
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorinated β-diketonate complexes M(tfac)2(TMEDA) (M = Fe, Ni, Cu, Zn) as precursors for the MOCVD growth of metal and metal oxide thin films†
Christian Stienen,
Julian Grahl,
Christoph Wölper,
Stephan Schulz and
Georg Bendt*
and
Georg Bendt*
Institute for Inorganic Chemistry, University of Duisburg-Essen, Universitätsstraße 5-7, 45117 Essen, Germany. E-mail: georg.bendt@uni-due.de
First published on 16th August 2022
Abstract
Partially fluorinated β-diketonate complexes M(tfac)2(TMEDA) (M = Fe 1, Ni 2, Cu 3, Zn 4; tfac = 1,1,1-trifluoro-2,4-pentanedionate; TMEDA = N,N,N′,N′-tetramethylethylenediamine) were synthesized and structurally (sc-XRD) and thermochemically (TGA) characterised. A new polymorph of Fe(tfac)2(TMEDA) was found. The structural and physicochemical properties of 1–4 were compared with related M(acac)2(TMEDA) and M(hfac)2(TMEDA) (acac = 2,4-pentanedionate, hfac = 1,1,1,5,5,5-hexafluoro-2,4-pentanedionate) β-diketonate complexes to evaluate the effect of the degree of fluorination. A positive effect on the thermal behaviour of the metal acetylacetonates was observed, but no discernible trends. Application of complexes 1–4 as precursors in a MOCVD process yielded either metal (Ni, Cu) or metal oxide thin films (Fe3O4, ZnO), which were further oxidized to NiO, CuO and α-Fe2O3 films by calcination in air at 500 °C.
Introduction
Metal β-diketonate complexes are among the most widely used classes of precursors for both the synthesis of metal and metal oxide nanoparticles in liquid phase and the deposition of thin films via gas phase processes. They are available with virtually every metal in the periodic table, and their chemical and physical properties can be largely modified by varying the size of the ligand backbone and the steric and electronic properties of the substituents.1 Metal β-diketonate complexes play an important role in the synthesis of materials based on first row transition metals such as the bare metals as well as their oxides and sulfides.For the wet chemical synthesis of these materials, commercially available metal acetylacetonate complexes M(acac)x (acac = 2,4-pentanedionate; x = 2, 3) are preferably used due to their high solubility in organic solvents and their moderate decomposition temperatures.2a–f In contrast, gas-phase-based processes, e.g. atomic layer deposition (ALD) and (metal organic) chemical vapour deposition ((MO)CVD), heavily rely on the controlled, reproducible and decomposition-free transfer of the precursors into the gas phase, hence a reasonable high vapor pressure is crucial. Unfortunately, homoleptic complexes of trivalent metals M(acac)3 (M = Cr, Mn, Fe, Co) tend to decompose into M(acac)2 and various by-products upon heating,3 whereas M(acac)2 complexes often oligomerize in the solid state with formation of trinuclear (Ni(acac)2)4 or tetranuclear (Co(acac)2)5 complexes as was proven by single crystal X-ray diffraction. Such oligomeric complexes not only exhibit low volatilities but often also contain variable amounts of water, resulting in poor sublimation behavior and hence limited suitability as precursor in gas phase-based processes. The thermochemical disadvantages of M(acac)2 and M(acac)3 were resolved by injecting these precursors into the gas-phase reactor in the form of solutions as is applied in direct liquid injection (DLI-CVD), aerosol assisted (AA-CVD), and pulsed spray evaporation (PSE-CVD) CVD processes. For example, an ethanolic solution of Fe(acac)3 and Co(acac)3 was successfully used as precursor for the growth of CoFe2O4 thin films,6 while a solution of Cu(acac)2 in ethanol was used for the growth of copper oxide films by PSE-CVD.7 The stable and reliable production of aerosols is challenging and increases the overall complexity of the CVD process compared to the thermal evaporation of precursors and requires special equipment. Moreover, solvent molecules in the gas phase are a possible source of unwanted impurities in the resulting material film.8
The addition of a Lewis base is also a well-established strategy to suppress the tendency for oligomerization and to prevent the coordination of water. Monomeric M(acac)2(TMEDA) complexes9a–c have shown significantly better evaporation properties compared to M(acac)2. The TGA curve of Ni(acac)2 revealed four distinctive mass loss events and a high amount of non-volatile residue, whereas the Ni(acac)2(TMEDA) adduct exhibited a single mass loss event without significant residue occurring roughly 100 °C below the sublimation temperature of Ni(acac)2.10 A comparable thermal behaviour was recently reported for Co(acac)2 and Co(acac)2(TMEDA).11 The volatility of these compounds can be further improved by replacing the CH3 groups with electron-withdrawing CF3 groups. A large number of first-row transition metal complexes of the type M(hfac)2(TMEDA) have been prepared and used as precursors for the deposition of transition metal oxide films.12a–g In remarkable contrast to widely applied M(acac)2(TMEDA) and M(hfac)2(TMEDA) complexes, the corresponding M(tfac)2(TMEDA) complexes have received far less attention, and within the first-row transition metals only manganese, iron and cobalt complexes have been applied for the deposition of Mn3O4,13 CoO,11 Co3O4 (ref. 14) and Fe2O3 (ref. 15) thin films. We recently demonstrated that the sublimation temperature of different fluorinated cobalt acetylacetonate complexes largely depends on the degree of fluorination. In addition, using these complexes as precursors in MOCVD processes demonstrated that the amount of fluorine-containing impurities in the resulting material films can be tailored by the degree of fluorination.11 These fluorine-containing impurities can have either positive16a,b or have no11 effect on the physical and chemical properties of the resulting material films. In addition to the impurity content, the ligand design directly influences the possible formation of corrosive or toxic decomposition products such as hydrogen fluoride.17
We herein report on the solid-state structures and the thermochemical properties of metal β-diketonate complexes M(tfac)2(TMEDA) (M = Fe 1, Ni 2, Cu 3, Zn 4). To reveal the influence of the fluorine substituents on the thermochemistry, we also compared 1–4 with the corresponding acac and hfac complexes. In addition, the complexes were used as precursors for the MOCVD growth of metal and metal oxide thin films.
Results and discussion
Structural and thermal properties of M(tfac)2(TMEDA) (M = Fe 1, Ni 2, Cu 3, Zn 4)
M(tfac)2(TMEDA) (M = Fe 1, Ni 2, Cu 3, Zn 4) was prepared by the reaction of MCl2 with two equivalents Htfac in an alkaline aqueous medium at ambient temperature, followed by the slow addition of one equivalent of TMEDA (Scheme 1). The product was extracted from the reaction mixture with dichloromethane, and the isolated solids were purified by evaporation at 100 °C under reduced pressure (1 × 10−3 mbar).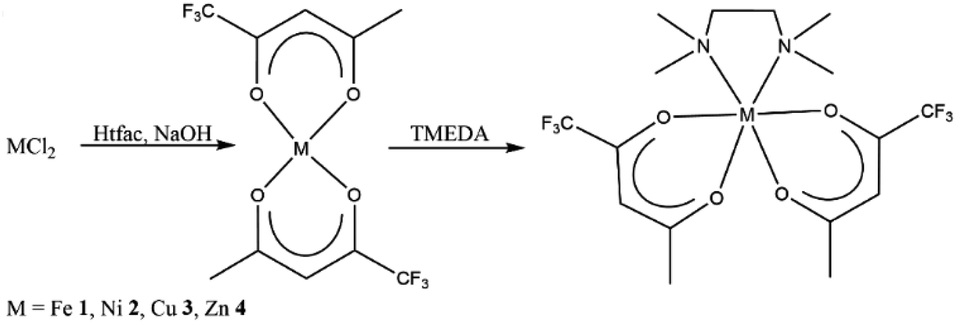 | ||
| Scheme 1 Synthesis of 1–4. | ||
Single crystals of 1–4 were obtained by the slow evaporation of the solvent from concentrated solutions in dichloromethane at ambient temperature. Complexes M(tfac)2(TMEDA) crystallize in the monoclinic space group P21/c (1, 2, 4) and P21/n (3) with one independent molecule in the asymmetric unit (Fig. 1). The central metal cations in complexes 1–4 are sixfold coordinated by four oxygen atoms of the tfac anions and two nitrogen atoms of the TMEDA base. The central metal cation consequently adopts a distorted octahedral coordination geometry. The M1–N1 bond lengths are shorter than those in M(acac)2(TMEDA) (the structure of Cu(acac)2(TMEDA) has not been reported to date). Even shorter M–N bonds were found in M(hfac)2(TMEDA), due to the increasing electron withdrawing effect of the acetylacetonate ligand with increasing degree of fluorination whereas the effect on the M–N bond length is less pronounced between M(acac)2(TMEDA) and M(tfac)2(TMEDA) than between M(tfac)2(TMEDA) and M(hfac)2(TMEDA). As was observed for Co(acac)2(TMEDA) and Fe(acac)2(TMEDA) complexes, the M1–O bonds in 1, 2 and 4 in trans position to the nitrogen atoms of the TMEDA ligand are slightly longer than the other M1–O bonds, whereas the opposite case was observed for 3. In 3, the Cu–O bonds are significantly elongated (2.318(1) Å) compared to the Cu–O bonds in trans position (1.973(5) Å) as a result of the Jahn–Teller distortion. This effect was also, slightly less pronounced, observed in both polymorphs reported for Cu(hfac)2(TMEDA).18 The N1–M1–N1′ bite angles increase with increasing degree of fluorination for all the complexes studied. Comparable to the observed effect on the M–N bond length, the increase of the bite angle between M(acac)2(TMEDA) and M(tfac)2(TMEDA) is less pronounced than between M(tfac)2TMEDA and M(hfac)2TMEDA. For example, the N1–M1–N1′-bite angle between Zn(acac)2(TMEDA) and Zn(tfac)2(TMEDA) increases by 2.01° and by 3.49° between Zn(tfac)2(TMEDA) and Zn(hfac)2(TMEDA) (Table 1).
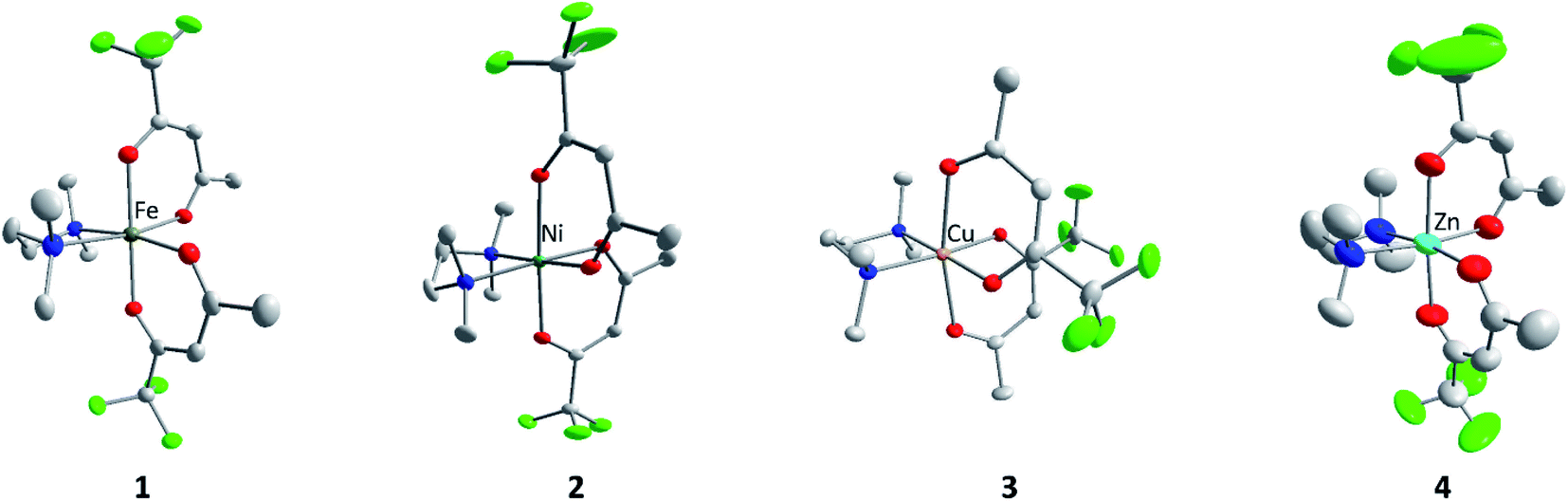 | ||
| Fig. 1 Crystal structure of 1–4. Hydrogen atoms are not shown for clarity and displacement ellipsoids are displayed at 50% probability levels. Oxygen atoms in red, nitrogen in blue, fluorine in green and carbon in grey. Minor components of disordered part have been omitted for clarity. | ||
| M1–O(trans) | M1–O | O1–M1–O1′ | N1–M1–O2′ | N1–M1–N1′ | M1–N1 | |
|---|---|---|---|---|---|---|
| Fe(acac)2(TMEDA)9a | 2.093(1) | 2.051(1) | 174.86(4) | 92.43(4) | 79.36(5) | 2.310(1) |
| Fe(tfac)2(TMEDA) 1 | 2.088(1) | 2.051(1) | 178.84(5) | 93.25(5) | 80.64(5) | 2.275(1) |
| Fe(hfac)2(TMEDA)23 | 2.094(1) | 2.056(2) | 175.01(1) | 94.18(1) | 82.02(9) | 2.229(9) |
| Ni(acac)2(TMEDA)20 | 2.030(4) | 2.034(4) | 176.9(2) | 91.9(2) | 83.9(2) | 2.183(6) |
| Ni(tfac)2(TMEDA) 2 | 2.047(7) | 2.065(1) | 176.77(3)° | 92.15(3) | 84.65(3) | 2.156(8) |
| Ni(hfac)2(TMEDA) | 2.052(9) | 2.035(2) | 175.0(1) | 93.0(1) | 86.03(3) | 2.1218(10) |
| Cu(tfac)2(TMEDA) 3 | 1.973(5) | 2.318(1) | 167.61(3) | 95.61(4) | 86.46(4) | 2.065(1) |
| Cu(hfac)2(TMEDA)18a | 1.990(5) | 2.300(2) | 166.62(7) | 96.76(7) | 87.09(9) | 2.052(2) |
| Zn(acac)2(TMEDA)9a | 2.077(1) | 2.061(1) | 175.16(4) | 89.28(5) | 80.27(5) | 2.263(1) |
| Zn(tfac)2(TMEDA) 4 | 2.119(5) | 2.099(5) | 174.53(9) | 88.56(13) | 82.28(9) | 2.234(5) |
| Zn(hfac)2(TMEDA)12e | 2.119(1) | 2.099(1) | 173.65(4) | 95.71(5) | 85.74(5) | 2.148(1) |
The solid-state structure of 1 is a polymorph of the recently reported structure of Fe(tfac)2(TMEDA).15 Both polymorphs crystallize in the same monoclinic space group with a similar coordination geometry. According to powder XRD the synthesis protocol reported in this work results in a mixture of both polymorphs, while no evidence of polymorphs was found in the case of 2–4 (Fig. S14–S17†). The formation of polymorphic structures was also reported for Mg(tfac)2(TMEDA).19
The sublimation behaviour and thus the suitability of precursors for MOCVD processes is largely determined by intermolecular interactions in the solid state. In the investigated metal acetylacetonates with different degrees of fluorination, mainly van der Waals interactions were observed, whereas the sum of the van der Waals radii (H 1.20 Å, F 1.47 Å)19 was used as a criterion. The only exception is Fe(acac)2(TMEDA), were also weak CH⋯O hydrogen bridges were reported. Regarding the type and number of contacts within the investigated complexes, no clear trend was observed. For example, for 1, only two H⋯F contacts were found, while 2 has one H⋯H, three H⋯F and one F⋯F contacts. In 3, one H⋯H and four H⋯F interactions exists, while 4 is the only investigated complex without intermolecular interactions. All intermolecular contacts are summarized in Table S5.†
The thermal behaviour of the different M(tfac)2(TMEDA) (M = Fe 1, Ni 2, Cu 3, Zn 4) complexes in comparison with the non-fluorinated M(acac)2(TMEDA) and the higher fluorinated M(hfac)2(TMEDA) were studied by thermogravimetric analysis TGA under inert gas conditions at atmospheric pressure (Fig. 2). For M = Fe, the dynamic TGA curve of the iron precursors showed a decreasing onset temperature for the mass loss event indicating sublimation with increasing degree of fluorination. However, a high amount of non-volatile residues remained for Fe(acac)2 and Fe(acac)2(TMEDA), whereas complexes 3 and Fe(hfac)2(TMEDA) almost completely evaporated (residues <1%). The TGA curve of [Ni(acac)2]n showed several mass loss events and a high amount of non-volatile residues, whereas the dynamic TGA curves for the TMEDA adducts showed only a single mass loss event starting at remarkably lower temperatures. Complexes Ni(acac)2(TMEDA), 2, and Ni(hfac)2(TMEDA) almost completely evaporated with nearly no residue remaining. Cu(acac)2 and Cu(hfac)2(TMEDA) sublimed in a single mass loss event and without residue. The onset of sublimation for Cu(hfac)2(TMEDA) is reduced by about 50 °C compared to Cu(acac)2. The observed mass loss of 3 in the TGA trace is due to the sublimation of 3. In addition, a significant amount of non-volatile, highly viscous and dark coloured residue was observed. The decomposition pathway and products at atmospheric pressure remain unclear, although the loss of TMEDA and the formation of uncoordinated Cu(tfac)2, a light blue crystalline substance, can be excluded by 1H-NMR spectroscopy (Fig. S1†). Under high vacuum conditions, 3 sublimes without decomposition. The TGA of [Zn(acac)2]n showed two mass loss events, with the initial event starting at around 75 °C which corresponds to the loss of water. The second mass loss indicates the sublimation of [Zn(acac)2]n and started at around 120 °C with a high amount of non-volatile residue. In contrast, the TMEDA adducts Zn(acac)2(TMEDA), 4, and Zn(hfac)2(TMEDA) sublimed without residues, with on-set temperatures for the sublimation higher than for uncoordinated [Zn(acac)2]n. Except for 3, the isothermal TGA curves of all compounds investigated (125 °C, Fig. S13†) show a linear mass loss behaviour, which is an important prerequisite for the reliable evaporation of solids in gas phase-based processes. Compared to the uncoordinated [M(acac)2]n, the TMEDA adducts show a significantly higher evaporation rate.
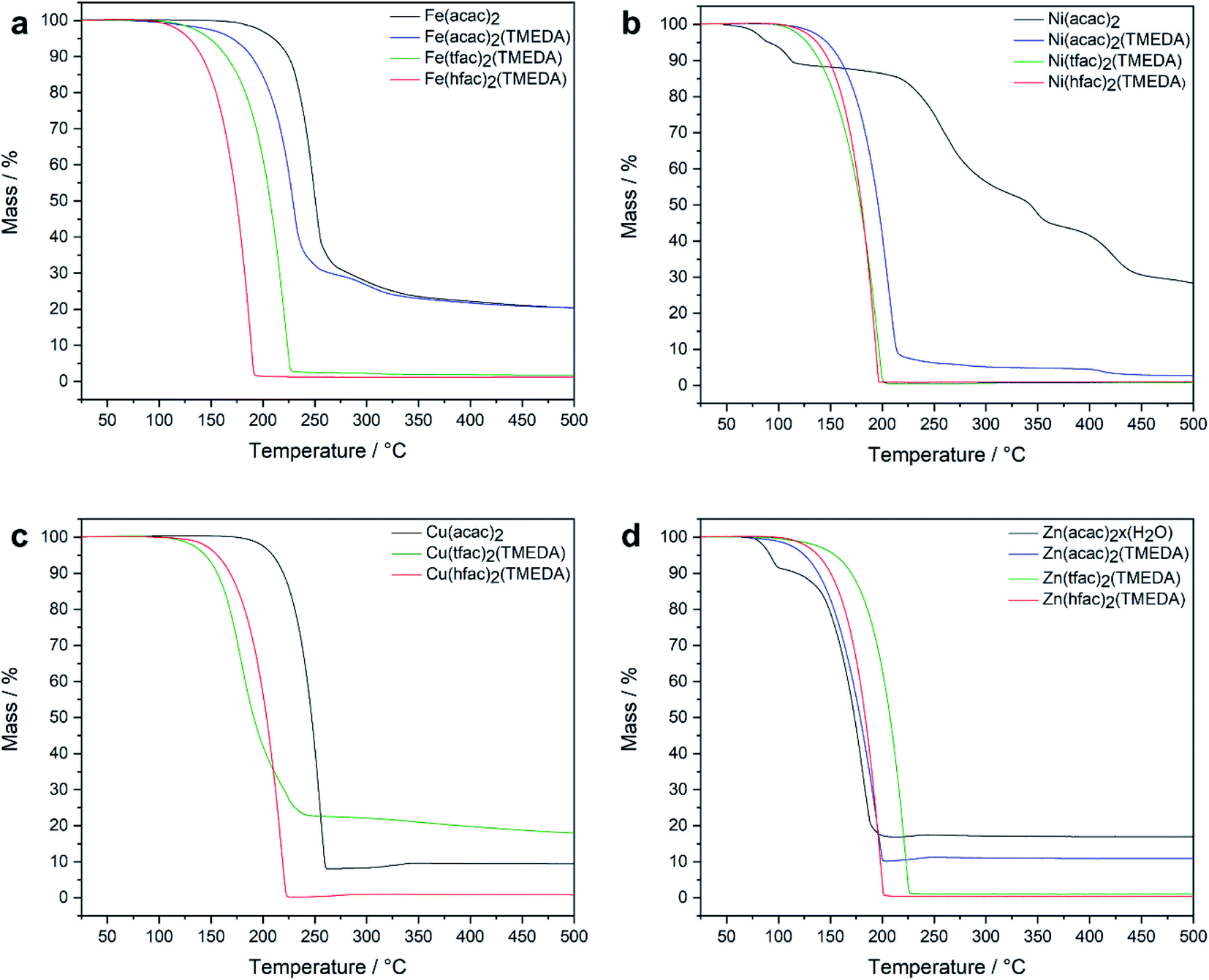 | ||
| Fig. 2 Dynamic TGA curves for [M(acac)2]n, M(acac)2(TMEDA), M(tfac)2(TMEDA) and M(hfac)2(TMEDA) (M = Fe (a), Ni (b), Cu (c) and Zn (d)). | ||
A positive effect of the degree of fluorination on the thermal behaviour of the investigated compounds, but no discernible trends were observed. While the related cobalt complexes Co(acac)2TMEDA, Co(tfac)2TMEDA and Co(hfac)2TMEDA showed in a recent study11 a successive reduction of the on-set temperature of sublimation with increasing degree of fluorination, this is not the case for the investigated complexes with M = Fe, Ni, Cu and Zn.
MOCVD growth
To evaluate the behaviour of 1–4 as MOCVD precursors, deposition studies were performed using a custom-made hot-wall low-pressure reactor (Fig. 3) at 500 °C on Si(001) and Al2O3(0001) substrates with synthetic air (80% N2 and 20% O2), which served as carrier gas and oxidizer. The Si(001) substrate (10·110 mm) was divided into eleven sections and placed into the natural temperature gradient along the reactor axis with substrate temperatures ranging from 300 °C to 500 °C. This method allowed to investigate the influence of different substrate temperatures on the precursor decomposition and film growth in one deposition run. The Al2O3(0001) substrate (10·10 mm) was placed into the isothermal zone in the centre of the furnace. 0.3 mmol precursor were used for each deposition run. 25 sccm dried synthetic air was used as carrier gas and oxidizer. For reasons of clarity, the samples are referred as X_Y where X refers to the precursor and Y refers to the substrate.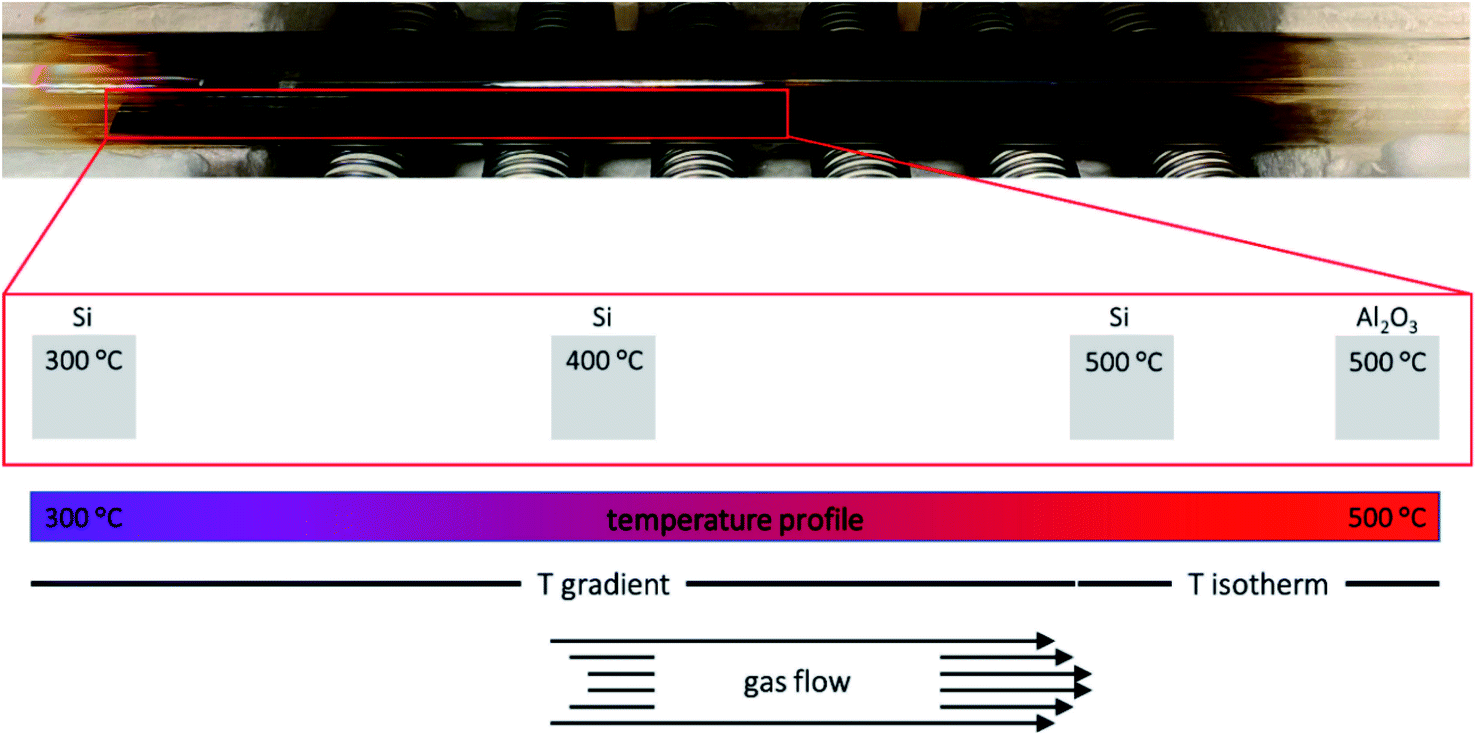 | ||
| Fig. 3 Photograph of the reactor tube after film deposition with 1 at 500 °C. Schematic representation of the temperature profile along the reactor tube and the position of the different substrates inside the MOCVD reactor. | ||
The chemical composition of the resulting material films was analysed by energy-dispersive X-ray spectroscopy (SI, Fig. S23–S26†) showing the expected signals for oxygen (O Kα = 0.53 keV), the transition metals (M Kα = 6.39 keV (Fe), 7.47 keV (Ni), 8.04 keV (Cu), 8.63 keV (Zn)) and aluminium (Al Kα = 1.49 keV) from the substrate. No signal for nitrogen (N Kα = 0.39 keV) or fluorine (F Kα = 0.68 keV) was detected.
| Precursor | Substrate | Temperature | Phase |
|---|---|---|---|
| 1 | Si(001) | 300 °C | Amorphous |
| 1 | Si(001) | 400 °C | Fe3O4 |
| 1 | Si(001) | 500 °C | α-Fe2O3 |
| 2 | Si(001) | 300 °C | Amorphous |
| 2 | Si(001) | 400 °C | Ni |
| 2 | Si(001) | 500 °C | Ni |
| 3 | Si(001) | 300 °C | Amorphous |
| 3 | Si(001) | 400 °C | Cu |
| 3 | Si(001) | 500 °C | Cu |
| 4 | Si(001) | 300 °C | Amorphous |
| 4 | Si(001) | 400 °C | ZnO |
| 4 | Si(001) | 500 °C | ZnO |
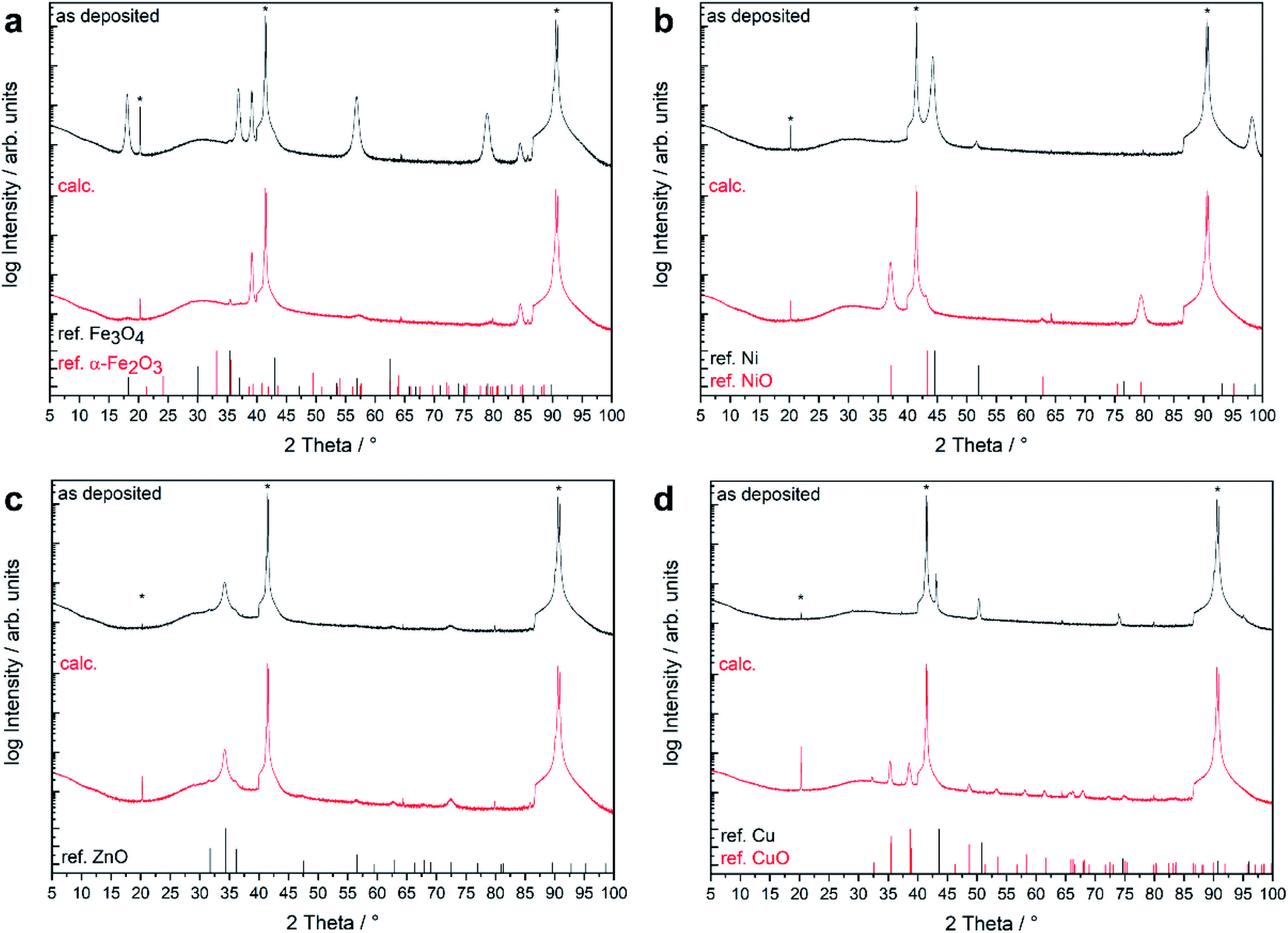 | ||
| Fig. 4 θ–2θ X-ray diffraction pattern of thin films grown on Al2O3(0001) at 500 °C using the different precursors 1–4 before (as deposited) and after calcination (calc.). The vertical lines mark the reference values for the identified crystalline phases. | ||
The native film obtained from 2 showed two distinct reflexes at 44.5 and 51.9° which were assigned to metallic nickel with strong 111 orientation. Reflexes corresponding to other crystalline phases such as NiO or Ni(OH)2 were not observed. The nickel film was converted to phase-pure NiO while maintaining the 111 preferred orientation during calcination.
For the as-deposited film from precursor 3, Bragg reflections corresponding to fcc metallic copper were detected as the only crystalline phase aside from the substrate reflections. The metallic copper film was oxidized into phase-pure CuO during the calcination treatment. Neither the Cu nor the CuO film showed any significant preferential growth. The use of 4 as MOCVD precursor yielded ZnO thin films without the presence of additional crystalline phases. Wurtzite type ZnO adopt the same hexagonal crystal structure as the Al2O3 substrate with a very large lattice mismatch of 19%.21 The ZnO film exhibited a strong (0001)-preferred orientation, as shown from the very dominant Bragg reflections at 2θ = 34.4° (0002) and 72.7° (0004). The XRD pattern was not affected by the calcination treatment (Table 3).
| Precursor | Substrate | Temperature | Phase | Phase after calcination |
|---|---|---|---|---|
| 1 | Al2O3(0001) | 500 °C | Fe3O4(111) | α-Fe2O3(0001) |
| α-Fe2O3(0001) | ||||
| 2 | Al2O3(0001) | 500 °C | Ni(111) | NiO(111) |
| 3 | Al2O3(0001) | 500 °C | Cu | CuO |
| 4 | Al2O3(0001) | 500 °C | ZnO(0001) | ZnO(0001) |
SEM photographs of the MOCVD grown thin films on Al2O3(0001) at 500 °C using precursors 1–4 are displayed in Fig. 5. The iron oxide film 1_Al grown from precursor 1 consisted of densely packed particles. The smooth film is overgrown with angular particles, many of which were identified as octahedra. The 2_Al film exhibited a metallic lustre. According to the XRD pattern, the deposited film consisted of phase-pure metallic Ni. The surface morphology of this film (Fig. 5b) is made of ∼100 nm sized irregular shaped particles. The 3_Al film showed a dull surface without metallic lustre. The SEM photograph (Fig. 5c) showed a very rough surface morphology consisting of interconnected structures up to 1 μm in size which are built up from visibly smaller particles. The SEM photograph of the surface morphology of the 4_Al film (Fig. 5d) showed that the film consists of densely packed and uniform nanosized (50 nm) grains. Aside from the granular surface morphology, the formation of 500 nm long nanowires measuring ∼50 nm in diameter was observed. The grow of nanowires and columnar grains during the MOCVD growth of ZnO was previously reported by several authors.25 Cross section photographs of the thin films grown on Al2O3 substrates are available in the ESI (Fig. S32–S36†).
 | ||
| Fig. 5 SEM photographs of thin films grown on Al2O3(0001) at 500 °C using the different precursors 1–4. | ||
To gain deeper insight into the surface composition and the valence states of the respective elements, X-ray photoelectron spectroscopy (XPS) (Fig. 6) was performed on the MOCVD grown thin films in their native state and after calcination. C 1s signals were observed for all samples. The binding energies of these signals are associated with C–C bond (285 eV) and C–O bond carbon (288 eV). This is the typically signature of adventitious carbon and found on all surfaces handled at ambient conditions. The intensity of the C 1s signal decreased strongly after calcination due to the removal of near-surface contaminations and decomposition products of the acetylacetonate ligand. No F 1s signals were found. The Fe 2p high-resolution spectra consisted of two main Fe 2p3/2 and Fe 2p1/2 peaks located at 710.9 and 724.3 eV with a binding energy of 13.4 eV, which confirmed the presence of iron in a +III oxidation state. This assignment is further supported by the characteristic satellite peaks of Fe3+ at 718.4 and 724.9 eV. The Ni 2p spectra for the as-deposited film showed the characteristic signature of metallic nickel with the Ni 2p3/2 peak located at 853.0 eV and the Ni 2p1/2 peak at 870.2 eV. After calcination, the Ni 2p3/2 and Ni 2p1/2 peaks are shifted to higher binding energies (853.7 and 873.3 eV) and show multiplet splitting, yet the peak position and shape of the Ni 2p signal are in good agreement with the reported values of NiO. The Cu 2p spectra of the native film exhibited peaks at 933.0 and 952.8 eV and no satellite signals at around 945 eV, which excludes the presence of CuO. From the position of the Cu 2p signal and the Cu LMM Auger signal at 916.4 eV (ESI, Fig. S41†) a modified Auger parameter of 1849.4 eV can be calculated, which is in good agreement with the reported reference value for Cu2O.22 In contrast, a shift to higher binding energies and peak broadening of the Cu 2p signals can be observed for the oxidized film. The satellite signals along with multiplet splitting at 945.5 eV indicate the formation of CuO as a result of calcination, which can also be observed in the XRD pattern.
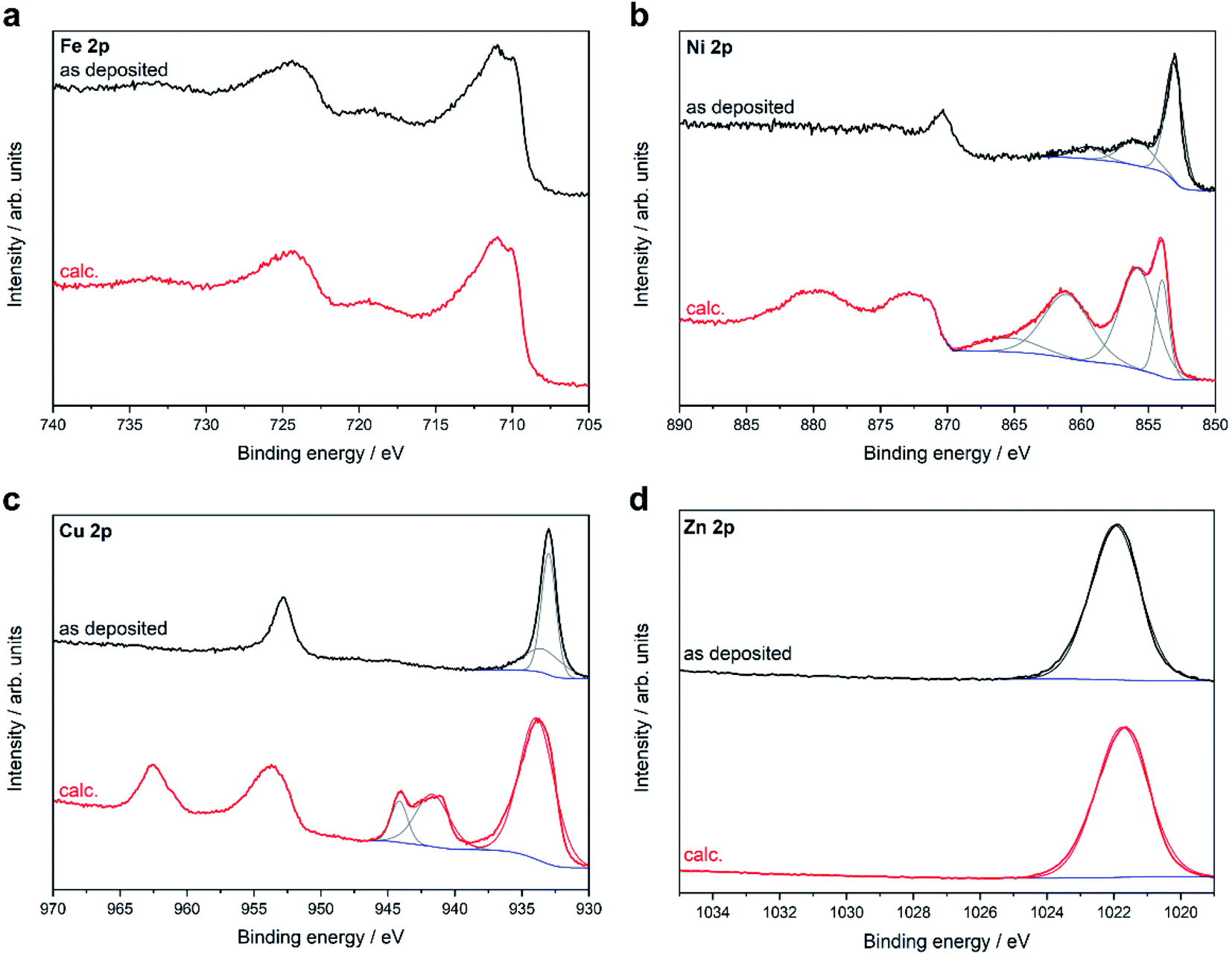 | ||
| Fig. 6 XPS core level metal 2p spectra of thin films grown on Al2O3(0001) at 500 °C using the different precursors 1–4 before (as deposited) and after calcination (calc.). | ||
The high-resolution Zn 2p spectrum shows the Zn 2p3/2 peak at 1021.5 eV. The Zn 2p spectra show no changes in the peak position or peak shape after calcination, which is expected since the Zn 2p signal is insensitive to different zinc species and does not show any characteristic satellite peaks. By calculating the modified Auger parameter (ESI, Fig. S44†), the presence of ZnO was confirmed in good agreement with the XRD results in both the native and calcined films. The O 1s signal was deconvoluted into three peaks. The peak at low binding energy originates from lattice oxygen, whereas the peak at slightly higher binding energy is related to oxygen vacancies or defects. The peak at high binding energy corresponds to chemi- or physiosorbed oxygen species (ESI, Fig S42†).
Conclusions
The fluorinated transition metal β-diketonate complexes M(tfac)2(TMEDA) (M = Fe 1, Ni 2, Cu 3, Zn 4) were synthesized and structurally characterized by single crystal X-ray diffraction. The thermochemical properties (TGA) of the trifluoroacetylacetonate complexes were studied and compared with those of the non-fluorinated acetylacetonate complexes M(acac)2(TMEDA) and the higher fluorinated complexes M(hfac)2(TMEDA). In the series M(acac)2(TMEDA)–M(ftac)2(TMEDA)–M(hfac)2(TMEDA), a successive decrease in evaporation temperature with increasing degree of fluorination was observed only for M = Fe, while no clear trend could be derived for M = Ni, Cu, Zn. 1, 2 and 4 are suitable MOCVD precursors, whereas complex 3 shows signs of decomposition upon heating under low vacuum conditions, which limits the use of 3 as a MOCVD precursor.1–4 were employed as precursors for the growth of thin films on Al2O3(0001) in the presence of oxygen as the carrier gas, resulting in the growth of highly oriented metal (Ni(111), Cu) or metal oxide thin films (α-Fe2O3(0001)/Fe3O4(111), ZnO(0001)). The material films were further oxidized to NiO, CuO and·α-Fe2O3 films by post deposition calcination in air at 500 °C. The deposition of Ni and Cu films usually requires the presence of hydrogen. 2 and 3 are rare examples of precursors that allow the deposition of metallic Ni and Cu films under oxidizing conditions.
For the MOCVD growth of complex materials containing more than one metal, such as multimetallic oxides like CoFe2O4, suitable precursors are required whose thermal properties can be adjusted to each other so that they evaporate simultaneously under the given conditions. The growth of complex oxides from these precursors is currently under investigation.
Experimental
Synthetic procedures, materials and methods
1,1,1-Trifluoro-2,4-pentanedione (Htfac) and N,N,N′,N′-tetramethylethylenediamine (TMEDA) were obtained from abcr and used as received without further purification.Synthesis of M(tfac)2(TMEDA) (M = Fe 1, Ni 2, Cu 3, Zn 4)
1–4 were synthesized according to a previously reported synthesis protocol for Co(tfac)2(TMEDA) with minor modifications.112.8 mL (23.3 mmol) of Htfac was added dropwise to a solution of 0.94 g NaOH in 50 mL deionized water and stirred for 20 min 11.73 mmol of the corresponding metal salt (2.33 g FeCl2·4H2O, 2.79 g NiCl2·6H2O, 2.00 g CuCl2·2H2O, 1.60 g ZnCl2) was dissolved separately in 75 mL water and then added dropwise to the previously prepared aqueous solution of Na(tfac). The reaction mixture was stirred for another 30 min and then 1.8 mL (11.93 mmol) of TMEDA was added. After 12 h reaction time, the reaction mixture was repeatedly extracted with dichloromethane until the aqueous phase is decolorized. The solvent was removed from the combined organic phases using a rotary evaporator and the resulting solid was purified by evaporation (1 × 10−3 mbar, 100 °C). For single crystal X-ray structural analysis, the product was dissolved in dichloromethane and the solvent was allowed to evaporate slowly at room temperature.
1H-, 13C- and 19F-NMR data of 4 are available in the experimental section. In contrast, 1–3 showed complex spectra with broad signals which could not be clearly assigned. Not all expected signals could be observed in the range from −10 to 20 ppm. No signals were found in the 13C and 19F NMR spectra of 1–3 in a range of −15 to 215 ppm and −30 to −130 ppm, respectively.
1 Fe(tfac)2(TMEDA): M = 478.21 g mol−1. Yield: 4.85 g (10.14 mmol/87%). Mp. 86.7 °C (by DSC). Anal. calcd for C16H24F6N2FeO4: C 40.2%, H 5.1% N 5.9% Fe 11.7%. Found: C 40.1, H 4.8%, N 5.8%, Fe 11.8%. IR (ATR): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 2865, 1617, 1549, 1497, 1453, 1436, 1400, 1354, 1275, 1166, 1121, 1022, 948, 847, 791, 774, 723, 560 cm−1.
= 2865, 1617, 1549, 1497, 1453, 1436, 1400, 1354, 1275, 1166, 1121, 1022, 948, 847, 791, 774, 723, 560 cm−1.
2 Ni(tfac)2(TMEDA): M = 481.06 g mol−1. Yield: 5.21 g (10.83 mmol/93%). Mp. 81.3 °C (by DSC). Anal. calcd for C16H24F6N2NiO4: C 40.0%, H 5.0% N 5.8% Ni 12.2%. Found: C 40.1, H 5.0%, N 6.1%, Ni 12.0%. IR (ATR): = 2883, 1623, 1537, 1504, 1454, 1436, 1400, 1354, 1274, 1166, 1119, 1020, 950, 850, 799, 767, 724, 571 cm−1.
3 Cu(tfac)2(TMEDA): M = 485.91 g mol−1. Yield: 5.04 g (10.37 mmol/89%). Mp. 93.2 °C (by DSC). Anal. calcd for C16H24F6N2CuO4: C 39.6%, H 5.0% N 5.8% Cu 13.1%. Found: C 39.6, H 4.7%, N 5.7%, Cu 13.6%. IR (ATR): = 2885, 1633, 1516, 1457, 1434, 1404, 1354, 1270, 1164, 1118, 1019, 950, 847, 806, 766, 721, 554 cm−1.
4 Zn(tfac)2(TMEDA): M = 487.75 g mol−1. Yield: 5.23 g (10.72 mmol/92%). Mp. 85.2 °C (by DSC). Anal. calcd for C16H24F6N2ZnO4: C 39.4%, H 5.0% N 5.7% Zn 13.4%. Found: C 39.5, H 5.1%, N 5.6%, Zn 13.0%. IR (ATR): = 2867, 1623, 1543, 1497, 1454, 1436, 1401, 1353, 1274, 1166, 1119, 1022, 948, 849, 796, 769, 723, 564 cm−1. 1H NMR: (300 MHz, CDCl3) δ = 1.95 (s, 6H, CH3), 2.33 (s, 12H, CH3), 2.58 (s, 4H, CH2), 5.49 (s, 2H, CH). 13C{1H} NMR:29.3, 47.0 56.4, 93.8, 99.6. 19F{1H} NMR: −76.4 (s, CF3).
Film deposition
The MOCVD deposition experiments were performed in a custom-made hot-wall low-pressure MOCVD reactor (Fig. S18†). 300 mg of the precursor were weighed into a ceramic boat and placed in the evaporation zone of a quartz tube heated by a heating tape. The evaporation temperature was chosen as 100 °C for all precursors. Dried synthetic air (25 sccm) was used as carrier gas and oxidizer. Cleaned Si(001) and Al2O3(0001) was used as substrate and placed in the isothermal deposition zone of the quartz tube, which was heated by a tube furnace. Material films were deposited within 20 min at a working pressure of 0.35 mbar and a substrate temperature of 500 °C. After the film deposition was complete, the system was slowly cooled to ambient temperature within 3 h under vacuum.For the post-deposition calcination, the films were heated to 500 °C with a heating rate of 500 K h−1. After 3 h at 500 °C the films were slowly cooled to ambient temperature.
Characterization
Crystals were mounted on nylon loops in inert oil for single-crystal X-ray analyses. The data was collected on a Bruker AXS D8 Kappa diffractometer with APEX2 detector (mono-chromated Mo Kα radiation, λ = 0.71073 Å) at 100(2) K. Details regarding the structure solution are available in the ESI.†CCDC-2099616 (1), −2099619 (2), −2099618 (3) and −2099617 (4), contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/data_request/cif.
XRD patterns were measured using a Bruker D8 Advance powder diffractometer with Cu Kα radiation with λ = 1.5418 Å in the range of 2θ = 5–100° with a step size of 0.01° and a counting time of 0.6 s. For the XRD measurements of 1–4, powdered samples were dispersed with water on a Si single crystal.
X-ray photoelectron spectroscopy was performed using a Versaprobe II™ from Ulvac-Phi with monochromatic Al Kα light at 1486.6 eV photon energy. The emission angle between analyser and sample was 45°. The C 1s signal at 284.8 eV binding energy was used as the binding energy reference.
The surface morphology of the thin films was investigated by scanning electron microscope (SEM) using an Apreo S LoVac equipped with an energy dispersive X-ray spectroscopy (EDX) device. For the cross-sectional photographs, the thin films were mechanically fragmented and sputtered with 5 nm Pd.
XP spectra of the thin films were recorded without any pre-treatment. Surface charging effects were corrected by adjusting the binding energy of the adventitious C 1s core level signal to 284.8 eV. CasaXPS software version 2.3.19 (Casa Software Ltd) was used for peak fitting by applying a Shirley background and Gaussian-Lorenzian peaks. The Handbook of X-ray Photoelectron Spectroscopy26 was used for the identification of the oxidation states of the different transition metals.
IR spectra were recorded with an ALPHA-II FTIR spectrometer equipped with a single-reflection ATR sampling module. Crystals of 1–4 were grinded into powders before the measurement.
TGA measurements were performed using a TGA/DSC 1 Star System from Mettler Toledo with 9.5 mg substance for each run. A heating rate of 5 K min−1 were used for dynamic measurement in the temperature range from 25–500 °C. Argon was used as carrier gas with a flow rate of 20 mL min−1. Static TGA measurements were done at 125 °C.
Elemental analyses were performed using a EURO EA Elemental Analyzer (Euro Vector). The metal content was determined by atomic absorption spectroscopy using a M-Series spectrometer from Thermo Electron.
1H, 13C NMR and 19F spectra were recorded at 297 K in CDCl3 solution using a Bruker Avance 300 spectrometer (1H: 300 MHz, 13C: 75 MHz, 19F: 75 MHz). 1H and 13C NMR spectra were referenced to internal CDCl3 (1H = 7.26 ppm, 13C = 77.16 ppm).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
GB is thankful for funding by the “Programm zur Förderung des exzellenten wissenschaftlichen Nachwuchses” from the University Duisburg-Essen. Support by the Interdisciplinary Center for Analytics on the Nanoscale (ICAN) of the University of Duisburg-Essen, a DFG funded core facility (DFG RIsources reference: RI_00313), is gratefully acknowledged (XPS measurements, service operation).Notes and references
- Y. C. Tsai, Coord. Chem. Rev., 2012, 256, 722–758 CrossRef CAS.
- (a) G. Bendt, S. Saddeler and S. Schulz, Eur. J. Inorg. Chem., 2019, 602–608 CrossRef CAS; (b) K. Chakrapani, G. Bendt, H. Hajiyani, I. Schwarzrock, T. Lunkenbein, S. Salamon, J. Landers, H. Wende, R. Schlögl, R. Pentcheva, M. Behrens and S. Schulz, ChemCatChem, 2017, 9, 2988–2995 CrossRef CAS; (c) K. Chakrapani, G. Bendt, H. Hajiyani, T. Lunkenbein, M. T. Greiner, L. Masliuk, S. Salamon, J. Landers, R. Schlögl, H. Wende, R. Pentcheva, S. Schulz and M. Behrens, ACS Catal., 2018, 8, 1259–1267 CrossRef CAS; (d) S. Saddeler, U. Hagemann and S. Schulz, Inorg. Chem., 2020, 59, 10013–10024 CrossRef CAS PubMed; (e) S. Saddeler, G. Bendt, S. Salamon, F. T. Haase, J. Landers, J. Timoshenko, C. Rettenmaier, H. S. Jeon, A. Bergmann, H. Wende, B. Roldan Cuenya and S. Schulz, J. Mater. Chem. A, 2021, 9, 25381–25390 RSC; (f) R. K. Sodhi and S. Paul, Catal. Surv. Asia, 2018, 22, 31–62 CrossRef CAS.
- R. A. Lalancette, D. Syzdek, J. Grebowicz, E. Arslan and I. Bernal, J. Therm. Anal. Calorim., 2019, 135, 3463–3470 CrossRef CAS.
- G. J. Bullen, R. Mason and P. Pauling, Nature, 1961, 189, 291–292 CrossRef CAS.
- (a) F. A. Cotton and R. C. Elder, J. Am. Chem. Soc., 1964, 86, 2294–2295 CrossRef CAS; (b) V. D. Vreshch, J. H. Yang, H. Zhang, A. S. Filatov and E. V. Dikarev, Inorg. Chem., 2010, 49, 8430–8434 CrossRef CAS PubMed.
- P. M. Kouotou and Z. Y. Tian, Phys. Status Solidi A, 2015, 212, 1508–1513 CrossRef CAS.
- G. F. Pan, S. Bin Fan, J. Liang, Y. X. Liu and Z. Y. Tian, RSC Adv., 2015, 5, 42477–42481 RSC.
- J. P. Sénateur, C. Dubourdieu, F. Weiss, M. Rosina and A. Abrutis, Adv. Funct. Mater., 2000, 10, 155–161 Search PubMed.
- (a) J. H. Halz, C. Heiser, C. Wagner and K. Merzweiler, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2020, 76, 66–71 CrossRef CAS PubMed; (b) A. Zeller, E. Herdtweck and T. Strassner, Inorg. Chem. Commun., 2004, 7, 296–301 CrossRef CAS; (c) S. Pasko, L. G. Hubert-Pfalzgraf, A. Abrutis and J. Vaissermann, Polyhedron, 2004, 23, 735–741 CrossRef CAS.
- Y. Zhang, L. Du, X. Liu and Y. Ding, Nanoscale, 2019, 11, 3484–3488 RSC.
- C. Stienen, D. Rogalla, O. Prymak and G. Bendt, Eur. J. Inorg. Chem., 2021, 4298–4306 CrossRef CAS.
- (a) L. Bigiani, M. Hassan, D. Peddis, C. Maccato, G. Varvaro, C. Sada, E. Bontempi, S. Martí-Sánchez, J. Arbiol and D. Barreca, ACS Appl. Nano Mater., 2019, 2, 1704–1712 CrossRef CAS; (b) M. E. A. Warwick, D. Barreca, E. Bontempi, G. Carraro, A. Gasparotto, C. Maccato, K. Kaunisto, T. P. Ruoko, H. Lemmetyinen, C. Sada, Y. Gönüllü and S. Mathur, Phys. Chem. Chem. Phys., 2015, 17, 12899–12907 RSC; (c) N. A. Bailey, D. E. Fenton and M. S. Leal Gonzalez, Inorg. Chim. Acta, 1984, 88, 125–134 CrossRef CAS; (d) C. I. M. A. Spee, H. L. Linden, A. Mackor, K. Timmer and H. A. Meinema, Mater. Res. Soc. Symp. Proc., 1996, 415, 93–98 CrossRef CAS; (e) J. Ni, H. Yan, A. Wang, Y. Yang, C. L. Stern, A. W. Metz, S. Jin, L. Wang, T. J. Marks, J. R. Ireland and C. R. Kannewurf, J. Am. Chem. Soc., 2005, 127, 5613–5624 CrossRef CAS PubMed; (f) S. Delgado, A. Muñoz, M. E. Medina and C. J. Pastor, Inorg. Chim. Acta, 2006, 359, 109–117 CrossRef CAS; (g) D. Barreca, G. Carraro, A. Gasparotto, C. Maccato, O. I. Lebedev, A. Parfenova, S. Turner, E. Tondello and G. Van Tendeloo, Langmuir, 2011, 27, 6409–6417 CrossRef CAS PubMed.
- D. Barreca, G. Carraro, E. Fois, A. Gasparotto, F. Gri, R. Seraglia, M. Wilken, A. Venzo, A. Devi, G. Tabacchi and C. Maccato, J. Phys. Chem. C, 2018, 122, 1367–1375 CrossRef CAS.
- M. Klotzsche, D. Barreca, L. Bigiani, R. Seraglia, A. Gasparotto, L. Vanin, C. Jandl, A. Pöthig, M. Roverso, S. Bogialli, G. Tabacchi, E. Fois, E. Callone, S. Dirèe and C. Maccato, Dalton Trans., 2021, 50, 10374–10385 RSC.
- D. Barreca, L. Bigiani, M. Klotzsche, A. Gasparotto, R. Seraglia, C. Jandl, A. Pöthig, E. Fois, L. Vanin, G. Tabacchi, M. Roverso, S. Bogialli, E. Callone, S. Dirè and C. Maccato, Mater. Chem. Phys., 2022, 277, 125534 CrossRef CAS.
- (a) A. Gasparotto, D. Barreca, D. Bekermann, A. Devi, R. A. Fischer, P. Fornasiero, V. Gombac, O. I. Lebedev, C. MacCato, T. Montini, G. Van Tendeloo and E. Tondello, J. Am. Chem. Soc., 2011, 133, 19362–19365 CrossRef CAS PubMed; (b) G. Carraro, A. Gasparotto, C. Maccato, E. Bontempi, O. I. Lebedev, S. Turner, C. Sada, L. E. Depero, G. Van Tendeloo and D. Barreca, RSC Adv., 2013, 3, 23762–23768 RSC.
- S. Mishra and S. Daniele, Chem. Rev., 2015, 115, 8379–8448 CrossRef CAS PubMed.
- (a) S. Delgado, A. Muñoz, M. E. Medina and C. J. Pastor, Inorg. Chim. Acta, 2006, 359, 109–117 CrossRef CAS; (b) G. Bandoli, D. Barreca, A. Gasparotto, R. Seraglia, E. Tondello, A. Devi, R. A. Fischer, M. Winter, E. Fois, A. Gamba and G. Tabacchi, Phys. Chem. Chem. Phys., 2009, 11, 5998–6007 RSC.
- (a) E. S. Vikulova, K. V. Zherikova, D. A. Piryazev, I. V. Korol'kov, N. B. Morozova and I. K. Igumenov, J. Struct. Chem., 2017, 58, 1681–1684 CrossRef CAS; (b) A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- A. Zeller, E. Herdtweck and T. Strassner, Inorg. Chem. Commun., 2004, 7, 296–301 CrossRef CAS.
- P. Fons, K. Iwata, S. Niki, A. Yamada and K. Matsubara, J. Cryst. Growth, 1999, 201, 627–632 CrossRef.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- D. Barreca, G. Carraro, A. Devi, E. Fois, A. Gasparotto, R. Seraglia, C. Maccato, C. Sada, G. Tabacchi, E. Tondello, A. Venzo and M. Winter, Dalton Trans., 2012, 41, 149–155 RSC.
- C. M. Tian, W. W. Li, Y. M. Lin, Z. Z. Yang, L. Wang, Y. G. Du, H. Y. Xiao, L. Qiao, J. Y. Zhang, L. Chen, J. L. MacManus-Driscoll and K. H. L. Zhang, J. Phys. Chem. C, 2020, 124, 12548–12558 CrossRef CAS.
- (a) G. Perillat-Merceroz, H. Jouneau, G. Feuillet, R. Thierry, M. Rosina and P. Ferret, J. Phys.: Conf. Ser., 2010, 209, 012034 CrossRef; (b) S. Muthukumar, H. Sheng, Z. Zhang, J. Zhong, N. W. Emanaetoglu and Y. Lu, IEEE Trans. Nanotechnol., 2002, 2, 50–54 CrossRef.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Eden Prairie, Perkin-Elmer Corp., 1992 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2099616–2099619. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra01338j |
| This journal is © The Royal Society of Chemistry 2022 |