
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA concise and efficient total synthetic route of active stilbene dimer (±)-ε-viniferin†
Qibin Zhu‡
 ,
Binhao Teng‡,
Ying Chen‡,
Fubao Su,
Yanqiu Li,
Qingyun Yang and
Chunsuo Yao*
,
Binhao Teng‡,
Ying Chen‡,
Fubao Su,
Yanqiu Li,
Qingyun Yang and
Chunsuo Yao*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, P. R. China. E-mail: yaocs@imm.ac.cn
First published on 8th April 2022
Abstract
A concise and efficient procedure for the total synthesis of natural stilbene dimer (±)-ε-viniferin was accomplished with high overall yield. Demethylation of the key intermediate methyl 3-arylbenzofuran-4-carboxylate was achieved successfully through bromination followed by BBr3-or BCl3/TBAI-mediated ether cleavage reaction. Pd/C and bromobenzene-catalyzed MOM ether cleavage was successfully carried out to aquire (±)-ε-viniferin.
Oligostilbenes are highly oxygenated natural products possessing more than two stilbene monomers and complex structures. The total synthesis of several oligostilbenes isolated from natural sources has been investigated in the past three decades due to their interesting structures and different biological activities.1–7 (±)-ε-Viniferin (1), a natural resveratrol dimer containing the 2,3-diaryl-4-styryldihydrobenzofuran skeleton (Fig. 1), has been isolated from Vitaceaeous plants and determined to have potent activities, such as anti-inflammatory, antioxidant, antifungal, and anticancer.8–13 However, only a few chemical approaches for this compound have been reported because of their unique carbon framework and structural instability.14,15 Most of the attempts to prepare (±)-ε-viniferin derived from its presumed biogenesis, that is, oxidative dimerization of resveratrol, which led to the generation of its dimer (±)-ε-viniferin.16–20 Several well-designed cascadse reactions and total synthesis methods for the chemically controlled synthetic routes are not readily applicable to the synthesis of (±)-ε-viniferin because of the insufficient regioselectivity and low yield.6,7,21,22 Therefore, the development of an effective synthetic route for the preparation of (±)-ε-viniferin is of great important.
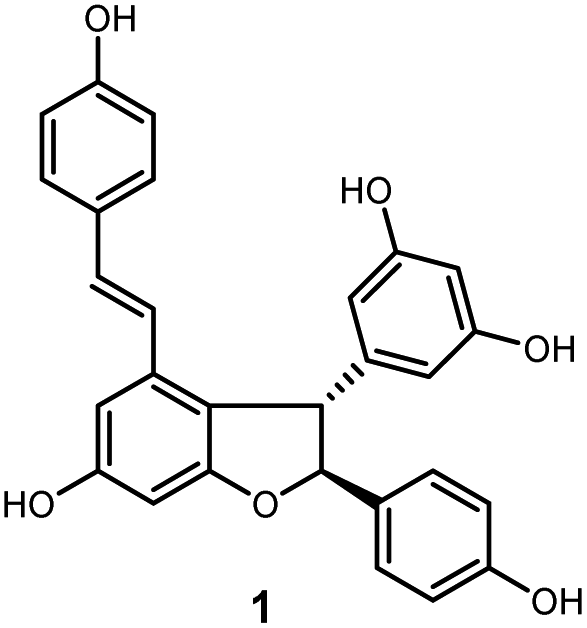 | ||
| Fig. 1 Structure of (±)-ε-viniferin (1). | ||
In 2009, Ikyon Kim and Jihyun Choi reported a versatile synthetic route to permethylated vinferifuan.23 Encouraged by their work, Elofsson et al. investigated the total synthesis of (±)-ε-viniferin and reported a synthetic route with methyl, cyclopropylmethyl and acetyl as protecting group.21 However, the long reaction steps and multiple protecting group switch make their synthetic route unsuitable for specific alterations of substitution patterns. We also failed in our investigation on the total synthesis of (±)-ε-viniferin due to the unsuccessful demethylation of pentamethylated (±)-ε-viniferin in the last step.22
Considering the importance of (±)-ε-viniferin as an active lead compound and existing synthesis challenges, we continuously focused our attention on the total synthesis of (±)-ε-viniferin and its analogues. In this study, we report a practical total synthetic route of (±)-ε-viniferin on the basis of the exploration of the demethylation of methylated methyl 3-arylbenzofuran-4-carboxylate mediated by BBr3 or BCl3 under the assistance of bromination.
In 2016, Elofsson and our group described two approaches to the synthesis of (±)-ε-viniferin with phenols protected as methyl or cyclopropylmethyl (cPrMe) ethers.21,22 As outlined in Scheme 1, etherification of compounds 2 (or 2a) and 3 followed by dehydrative cyclization (or alkoxycarbonylation) generated the key intermediate 3-arylbenzofuran 4 (or 4a). Then, direct arylation of 4 (or 4a) produced the key intermediate 5 (or 5a), which was converted to methyl or cyclopropylmethyl ethers of (±)-ε-viniferin 6 (or 6a) through further elaboration. However, when 6 (or 6a) was treated with well-established techniques for ether cleavage, it was unexpectedly directly converted into (±)- ampelopsin F (7) or (±)-ampelopsin B (8) rather than into the desired target (±)-ε-viniferin (1). This result indicates that the conditions for removing the alkyl ether-protecting group were incompatible with the structure of 2,3-diaryl-4-styryldihydrobenzofuran. Hence, we realized that a different protecting group was necessary for the synthesis. Recently, we found that MOM as protecting group is easy to remove under the presence of Pd/C, bromobenzene, and hydrogen, but it has no effect on the 2,3-diaryl-4-styryldihydrobenzofuran skeleton. Comparison with acetyl group, MOM is able to tolerate the strong alkali conditions in the Horner–Wadsworth–Emmons-type olefination reaction. Its removal conditions is compatible with the 2,3-diaryl-4-styryldihydrobenzofuran skeleton. So, we envisioned that the MOM protecting group is suitable in the total synthesis of (±)-ε-viniferin.
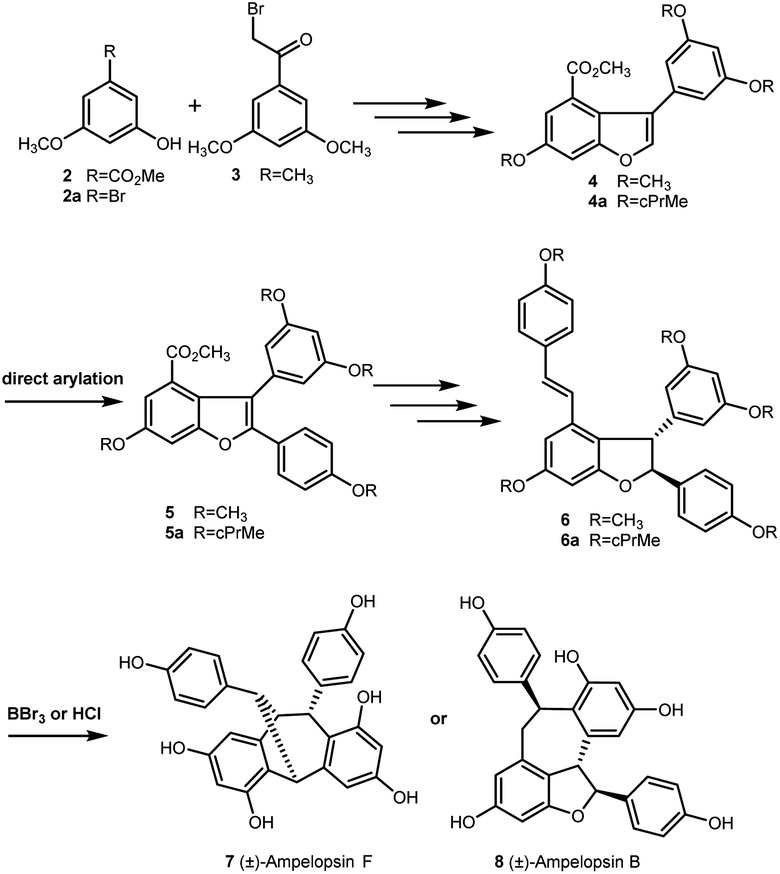 | ||
| Scheme 1 Previous synthetic approach to stilbene dimers. | ||
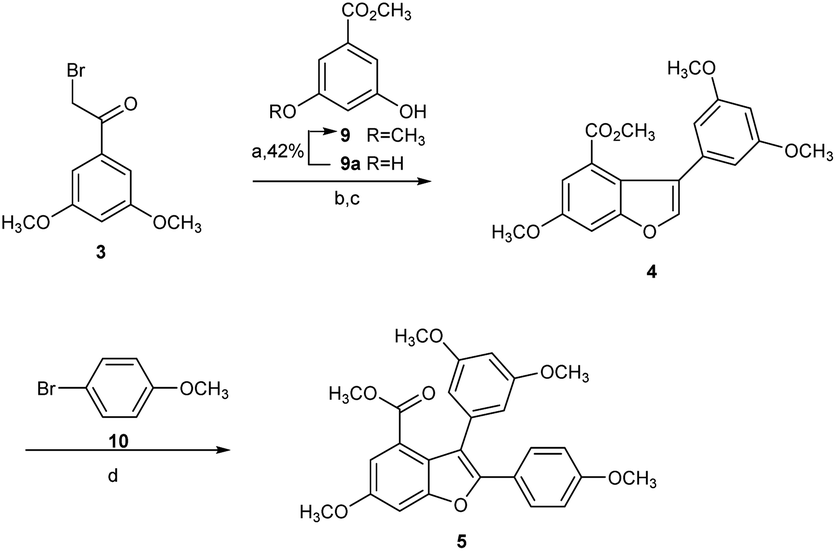 | ||
| Scheme 2 Preparation of methylated arylbenzofuran 4 and diarylbenzofuran 5. Reagents and conditions: (a) K2CO3, acetone, CH3I, r.t., 18 h, 42%; (b) K2CO3, acetone, reflux, 3 h, 98%; (c) Bi(OTf)3, DCM, reflux, 23 h, 81%; (d) Pd(OAc)2, PCy3·HBF4, Cs2CO3, Pivalic acid, DMA, 140 °C, 20 h, 87.0%. | ||
Hence, we commenced our synthesis with the preparation of 2,3-diarylbenzofuran (5), which was prepared from the etherification of α-bromoketone 3 and phenol 9 with 98% yield in the presence of K2CO3, followed by Bi(OTf)3-catalyzed dehydrative cyclization to produce 2-arylbenzofuran intermediate 4 with 81% yield, which was then converted to 5 via direct arylation with 10 at the C2 position in 87% yield (Scheme 2).22,23
Next, we focused our effort on the demethylation of the key intermediate 2,3-diarylbenzofuran 5. When 5 was treated with BBr3 in dichloromethane at −45 °C, followed by slow warming to room temperature overnight under nitrogen atmosphere, it was unexpectedly converted into Friedel–Crafts acylation product 11 rather than into the desired demethylation target molecule 15. Most well-established techniques for ether cleavage have been investigated, but none has been found to remove methyl groups with acceptable yield without Friedel–Crafts cyclization.24–26 Even with the more stable tert-butyl ester 5b instead of methyl ester 5, the same results were obtained. In line with literature,27 the acylation reaction occurred easily at the ortho-positions of 3,5-dimethoxyphenyl, which were prone to bromination. Moreover, the addition of halogen electrophile to alkenes was reversible. Initially, we envisaged that demethylation of 5 could be realized from bromide 12 through global demethylation and dehalogenation reactions. Exposure of 5 to 10 equivalent of Br2 in CH2Cl2 at 0 °C provided 12 with three extra halogen attached in 80% yield, and subsequent demethylation of 12 with BBr3 in CH2Cl2 under nitrogen atmosphere was achieved successfully to provide 13 in 39% yield. To our surprise, debromination of 13 in the presence of Pd/C under hydrogen atmosphere for 16 h produced only the monobrominated product 14 in 79% yield, but not the desired global dehalogenation product 15. The large steric hindrance at ortho-positions of 3,5-dimethoxyphenyl caused by the aromatic ring at C2 in 14 could be responsible for the results (Scheme 3).
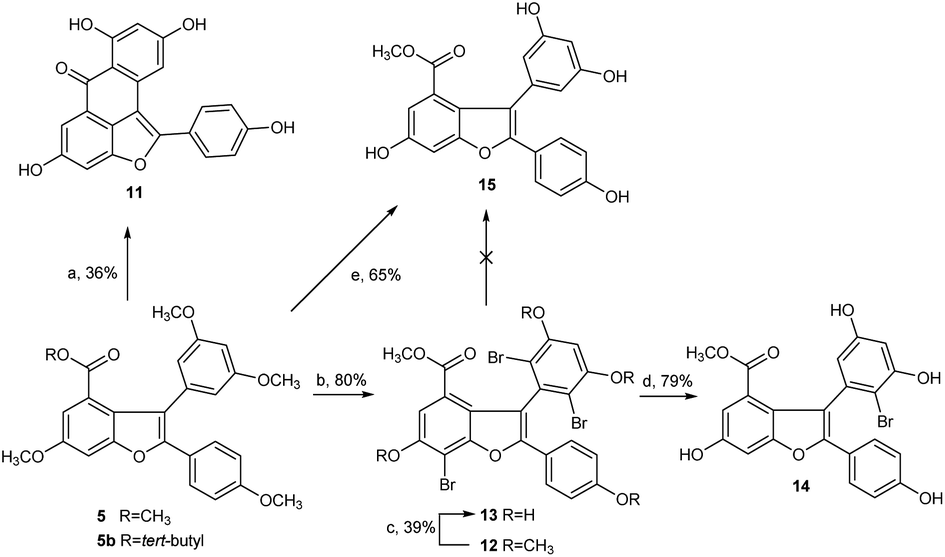 | ||
| Scheme 3 Demethylation of diarylbenzofuran 5. Reagents and conditions: (a) BBr3, DCM, N2,−45 °C -r.t, overnight, 36%; (b) Br2, DCM, 0 °C, 6 h, 80%; (c) BBr3, DCM, N2, −45 °C -r.t, overnight, 39%; (d) 10% Pd–C, H2 (0.6 MPa), 16 h, 79%; (e) BCl3, TBAI, Ar, 0 °C, 6 h, 65%. | ||
Afterward, we turned our attention to another intermediate 3-arylbenzofuran 4, which is theoretically the most likely to generate demethylated product 15. Exposure of 4 to 10 equivalent of bromine source in dichloromethane at 0 °C for 6 h afforded the desired bromide 16 with four extra halogen attached in 86% yield. From this bromide, BBr3-catalyzed demethylation in CH2Cl2 at −45 to 0 °C and subsequent Pd/C-catalyzed hydrogenative debromination in methanol under hydrogen atmosphere were achieved smoothly to provide 17 and 18 with yields of 42% and 67%, respectively, as expected. When 18 was exposed to the conditions [Pd(OAc)2, PCy3·HBF4Cs2CO3, pivalic acid, 19, DMA, 140 °C, 20 h] as reported by Zhang et al.,22 followed by Pd/C-catalyzed debenzylation reaction in ethanol under hydrogen atmosphere, the key intermediate 15 was obtained successfully with an overall yield of 44% in two steps (Scheme 4). Obviously, C2 arylation reaction of 18 also proceeded smoothly in the absence of protecting group for phenolic hydroxyls, although the yield is lower.
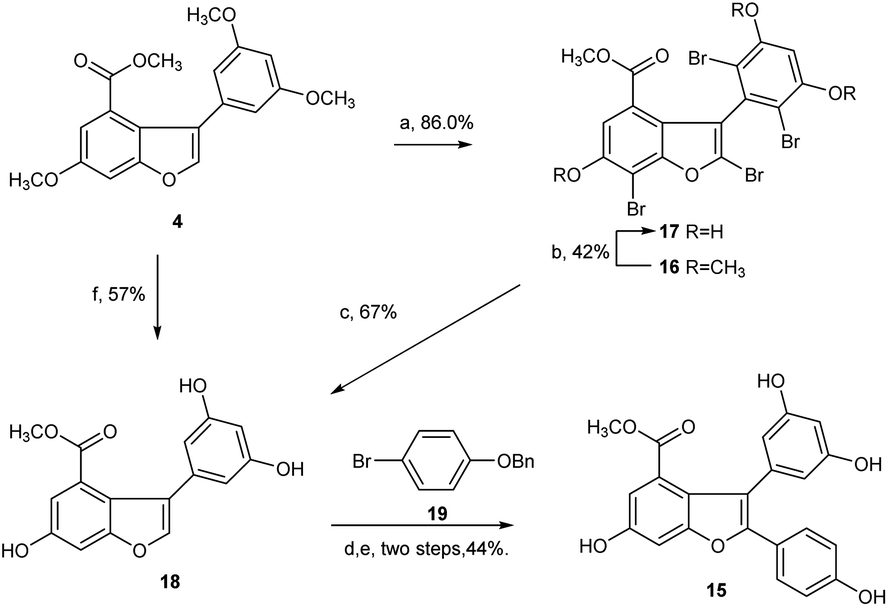 | ||
| Scheme 4 Demethylation of arylbenzofuran 4 and preparation of diarylbenzofuran 15. Reagents and conditions: (a) Br2, DCM, 0 °C, 6 h, 86%; (b) BBr3, DCM, −45 to 0 °C, overnight, 42%; (c) 10% Pd–C, H2 (0.5 MPa), MeOH, 6 h, 67%; (d) Pd(OAc)2, PCy3·HBF4, Cs2CO3, pivalic acid, DMA, 140 °C, 20 h; (e) 10% Pd–C, H2 (0.5 MPa), EtOH, 6 h. 44% for two steps; (f) BBr3, DCM, Ar, −20 to 0 °C, 6 h, 57%. | ||
However, the overall yield of 11% for 15 from 4 in four steps was too low to be acceptable. Through a series of investigations, combined with the methods reported in literature,25,26 we found that under high-purity argon atmosphere, direct demethylation of 4 mediated by BBr3 in CH2Cl2 at −20 °C to 0 °C produced the target 18 successfully with 57% yield (Scheme 4). Similarly, direct demethylation of 5 mediated by BCl3/TBAI in CH2Cl2 at 0 °C under argon atmosphere produced the target 15 with 65% yield (Scheme 3). Evidently, both global demethylation reactions were implemented at high yields, and high-purity argon played an important role in the two reactions.
With the key intermediate diarylbenzofuran 15 on hand, we explored the total synthesis of (±)-ε-viniferin. As mentioned above, identifying an applicable protecting group for phenolic hydroxyls is the critical issue in this synthesis. The group should be able to tolerate the strong alkali conditions in the Horner–Wadsworth–Emmons-type olefination reaction, and its removal conditions should be compatible with the 2,3-diaryl-4-styryldihydrobenzofuran skeleton. The protecting group MOM, which we found recently and proved to be easy to remove under mild conditions using Pd/C and bromobenzene under hydrogen atmosphere, should be suitable for this procedure. More importantly, its removing conditions has no effect on the reducible group (double bond) in (±)-ε-viniferin 1.
Similar to literature,6,22 when 15 was treated with triethylsilane in trifluoroacetic acid at 0 °C to room temperature overnight, dihydrobenzofuran 20 was obtained with 65% yield. Product 20 was then treated with MOMCl in the presence of Cs2CO3 in dry acetone at room temperature for 20 h to achieve MOM ether 21 with 97% yield. The methyl ester in 21 was subsequently reduced using LiAlH4 in THF at room temperature, then reoxidized with Dess–Martin periodinane in dichloromethane overnight to produce aldehyde 22 with an overall yield of 66% in two steps. As expected, Horner–Wadsworth–Emmons-type olefination of 22 with diethyl 4-methoxymethoxy benzylphosphonate 23 in the presence of t-BuOK in THF at room temperature for 24 h successfully generated MOM ether 24 with 93% yield. Final deprotection of MOM ether 24 catalyzed by Pd/C and bromobenzene in MeOH under hydrogen atmosphere led to the production of (±)-ε-viniferin 1 in quantitative yield (97%, Scheme 5).
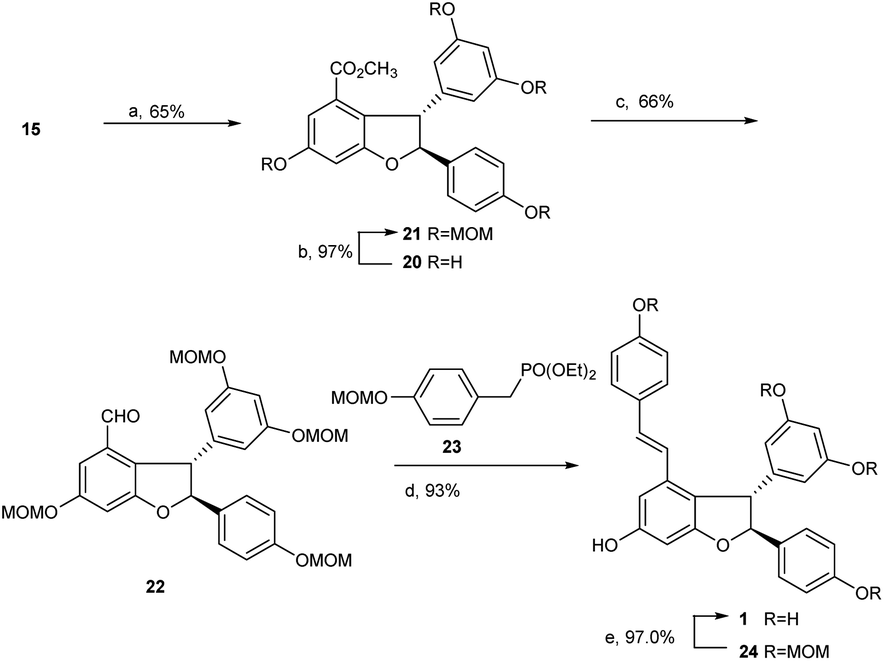 | ||
| Scheme 5 Synthesis of (±)-ε-viniferin. Reagents and conditions: (a) TFA, TES, 0 °C-r.t., overnight, 65%; (b) MOMCl, Cs2CO3, r.t., 20 h, 97%; (c) LiAlH4, THF, MeOH, r.t., 6 h; Dess-Martin, DCM, r.t., overnight, 66% for two steps; (d) t-BuOK, THF, r.t., 24 h, 93%; (e) 10% Pd–C, PhBr, H2 (0.5 MPa), MeOH, 5 h. 97%. | ||
In conclusion, we accomplished the total synthesis of resveratrol dimer (±)-ε-viniferin in 14 steps with an overall yield of 6.8%, through which a series of natural viniferin analogues could be prepared conveniently. Demethylation of the key intermediate methyl 3-arylbenzofuran-4-carboxylate was achieved through consecutive bromination, BBr3-mediated direct demethylation, and dehalogenation catalyzed by Pd/C and hydrogen or BBr3-and BCl3/TBAI-mediated direct demethylation under argon atmosphere. Pd/C and bromobenzene-catalyzed MOM ether cleavage was successfully carried out to acquire (±)-ε-viniferin for the first time. The efficient procedure for the removal of the MOM group in the presence of reducible and acid-sensitive groups was meaningful, and it will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the CAMS Innovation Fund for Medical Sciences (CIFMS 2021-I2M-1-028). We are grateful to the department of Instrumental Analysis, Institute of Material Medica, Chinese Academy of Medical Sciences and Peking Union Medical College for measuring the NMR and HRMS spectra.Notes and references
- M. H. Keylor, B. S. Matsuura and C. R. J. Stephenson, Chem. Rev., 2015, 115, 8976 CrossRef CAS PubMed.
- T. Shen, X.-N. Wang and H.-X. Lou, Nat. Prod. Rep., 2009, 26, 916 RSC.
- X.-F. Wang and C.-S. Yao, J Asian Nat. Prod. Res., 2016, 18, 376 CrossRef CAS PubMed.
- C.-H. Shang, Y.-L. Kang, Q.-Y. Yang, Q.-B. Zhu and C.-S. Yao, Adv. Synth. Catal., 2019, 361, 3768 CrossRef CAS.
- B.-H. Teng, Q.-B. Zhu, Y.-Y. Fan and C.-S. Yao, J Asian Nat. Prod. Res., 2020, 22, 947 CrossRef CAS PubMed.
- K. J. Romero, M. H. Keylor, M. Griesser, X. Zhu, E. J. Strobel, D. A. Pratt and C. R. J. Stephenson, J. Am. Chem. Soc., 2020, 142, 6499 CrossRef CAS PubMed.
- N. E. Wright and S. A. Snyder, Angew. Chem., Int. Ed., 2014, 53, 3409 CrossRef CAS PubMed.
- H. S. Cho, J.-H. Lee, S. Y. Ryu, S. W. Joo, M. H. Cho and J. Lee, J. Agric. Food Chem., 2013, 61, 7120 CrossRef CAS PubMed.
- C. Privat, J. P. Telo, V. Bernardes-Genisson, A. Vieira, J.-P. Souchard and F. Nepveu, J. Agric. Food Chem., 2002, 50, 1213 CrossRef CAS PubMed.
- B. Piver, F. Berthou, Y. Dreano and D. Lucas, Life Sci., 2003, 73, 1199 CrossRef CAS PubMed.
- J. Fu, J. Jin, R. H. Cichewicz, S. Hageman, T. Ellis, L. Xiang, Q. Peng, M. L. Jiang, N. Arbez, K. Hotaling, C. A. Ross and W.-Z. Duan, J. Biol. Chem., 2012, 287, 2446 CrossRef PubMed.
- C. Quiney, D. Dauzonne, C. Kern, J.-D. Fourneron, J.-C. Izard, R. M. Mohammad, J.-P. Kolb and C. Billard, Leuk. Res., 2004, 28, 851 CrossRef CAS PubMed.
- C. Billard, J.-C. Izard, V. Roman, C. Kern, C. Mathiot, F. Mentz and J.-P. Kolb, Leuk. Lymphoma, 2002, 43, 1991 CrossRef CAS PubMed.
- T. H. Jepsen, S. B. Thomas, Y. Lin, C. I. Stathakis, I. Miguel and S. A. Snyder, Angew. Chem., Int. Ed., 2014, 53, 6747 CrossRef CAS PubMed.
- Z. Wu, H. Li, X. Zhu, S. Li, Z. Wang, L. Wang, Z. Li and G. Chen, Catalysts, 2017, 7, 188 CrossRef.
- J.-Q. Zhang, G.-P. Li, Y.-L. Kang, B.-H. Teng and C.-S. Yao, Molecules, 2017, 22, 819 CrossRef PubMed.
- M. Liu, T. Dong, X. Guan, Z. Shao and W. Li, Tetrahedron, 2018, 74, 4013 CrossRef CAS.
- Y. Takaya, K. Terashima, J. Ito, Y.-H. He, M. Tateoka, N. Yamaguchi and M. Niwa, Tetrahedron, 2005, 61, 10285 CrossRef CAS.
- K.-S. Huang, M. Lin and Y.-H. Wang, Chin. Chem. Lett., 1999, 10, 817 CAS.
- R. Pezet, FEMS Microbiol. Lett., 1998, 167, 203 CrossRef CAS.
- A. E. G. Lindgren, C. T. Öberg, J. M. Hillgren and M. Elofsson, Eur. J. Org. Chem., 2016, 426 CrossRef CAS.
- J.-F. Zhang, J.-Q. Zhang, Y.-L. Kang, J.-G. Shi and C.-S. Yao, Synlett, 2016, 27, 1587 CrossRef CAS.
- I. Kim and J. Choi, Org. Biomol. Chem., 2009, 7, 2788 RSC.
- K. Kim and I. Kim, Org. Lett., 2010, 12, 5314 CrossRef CAS PubMed.
- D. D. Voa and M. Elofsson, Adv. Synth. Catal., 2016, 358, 4085 CrossRef PubMed.
- T. H. Jepsen, S. B. Thomas, Y. Lin, C. I. Stathakis, I. D. Miguel and S. A. Snyder, Angew. Chem., Int. Ed., 2014, 53, 1 CrossRef PubMed.
- S. A. Snyder, S. P. Breazzano, A. G. Ross, Y. Lin and A. L. Zografos, J. Am. Chem. Soc., 2009, 131, 1753 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data for compounds. See https://doi.org/10.1039/d2ra01385a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |