate the photocatalysts suspension. The gaseous product was sampled and analyzed directly by gas chromatography (Techcomp, GC-7900) with an argon gas carrier. The reaction system was kept at 5 °C by recirculating water.2.6 Photocatalytic degradation
The photodegradations of RhB (10 ppm), MB (10 ppm), and phenol (5 ppm) were employed to characterize the catalytic activity of the photocatalysts under visible light irradiation. Generally, 50 mg catalyst was added to 50 mL RhB (MB and phenol) solution. Before irradiation, the reaction solution was continuously stirred at 25 °C for 60 min in the dark to establish adsorption–desorption equilibrium. Next, 3.5 mL of RhB (MB and phenol) solution was extracted periodically to record the concentration change of RhB by the UV-vis absorbance spectrum at 553 nm (664 nm for MB and 270 nm for phenol). C/C0 was calculated, where C0 represents the initial concentration of RhB solution after the adsorption–desorption process, C stands for the real-time RhB concentration under visible light. The photocatalyst was recovered and reused in repeat cycles to investigate the stability. Moreover, different scavengers were used to explore the photocatalytic degradation mechanism of the core/shell PTCN composite. Before the reaction, p-BQ (0.25 mM), IPA (1 mM), and TEOA (1 mM) were injected in to the RhB solution, respectively, to trap superoxide radicals (˙O2−), hydroxyl radicals (˙OH), and holes (h+) species.3 Results and discussion
Scheme 1 exhibits a diagrammatic sketch for the preparation of porous TiO2@g-C3N4 (PTCN) fibers with the core/shell structure. The P-TiO2 fibers were constructed via a carbon nanoparticles (CNPs) template-assisted electrospinning approach. The electrospinning fluid was a mixture of home-made candle soot CNPs, PVP, and TBOT. The CNPs with an average diameter of 30 nm served as a template to produce the porous structure (Fig. S1a†). During the first calcination step, the organics and CNPs were removed and thus porous TiO2 (P-TiO2) fibers were obtained. After immersing the P-TiO2 fibers in a certain concentration of cyanamide (CY) solution, followed by second calcination step, PTCN fibers with a core/shell structure were obtained. | ||
| Scheme 1 Scheme for the preparation of porous TiO2@g-C3N4 (PTCN) core/shell fibers. Porous TiO2 (P-TiO2) fibers were produced via a template-assisted electrospinning technique with subsequent calcination. Then, the P-TiO2 fibers were immersed in cyanamide (CY) solution followed by a second step of calcination to prepare PTCN with a core/shell structure. | ||
Fig. 1a–f display the evolution process from electrospinning the precursor fibers to PTCN core/shell fibers with different g-C3N4 contents. The precursor fibers had a relatively smooth appearance with a few CNPs on the surface (Fig. 1a). The calcinated fibers exhibited an incompact and porous surface after removing PVP and CNPs (Fig. 1b and S1b†). The pore size of the P-TiO2 fibers matched well with the particle size of the CNPs template (Fig. S1c and d†) and the diameter of the P-TiO2 fiber was approximately 1.08 μm (Fig. S1f†). After the secondary calcination step, the CY underwent a thermal polymerization process and formed a continuous g-C3N4 thin layer on the PTCN fibers. The cross-section SEM and TEM images revealed the detailed structures of the P-TiO2 and PTCN fibers. The P-TiO2 fibers were composed of small nanoparticles (Fig. 1c and g). Layered g-C3N4 could be observed from all the PTCN samples (PTCN-30%, 70%, and 90%). When the concentration of CY solution was 30%, a very thin layer about 36 nm thick was detected around the outside of the P-TiO2 fibers (Fig. 1d and h). As the CY concentration increased to 70%, the outer shell of the fiber became distinct with a thickness of about 50 nm (Fig. 1e and i). When the CY concentration was up to 90%, a dense and continuous shell of approximately 85 nm thickness was wrapped around the P-TiO2 fiber tightly (Fig. 1f and j). The surface SEM image also showed the g-C3N4 shell covers around the P-TiO2 fiber became thicker and denser as the CY concentration increased (Fig. S2†). The digital photos of all samples presented a gradual color change from white to yellow with the increase in CY concentration (Fig. S3†). Fig. 1k shows the high-resolution TEM (HRTEM) image of the PTCN-90% composite. The boundary of a TiO2 crystalline grain with a lattice fringe of 0.35 nm was coherent with the anatase TiO2(101) lattice plane (Fig. 1k and S1e†).32 A thin g-C3N4 layer could be observed around the TiO2 grain, which was the g-C3N4 phase.42 In the inset of Fig. 1k, the selected area electron diffraction pattern (SAED) of HPCN-90% revealed the polycrystalline configuration of the TiO2/g-C3N4 heterojunction. The HAADF-STEM image and EDX elemental mapping images of PTCN-90% are shown in Fig. 1l. The broader distribution range of C and N agreed well with the TiO2@g-C3N4 core/shell structure, while the overlapping distribution range of Ti, O, C, and N indicated that an intimate TiO2/g-C3N4 heterojunction was also constructed in the P-TiO2 fiber together with the continuous g-C3N4 coating.
 | ||
| Fig. 1 SEM images of (a) pristine electrospinning fibers and (b) P-TiO2 fibers. (c–f) Cross section images of P-TiO2, PTCN-30%, PTCN-70%, and PTCN-90%. (g–j) TEM images of P-TiO2, PTCN-30%, PTCN-70%, and PTCN-90%. The g-C3N4 shell of PTCN became more contiguous and thicker apparently with the increase in CY concentration. (k) HRTEM image of PTCN-90% (the inset exhibits the SAED pattern of PTCN-90%, indicating the polycrystalline configuration). (l) HAADF-STEM image and EDX mapping images of elemental Ti, O, C, N and a layer image of a single PTCN-90% fiber. Mesopores homogeneously distributed in the PTCN-90% core/shell fiber. The distribution of four elements in the fiber further confirmed the formation of the TiO2@g-C3N4 core/shell structure. | ||
As contrast, solid TiO2 (S-TiO2) fiber@g-C3N4 (STCN) nanostructures were also prepared to investigate the effect of the nanopores in TiO2 fiber for the growth of the g-C3N4 coating. S-TiO2 fibers were fabricated by almost the same electrospinning method but without the CNPs template. After the first calcination step, only tiny holes could be observed on the surface of the S-TiO2 fibers (Fig. S4a†). After immersing the S-TiO2 fibers in a certain concentration of CY solution and followed by a second calcination step, the STCN composite was obtained. However, plenty of separate g-C3N4 nanosheets could be observed by the side of the S-TiO2 fiber in the STCN composite (Fig. S4b†). Although a small amount of g-C3N4 was coated on the S-TiO2 fibers, a continuous g-C3N4 layer was not formed like in PTCN (Fig. S4c†). This significant difference was because g-C3N4 is easy to agglomerate on S-TiO2 fibers during calcination at high temperature, while the porous structure of P-TiO2 makes CY solution easily infiltrate into the internal structure of the TiO2 fibers and hence forms a continuous g-C3N4 shell layer around the P-TiO2 fibers core. Apparently, the porous structure of P-TiO2 fibers is not only crucial for the intimate contact between TiO2 fibers and g-C3N4, but also significant for the continuous g-C3N4 coating on the TiO2 fibers.
X-ray diffraction (XRD) patterns were measured to investigate the crystal phases of TiO2, g-C3N4, and PTCN-90% (Fig. 2a). The diffraction peaks at 25.1°, 37.7°, and 47.9° corresponded to the (101), (004), and (221) lattice planes of anatase TiO2 (JCPDS no. 21-1272). The characteristic peak of g-C3N4 was located at 27.5°, corresponding to the (002) diffraction plane.23 The peak intensity of g-C3N4 in the PTCN became stronger with the increase in CY concentration from 30% to 90%, indicating a more crystalline g-C3N4 was grown in the P-TiO2 fibers (Fig. S5a†). The FT-IR spectra of the TiO2, g-C3N4, and PTCN-90% heterojunction photocatalysts are presented in Fig. 2b. In the range of 700–900 cm−1, TiO2 exhibited a wide band because of the Ti–O–Ti stretching vibration. Pure g-C3N4 displayed three feature adsorption regions located at 3000–3500, 1200–1700, and 810 cm−1, respectively. The broad adsorption region around 3000–3500 cm−1 was indexed to the N–H typical stretching vibration. The strong feature peaks between 1200 to 1700 cm−1 were attributed to the C–N heterocycles stretching vibration. Moreover, the sharp absorption peak at 810 cm−1 was related to the specific triazine units breathing mode.43 In the FT-IR spectra of PTCN-90%, we could easily find the characteristic peaks for TiO2 and g-C3N4 in the relevant positions, which indicated that g-C3N4 was formed in PTCN fiber. With the concentration of CY increasing from 30% to 90%, the peak intensity of g-C3N4 became stronger in the PTCN heterojunction photocatalysts (Fig. S5b†). N2 adsorption–desorption isotherms were obtained and showed that all the samples presented type IV isotherms (Fig. 2c and S7a†). S-TiO2 exhibited an H2 hysteresis loop, illustrating that the S-TiO2 fiber possessed uniform intergranular pores (Fig S6a†). P-TiO2 and PTCN-90% showed H2 and H3 hysteresis loops, indicating the existence of inkbottle pores and slit-shaped pores, resulting from interconnected pore channels and plate-like particles aggregation. Only a type H3 hysteresis loop was observed in g-C3N4, owing to its layered structure.44,45 P-TiO2 had a larger SBET of 53.71 m2 g−1, which was 1.48 times that of S-TiO2 (36.25 m2 g−1) owing to the introduction of mesopores in the electrospun TiO2 fibers. Compared to P-TiO2, the SBET of the PTCN-90% sample (22.85 m2 g−1) clearly dropped, ascribed to the coating of the g-C3N4 layer on the P-TiO2 nanopores. The coated g-C3N4 layer blocks part of the nanopores and nanocavities of PTCN-90%. By comparison, pure g-C3N4 presented the lowest SBET of 13.20 m2 g−1 (Table S1†). A main pore-size distribution around 10–15 nm was detected in S-TiO2, ascribed to the thermal decomposition of organic substances in the process of the calcination. P-TiO2 exhibited a larger pore diameter and wider pore distribution than S-TiO2, owning to the abundant pore structure in the fibers (Fig. S6b†). As shown in Fig. 2d and S7b,† PTCN-90% showed a much lower pore volume than P-TiO2, PTCN-30%, and PTCN-90%, on account of filling up the plentiful g-C3N4 nanosheets into the nanopores of P-TiO2. The pore-size distribution results coincided with the SBET and morphology characterization results.
 | ||
| Fig. 2 (a and b) XRD patterns and FT-IR spectra for P-TiO2, g-C3N4, and PTCN-90%. The characteristic peaks of TiO2 and g-C3N4 were both found in PTCN-90%, indicating g-C3N4 was successfully coupled on the TiO2 fibers. (c and d) The N2 adsorption–desorption isotherms and corresponding pore-size distributions of P-TiO2, PTCN-90%, and g-C3N4. Both SBET and pore volume of PTCN-90% decrease apparently compared with P-TiO2, due to the filling of g-C3N4 in to the nanopores of the P-TiO2 fibers. (e and f) High-resolution XPS spectra of N 1s and O 1s of PTCN-90%, g-C3N4, and P-TiO2. | ||
X-ray photoelectron spectroscopy (XPS) was performed to further explore the compositions and surface chemical status of the obtained photocatalysts. C, N, Ti, and O elements were all detected in the spectra of PTCN-90%, which was consistent with the EDX results (Fig. S8a†). Fig. 2e displays the N 1s high-resolution spectrum of PTCN-90% with g-C3N4 for comparison. N 1s peaks at 398.4, 398.9, and 400.9 eV were ascribed to the sp2-hybridized nitrogen of triazine rings (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C), tertiary nitrogen ((C)3–N), and nitrogen in the amino functional groups (C–N–H), respectively. The isolated peak at 403.3 eV was attributed to the charging effect or a positive charge localization in the heterocycles.30 The binding energy of N–(C)3 (398.9 eV) showed a slight negative shift of 0.2 eV by contrast with g-C3N4 (399.1 eV), confirming the strong interfacial interaction between the TiO2 fiber and g-C3N4 layer rather than a simple physical mixing.44 The C 1s spectral peaks in PTCN-90% at 284.8 and 288.0 eV represented the sp2-hybridized carbon (N–CN) groups and graphitic carbon (C–C), respectively (Fig. S8b†). A negative shift of 0.1 eV compared with pure g-C3N4 was observed, attributed to the interaction effect of O–Ti–O–C–N bonds between g-C3N4 and TiO2.21 The O 1s peaks at 530.2 and 531.6 eV in HPCN-90% were associate with Ti–O and Ti–OH (Fig. 2e). Fig. S8c† displays the Ti 2p high-resolution spectrum. The two peaks at 459.0 and 464.7 eV corresponded to Ti 2p3/2 and Ti 2p1/2 and originated from the spin orbit interaction, indicating that the Ti ions were tetravalent.46 Apparently positive shifts in the binding energies in Ti 2p and O 1s were observed, confirming the intimate contact of the g-C3N4 layer and P-TiO2 fiber in HPCN-90%. The high-quality TiO2/g-C3N4 heterojunction is crucial to accelerate the separation and transport of photo-induced electrons and holes.46,47
N–C), tertiary nitrogen ((C)3–N), and nitrogen in the amino functional groups (C–N–H), respectively. The isolated peak at 403.3 eV was attributed to the charging effect or a positive charge localization in the heterocycles.30 The binding energy of N–(C)3 (398.9 eV) showed a slight negative shift of 0.2 eV by contrast with g-C3N4 (399.1 eV), confirming the strong interfacial interaction between the TiO2 fiber and g-C3N4 layer rather than a simple physical mixing.44 The C 1s spectral peaks in PTCN-90% at 284.8 and 288.0 eV represented the sp2-hybridized carbon (N–CN) groups and graphitic carbon (C–C), respectively (Fig. S8b†). A negative shift of 0.1 eV compared with pure g-C3N4 was observed, attributed to the interaction effect of O–Ti–O–C–N bonds between g-C3N4 and TiO2.21 The O 1s peaks at 530.2 and 531.6 eV in HPCN-90% were associate with Ti–O and Ti–OH (Fig. 2e). Fig. S8c† displays the Ti 2p high-resolution spectrum. The two peaks at 459.0 and 464.7 eV corresponded to Ti 2p3/2 and Ti 2p1/2 and originated from the spin orbit interaction, indicating that the Ti ions were tetravalent.46 Apparently positive shifts in the binding energies in Ti 2p and O 1s were observed, confirming the intimate contact of the g-C3N4 layer and P-TiO2 fiber in HPCN-90%. The high-quality TiO2/g-C3N4 heterojunction is crucial to accelerate the separation and transport of photo-induced electrons and holes.46,47
Photocatalytic H2 production performance measurements of all the samples were conducted under a xenon lamp (300 W, λ ≥ 420 nm). A 1 wt% platinum (Pt) loading on the photocatalysts was performed by a photodeposition method to serve as a cocatalyst. As shown in Fig. 3a and b, P-TiO2 had almost no photocatalytic activity for H2 production under visible light irradiation, due to its limited photoresponse to visible light. Pure g-C3N4 showed a lower H2 evolution rate on account of the fast recombination of photo-induced carriers.48 All the PTCN samples showed a higher H2 production rate when compared with that of P-TiO2 and g-C3N4 under visible light. The H2-production efficiency gradually improved with the increase in CY concentration. To further investigate the influence of the photocatalyst structure on H2 evolution, the photocatalytic H2-generation performances of g-C3N4, P-TiO2, STCN-90%, and PTCN-90% were assessed and are depicted in Fig. 3c. STCN-90% and PTCN-90% showed evidently higher H2 performances compared to g-C3N4 and TiO2. The photocatalytic H2-evolution rate of STCN-90% was 310 μmol g−1 h−1, which was 4.4 times as much as that of g-C3N4 (70 μmol g−1 h−1). This demonstrated that the combination of TiO2 and g-C3N4 favored improving the photocatalytic H2-evolution rate. PTCN-90% presented the highest rate of 436 μmol g−1 h−1, which was 6.2 times higher than that of pure g-C3N4. The H2-evolution rate of PTCN-90% was improved by 40.6% when compared with STCN-90%, indicating that the intimate core/shell heterojunction with a continuous g-C3N4 layer played an important role in the photocatalytic H2-evolution performance. The reductions of the H2-production efficiency and structures were not significantly impacted after four cycles tests, demonstrating the great recyclability of PTCN-90% (Fig. 3d and S9†). The g-C3N4 layer still adhered to the TiO2 fibers firmly, which confirmed the high stability of the PTCN-90% photocatalyst. When comparing the different TiO2/g-C3N4 composite photocatalysts with other structures, such as nanowires, nanospheres, and nanosheets, the PTCN photocatalyst still exhibited an excellent H2-evolution performance (Table S2†).
 | ||
| Fig. 3 (a and b) Time courses of H2 generation and average generation rate of P-TiO2, g-C3N4, PTCN-30%, PTCN-70%, and PTCN-90% under visible-light irradiation. With the increase in CY concentration, the photocatalytic hydrogen generation efficiency of PTCN was improved. (c) Time courses of H2 generation of P-TiO2, g-C3N4, STCN-90%, and PTCN-90%. The intimate core/shell in PTCN-90% was beneficial to enhancing the H2-generation efficiency. (d) Cyclic runs for the photocatalytic H2 evolution of PTCN-90%. | ||
The photocatalytic RhB degradation activities of all the samples were characterized under visible light irradiation with a wavelength longer than 420 nm. The photodegradation of RhB was negligible in the absence of the photocatalyst. All the PTCN photocatalysts exhibited higher activity for RhB degradation than P-TiO2 and g-C3N4. Especially, PTCN-90% showed the best RhB photodegradation performance, with the complete degradation of RhB in 30 min (Fig. 4a). The photocatalytic degradation reaction conformed to pseudo-first-order kinetics described as −ln(C/C0) = kt, where k stands for the pseudo-first-order rate constant, which corresponds to the fitting line slope, and C0 and C represent the solution concentrations at time 0 and t, respectively. The kinetic curves of all the samples for photocatalytic RhB degradation are demonstrated in Fig. 4b. PTCN-90% had the highest k value of 0.1674 min−1, which was 54 times that of TiO2 (0.0030 min−1) and 6.3 times that of g-C3N4 (0.0267 min−1). In order to verify the effect of the intimate core/shell structure in PTCN, the photocatalytic RhB photodegradation performance and degradation kinetic curves for P-TiO2, g-C3N4, STCN-90%, and PTCN-90% were also assessed (Fig. 4c and d). The degradation rate of PTCN-90% was significantly higher than for STCN-90% (0.0524 min−1) due to the specific core/shell structure and strong heterojunction between TiO2 and g-C3N4. In 30 min, RhB was degraded almost completely by PTCN-90%; however, it could only be degraded 72% by STCN-90% (Fig. 4e). The corresponding changes in the UV-vis absorption spectra and optical images of RhB solution are exhibited in Fig. S10.† The absorption peak of the PTCN-90% sample dropped rapidly with prolonging the irradiation time and disappeared completely after 30 min. The inset shows the color change of RhB solution. The stability analysis of PTCN-90% for RhB degradation was also conducted by the recycling experiment (Fig. 4f). The performance of RhB degradation did not change obviously within four cycles, demonstrating the stability of the PTCN heterojunction photocatalyst. The core/shell PTCN-90% photocatalyst also exhibited good degradation performance for MB, in addition to RhB (Fig. S11†), where 10 ppm MB solution was completely degraded within 50 min by PTCN-90% under visible light irradiation. In consideration of the decolorization of dyes caused by a self-sensitization mechanism,49,50 phenol, which has no photosensitization, was selected as the substrate to further investigate the photocatalytic performance of the core/shell PTCN-90% photocatalyst (Fig. S12†). The core/shell PTCN-90% photocatalyst also exhibited good degradation performance for phenol, which was completely degraded within 3.5 h under visible light irradiation. This proved that the core/shell PTCN photocatalyst has wide application prospects in the field of photocatalysis for organic pollutant degradation.
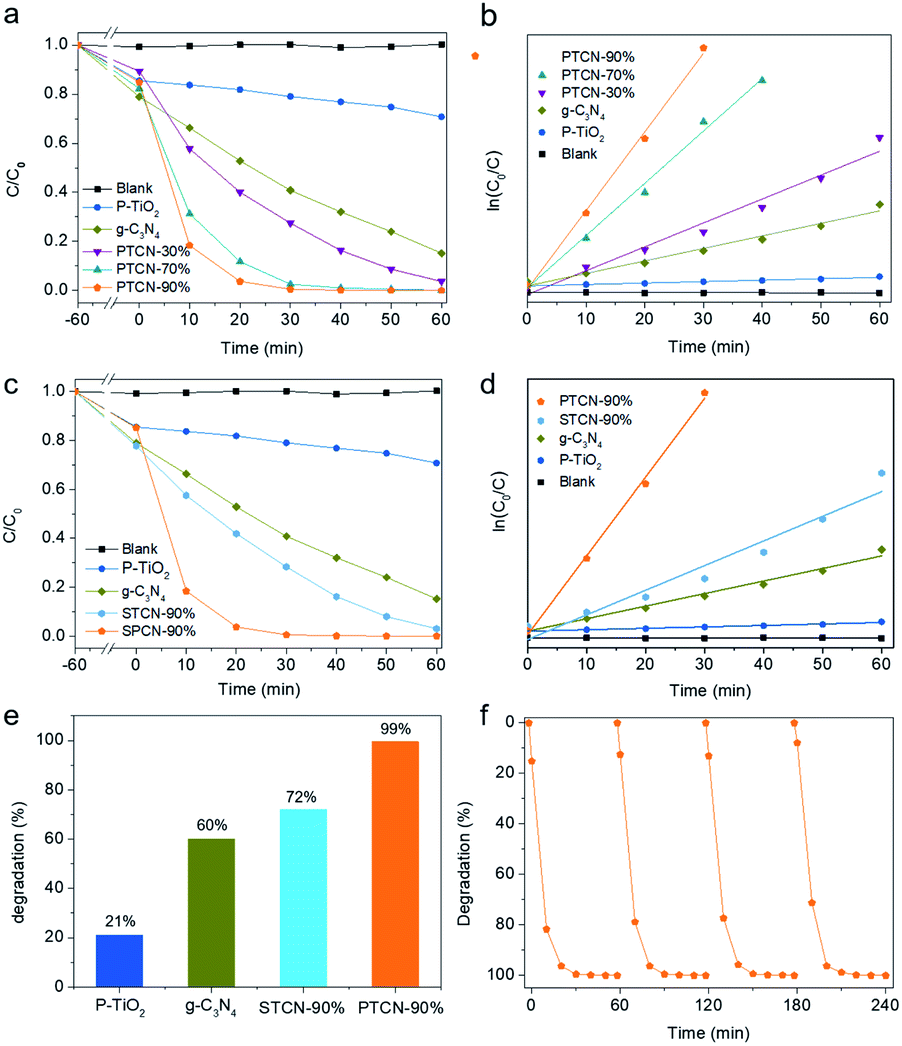 | ||
| Fig. 4 (a and b) Photocatalytic RhB degradation performance and degradation kinetic curves of P-TiO2 g-C3N4, PTCN-30%, PTCN-70%, and PTCN-90% under visible light irradiation. The RhB degradation rate was improved by raising the content of g-C3N4. PTCN-90% possessed the highest RhB degradation rate. (c and d) RhB photocatalytic degradation performance and degradation kinetic curves for P-TiO2, g-C3N4, STCN-90%, and PTCN-90%. (e) RhB photocatalytic degradation efficiency of P-TiO2, g-C3N4, STCN-90%, and PTCN-90% in 30 min. PTCN-90% had a much higher RhB degradation rate than STCN-90% due to the continuous core/shell structure advantage in PTCN. (f) Cyclic runs for RhB degradation over PTCN-90%. | ||
To further explore the photodegradation mechanism of RhB, active material trapping experiments were conducted (Fig. 5a). Superoxide radicals (˙O2−), photoexcited holes (h+), and hydroxyl radicals (˙OH) generated in the photocatalytic system could be shielded by p-BQ, TEOA, and IPA, respectively.12 The RhB degradation decline of PTCN-90% was not significant with the presence of IPA. However, the degradation effect dramatically decreased when TEOA was added. This demonstrated that h+ was the dominant active species in the photodegradation reaction of RhB. Moreover, the ˙O2− derived from dissolved O2 also acted as a partial player in the photocatalytic system. Photoluminescence (PL) emission spectroscopy is the most intuitive detection method to determine the separation efficiency of photo-induced carriers. The PL spectra for P-TiO2, g-C3N4, and PCN-90% were evaluated with an excitation wavelength of 320 nm (Fig. 5b).51 It was revealed that g-C3N4 had a strong peak at 470 nm related to the band–band PL phenomenon.52 The bandgap energy was almost equal to the emission light energy of g-C3N4.53 The emission peak of the PTCN-90% heterojunction photocatalyst was weaker than for g-C3N4, which proved that PTCN-90% had a higher photo-induced carriers separation rate by virtue of the heterojunction between g-C3N4 and P-TiO2. These results explain well its enhanced photocatalytic performance in photocatalytic H2 generation and dye degradation.54 Next, UV-visible diffuse reflectance spectroscopy (DRS) was performed to understand the optical absorption property of the photocatalysts. The DRS absorption edge of pure TiO2 was 398 nm, implying that pure TiO2 only has a response in the UV region; while g-C3N4 presented a wider absorption in the visible and UV region with the absorption edge of 450 nm. PTCN-90% showed an obvious absorption extension to the visible region compared to that of pure TiO2 (Fig. 5c).13 The absorption extension of PTCN was ascribed to the strong heterojunction between g-C3N4 and P-TiO2 in the composites. A close-contacted core–shell structure and heterojunction in PTCN catalysts are favorable for separating the photogenerated electrons rapidly, which is highly conducive to accelerating photocatalytic reactions. The band gap energy of photocatalysts could be calculated by Eg = 1240/λg, where Eg is the bandgap energy and λg is the edge of optical absorption.55 The calculated band gap energy of TiO2, g-C3N4 and PTCN-90% were 3.08, 2.60, and 2.37 eV, respectively (Fig. 5d). The PTCN-90% core/shell photocatalyst presented the narrowest band gap compared with P-TiO2 and g-C3N4, indicating its higher responsive to visible light.
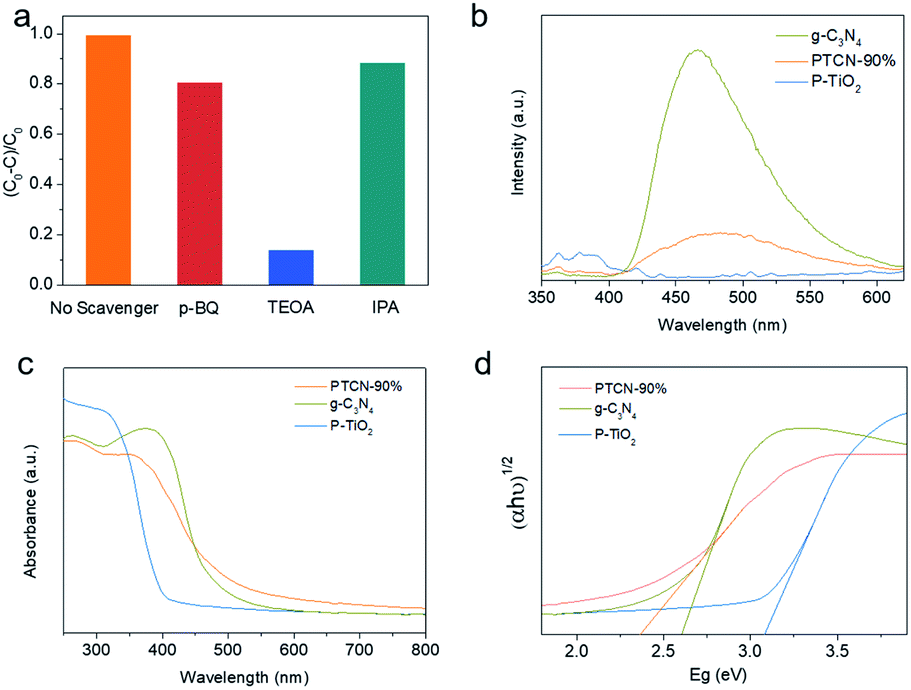 | ||
| Fig. 5 (a) Active species trapping during RhB photodegradation over PTCN-90% under visible light irradiation for 30 min. (b) PL spectra of PTCN-90%, g-C3N4, and P-TiO2. The strong heterojunction between g-C3N4 and TiO2 accelerated the separation and transportation rate of photo-induced carriers. (c) DRS spectra and (d) corresponding band gap energy of PTCN-90%, g-C3N4, and P-TiO2. A significant red-shift of absorption was observed in PTCN-90%, suggesting the close mutual effect between the g-C3N4 layer and P-TiO2 fibers. | ||
A reasonable mechanism is proposed to illustrate the improved performance of the PTCN core/shell photocatalyst (Scheme 2). The ECB and EVB values of TiO2 were calculated to be −0.23 and 2.85 eV. Correspondingly, the ECB and EVB values of g-C3N4 were −1.08 and 1.52 eV, respectively.56 TiO2 had a limited absorption range in the UV light area and no response in the visible region due to the broad band gap. However, the coating of a continuous g-C3N4 layer on P-TiO2 fibers to construct the PTCN heterojunction could broaden its light absorption range. Under visible light conditions, the photo-induced electrons separate, with the holes rapidly leaping to the g-C3N4 CB, and then transiting to the CB of TiO2 readily through the TiO2/g-C3N4 heterojunction in PTCN. The fast separate and transfer of the electrons inhibit the recombination of carriers efficiently. As the reaction progresses, more electrons gather in the TiO2 CB and more holes stay in g-C3N4 VB. In the process of photocatalytic water splitting, electrons accumulate on the cocatalyst and reduce H+ to H2. Photogenerated holes in g-C3N4 VB oxidize TEOA to CO2 and H2O. Moreover, during the RhB photodegradation process, a portion of dissolved O2 is reduced by electrons to create superoxide radicals with a high oxidation ability in the CB of g-C3N4. According to the active species trapping experiments, the photoexcited holes together with superoxide radicals acted as reactive species to decompose RhB into its degradation products. As a consequence, the strong heterojunction between g-C3N4 and TiO2 together with the intimate core/shell fiber structure facilitate the transport of photoexcited carriers. The unique TiO2/g-C3N4 core/shell structure and the TiO2/g-C3N4 heterojunction produce a synergistic effect to the photocatalytic efficiency in the PTCN composite.
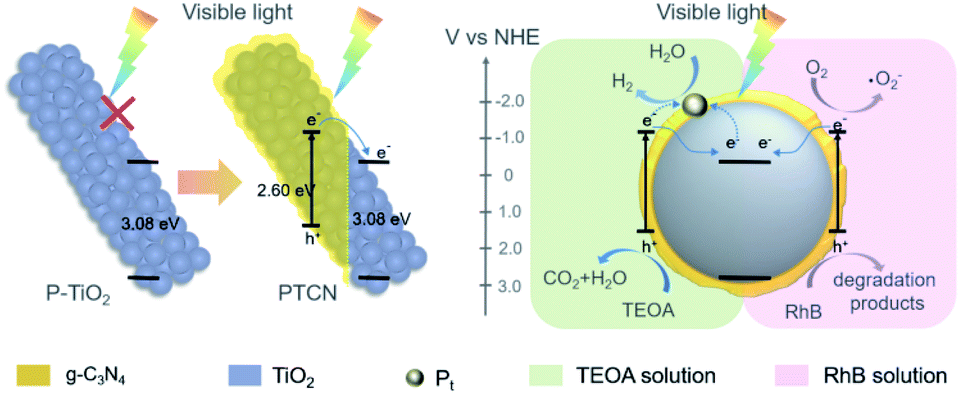 | ||
| Scheme 2 Proposed mechanism in PTCN fibers for photocatalytic H2 evolution and RhB degradation. Under visible light irradiation, P-TiO2 had no photocatalytic response, due to its broad band gap. While in the core/shell PTCN composite, photogenerated electrons in the g-C3N4 shell could transfer to the P-TiO2 core rapidly though the heterojunction, thereby realizing effective electron–hole separation. For the H2-evolution process, electrons accumulate on Pt particles and participate in the water splitting reaction. For the RhB degradation reaction, the photogenerated holes and the superoxide radicals derived from the dissolved O2 are responsible for RhB degradation. | ||
4 Conclusions
TiO2@g-C3N4 core/shell fibers were fabricated by a template-assisted electrospinning method together with a solution infiltration process. In the synthesis process, the porous structure of the TiO2 fibers enabled CY solution to readily immerse into the abundant nanopores and nanocavities to build a continuous and close-contacted TiO2 core/g-C3N4 shell structure. The strong heterojunction between TiO2 and g-C3N4 accelerated the separation and transfer of photoexcited electrons and holes rapidly. In addition, the intimate core/shell structure provided a rapid transmission channel for carriers as well. The synergistic effect of the strong heterojunction and intimate core/shell structure of the PTCN composite photocatalyst led to a significant improvement in the photocatalytic performance. PTCN-90% exhibited the highest performance in H2 generation and RhB degradation. This work offers a fresh insight in designing and synthesizing multi-structure heterojunction photocatalysts for extensive applications in the field of green and sustainable new energy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the National Natural Science Foundation of China (NSFC) (Grant No. 22175007, 52172080 and 21975007), National Natural Science Foundation for Outstanding Youth Foundation, the Fundamental Research Funds for the Central Universities, the National Program for Support of Top-notch Young Professionals, the 111 project (Grant No. B14009).Notes and references
- Q. Wang and Z. Yang, Environ. Pollut., 2016, 218, 358 CrossRef CAS PubMed.
- X. Zhang, X. Chen and X. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9193 CrossRef CAS PubMed.
- J. Chow, R. Kopp and P. Portney, Science, 2003, 302, 1528 CrossRef PubMed.
- A. Ahmed, E. Abdalla and M. Shaban, J. Phys. Chem. C, 2020, 124, 22347 CrossRef CAS.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611 CrossRef CAS PubMed.
- W. Li, X. Huang, T. Zeng, Y. Liu, W. Hu, H. Yang, Y. Zhang and K. Wen, Angew. Chem., Int. Ed. Engl., 2021, 60, 1869 CrossRef CAS PubMed.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. Howells, M. Sachs, D. Anjum, S. Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. DeLongchamp, J. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559 CrossRef CAS PubMed.
- Y. Wei, J. Wang, R. Yu, J. Wan and D. Wang, Angew. Chem., Int. Ed. Engl., 2019, 58, 1422 CrossRef CAS PubMed.
- K. K. Khaing, D. Yin, S. Xiao, L. Deng, F. Zhao, B. Liu, T. Chen, L. Li, X. Guo, J. Liu and Y. Zhang, J. Phys. Chem. C, 2020, 124, 11831 CrossRef CAS.
- T. Senasu, S. Nijpanich, S. Juabrum, N. Chanlek and S. Nanan, Appl. Surf. Sci., 2021, 567, 150850 CrossRef CAS.
- S.-G. Xia, Z. Zhang, J.-N. Wu, Y. Wang, M.-J. Sun, Y. Cui, C.-L. Zhao, J.-Y. Zhong, W. Cao, H. Wang, M. Zhang, Y.-C. Zheng and X.-B. Li, Appl. Catal., B, 2021, 284, 119703 CrossRef CAS.
- W. Tao, M. Wang, R. Ali, S. Nie, Q. Zeng, R. Yang, W.-M. Lau, L. He, H. Tang and X. Jian, Appl. Surf. Sci., 2019, 495, 143435 CrossRef CAS.
- Z. Lin, B. Yu and J. Huang, Langmuir, 2020, 36, 5967 CrossRef CAS PubMed.
- Z. Niu, S. Yi, C. Li, Y. Liu, Q. Pang, Z. Liu and X. Yue, Chem. Eng. J., 2020, 390, 124602 CrossRef CAS.
- J. Liu, G. Liu, M. Li, W. Shen, Z. Liu, J. Wang, J. Zhao, L. Jiang and Y. Song, Energy Environ. Sci., 2010, 3, 1503 RSC.
- Y. Liu, G. Xu and H. Lv, J. Mater. Sci.: Mater. Electron., 2018, 29, 10504 CrossRef CAS.
- S. Cho, C. Ahn, J. Park and S. Jeon, Nanoscale, 2018, 10, 9747 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- X. W. Shi, M. Fujitsuka, Z. Z. Lou, P. Zhang and T. Majima, J. Mater. Chem. A, 2017, 5, 9671–9681 RSC.
- H. Wei, W. A. McMaster, J. Z. Y. Tan, D. H. Chen and R. A. Caruso, J. Mater. Chem. A, 2018, 6, 7236–7245 RSC.
- C. Marchal, T. Cottineau, M. G. Méndez-Medrano, C. Colbeau-Justin, V. Caps and V. Keller, Adv. Energy Mater., 2018, 8, 1702142 CrossRef.
- X. Wang, F. Wang, B. Chen, K. Cheng, J. Wang, J. Zhang and H. Song, Appl. Surf. Sci., 2018, 453, 320 CrossRef CAS.
- X. Chen, J. Wei, R. Hou, Y. Liang, Z. Xie, Y. Zhu, X. Zhang and H. Wang, Appl. Catal., B, 2016, 188, 342 CrossRef CAS.
- T. Zhao, Z. Liu, K. Nakata, S. Nishimoto, T. Murakami, Y. Zhao, L. Jiang and A. Fujishima, J. Mater. Chem., 2010, 20, 5095 RSC.
- N. Wang, Y. Gao, Y. Wang, K. Liu, W. Lai, Y. Hu, Y. Zhao, S. Chou and L. Jiang, Adv. Sci., 2016, 3, 1600013 CrossRef PubMed.
- G. Yue, S. Li, D. Li, J. Liu, Y. Wang, Y. Zhao, N. Wang, Z. Cui and Y. Zhao, Langmuir, 2019, 35, 4843 CrossRef CAS PubMed.
- L. Lang, D. Wu and Z. Xu, Chem.–Eur. J., 2012, 18, 10661 CrossRef CAS PubMed.
- D. Li, F. Guo, Z. Cui, J. Zhou, Y. Zhai, Y. Du, J. Liu, N. Wang and Y. Zhao, ACS Appl. Mater. Interfaces, 2020, 12, 53503 CrossRef CAS PubMed.
- D. Li, H. Li, S. Zheng, N. Gao, S. Li, J. Liu, L. Hou, J. Liu, B. Miao, J. Bai, Z. Cui, N. Wang, B. Wang and Y. Zhao, J. Colloid Interface Sci., 2022, 607, 655 CrossRef CAS PubMed.
- C. Han, Y. Wang, Y. Lei, B. Wang, N. Wu, Q. Shi and Q. Li, Nano Res., 2015, 8, 1199 CrossRef CAS.
- S. P. Adhikari, G. P. Awasthi, H. J. Kim, C. H. Park and C. S. Kim, Langmuir, 2016, 32, 6163 CrossRef CAS PubMed.
- C. Wang, L. Hu, B. Chai, J. Yan and J. Li, Appl. Surf. Sci., 2018, 430, 243 CrossRef CAS.
- T. Wang, J. Xu, Z. Zhang, H. Bian, H. Xiao and T. Sun, J. Mater. Sci.: Mater. Electron., 2020, 32, 1178 CrossRef.
- H. Wei, W. A. McMaster, J. Z. Y. Tan, L. Cao, D. H. Chen and R. A. Caruso, J. Phys. Chem. C, 2017, 121, 22114 CrossRef CAS.
- H. L. Hou, L. Wang, F. M. Gao, X. F. Yang and W. Y. Yang, J. Mater. Chem. C, 2019, 7, 7858 RSC.
- C. B. Liu, L. L. Wang, Y. H. Tang, S. L. Luo, Y. T. Liu, S. Q. Zhang, Y. X. Zeng and Y. Z. Xu, Appl. Catal., B, 2015, 164, 1 CrossRef CAS.
- J. Liu, D. Li, X. Liu, J. Zhou, H. Zhao, N. Wang, Z. Cui, J. Bai and Y. Zhao, New J. Chem., 2021, 45, 22123 RSC.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed. Engl., 2019, 58, 6164 CrossRef CAS PubMed.
- M. Rahman and C. Mullins, Acc. Chem. Res., 2019, 52, 248 CrossRef CAS PubMed.
- A. Khan and M. Tahir, Appl. Catal., B, 2021, 285, 119777 CrossRef CAS.
- S. Kumar, A. Baruah, S. Tonda, B. Kumar, V. Shanker and B. Sreedhar, Nanoscale, 2014, 6, 4830 RSC.
- C. Hu, L. E, K. Hu, L. Lai, D. Zhao, W. Zhao and H. Rong, J. Mater. Sci., 2019, 55, 151 CrossRef.
- Z. Huang, Q. Sun, K. Lv, Z. Zhang, M. Li and B. Li, Appl. Catal., B, 2015, 164, 420 CrossRef CAS.
- J. Wang, G. Wang, X. Wang, Y. Wu, Y. Su and H. Tang, Carbon, 2019, 149, 618 CrossRef CAS.
- W. Li, Q. Ma, X. Wang, X. Chu, F. Wang, X. Wang and C. Wang, J. Mater. Chem. A, 2020, 8, 19533 RSC.
- F. Li, X. Xiao, C. Zhao, J. Liu, Q. Li, C. Guo, C. Tian, L. Zhang, J. Hu and B. Jiang, J. Colloid Interface Sci., 2020, 572, 22 CrossRef CAS PubMed.
- L. Y. Lu, G. H. Wang, M. Zou, J. Wang and J. Li, Appl. Surf. Sci., 2018, 441, 1012 CrossRef CAS.
- H. Zhao, Y. Dong, P. Jiang, H. Miao, G. Wang and J. Zhang, J. Mater. Chem. A, 2015, 3, 7375 RSC.
- T. Wu, G. Liu and J. Zhao, J. Phys. Chem. B, 1998, 102, 5845 CrossRef CAS.
- M. Rochkind, S. Pasternak and Y. Paz, Molecules, 2014, 20, 88–110 CrossRef PubMed.
- R. Hao, G. Wang, H. Tang, L. Sun, C. Xu and D. Han, Appl. Catal., B, 2016, 187, 47 CrossRef CAS.
- L. Huang, H. Xu, Y. Li, H. Li, X. Cheng, J. Xia, Y. Xu and G. Cai, Dalton Trans., 2013, 42, 8606 RSC.
- Z. Tong, D. Yang, T. Xiao, Y. Tian and Z. Jiang, Chem. Eng. J., 2015, 260, 117 CrossRef CAS.
- Y. Li, H. Zhang, P. Liu, D. Wang, Y. Li and H. Zhao, Small, 2013, 9, 3336 CAS.
- B. Chai, J. Yan, C. Wang, Z. Ren and Y. Zhu, Appl. Surf. Sci., 2017, 391, 376 CrossRef CAS.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra01093c |
| This journal is © The Royal Society of Chemistry 2022 |