
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox behavior of potassium doped and transition metal co-doped Ce0.75Zr0.25O2 for thermochemical H2O/CO2 splitting†
Maria Portarapilloa,
Gianluca Landi *b,
Giuseppina Luciani*a,
Claudio Imparatoa,
Giuseppe Vitielloac,
Fabio A. Deorsolad,
Antonio Aronnea and
Almerinda Di Benedettoa
*b,
Giuseppina Luciani*a,
Claudio Imparatoa,
Giuseppe Vitielloac,
Fabio A. Deorsolad,
Antonio Aronnea and
Almerinda Di Benedettoa
aDipartimento di Ingegneria Chimica, dei Materiali e della Produzione Industriale, Univ. of Naples Federico II, P.le Tecchio 80, 80125, Naples, Italy. E-mail: luciani@unina.it
bInstitute of Sciences and Technologies for Sustainable Energy and Mobility, CNR, P.le Tecchio 80, 80125, Naples, Italy. E-mail: gianluca.landi@cnr.it
cCSGI, Center for Colloids and Surface Science, 50019, Sesto Fiorentino (FI), Italy
dDepartment of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Turin, Italy
First published on 16th May 2022
Abstract
CeO2 slow redox kinetics as well as low oxygen exchange ability limit its application as a catalyst in solar thermochemical two-step cycles. In this study, Ce0.75Zr0.25O2 catalysts doped with potassium or transition metals (Cu, Mn, Fe), as well as co-doped materials were synthesized. Samples were investigated by X-ray diffraction (XRD), N2 sorption (BET), as well as by electron paramagnetic resonance (EPR) and X-ray photoelectron spectroscopy (XPS) to gain insight into surface and bulk features, which were connected to redox properties assessed both in a thermogravimetric (TG) balance and in a fixed bed reactor. Obtained results revealed that doping as well as co-doping with non-reducible K cations promoted the increase of both surface and bulk oxygen vacancies. Accordingly, K-doped and Fe-K co-doped materials show the best redox performances evidencing the highest reduction degree, the largest H2 amounts and the fastest kinetics, thus emerging as very interesting materials for solar thermochemical splitting cycles.
Introduction
Solar thermochemical splitting cycles have been gaining scientific and technological relevance as an environmentally sustainable route to produce synthetic fuels from H2O and CO2. Actually, they enable straight solar energy harvesting and conversion into synthesis gas (syngas), a CO/H2 mixture, that can be further processed to gaseous or liquid fuels.1–5Among available technologies, two-step thermochemical redox cycles based on non-stoichiometric metal oxides (MOx) hold huge promise, exhibiting the lowest complexity as well as the highest theoretical solar-to-fuel energy conversion efficiency (ηsolar-to-fuel) as a result of the use of the whole solar spectrum.
In the first step of a typical red-ox cycle, MOx undergoes high temperature self-reduction to MO(x−δ), according to the following reaction:
| MOx → MO(x−δ)+ δ/2O2 |
Then, in the second step, MO(x−δ) is oxidized by CO2 and/or H2O to form CO and/or H2 as described by the following equations:
| MO(x−δ) + δCO2 → MOx + δCO |
| MO(x−δ) + δH2O → MOx + δH2 |
Ceria (CeO2) is considered the most attractive material for these processes, combining good thermal stability, high oxygen storage capacity (OSC) without any structural changes as well as faster reduction and splitting kinetics than other oxides.1,6 Notably, CeO2 based materials, particularly doped samples, often can outperform perovskites, affording higher solar-to-fuel efficiency above all in the CO2 splitting reaction.7
However, several major drawbacks still prevent their technological application. These include the limited amount of oxygen vacancies as well as high reduction temperature, which involves poor cyclability due to structural modifications.
Partial substitution of Ce(IV) species and doping with metal cations in the fluorite structure of ceria is a successful strategy to address these issues, enhancing the redox activity of the material.8–19
Density-functional-theory (DFT) calculations13 support experimental results20 indicating that Ce(IV) substitution by Zr(IV) ions could improve ceria splitting properties and particularly its self-reduction behavior. Among the investigated compositions, Ce0.75Zr0.25O2 showed the best catalytic cyclability.21–25 Moreover, bi- or trivalent cations doping, including Cu(II), Mn(III), Fe(III), can improve both self-reducibility and splitting activity of bare CeO2.16–18 Notably, Terribile et al. (1999) analyzed the reduction and catalytic behavior of Mn- and Cu-doped ceria–zirconia solid solutions. They found that manganese and copper substitution created ionic defects in ceria fluorite structure, by a charge compensation mechanism, which promoted low-temperature redox activity, involving reduction of Cu2+ to Cu0 and of Mn3+/4+ to Mn2+.26
Recently, Takalkar et al. (2020) showed that doping with alkali (Li), alkaline earth (Mg, Ca, Sr, Ba), and post-transition (Sn) metal improved both O2 release and CO production during CO2 red-ox splitting cycles, due to the formation of mixed phases neglected by the DFT calculations.27 Moreover, Ruan et al. (2017) reported that the formation of Ce2Sn2O7 pyrochlore phase in CeO2–SnO solid solution, boosted both self-reduction and splitting activity.28 Similarly, according to Pappacena et al. (2016 and 2017), the formation of nanostructured Zr2ON2-like phase in Ce0.15Zr0.85O2 promoted splitting.29,30
An intimate mixing at the nanoscale of two different active sites involved in oxygen evolution and splitting reactions has been emerging as successful strategy for the design of high-performance catalysts.31,32
In order to improve ceria thermochemical red-ox performance, Charvin et al. (2009) proposed a three-step cycle.33 After the usual solar-driven self-reduction step, the process involved reaction between reduced oxide and an alkali hydroxide (NaOH or KOH), producing hydrogen and a mixed oxide. Its subsequent hydrolysis regenerated the starting oxide and the alkali hydroxide. Furthermore, potassium cation doping has been reported to improve perovskite-based catalysts performance towards water gas shift (WGS) reaction.34 Moreover, for the same process, Zugic et al. (2014) suggested that the addition of the electropositive alkali metal cations promoted the formation of active –OH species, thus increasing adsorption active sites number with high thermal stability.35
Inspired by literature data, in this study, we investigated the effect of K(I) doping on Ce0.75Zr0.25O2 (CeZr) red-ox activity towards both CO2 and H2O splitting. K was also investigated as co-dopant with transition metal cations (M = Cu, Mn, Fe) which were found to improve CeZr thermochemical performance and were also used as reference to compare the performances of K-doped samples. Prepared materials were submitted to in-depth physical-chemical characterization using different techniques. Furthermore, thermogravimetric analysis (TGA) was carried out to assess redox activity towards CO2 splitting during repeated cycles, whereas flow reactor tests were performed to investigate catalytic activity towards H2O splitting. Particularly, the outcomes of catalytic experiments were combined with XPS and EPR spectroscopy results, providing key information on the molecular features underlying redox behaviour, thus drawing strategic guidelines for the design of high-performance catalysts for thermochemical splitting cycles.
Experimental section
Materials
The following reagents were employed for the catalyst synthesis: cerium(III) nitrate hexahydrate, Ce(NO3)3·6H2O; zirconium(IV) oxynitrate hydrate, ZrO(NO3)2·xH2O; iron(III) nitrate nonahydrate, Fe(NO3)3·9H2O; potassium nitrate, KNO3; copper(II) nitrate tetrahydrate, Cu(NO3)2·4H2O; manganese(II) nitrate tetrahydrate, Mn(NO3)2·4H2O. All products were purchased from Sigma-Aldrich and used as received.Catalyst synthesis
To achieve high reduction behaviour, Ce/Zr molar ratio was kept constant at 3 (Ce0.75Zr0.25O2 as general formula), since it ensured high reducible behaviour.36Bare ceria–zirconia, M-doped (M = Cu, Fe, Mn) and K-doped ceria–zirconia materials, as well as co-doped samples were prepared according to the co-precipitation method described elsewhere.32
Briefly, stoichiometric amounts of cerium and zirconium precursors were dissolved in 75 ml bi-distilled water and then stoichiometric quantity of the doping metal was added and stirred for 3 h. Afterwards, solutions were heated in a microwave oven (CEM SAM-155) and the resulting gel was calcined in air at 1100 °C for 4 h. Table 1 reports obtained material with their nominal and ICP measured compositions as well as the label used thereof, whereas Fig. S1† shows the co-precipitation synthesis steps of the doped ceria materials.
| Sample | Theoretical | ICP | General formula | nO2,max | |||
|---|---|---|---|---|---|---|---|
| M/(Ce + Zr) | K/(Ce + Zr) | M/(Ce + Zr) | K/(Ce + Zr) | Ce/Zr | |||
| CeZr | — | — | 3.02 | Ce0.75Zr0.25O2 | 1.17 | ||
| Fe-CeZr | 0.05 | — | 0.0523 | 3.14 | Fe0.05Ce0.71Zr0.24O1.98 | 1.22 | |
| Mn-CeZr | 0.05 | — | 0.0479 | 2.97 | Mn0.05Ce0.71Zr0.24O1.98 | 1.29 | |
| Cu-CeZr | 0.05 | — | 0.0521 | 2.89 | Cu0.05Ce0.71Zr0.24O1.95 | 1.3 | |
| K-CeZr | — | 0.05 | 0.0518 | 3.11 | K0.05Ce0.71Zr0.24O1.93 | 1.16 | |
| K-Fe-CeZr | 0.05 | 0.05 | 0.0485 | 0.0522 | 3.05 | K0.045Fe0.045Ce0.68Zr0.23O1.91 | 1.2 |
| K-Cu-CeZr | 0.05 | 0.05 | 0.0509 | 0.0488 | 2.94 | K0.045Cu0.045Ce0.68Zr0.23O1.88 | 1.28 |
Materials characterization
ICP-MS analysis (Agilent 7500CE instrument) was used to assess the actual composition of the samples. The measured compositions differed from the nominal ones within the experimental error (±5%).X-ray diffraction (XRD) measurements were performed with a PANalytical X'Pert Pro XRD diffractometer (step size: 0.02°; counting time: 80 s per step). Scherrer equation was employed to calculate the average crystal size (τ):
 | (1) |
nλ = 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(θ) sin(θ)
| (2) |
Interplanar spacing values were employed to evaluate the cell parameter (a), using the equation:
 | (3) |
N2 adsorption at 77 K with a Quantachrome Autosorb-1C instrument was employed to measure BET specific surface areas (SSA); samples were degassed at 150 °C for 1.5 h before each measurement.
X-ray photoelectron spectroscopy (XPS; XPS PHI 5000 Versa probe instrument) analyses were performed on both pristine and used samples. The band-pass energy, the take-off angle, and the X-ray spot size for survey spectra were set at 187.85 eV, 45°, and 100.0 μm diameter respectively. High-resolution XPS spectra were recorded using the following experimental conditions: pass energy 20 eV, resolution 1.1 eV and step 0.2 eV. The C 1s line with a binding energy (BE) value of 284.8 eV was used as internal reference in order to compensate sample charging effects. XP-spectra were deconvoluted by means of a commercial software (CasaXPS, version 2.3.17), using mixed Gaussian–Lorentzian (70–30%) profiles.
Electron paramagnetic resonance (EPR; X-band (9 GHz) Bruker Elexys E-500 spectrometer (Bruker, Rheinstetten, Germany), equipped with a super-high sensitivity probe head) measurements were carried out on selected as-prepared and used samples. Solid samples were placed into flame-sealed glass capillaries which, in turn, were coaxially put into a standard 3 mm quartz sample tube. EPR spectra were recorded with an attenuation of 15 dB and by accumulating 128 scans to improve the signal-to-noise ratio. Details of the measurements settings are reported elsewhere.37 A quantitative analysis was performed by determining the g-factor and ΔB values, as previously described.37
Thermochemical CO2 splitting tests
Thermochemical CO2 splitting cycles were carried out in a TGA/DSC apparatus (TA Q600SDT), according to the procedure reported elsewhere32 and briefly described hereafter. Fine powdered samples (about 30 mg) were put into an alumina crucible, first pre-treated in air at 1200 °C (heating rate: 20 °C min−1) to remove any chemisorbed CO2, cooled down to room temperature and purged under a nitrogen flow for 1 h. Then, the samples were submitted to five consecutive reduction (1350 °C) and CO2 splitting steps (1000 °C). During each self-reduction, the sample was heated up to 1350 °C (heating rate: 20 °C min−1), and then kept at this temperature for 20 min under a nitrogen flow (0.1 l (STP) min−1). Afterwards, the sample was cooled down to 1000 °C (cooling rate: 40 °C min−1), the gas flow was fast changed to pure CO2 and kept for 60 min.The following equations were used to evaluate the amount of gaseous products (mol g−1) released during the self-reduction and oxidation step respectively:
 | (4) |
 | (5) |
The performance of the samples was assessed by evaluating the reduction degree (xred,i) after each reduction or oxidation step (i), the oxidation yield (αi) and the reduction yield (βi) as follows:
 | (6a) |
 | (6b) |
 | (7) |
 | (8) |
Thermochemical H2O splitting tests
H2O splitting activity of prepared samples was assessed following an experimental already employed in previous papers.32,37,38 Briefly, powdered samples (500 mg; 170–300 μm) were submitted to consecutive temperature programmed reduction (TPR) and oxidation (TPO) runs in a tubular quartz reactor externally heated by an electric tubular furnace (Lenton). Temperature was measured by a K-type thermocouple hosted inside a co-axial tube.Evolved gases were continuously analysed by a Fisher-Rosemount NGA2000 analyser which analyses CO and CO2 by infrared detectors, O2 by a paramagnetic detector and hydrogen by a thermal conductivity detector. The analyser is provided with a cross-sensitivity system.
Samples were heated up to about 1000 °C at 10 °C min−1 in H2 mixture flow (TPR; 10 l (STP) h−1; 5 vol% H2/N2)37,38 and then cooled down to 60 °C under N2 flow. TPO was then performed switching the atmosphere to 3 vol% H2O/N2 mixture (10 l (STP) h−1), heating up to 1000 °C (heating rate: 10 °C min−1) and keeping the sample at this temperature for 20 min. The use of diluted water mixture was previously reported39 to enable reaction rate control, particularly in the case of H2-reduced samples. Reduction and oxidation profiles obtained with the gas analyser were deconvoluted using OriginPro 8.5 software.
Oxygen and hydrogen amounts (mol g−1), nO2 and nH2, respectively, were evaluated as follows:
 | (9) |
 | (10) |
Results and discussion
Materials characterization
Prepared samples were submitted to an in-depth physico-chemical characterization to elucidate the microscopical features accounting for their red-ox behaviour.XRD analysis was carried out on investigated samples both before (fresh samples, treated up to 1100 °C) and after a redox cycle in the TG apparatus (used samples, treated up to 1350 °C). Fig. 1 shows the XRD patterns of the used samples, while lattice parameters are reported in Table S1.† All samples show a fluorite structure typical of CeO2 lattice, no additional phases due to doping species being detectable. Moreover, the absence of any other significant diffraction peak in the XRD profiles confirms the formation of a solid solution, through the incorporation of Zr4+ into CeO2 lattice. In Fe-CeZr and Mn-CeZr materials the reflection of the (111) plane at 2θ = 28.4° shifts to lower angles than bare CeZr; this could indicate the formation of solid solutions. However, it is known that lower valence ions such as Fe3+ and Mn3+ are extremely difficult to dissolve into the ceria–zirconia lattice, especially when treating at high temperature.40 Thus, some iron and manganese ions could also be encapsulated in ceria–zirconia lattice as free mixed (Fe,Mn)2O3 or (Fe,Mn)O nanostructured oxides, not detectable by XRD analysis.41 Due to their smaller size than Ce4+ (ionic radius = 0.97 Å), Fe3+ (ionic radius = 0.55 Å) and Mn2+ (ionic radius = 0.67 Å) species are expected to be incorporated as interstitial defects, which should produce an expanded lattice with a consequent shift of XRD peaks to lower angles, as appreciated in XRD patterns of doped samples. Conversely, potassium doping induces a decrease in cell parameter, as confirmed by the shift of (111) reflection at higher angles. This could be due to the formation of K2O species segregated into CeO2–ZrO2 lattice.42
 | ||
| Fig. 1 XRD profiles of samples after a redox cycle in TG (used samples). Miller indices relative to fluorite CeO2 from Terribile et al.26 | ||
Furthermore, a small peak at 2θ = 29.4° can be appreciated in CeZr, Fe-CeZr and Mn-CeZr XRD patterns probably due to the presence of some segregate tetragonal ZrO2 phase. XRD spectra of fresh samples are quite similar to those of used materials. However, some of them, particularly fresh K-Fe-CeZr, show wider and less intense peaks (Fig. S4†), indicating that the reduction step at 1350 °C causes an increase in the average crystal size.
BET surface areas (Table S1†) are very low, as expected for samples treated at high temperature.
XPS and EPR spectroscopies were carried out on prepared samples in order to assess surface and bulk oxygen vacancies, respectively, which determine oxygen exchange ability, governing CeO2 catalytic performance.43 Notably, surface properties of fresh and used samples were investigated by means of XPS to assess any changes in surface composition after splitting tests. Table 2 reports Ce/Zr ratio, Ce oxidation states as evaluated from high-resolution XPS spectra and the curve-fitting of the Ce 3d region, respectively. The relative amount of surface labile oxygen (expressed as ratio between labile oxygen (Oα) and bulk oxygen (Oβ) moles) are reported in Table S2.†
| Sample | Fresh | Used | ||||||
|---|---|---|---|---|---|---|---|---|
| Ce/Zr | M/(Ce + Zr) | K/(Ce + Zr) | Ce3+/Ce4+ | Ce/Zr | M/(Ce + Zr) | K/(Ce + Zr) | Ce3+/Ce4+ | |
| CeZr | 1.84 | 0.24 | 2.80 | 0.30 | ||||
| Fe-CeZr | 2.46 | 0.31 | 0.31 | 1.93 | 0.28 | 0.73 | ||
| Mn-CeZr | 2.29 | 0.32 | 0.59 | 1.36 | 0.26 | 0.44 | ||
| Cu-CeZr | 2.73 | 0.60 | 0.70 | 1.41 | 0.47 | 0.39 | ||
| K-CeZr | 2.86 | 0.15 | 0.36 | 2.70 | 0.06 | 0.41 | ||
| K-Fe-CeZr | 3.92 | 0.30 | 0.06 | 0.55 | 2.17 | 0.40 | 0.15 | 0.58 |
| K-Cu-CeZr | 2.48 | 0.08 | 0.14 | 0.53 | 1.43 | 0.37 | 0.35 | 1.01 |
The nominal bulk Ce/Zr atomic ratio is equal to 3. In general, all fresh samples show a decrease in this ratio, indicating a zirconium surface enrichment. This segregation is more evident for the CeZr sample and gradually decreases in the doped samples, with the following order: CeZr > Mn-CeZr > Fe-CeZr > K-Cu-CeZr > Cu-CeZr > K-CeZr > K-Fe-CeZr. This can be attributed to the surface dispersion of the metal cation species which partly replace the Zr atoms.44 The highest depletion in Zr content is disclosed by K-Fe-CeZr sample, where the Ce/Zr ratio is almost 4, thus suggesting a Zr depletion of the surface. After the splitting tests, the Ce/Zr ratio increases for CeZr sample, in agreement with our previous work,37 whilst it is considerably smaller than the fresh catalyst for the doped samples, in accordance with Trovarelli et al.30 From the analysis of XPS data it can be inferred a zirconium enrichment, which grows in the following order: K-CeZr < Fe-CeZr < Mn-CeZr < K-Cu-CeZr < Cu-CeZr < K-Fe-CeZr. In particular, the K-Fe-CeZr sample shows the biggest decrease in surface Ce content, since it passes from an initial superficial Zr depletion to a final remarkable enrichment. As reported in the literature, the formation of Zr-rich phases enhances the splitting activity.30 Eventually, this could explain better red-ox performance of doped samples than bare CeZr that will be investigated in the following sections.
The high-resolution XPS spectra of the Ce 3d region and the relative curve-fitting are illustrated in Fig. S5.† According to the literature, 10 components were selected, “u” and “v”, corresponding to Ce 3d3/2 and Ce 3d5/2 spin-orbits, respectively.45 The u′′′, u′′, u, v′′′, v′′, v peaks were associated to the Ce4+ state while the u′, u0, v′ and v0 peaks were attributed to the Ce3+ state.46–48 As can be appreciated in Table 2, the fresh doped samples show a significant increase in superficial Ce3+ with respect to the CeZr sample. This suggests that the atoms of the transition metals can enter into the CeZr lattice and then Ce4+ can be partially converted to Ce3+.44 As reported in previous studies, Ce3+ ions concentration is associated to the formation of oxygen vacancies,49 which can promote oxygen migration during self-reduction as well as the dissociation of the O–H bonds in the H2O splitting reaction.43 Ce3+/Ce4+ atomic ratio after the splitting tests follows different trends: it keeps constant in CeZr, K-CeZr and K-Fe-CeZr, and significantly decreases in Cu-CeZr and Mn-CeZr samples. On the other hand, Fe-CeZr and K-Cu-CeZr show a marked increase of this ratio, evidencing the ability to keep a high concentration of surface Ce3+ ions that can be available for next cycles. This behavior is envisaged to promote CO/H2 production. Indeed, surface enrichment in Zr species (decrease in the Ce/Zr ratio) often determines an increase of the Ce3+/Ce4+ atomic ratio since smaller Zr4+ ions could itself promote the conversion of Ce4+ ions into larger Ce3+ species in order to compensate for lattice distortion.
Since splitting reaction often involves bulk sites, bulk properties were investigated through electron paramagnetic resonance (EPR) analysis. EPR spectra of CeZr samples doped with different metals were recorded at room temperature and are shown in Fig. 2. Different behaviors were observed, also due to the presence of various paramagnetic species. For the fresh K-Cu-CeZr, a single asymmetric broad peak with a g-factor value of ∼1.968 ± 0.003 is observed (Fig. 2a) and presents a line-shape amplitude ΔB = 8.4 ± 0.2 G. This peak is commonly assigned to surface Ce3+ ions that contain unpaired electrons.50–52 After the treatment at 1350 °C, a slight increase in the peak intensity is observed confirming increase of Ce3+ species (Fig. 2b), in accordance with XPS results. For the fresh K-CeZr sample no signal is appreciated (spectra not shown), suggesting that no paramagnetic species can be clearly revealed. On the other hand, a weak signal at g-factor value of ∼1.971 ± 0.004, ascribable to Ce3+ ions,50 is observed for fresh Fe-CeZr (Fig. 2e), which gets slightly more intense after treatment (Fig. 2f), confirming an increase in the paramagnetic species as also indicated by XPS results. For the Mn-CeZr sample, the EPR spectrum shows the characteristic six-line hyperfine splitting pattern of Mn2+ at g-factor value of 2.004 ± 0.003 (Fig. 2c), due to the interaction of electron spin (S = 5/2) of Mn2+ ions with nuclear spin I = 5/2.53 Being the EPR signal of Mn2+ characterized by a high intensity, it is not possible to discriminate the presence of any Ce3+ signal. However, after treatment at 1350 °C, a strong decrease in Mn2+ peaks intensity is detected (Fig. 2d), suggesting a change in its oxidation state.
 | ||
| Fig. 2 EPR spectra of K-Cu-CeZr, Mn-CeZr and Fe-CeZr samples, fresh (a, c and e) and treated at 1350 °C (b, d and f). | ||
Interestingly, even K+ species which cannot undergo any reduction, are able to influence the Ce3+/Ce4+ ratio, once employed as dopants. This behavior has been more extensively investigated in a previous study.54
Thermochemical CO2 splitting
Redox activity towards CO2-splitting was investigated by repeated thermochemical cycles performed in TG apparatus.37,38 The reduction of samples was appreciated through a mass loss in TG curve due to evolved O2 (Fig. S6†), and the corresponding onset temperatures are displayed in Table 3.| CeZr | Fe-CeZr | Mn-CeZr | Cu-CeZr | K-CeZr | K-Fe-CeZr | K-Cu-CeZr | ||
|---|---|---|---|---|---|---|---|---|
| a Expected error (≤10%). In the case of K-CeZr and K-Fe-CeZr during the II cycle, the CO is more than twice the released oxygen, probably due to a reorganization of the catalytic active site during the first cycle. In the case of K-Cu-CeZr during the III–V cycles, the β value is abnormal due to the baseline drift. In these conditions, the evaluation of this parameters is quite useless. | ||||||||
| TOR | 1150 | 1150 | 1100 | 750, 1100 | 870 | 900 | 725, 880 | |
| I cycle | nO2 | 300 | 313 | 297 | 406 | 307 | 279 | 553 |
| nCO | 83 | 219 | 237 | 125 | 332 | 294 | 331 | |
| II cycle | nO2 | 63 | — | — | — | 72 | 46 | 161 |
| nCO | 25 | — | — | — | 160 | 122 | 59 | |
| III cycle | nO2 | — | — | — | — | 73 | 54 | 109 |
| nCO | — | — | — | — | 134 | 99 | 19 | |
| IV cycle | nO2 | — | — | — | — | 71 | 51 | 65 |
| nCO | — | — | — | — | 110 | 97 | 0 | |
| V cycle | nO2 | — | — | — | — | 70 | 56 | 81 |
| nCO | — | — | — | — | 107 | 90 | 12 | |
Bare CeZr and Fe-CeZr samples show an onset temperature of about 1150 °C, whereas the reduction process starts at lower temperatures for all the other investigated materials. Notably, Cu-doped samples are reduced in two steps the former of which occurs at about 750 °C, producing 156 μmol gcat−1 of oxygen. TG curves of all doped samples display higher slope change than bare CeZr in the reduction step suggesting faster self-reduction kinetics (DTG curves, Fig. S7†). Overall, Cu containing materials featured higher weight losses than bare CeZr indicating both larger evolved oxygen amount and higher reduction degree (Table 3).
After reduction, the samples underwent a consecutive oxidation step in TG apparatus, that was carried out at 1000 °C under a CO2 flow (Fig. S8†). The amount of produced CO was driven from the recorded mass gain in TG curve, due to material oxidation through eqn (5) (Table 3).
Cu-CeZr showed poor activity towards CO2 splitting and overall oxidation yield comparable to bare CeZr in spite of its high self-reduction extent, the highest among transition metal doped samples. Conversely, the other doped materials have better overall redox performance than bare CeZr, resulting into higher oxidation yield (Table 3). Actually, K-containing samples (K-CeZr, K-Fe-CeZr, K-Cu-CeZr) showed the best activity in the redox cycle, featuring both the lowest reduction onset temperature and the highest oxidation yield (Table 3). K and Cu appear to have a synergistic effect, providing an outstanding reduction degree, although it results partially irreversible giving an oxidation yield of about 40%.
Red-ox activity of the most promising catalysts was also evaluated in five consecutive cycles in the TG apparatus, to assess their cyclability. Fig. 3 reports obtained TG curves for K-Fe-CeZr, K-Cu-CeZr and K-CeZr samples. The amounts of oxygen and carbon oxide production were evaluated by mass changes in TG curves and are reported in Table 3. Since bare CeZr sample showed a marked decrease of redox activity after two redox cycles (Fig. S6†), it was not investigated over 5 repeated cycles. K-Fe-CeZr, and K-CeZr samples show very good and stable activity during five consecutive cycles (Fig. 3a), displaying high oxidation yield, about 60%, as well as complete reversibility, confirmed by a reduction yield of about 100%, whereas K-Cu-CeZr sample evidences a poor stability due to a heavy decrease of mass changes in TG apparatus over repeated redox cycles (Fig. 3b). Reduction degree after each step (xred, %), oxidation yield (α, %), and reduction yield (β, %) are reported in ESI (Table S3†).
 | ||
| Fig. 3 TG profiles of five consecutive CO2 splitting cycles on (a) K-Fe-CeZr and K-CeZr samples and (b) K-Cu-CeZr sample. | ||
H2O splitting tests
In order to further assess the splitting properties of the investigated materials, these were tested towards H2O splitting following a procedure, which submitted the samples to alternated flows of H2 and H2O mixtures.29,30 Actually, H2 driven reduction is expected to lead to higher reduction degrees than self-reduction. However, recent studies proved that the qualitative trends of red-ox performance were closely related to doping and not influenced by reduction pathway, thus allowing for effective comparison between H2O splitting (lab-reactor) tests and CO2 oxidation measurements in TG apparatus.32 Fig. 4 reports a typical TPR profile together with its deconvolution by Gaussian functions obtained for K-Fe-CeZr sample, which can be considered as representative of TPR curves of the other samples (Fig. S9†). | ||
| Fig. 4 TPR profile obtained on the K-Fe-CeZr during the first cycle and related curve-fitting. | ||
Overall amounts of released oxygen and produced hydrogen were assessed by integrating the obtained peaks in TPR (Fig. S9†) and TPO profiles (Fig. S10†). Fig. 5 reports evolved oxygen and hydrogen amounts at each main peak vs. peak temperature in two consecutive reduction–oxidation cycles. Overall oxygen and hydrogen amounts produced in each cycle are reported in Table 4. Reduction degree after each step (xred, %), oxidation yield (α, %), and reduction yield (β, %) are reported in ESI (Table S4†).
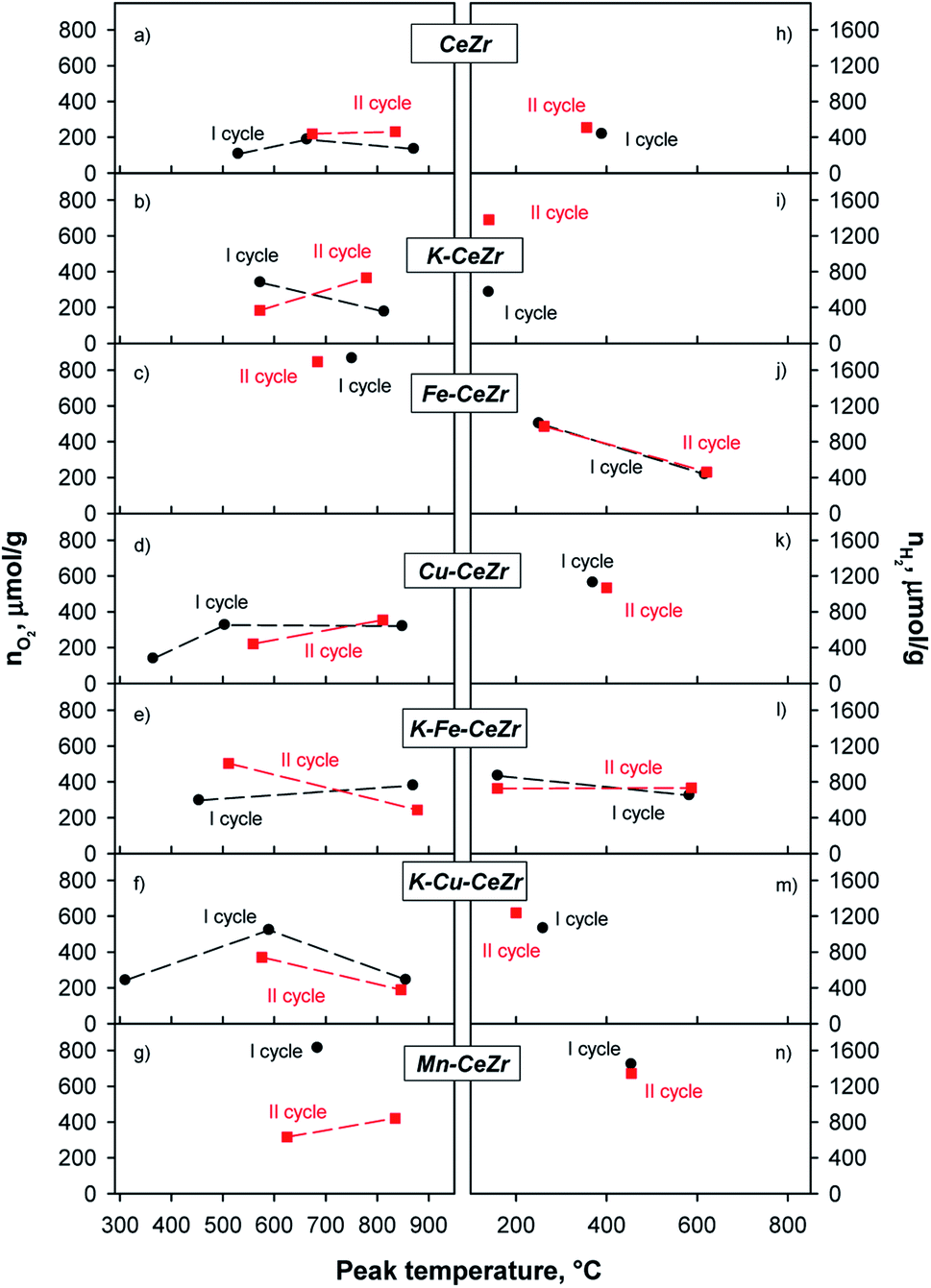 | ||
| Fig. 5 Oxygen evolved during H2 treatments (a–g) and hydrogen produced during H2O splitting tests (h–n) in two reduction–oxidation cycles for investigated materials. | ||
| Sample | CeZr | Fe-CeZr | Mn-CeZr | Cu-CeZr | K-CeZr | K-Fe-CeZr | K-Cu-CeZr | |
|---|---|---|---|---|---|---|---|---|
| I cycle | nO2 | 428 | 866 | 814 | 785 | 516 | 676 | 1008 |
| nH2 | 436 | 1440 | 1445 | 1127 | 573 | 1517 | 1064 | |
| II cycle | nO2 | 450 | 847 | 738 | 576 | 549 | 747 | 559 |
| nH2 | 509 | 1437 | 1344 | 1070 | 1380 | 1458 | 1237 |
During the first reduction cycle the TPR profile of CeZr sample shows three peaks at about 530, 660, and 870 °C, while only two peaks, at about 670 and 835 °C can be detected in the second reduction run (Fig. 5a and S9†). For CeZr low temperature reduction peaks in the range 300–600 °C are usually ascribed to surface sites, whereas those occurring at high temperature, T ≥ 800 °C, are expected to involve bulk sites.55 The decrease in the number of reduction peaks after the first run could be due to sintering phenomenon, which might likely convert surface sites into bulk ones.41,55
K, Fe, Cu and Mn doping significantly modifies the reduction profiles, which disclose at least two superimposed (Fe-CeZr sample) or well-resolved peaks involving surface and bulk sites reduction, respectively. Notably, the changes of both peak temperature and peak shape recorded in the second run for most samples suggest that they undergo a restructuring during thermal treatments (Fig. 5b–g).
TPR profile of Cu-CeZr shows a peak at low temperature (about 365 °C) that is no longer evident in the second cycle (Fig. 5d). This could be due to reduction of some copper (or copper-ceria) species, which are not re-oxidized by H2O during the splitting step, in accordance with the red-ox behavior assessed through TG analysis. In fact, it has been reported that different copper species in CuO/CeO2-based catalysts can be reduced at low temperature (100–250 °C).55,56 K-Cu-CeZr shows a similar behavior in which the reduction peak is further shifted to lower temperatures. Actually, the low temperature peak (about 310 °C) exhibits very similar shape and area in Cu-doped and K-Cu co-doped samples, which share the same Cu content, confirming its attribution to reduction of copper (or copper-containing) species. In general, all doped samples show better reduction performance than bare CeZr, as already evidenced by TG measurements. Indeed, peak edge in TPR profiles correspond to the highest reaction rate. Thus, since in copper and iron doped samples, these maxima occur at lower temperature than any other investigated material (Fig. S9†), it can be driven that copper and iron doping results in faster reduction kinetics (especially for Fe starting from the second cycle), in agreement with previous studies. On the contrary, Mn-CeZr does not show any improvement in reduction kinetics after its re-organization during the first redox cycle, as driven from the shift of the larger reduction effect towards higher temperatures in the second cycle (Fig. S9† and 5g).
Moreover, K-containing samples (K-CeZr, K-Fe-CeZr) show stable TPR profiles during two consecutive red-ox cycle (Fig. 5b), evidencing comparable peak temperature (Fig. 5) as well as evolved O2 amount (Table 4), differences lying within expected range of uncertainty (≤10%). These features suggest that K-doping improves thermal stability of CeZr samples preventing the sintering phenomenon, in accordance with TG results.
Re-oxidation occurs through one or two phenomena which can involve surface and sub-surface sites or bulk sites. TPO profiles of bare CeZr (Fig. 5h and S10†), Cu-CeZr (Fig. 5k and S10†) and Mn-CeZr (Fig. 5n and S10†) samples evidence a broad oxidation effect, at 350 °C, 400 °C and 450 °C, respectively. This suggests that Mn doping gets splitting kinetics decreased (Fig. 5n). On the contrary, two effects characterize the oxidation profiles of Fe-CeZr in both cycles (Fig. 5j). The first oxidation phenomenon occurs in a wide temperature range, with a main peak at about 100 and 300 °C. K-CeZr, K-Fe-CeZr. Moreover K-Cu-CeZr samples show faster oxidation kinetics than the corresponding K-free materials. In particular, H2 production occurs at lower temperature; only K-Fe-CeZr shows a second oxidation phenomenon at intermediate temperature (at about 590 °C, Fig. 5l).
Doping by transition metals significantly improves the reducibility of the ceria–zirconia, as well as hydrogen production and oxidation yield (α). Interestingly, both reduction (β) and oxidation (α) yields of the second cycle are very high and close to 100% (Table 4).
Notably, K-doping promotes red-ox performance, since it enables reduction at lower temperature than bare CeZr and improves water splitting activity, as evidenced by the marked increase of evolved H2, starting from the second red-ox cycle (Fig. 3b and i).
K addition to the lattice does not significantly affect the quantitative redox performance of the Fe and Cu doped materials (Fig. 5 and Table 4). Interestingly, K-Fe-CeZr shows negative reduction degrees after TPO; this means that the atomic oxygen captured by the sample during the first TPO is larger than the oxygen evolved during first TPR. This behavior can be addressed to the presence of oxygen vacancies after the high temperature calcination that can be fully oxidized by water during TPO.
The effect of doping and co-doping on hydrogen production and oxidation temperature is better illustrated in Fig. 6, where average temperatures are reported for the best performing samples, i.e. those containing either Fe(III) or Cu(II) and/or K(I) species. Fig. 6 clearly evidences that hydrogen production is markedly influenced by doping either with transition metals (Fe(III), Cu(II)) or potassium ions. Moreover, K-addition to the lattice improves the oxidation kinetics, as evidenced by lower reaction temperature (Fig. 6b). The positive influence of K-doping on oxidation kinetics is confirmed by results of a mathematical model which recently formulated and evidenced that K species can increase the number of active sites, promoting redox performances.54
 | ||
| Fig. 6 H2 production (top) and average oxidation temperature (bottom) during the oxidation steps of the second cycle for transition metals (Cu and Fe) co-doped and K-doped materials. | ||
K-doped and co-doped materials showed the best redox performances in terms of (i) higher reduction degrees, reduction and oxidation yields (i.e. larger H2 amounts), (ii) lower reduction and oxidation temperatures. These results are in accordance with red-ox performance towards CO2 splitting evaluated through TG analysis. Obtained materials were further cycled showing stable performance, especially the co-doped samples; Fig. S11† shows the H2 production measured during five consecutive cycles. It can be noticed that from the second cycle H2 production keeps the same value within the 6% error range.
Combining results of thermochemical splitting tests towards CO2 and H2O with physico-chemical features driven from EPR and XPS analysis, it can be inferred that red-ox activity is positively affected by surface segregation of Zr species, as well as by a high fraction of Ce3+ both on the surface and in the bulk, which should account for a high concentration of oxygen vacancies. Notably, Ce3+/Ce4+ surface amount and related oxygen vacancies exert a heavy influence on overall redox behavior; actually, this parameter either increases after use or keeps constant and higher than bare CeZr in all the best performing compositions: K-CeZr and K-Fe-CeZr, enabling relevant and stable activity towards both CO2 and H2O splitting.
Conclusions
In this study, Ce0.75Zr0.25O2 mixed oxides were doped with both transition metal (Cu, Mn, Fe) and potassium cations and tested in CO2 and H2O thermochemical splitting cycles. Furthermore, on the basis of XPS and EPR spectroscopic evidence bulk and surface properties accounting for red-ox behavior were enlightened. K doping enhances splitting properties towards both CO2 and H2O improving evolved oxygen amounts and lowering activation temperature (i.e. faster kinetics) during reduction step. Even Fe and Cu doping and Fe/K or Cu/K co-doping improves redox performance, reducing reduction/oxidation temperatures.Physico-chemical characterization evidences a higher fraction of both surface and bulk oxygen vacancies (OV) in K-doped and co-doped samples. Both govern the catalytic activity, promoting oxygen diffusion through the sample during self-reduction as well as the dissociation of the O–H bonds in H2O splitting. The improved OV migration in the bulk phase and between the bulk and the surface, account for enhanced thermo-catalytic properties.
Oxygen vacancies can be tuned by modifying the catalyst with reducible as well as non-reducible cations. In particular, the use of alkali metals appears a promising route to enhance the redox properties. Moreover, co-doping can produce a synergistic effect as shown by K-Fe-CeZr and K-Cu-CeZr materials.
Overall this study provides strategic guidelines to tailor catalytic performance in thermochemical splitting cycles.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Mr Andrea Bizzarro for BET analysis, and Mr Fernando Stanzione for ICP-MS measurements.References
- G. Takalkar, R. R. Bhosale and F. AlMomani, Fuel, 2019, 256, 115834 CrossRef CAS.
- F. A. Costa Oliveira, M. A. Barreiros, S. Abanades, A. P. F. Caetano, R. M. Novais and R. C. Pullar, J. CO2 Util., 2018, 26, 552–563 CrossRef CAS.
- R. R. Bhosale, G. Takalkar, P. Sutar, A. Kumar, F. AlMomani and M. Khraisheh, Int. J. Hydrogen Energy, 2019, 44, 34–60 CrossRef CAS.
- Y. Mao, Y. Gao, W. Dong, H. Wu, Z. Song, X. Zhao, J. Sun and W. Wang, Appl. Energy, 2020, 267, 114860 CrossRef CAS.
- S. Chuayboon and S. Abanades, Int. J. Hydrogen Energy, 2020, 45(48), 25783–25810 CrossRef CAS.
- Y. Chen, X. Zhu, K. Li, Y. Wei, Y. Zheng and H. Wang, ACS Sustainable Chem. Eng., 2019, 7, 15452–15462 CrossRef CAS.
- C. L. Muhich, S. Blaser, M. C. Hoes and A. Steinfeld, Int. J. Hydrogen Energy, 2018, 43, 18814–18831 CrossRef CAS.
- B. Zhao, C. Huang, R. Ran, X. Wu and D. Weng, J. Mater. Sci., 2016, 51, 2299–2306 CrossRef CAS.
- R. R. Bhosale, A. Kumar, F. Almomani, U. Ghosh, S. Al-Muhtaseb, R. Gupta and I. Alxneit, Ceram. Int., 2016, 42, 9354–9362 CrossRef CAS.
- G. D. Takalkar, R. R. Bhosale, A. Kumar, F. AlMomani, M. Khraisheh, R. A. Shakoor and R. B. Gupta, Sol. Energy, 2018, 172, 204–211 CrossRef CAS.
- R. R. Bhosale and G. D. Takalkar, Ceram. Int., 2018, 44, 16688–16697 CrossRef CAS.
- T. Cooper, J. R. Scheffe, M. E. Galvez, R. Jacot, G. Patzke and A. Steinfeld, Energy Technol., 2015, 3, 1130–1142 CrossRef CAS.
- C. Muhich and A. Steinfeld, J. Mater. Chem. A, 2017, 5, 15578–15590 RSC.
- S. Mostrou, R. Büchel, S. E. Pratsinis and J. A. van Bokhoven, Appl. Catal., A, 2017, 537, 40–49 CrossRef CAS.
- M. Takacs, J. R. Scheffe and A. Steinfeld, Phys. Chem. Chem. Phys., 2015, 17, 7813–7822 RSC.
- N. Gokon, T. Suda and T. Kodama, Thermochim. Acta, 2015, 617, 179–190 CrossRef CAS.
- N. Gokon, T. Suda and T. Kodama, Energy, 2015, 90, 1280–1289 CrossRef CAS.
- F. Lin, V. A. Samson, A. O. Wismer, D. Grolimund, I. Alxneit and A. Wokaun, CrystEngComm, 2016, 18, 2559–2569 RSC.
- M. Kang, J. Zhang, C. Wang, F. Wang, N. Zhao, F. Xiao, W. Wei and Y. Sun, RSC Adv., 2013, 3, 18878–18885 RSC.
- R. Jacot, R. Moré, R. Michalsky, A. Steinfeld and G. R. Patzke, J. Mater. Chem. A, 2017, 5, 19901–19913 RSC.
- A. Le Gal, S. Abanades, N. Bion, T. Le Mercier and V. Harlé, Energy Fuel., 2013, 27, 6068–6078 CrossRef CAS.
- S. Abanades, A. Legal, A. Cordier, G. Peraudeau, G. Flamant and A. Julbe, J. Mater. Sci., 2010, 45, 4163–4173 CrossRef CAS.
- A. Le Gal and S. Abanades, Int. J. Hydrogen Energy, 2011, 36, 4739–4748 CrossRef CAS.
- S. Abanades and A. Le Gal, Fuel, 2012, 102, 180–186 CrossRef CAS.
- D. Arifin, A. Ambrosini, S. A. Wilson, B. Mandal, C. L. Muhich and A. W. Weimer, Int. J. Hydrogen Energy, 2020, 45, 160–174 CrossRef CAS.
- D. Terribile, A. Trovarelli, C. De Leitenburg, A. Primavera and G. Dolcetti, Catal. Today, 1999, 47, 133–140 CrossRef CAS.
- G. Takalkar, R. R. Bhosale, S. Rashid, F. AlMomani, R. A. Shakoor and A. Al Ashraf, J. Mater. Sci., 2020, 55, 11797–11807 CrossRef CAS.
- C. Ruan, Y. Tan, L. Li, J. Wang, X. Liu and X. Wang, AIChE J., 2017, 63, 3450–3462 CrossRef CAS.
- A. Pappacena, M. Boaro, L. Armelao, J. Llorca and A. Trovarelli, Catal. Sci. Technol., 2016, 6, 1–9 RSC.
- A. Pappacena, M. Rancan, L. Armelao, J. Llorca, W. Ge, B. Ye, A. Lucotti, A. Trovarelli and M. Boaro, J. Phys. Chem. C, 2017, 121, 17746–17755 CrossRef CAS.
- C.-K. Yang, Y. Yamazaki, A. Aydin and S. M. Haile, J. Mater. Chem. A, 2014, 2, 13612–13623 RSC.
- G. Luciani, G. Landi, A. Aronne and A. Di Benedetto, Sol. Energy, 2018, 171, 1–7 CrossRef CAS.
- P. Charvin, S. Abanades, E. Beche, F. Lemont and G. Flamant, Solid State Ionics, 2009, 180, 1003–1010 CrossRef CAS.
- T. Maneerung, K. Hidajat and S. Kawi, Int. J. Hydrogen Energy, 2017, 42, 9840–9857 CrossRef CAS.
- B. Zugic, D. C. Bell and M. Flytzani-stephanopoulos, Appl. Catal., B, 2014, 144, 243–251 CrossRef CAS.
- A. Le Gal and S. Abanades, J. Phys. Chem. C, 2012, 116, 13516–13523 CrossRef CAS.
- G. Luciani, G. Landi, C. Imparato, G. Vitiello, F. A. Deorsola, A. Di Benedetto and A. Aronne, Int. J. Hydrogen Energy, 2019, 44(33), 17565–17577 CrossRef CAS.
- M. Portarapillo, A. Aronne, A. Di Benedetto, C. Imparato, G. Landi and G. Luciani, Chem. Eng. Trans., 2019, 74, 43–48 Search PubMed.
- N. D. Petkovich, S. G. Rudisill, L. J. Venstrom, D. B. Boman, J. H. Davidson and A. Stein, J. Phys. Chem. C, 2011, 115, 21022–21033 CrossRef CAS.
- E. Aneggi, C. de Leitenburg, G. Dolcetti and A. Trovarelli, Catal. Today, 2006, 114, 40–47 CrossRef CAS.
- P. S. Barbato, S. Colussi, A. Di Benedetto, G. Landi, L. Lisi, J. Llorca and A. Trovarelli, Appl. Catal., A, 2015, 506, 268–277 CrossRef CAS.
- K. Asano, C. Ohnishi, S. Iwamoto, Y. Shioya and M. Inoue, Appl. Catal., B, 2008, 78, 242–249 CrossRef CAS.
- Z. Su, W. Yang, C. Wang, S. Xiong, X. Cao, Y. Peng, W. Si, Y. Weng, M. Xue and J. Li, Environ. Sci. Technol., 2020, 54, 12684–12692 CrossRef CAS PubMed.
- G. Li, Q. Wang, B. Zhao and R. Zhou, Fuel, 2012, 92, 360–368 CrossRef CAS.
- D. R. Mullins, Surf. Sci. Rep., 2015, 70, 42–85 CrossRef CAS.
- E. Bêche, P. Charvin, D. Perarnau, S. Abanades and G. Flamant, Surf. Interface Anal., 2008, 40, 264–267 CrossRef.
- Y. Polyak and Z. Bastl, Surf. Interface Anal., 2015, 47, 663–671 CrossRef CAS.
- F. Larachi, J. Pierre, A. Adnot and A. Bernis, Appl. Surf. Sci., 2002, 195, 236–250 CrossRef CAS.
- L. Zhou, X. Li, Z. Yao, Z. Chen, M. Hong, R. Zhu, Y. Liang and J. Zhao, Sci. Rep., 2016, 6, 23900 CrossRef CAS.
- M. Zhao, M. Shen and J. Wang, J. Catal., 2007, 248, 258–267 CrossRef CAS.
- J. Wang, J. Wen and M. Shen, J. Phys. Chem. C, 2008, 112, 5113–5122 CrossRef CAS.
- J. Xu, J. Harmer, G. Li, T. Chapman, P. Collier, S. Longworth and S. C. Tsang, Chem. Commun., 2010, 46, 1887–1889 RSC.
- P. I. Archer, S. A. Santangelo and D. R. Gamelin, Nano Lett., 2007, 7, 1037–1043 CrossRef CAS PubMed.
- M. Portarapillo, D. Russo, G. Landi, G. Luciani and A. Di Benedetto, RSC Adv., 2021, 11, 39420–39427 RSC.
- A. Di Benedetto, G. Landi and L. Lisi, Catalysts, 2018, 8, 209 CrossRef.
- T. Caputo, L. Lisi, R. Pirone and G. Russo, Appl. Catal., A, 2008, 348, 42–53 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01355j |
| This journal is © The Royal Society of Chemistry 2022 |