
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermining the inherent selectivity for carbon radical hydroxylation versus halogenation with high-spin oxoiron(IV)–halide complexes: a concerted rebound step†
Yaping Tao‡
 a,
Zixian Li‡b,
Yiman Zhanga,
kexi Suna and
Zhaojun Liu*a
a,
Zixian Li‡b,
Yiman Zhanga,
kexi Suna and
Zhaojun Liu*a
aCollege of Physics and Electronic Information & Henan Key Laboratory of Electromagnetic Transformation and Detection, Luoyang Normal University, Luoyang 471934, China. E-mail: zhaojunliu@gmail.com
bState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China
First published on 29th March 2022
Abstract
A synthetic iron model can process both halogenation and hydroxylation with vague selectivity, which is different from halogenase even though these structures are used for the simulation of halogenase. The key factor of the synthetic oxo-iron model mediated hydroxylation or the halogenation is still under debate. Herein density functional theory calculation is used to investigate the hydroxylation versus halogenation of propylene by the complex [FeIV(O)(TQA)(X)]+ (X = F, Cl, Br). Our results suggest that a concerted rebound mechanism (between the -X and the hydroxyl ligands after the hydrogen abstraction) leads to the formation of two different kinds of products.
1. Introduction
There are many enzymes that can functionalize C–H in organisms, which undertake important physiological functions.1 For example, αKG-dependent hydroxylase TauD can hydroxylate taurine,2 while αKG-dependent SyrB2,3 CytC3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 and WelO55,6 can halogenate many natural products.7 The first step of their catalytic mechanisms is similar, involving the hydrogen atom being extracted from the C–H substrate to generate carbon radicals, then undergoing a so-called “rebound” process.8 For hydroxylase, it is the hydroxyl group that rebounds in this process to produce hydroxylated products; while for halogenase, the rebound of this process is the halogen atom (support as the ligand of the Fe–O center) to produce halogenated products.9–14 The catalytic activity of halogenase comes from controlling the selective transfer of –OH or -X to nearby carbon radicals.
4 and WelO55,6 can halogenate many natural products.7 The first step of their catalytic mechanisms is similar, involving the hydrogen atom being extracted from the C–H substrate to generate carbon radicals, then undergoing a so-called “rebound” process.8 For hydroxylase, it is the hydroxyl group that rebounds in this process to produce hydroxylated products; while for halogenase, the rebound of this process is the halogen atom (support as the ligand of the Fe–O center) to produce halogenated products.9–14 The catalytic activity of halogenase comes from controlling the selective transfer of –OH or -X to nearby carbon radicals.
In recent years, many outstanding efforts have been used to simulate the structure of the active site of halogenase for the C–H halogenation reaction.15–19 However, the synthesized iron model can catalyze the simultaneous formation of halogenated and hydroxylated products, showing lower selectivity comparing with the natural halogenase.20–22 This has aroused great interest among researchers. Due to the lack of direct experimental observation of the short-lived rebound intermediates, the key factor of the synthetic oxo-iron model mediated hydroxylation reaction or the halogenation reaction is still under debate.14 A series of different suggestions try to describe these factors that affect the selectivity of the reaction process. Previous studies have shown that alcohol products are more thermodynamically stable than halogenated compounds,23–26 therefore, it is expected that halogenation is kinetically preferred.21,27 However, at present, there is no specific and definitive conclusion to explain the reactivity of [FeIV(OH)(X)]+ species.
In 2016, Que and his co-workers reported the first example of the synthesis of oxoiron(IV)-halide complexes to halogenate C–H bonds.15 The S = 2 complexes 2 and 3 not only exhibit excellent Mössbauer parameters, which are in good agreement with the halogenase S = 2 oxygenated iron(IV) intermediate, but also react much more rapidly. Therefore, 2 and 3 can support a variety of different spin state options for oxo-iron(IV) oxidants in halide enzymes15 acting as an excellent spectroscopic and functional models of CytC3 and SyrB2 intermediates.
Our previous studies have shown that the utilization of pseudo-octahedral structure is the source of the structure to ensure the S = 2 ground state of the central iron atom.28 The high-spin state, which is close to the natural spin state of the iron atom in biological enzymes, provides a guarantee for the catalytic activity of the entire system. It is important to explore how it catalyzes the hydroxylation and halogenation reactions of organic substrates.
It is world-wide known that propylene is the precursor for the production of various important key compounds.29–33 In order to investigate the influence of different halogen atoms on the catalytic activity of the oxo-iron system and the chemical selectivity of the entire reaction process, we selected three periodic halogen atoms (F, Cl, Br) as ligands for the catalytic system (Scheme 1). Herein we report the computational investigation of three [FeIV(TQA)(X)]+ (X = F, Cl, Br) complexes react with propylene, and one kind of synergistic reaction mechanism is proposed.
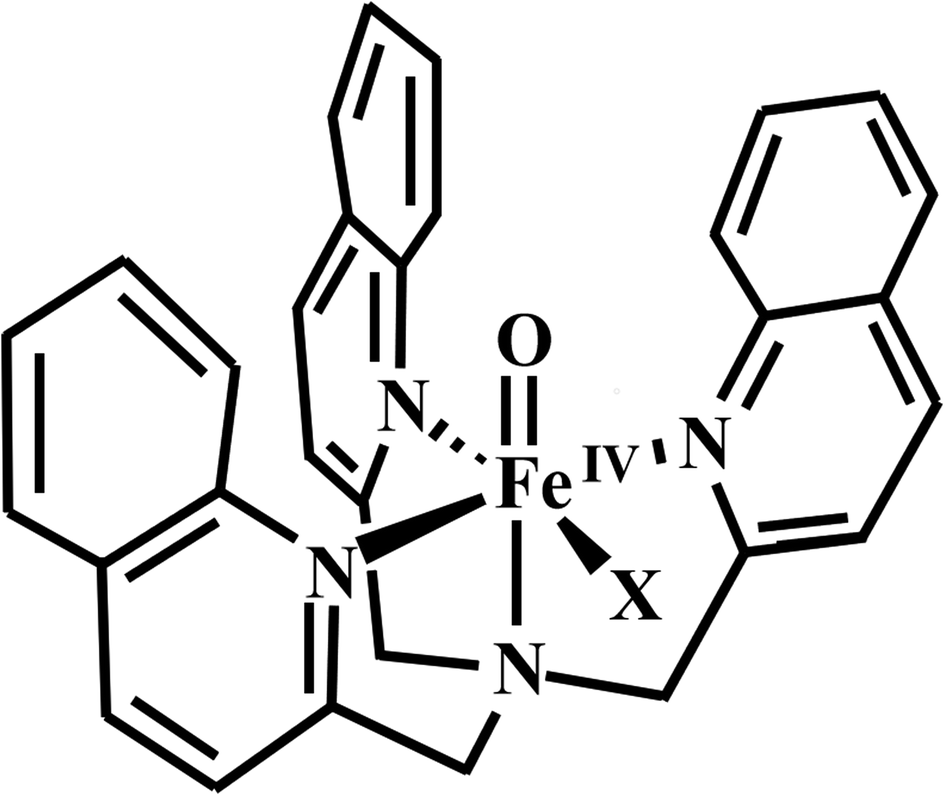 | ||
| Scheme 1 Structures of [FeIV(O)(TQA)(X)]+ (X = F, Cl, Br). | ||
2. Computational details
In this work, all the geometric and electronic structures presented were performed with the Gaussian 09 package34 with the unrestricted hybrid density functional method unrestricted Becke, three-parameter, Lee–Yang–Parr (UB3LYP).35–38 The geometries of the [FeIV(O)(TQA)(X)]+ were fully optimised without symmetry constraints. We used the double-ζ LACVP39,40 basis set for iron atom, the 6-311G** for halogen atom and the 6-31G** basis set for the C, H, O, and N (B1 in brief) in the structure optimization. Furthermore, the single-point energy of the optimised geometry was calculated with a higher basis set TZVP41,42 (B2 in brief) to achieve improvement of the energetics. Moreover, acetonitrile solvation including the self-consistent reaction field (SCRF) was used in optimizing the structures and calculating single-point energy, within polarizable continuum model (PCM).39We also used other functionals and basis sets to ascertain our UB3LYP functional and B1/B2 selection, and the test results shown in the ESI † were consistent with those obtained from UB3LYP/B1/B2.
3. Results and discussion
3.1 Electronic and structural properties
The S = 2 ground state spin state of the iron-oxygen complex can be rationalized according to its structure. Before discussing the reaction between the oxo-iron system and the substrate, we need to consider the influence of different halogen ligands on the structure.Table 1 displays the optimized geometries features of the [FeIV(O)(TQA)(X)]+ complexes in quintet spin state. In particular, the Fe–O distance does not considerably vary with the different halogen ligands (1.627 Å for X = F, 1.625 Å for X = Cl and 1.627 Å for X = Br), but the bond length between the iron atom and the halogen ligand obviously varies, which is reasonable due to the different atomic radius. From the group spin densities, it can be seen that Fe atom has the highest spin densities, which means that the high ground state of the whole complex origins from the metal atom. The Fe atom has four unpaired electrons leading to the quintet state.
| Fe–O | Fe–X | ρFe | ρO | ρX | |
|---|---|---|---|---|---|
| X = F | 1.627 | 1.949 | 3.128 | 0.599 | 0.099 |
| X = Cl | 1.625 | 2.405 | 3.079 | 0.647 | 0.093 |
| X = Br | 1.627 | 2.546 | 3.074 | 0.648 | 0.096 |
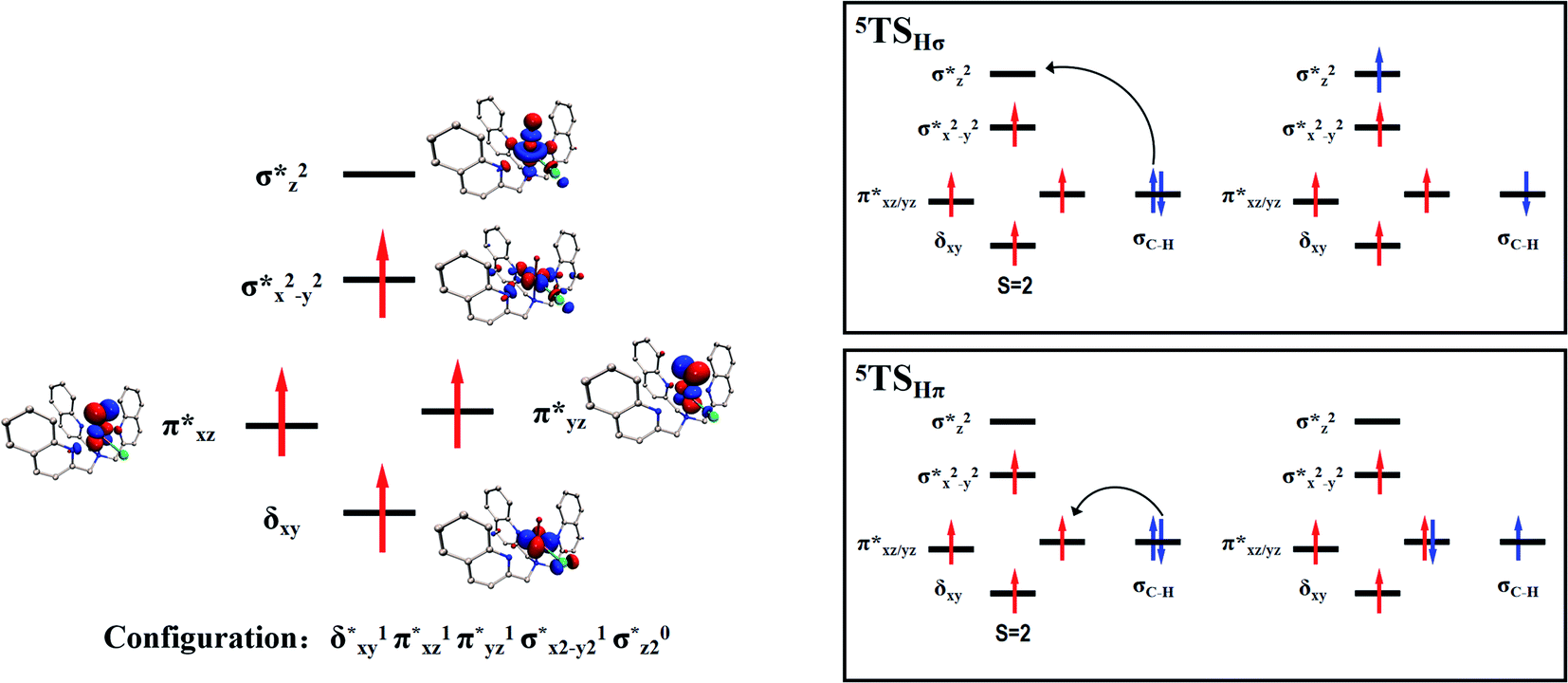
Scheme 2 (left-hand-side) shows the occupied and virtual orbitals of the iron(IV)-oxo species. The reactant state has a set of one-electron-occupied antibonding orbitals (π*) in the xz- and yz-planes which is the mixture of the metal 3d orbital with O2p orbital. In addition, there is another metal 3d orbital (xy) located in the plane of the ligands. Two high-lying orbitals (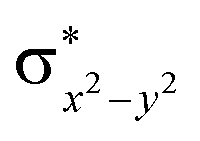 and
and 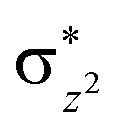 ) in the xy-plane and along the z-axis complement the series of metal-type orbitals. Thus, the configuration of the quintet spin state is
) in the xy-plane and along the z-axis complement the series of metal-type orbitals. Thus, the configuration of the quintet spin state is 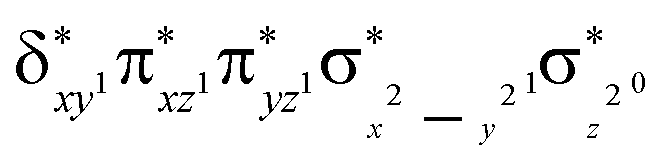
 | ||
| Scheme 2 Reactant orbital occupation and electron transfer processes in the hydrogen atom abstraction. | ||
3.2 Structures and reaction pathways of H atom abstraction
A series of structures along the reaction pathways of [FeIV(O)(TQA)(X)]+ with propylene were optimized via DFT studies. In general, when the substrate is close to the ferrite center, an electron is activated and transferred into the Fe–O orbital leading to the O–H bonding. In the quintet spin state, this electron can either transfer to the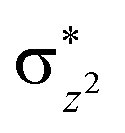 orbital (5σ-pathway) or the
orbital (5σ-pathway) or the  orbital (5π-pathway). The choice of electron between these two transfer pathways is the result of many complex factors, such as the nature of the orbital splitting, the electron-donating effect of ligand and environmental perturbations.19 Scheme 2 shows more orbital details of these two electron transfer pathways. Generally, the 5σ-pathway is more energetic preferred because it leads to a more even electron distribution.
orbital (5π-pathway). The choice of electron between these two transfer pathways is the result of many complex factors, such as the nature of the orbital splitting, the electron-donating effect of ligand and environmental perturbations.19 Scheme 2 shows more orbital details of these two electron transfer pathways. Generally, the 5σ-pathway is more energetic preferred because it leads to a more even electron distribution.
As illustrated in Fig. 1, the optimised geometries of the transition states vary a lot depending on the different halogen ligands. The C–H bond is the shortest in the reaction between [FeIV(O)(TQA)(F)]+ and propylene (1.287 Å), whereas the Fe–O and the O–H bonds of this reaction are the longest (1.726 Å for the Fe–O bond and 1.280 Å for the O–H bond). The transition states of other two iron(IV)-hydroxo complexes shows similar features in case of the pivotal bond length. Moreover, the value of ∠FeOH in reaction of [FeIV(O)(TQA)(F)]+ (110.2°) is close to 120°, and the values of other two systems are near 180° (168.5° for Cl and 168.4° for Br). Therefore, the substrate tends to attack the oxo-iron center from the side in case of the F atom as the halogen ligands when the catalytic reaction occurs. As we mentioned earlier, this is exactly when the electron is transferred through the 5σ-pathway. The substrates-attack-angle of the other two systems are close to 180°, corresponding to the 5π-pathway electron transfer path.
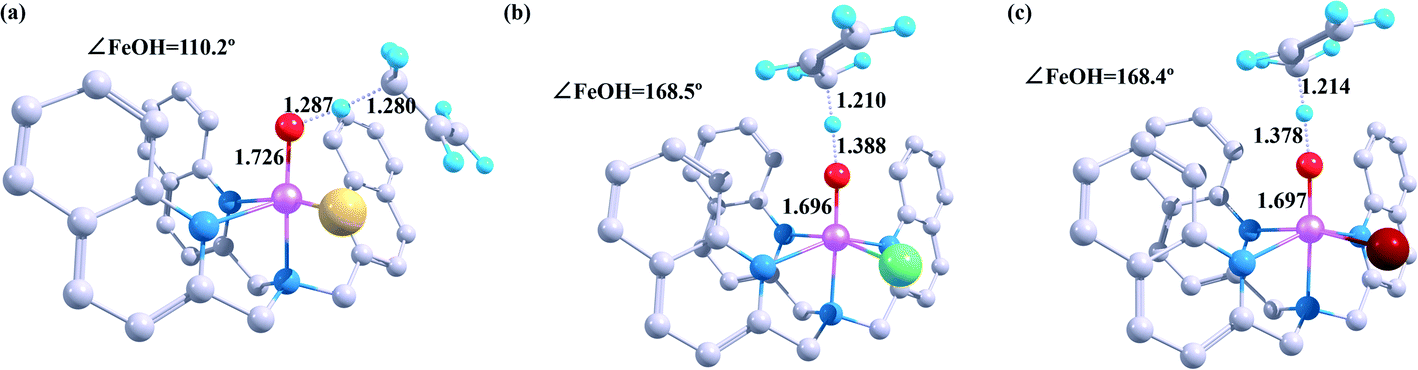 | ||
| Fig. 1 Geometric details of the hydrogen-abstraction transition states of the [FeIV(O)(TQA)(X)]+ react with propylene at the B2 level. (a) X = F (b) X = Cl (c) X = Br. Bond lengths are in Å, angles are in °. | ||
In order to further verify whether our conclusions are reliable, we analyzed the spin density of the iron atoms in the reactants, transition states and the intermediates systems. As shown in Table 2, the spin densities of the iron atoms in the oxo-iron complexes with Cl and Br as the ligands show an upward trend after hydrogen atom abstraction, and the spin densities of iron atoms in the intermediates are close to 4 (4.16 for X = Cl and 4.14 for X = Br). However, for the X = F system, the spin density of the iron atom in the intermediate after the catalytic reaction is reduced compared with the initial reactant. It is precisely because the  orbital accepts electron, which increases the number of electron-pairing-orbitals and the spin density of iron atom will decrease. This further proves that the electron transfer is carried out by 5π-pathway for the reaction of [FeIV(O)(TQA)(F)]+ with propylene.
orbital accepts electron, which increases the number of electron-pairing-orbitals and the spin density of iron atom will decrease. This further proves that the electron transfer is carried out by 5π-pathway for the reaction of [FeIV(O)(TQA)(F)]+ with propylene.
| Reactants | Transition states | Intermediates | |
|---|---|---|---|
| X = F | 3.13 | 3.79 | 2.80 |
| X = Cl | 3.09 | 3.79 | 4.16 |
| X = Br | 3.09 | 3.79 | 4.14 |
As illustrated in Fig. 2, the energy barriers of hydrogen atom abstraction by [FeIV(O)(TQA)(X)]+ (X = F, Cl, Br) with propylene are 14.30, 9.22 and 9.65 kcal mol−1, respectively. The oxo–iron complex with F atom as the ligand has noteworthy higher barrier. The different electron-transfer mechanisms have marked effects on the reaction barriers. These results suggest that oxidative ligand transfer may be influenced by electronic factors such as the oxidizability of the ligand, as Cl and Br.43
 | ||
| Fig. 2 Energy profile for the hydroxylation/halogenation reactions of substrate with [FeIV(O)(TQA)(X)]+. All energies are in kcal mol−1 relative to rc; relative energies are indicated in the order: B2(B2 + ZPE) level. Red stands for F, blue stands for Cl and green stands for Br. | ||
Fig. 1 shows optimized geometry structures of the transition states in the first step hydrogen atom abstraction. The O–H bond length ranges from 1.287 Å to 1.388 Å (1.287 Å for X = F, 1.210 Å for X = Cl and 1.214 Å for X = Br) at B2 level, and the O–H bond length is intriguingly related to the hydroxylation reactivity as can be seen from Fig. 2 (14.30 kcal mol−1 for X = F, 9.22 kcal mol−1 for X = Cl and 9.65 kcal mol−1 for X = Br at B2 level), which indicates that as the O–H bond lengthening, the hydroxylation reactivity decreases. Our date agrees well with Mayer theory.44
In addition to the O–H bond length, the reaction energy barrier is also affected by the Fe–O bond distance in the transition state structure. The reaction barrier decreases with the shortening of the Fe–O bond length. Our results support the theory proposed by Roy, that the enhancement of Fe–O bond along the z-axis promotes the orbital overlap of C–H and O- on the substrate, so that the oxyl radicals has high electrophilicity and ensures high reaction activity. Apart from the geometric bond length parameters, the imaginary frequencies are also associated with the reaction barrier.45,46 The imaginary frequencies values of i1634, i951, and i999 cm−1 are obtained for X = F, X = Cl, and X = Br in the hydrogen atom abstraction, which indicates sharp and narrow barriers. Data analysis of theoretical calculation shows that there is a delicate balance between the energy barriers of the reaction and the transition state structure.
3.3 Hydroxylation versus halogenation
The C–H bonds in the substrate are broken after hydrogen abstraction and lead to the formation of carbon radical. The research shows that both OH– and X–, as two different ligands of iron center, can bond with carbon radical through rebound mechanism, and then form hydroxylation or halogenation product. This is also the reason why the synthetic iron model does not show specific halogenation activity. The formation of hydroxylation products is still the bottleneck in the synthesis of selective halogenated compounds.20Recent studies showed that substrate positioning between halide and hydroxide plays a key role in ensuring selectivity for halogenation.47–51 In order to explore the reaction mechanism in the rebound process, we simultaneously scanned the bond lengths of C–O and C–X. In other words, we scanned a reaction potential energy surface. If there is no interaction between the hydroxide rebound process and the halide rebound process, we will observe two transition states structures on the potential energy surface, corresponding to the hydroxide rebound process and the halide transfer process. Intriguingly, as shown in the Fig. 3, there is only one peak on the potential energy surface, which is the highest energy point (transition state structure). This means that during the reaction between the [FeIV(O)(TQA)(X)]+ (X = Cl and Br) with propylene, for the second step of rebound process, the transfer of the halogen ligand and the rebound of the hydroxide group interfere with each other. The rebound process follows a cooperative reaction mechanism. There is only one transition state structure for these two reaction pathways, leading to two kinds of products. In order to support this conclusion, we have calculated the intrinsic reaction coordinate (Fig. S1†). Through the analysis of the intrinsic reaction coordinates, it can be identified that one transition state passes through different reaction coordinates to obtain halogenated products and hydroxylated products respectively, and the energy of hydroxylated products is lower, which is consistent with the product energy order shown in Fig. 2. Moreover, the intrinsic reaction coordinates support our previous conclusion that two different reaction pathways share the same transition state.
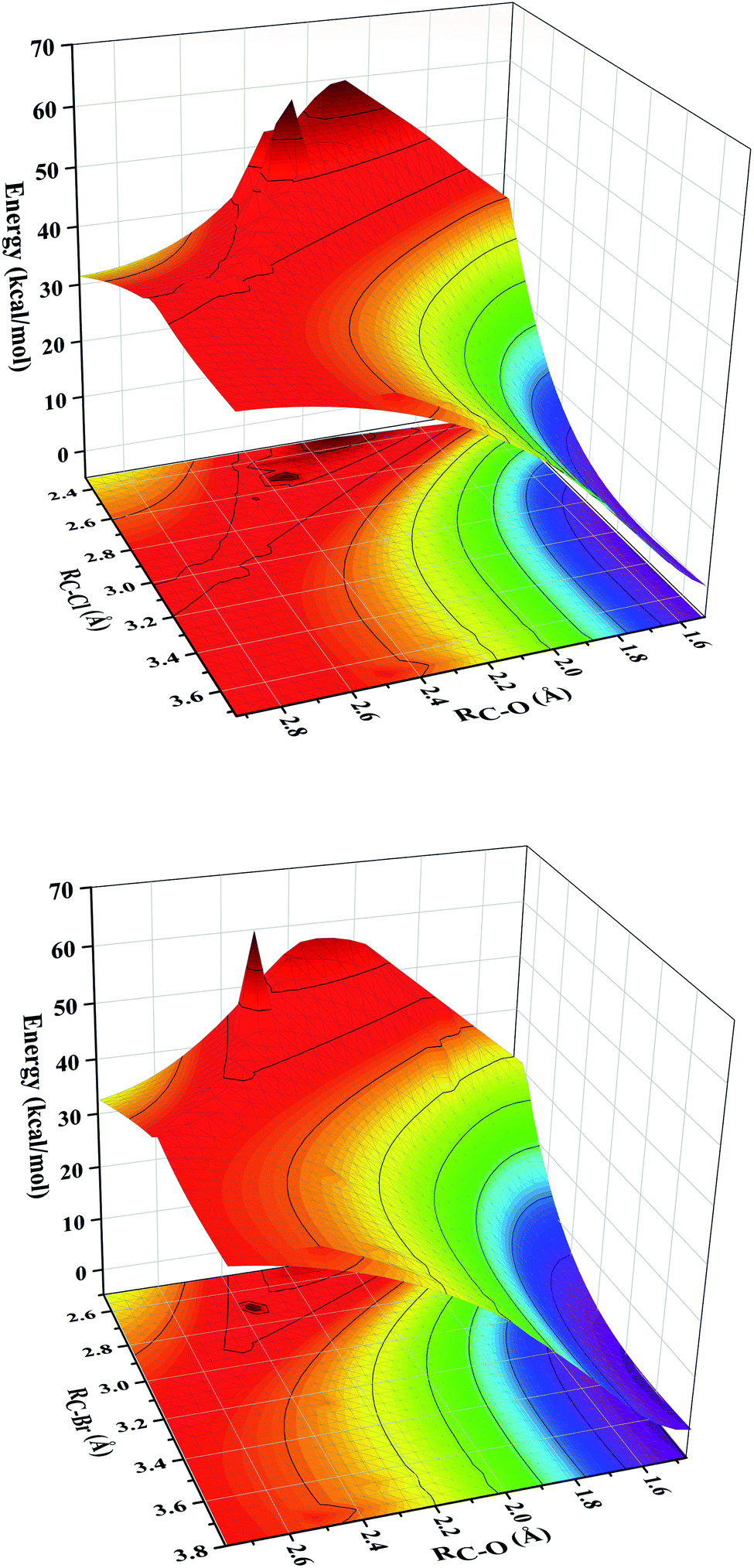 | ||
| Fig. 3 The potential energy surface of the bond lengths of C–O and C–X in the second –OH rebound/–X transfer process. | ||
In order to better explore the cooperative reaction mechanism, herein we show the transition state structures of the optimized rebound process at B2 level. As shown in Fig. 4, after the propylene substrate undergoes the hydrogen atom abstraction, the remaining three carbon atoms are connected to each other by a conjugated π bond, and the electrons dissociate uniformly among these three carbon atoms, which leads to the fact that there are actually two identical carbon radicals (C1 and C3) in the substrate at the same time. When reaching the appropriate position between the hydroxyl ligand and the halogen ligand, the two carbon radicals can interact with the hydroxyl ligand and the halogen atom ligand at the same time (TSOH–X). The existence of this interaction can cause two kinds of different products.
 | ||
| Fig. 4 Geometric details of the hydroxyl rebound/halogen transfer transition states of the [FeIV(O)(TQA)(X)]+ react with propylene at the B3 level. (a) X = F (b) X = Cl (c) X = Br. Bond lengths are in Å, angles are in °. | ||
Different halogen substituents have a significant impact on the transition state during the second rebound process. The law of the increase of the second energy barrier is consistent with the periodicity of halogen atoms, which suggest that halide ligand transfer maybe influenced by electronic factors such as the oxidizability of the ligand, as F, Cl and Br have gradually decreasing oxidation potentials.
As shown in Fig. 2, for X = F, Cl, Br, the energy of TSOH–X structures are 4.62, 1.94 and 1.71 kcal mol−1, respectively. The reason why the X = F complex has the highest barrier in the –OH rebound/-X transfer process depending on the valence bond modelling. Previously, valence bond modelling has been used to rationalize the ligand rebound/transfer preference by non-heme iron complexes.52,53 The energy for breaking the old bonds and forming the new ones in the transition states is the crucial determination refers to the barrier height. The 3-electron πyz/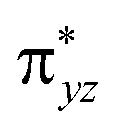 pair of orbitals would be broken during the transfer of the OH group (rebound) to the radical. One of these three electrons stays in the 3dyz orbital of Fe whilst the other two pairs up with the radical to form the C–O bond and pairs up with the δxy electron separately. Therefore, the level of OH rebound is dependent on the energy to break the πyz/
pair of orbitals would be broken during the transfer of the OH group (rebound) to the radical. One of these three electrons stays in the 3dyz orbital of Fe whilst the other two pairs up with the radical to form the C–O bond and pairs up with the δxy electron separately. Therefore, the level of OH rebound is dependent on the energy to break the πyz/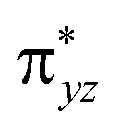 orbitals, the energy to form the C–O bond and the energy to promote an electron from πyz to δxy.
orbitals, the energy to form the C–O bond and the energy to promote an electron from πyz to δxy.
Since the rebound of the OH group is exactly the same here in our three complexes (all the same πyz/ orbitals to break, C–O bond to form and electron to promote), the energetic difference between the second transition state would be caused by breaking the Fe–X bond and forming the C–X bond. In fact, the stronger electron-withdrawing ability the substituent have, the higher energy gap between the πyz and
orbitals to break, C–O bond to form and electron to promote), the energetic difference between the second transition state would be caused by breaking the Fe–X bond and forming the C–X bond. In fact, the stronger electron-withdrawing ability the substituent have, the higher energy gap between the πyz and  orbitals exists.54 Because the F atom has the strongest electron-withdrawing ability, for X = F complex, the widening of the energy gap will make it harder to break, leading to a higher transition state energy.
orbitals exists.54 Because the F atom has the strongest electron-withdrawing ability, for X = F complex, the widening of the energy gap will make it harder to break, leading to a higher transition state energy.
4. Conclusions
In this work, we present a detailed study into the hydroxylation versus halogenation of propylene by [FeIV(O)(TQA)X]+, X = F, Cl and Br. A series of density functional theory calculations reveal the mechanisms of the subsequent pathways for OH/X concerted rebound following the hydrogen abstraction. Two electronically different pathways (5TSσ− and 5TSπ−) are considered for hydrogen atom abstraction. The spin densities of the iron atoms suggested the electronic configuration of the metal center and the electronic environment of the ligand as the determining factors. After the hydrogen abstraction, there are two indistinguishable carbon radicals. Thus, the mechanism of rebound step is synergistic reaction, and the barrier heights change depending on subtle electronic environment. As such, a combination of potential energy surface and transition state structure has been used to propose a potential reaction pathways leading to a mixture of hydroxylated products and halogenated products. It is quite possible that an analogous reaction can be catalyzed by nonheme iron model by supplying the system with the next halogen elements I. We hope there will be further experimental verification in future studies.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 51801093). This work was also supported by the Key Scientific Research Projects for Higher Education in Henan Province (Grant No. 21B140007) and General innovation project of college students in Henan Province (No. S202110482031).References
- E. Roduner, W. Kaim, B. Sarkar, V. B. Urlacher, J. Pleiss, R. Gläser, W. D. Einicke, G. A. Sprenger, U. Beifuß and E. Klemm, ChemCatChem, 2013, 5, 82–112 CrossRef CAS.
- S. Martinez and R. P. Hausinger, J. Biol. Chem., 2015, 290, 20702–20711 CrossRef CAS PubMed.
- F. H. Vaillancourt, J. Yin and C. T. Walsh, Proc. Natl. Acad. Sci. U.S.A, 2005, 102, 10111–10116 CrossRef CAS PubMed.
- C. Wong, D. G. Fujimori, C. T. Walsh and C. L. Drennan, J. Am. Chem. Soc., 2009, 131, 4872–4879 CrossRef CAS PubMed.
- M. L. Hillwig and X. Liu, Nat. Chem. Biol., 2014, 10, 921–923 CrossRef CAS PubMed.
- A. J. Mitchell, Q. Zhu, A. O. Maggiolo, N. R. Ananth, M. L. Hillwig, X. Liu and A. K. Boal, Nat. Chem. Biol., 2016, 12, 636–640 CrossRef CAS PubMed.
- J. Latham, E. Brandenburger, S. A. Shepherd, B. R. Menon and J. Micklefield, Chem. Rev., 2018, 118, 232–269 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, J. Biol. Inorg Chem., 2017, 22, 185–207 CrossRef CAS PubMed.
- K.-B. Cho, H. Hirao, S. Shaik and W. Nam, Chem. Soc. Rev., 2016, 45, 1197–1210 RSC.
- X. Wu, M. S. Seo, K. M. Davis, Y. M. Lee, J. Chen, K. B. Cho, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2011, 133, 20088–20091 CrossRef CAS PubMed.
- K.-B. Cho, S. Shaik and W. Nam, J. Phys. Chem. Lett., 2012, 3, 2851–2856 CrossRef CAS.
- K.-B. Cho, X. Wu, Y.-M. Lee, Y. H. Kwon, S. Shaik and W. Nam, J. Am. Chem. Soc., 2012, 134, 20222–20225 CrossRef CAS PubMed.
- K. B. Cho, H. Kang, J. Woo, Y. J. Park, M. S. Seo, J. Cho and W. Nam, Inorg. Chem., 2014, 53, 645–652 CrossRef CAS PubMed.
- S. N. Dhuri, K. B. Cho, Y. M. Lee, S. Y. Shin, J. H. Kim, D. Mandal, S. Shaik and W. Nam, J. Am. Chem. Soc., 2015, 137, 8623–8632 CrossRef CAS PubMed.
- M. Puri, A. N. Biswas, R. Fan, Y. Guo and L. Que, Jr., J. Am. Chem. Soc., 2016, 138, 2484–2487 CrossRef CAS PubMed.
- O. Planas, M. Clemancey, J. M. Latour, A. Company and M. Costas, Chem. Commun., 2014, 50, 10887–10890 RSC.
- P. Comba and S. Wunderlich, Chem.–Eur. J., 2010, 16, 7293–7299 CrossRef CAS PubMed.
- S. Chatterjee and T. K. Paine, Angew. Chem., 2016, 55, 7717–7722 CrossRef CAS PubMed.
- S. Rana, J. P. Biswas, A. Sen, M. Clemancey, G. Blondin, J. M. Latour, G. Rajaraman and D. Maiti, Chem. Sci., 2018, 9, 7843–7858 RSC.
- M. Srnec and E. I. Solomon, J. Am. Chem. Soc., 2017, 139, 2396–2407 CrossRef CAS PubMed.
- J. Huang, C. Li, B. Wang, D. A. Sharon, W. Wu and S. Shaik, ACS Catal., 2016, 6, 2694–2704 CrossRef CAS.
- M. G. Quesne and S. P. de Visser, J. Biol. Inorg Chem., 2012, 17, 841–852 CrossRef CAS PubMed.
- A. Timmins, N. J. Fowler, J. Warwicker, G. D. Straganz and S. P. de Visser, Front. Chem., 2018, 6, 513 CrossRef CAS PubMed.
- M. L. Matthews, C. S. Neumann, L. A. Miles, T. L. Grove, S. J. Booker, C. Krebs, C. T. Walsh and J. M. Bollinger, Proc. Natl. Acad. Sci. U.S.A, 2009, 106, 17723–17728 CrossRef CAS PubMed.
- S. Pandian, M. A. Vincent, I. H. Hillier and N. A. Burton, Dalton Trans., 2009, 6201–6207 RSC.
- S. P. De Visser and R. Latifi, J. Phy. Chem. B, 2009, 113, 12–14 CrossRef CAS PubMed.
- T. Borowski, H. Noack, M. Radon, K. Zych and P. E. Siegbahn, J. Am. Chem. Soc., 2010, 132, 12887–12898 CrossRef CAS PubMed.
- Z. Li, Y. Wang, W. Li, Q. Li, F. Li, Z. Gao, X. Fei, J. Tian and L. Dong, JBIC, J. Biol. Inorg. Chem., 2020, 25, 371–382 CrossRef CAS PubMed.
- H. Zimmermann, Propene-in Ullmann's Encyclopedia of Industrial Chemistry, John Wiley and Sons, 2013 Search PubMed.
- S. Ghosh, S. S. Acharyya, R. Tiwari, B. Sarkar, R. K. Singha, C. Pendem, T. Sasaki and R. Bal, ACS Catal., 2014, 4, 2169–2174 CrossRef CAS.
- J.-C. Liu, Y. Tang, C.-R. Chang, Y.-G. Wang and J. Li, ACS Catal., 2016, 6, 2525–2535 CrossRef CAS.
- J. Teržan, M. Huš, B. Likozar and P. Djinović, ACS Catal., 2020, 10, 13415–13436 CrossRef.
- E. Galitzenstein and C. Woolf, J. Soc. Chem. Ind., 1950, 69, 292–294 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz and J. L. S. A. F. Izmaylov, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, GAUSSIAN 09 (Revision D.01), Gaussian, Inc., C.T.Wallingford, 2013 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1992, 97, 9173–9177 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- R. A. Friesner, R. B. Murphy, M. D. Beachy, M. N. Ringnalda, W. T. Pollard, B. D. Dunietz and Y. Cao, J. Phys. Chem. A, 1999, 103, 1913–1928 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- M. Puri and L. Que Jr, Acc. Chem. Res., 2015, 48, 2443–2452 CrossRef CAS PubMed.
- C. T. Saouma and J. M. Mayer, Chem. Sci., 2014, 5, 21–31 RSC.
- S. P. De Visser, D. Kumar, S. Cohen, R. Shacham and S. Shaik, J. Am. Chem. Soc., 2004, 126, 8362–8363 CrossRef CAS PubMed.
- L. Ji, A. S. Faponle, M. G. Quesne, M. A. Sainna, J. Zhang, A. Franke, D. Kumar, R. van Eldik, W. Liu and S. P. de Visser, Chem.–Eur. J., 2015, 21, 9083–9092 CrossRef CAS PubMed.
- E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S.-K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y.-S. Yang and J. Zhou, Chem. Res., 2000, 100, 235–350 CAS.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed.
- L. C. Blasiak and C. L. Drennan, Acc. Chem. Res., 2009, 42, 147–155 CrossRef CAS PubMed.
- S. Shaik, G. D. Straganz, M. Costas, P. Comba, F. Neese, P. Siegbahn, T. Borowski, A. Mulholland, A. Dey and A. W. Munro, Iron-containing enzymes: Versatile catalysts of hydroxylation reactions in nature, Royal Society of Chemistry, 2011 Search PubMed.
- D. Buongiorno and G. D. Straganz, Coord. Chem. Rev., 2013, 257, 541–563 CrossRef CAS PubMed.
- S. Shaik, D. Kumar and S. P. De Visser, J. Am. Chem. Soc., 2008, 130, 10128–10140 CrossRef CAS PubMed.
- R. Latifi, M. Bagherzadeh and S. P. de Visser, Chem.–Eur. J., 2009, 15, 6651–6662 CrossRef CAS PubMed.
- A. Timmins, M. G. Quesne, T. Borowski and S. P. de Visser, ACS Catal., 2018, 8, 8685–8698 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra01384c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |