
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodine(I) complexes incorporating sterically bulky 2-substituted pyridines†
Jas S. Ward *a,
Rosa M. Gomilab,
Antonio Fronterab and
Kari Rissanena
*a,
Rosa M. Gomilab,
Antonio Fronterab and
Kari Rissanena
aUniversity of Jyvaskyla, Department of Chemistry, Jyväskylä 40014, Finland. E-mail: james.s.ward@jyu.fi
bDepartment of Chemistry, Universitat de les Illes Balears, Crts de Valldemossa km 7.6, 07122 Palma de Mallorca, Baleares, Spain
First published on 21st March 2022
Abstract
The silver(I) and iodine(I) complexes of the 2-substituted pyridines 2-(diphenylmethyl)pyridine (1) and 2-(1,1-diphenylethyl)pyridine (2), along with their potential protonated side products, were synthesised to investigate the steric limitations of iodine(I) complex formation. The complexes were characterised by 1H and 1H–15N HMBC NMR, X-ray crystallography, and DFT calculations. The solid-state structures for the silver(I) and iodine(I) complexes were extensively compared to the literature and analysed by DFT to examine the influence of the sterically bulky pyridines and their anions.
Introduction
Since their advent in the 1960s,1,2 halogen(I) (also known as halonium) ions, X+ (X = Br, I), stabilised by a pair of Lewis bases (L) in the form [L–X–L]+, have existed as examples of halogen group elements formally in the unusual +1 oxidation state, though it was not until the 1990s that they gained mainstream recognition due to the myriad of organic transformations they were deftly demonstrated to effect.3–5 In addition to this utility, halogen(I) ions possess other favourable properties arising from their σ-hole interaction,6 most notably the reliable high degree of linear directionality which has been fruitfully utilised in self-assembling supramolecular architectures,7–9 and recently in coordination polymers such as halogen-bonded organic frameworks (XOFs).10Halogen(I) complexes can be straightforwardly synthesised in a one pot reaction by addition of an elemental halogen, X2 (X = Br, I) to the analogous 2-coordinate silver(I) complex by Ag+ to X+ cation exchange,11,12,14 or as was recently shown, also from 3-coordinate silver(I) complexes via partial cation exchange.15,16
The use of substituted pyridines as the stabilising Lewis bases dominates the literature of halogen(I) complexes, and of those examples, it is pyridines substituted in the 4-position which overwhelmingly comprise the largest subset after pyridine itself.17 The 4-position of coordinating pyridines is one that can be described as only electronically affecting halogen(I) ion formation, and has been previously utilised to explore that relationship in halogen(I) complexes.11 With respect to the relationship of sterics toward halogen(I) formation, currently the iodine(I) complexes in the literature with the most steric bulk around the I+ ion are those incorporating 2,6-dimethylpyridine (2,6-lutidine) and 2,4,6-trimethylpyridine,18–22 as well as a single solid-state example of a bromine(I) complex with quinoline as the Lewis base,23 though this is not including the molecular clamps reported by Erdélyi and co-workers as those ligands would also provide an additional stabilising contribution via the chelate effect.9,11 Barluenga's reagent, [I(py)2]BF4 (py = pyridine), the ubiquitous iodination reagent for which iodine(I) chemistry owes its current renown, is commercially available and demonstrates a vast scope of utility, however, decomposition is observed over time. Therefore, an expansion of the pyridine scaffold would be an ideal starting point to explore the steric limitations of halogen(I) ions, and their potential applications toward a new generation of halogen(I) reagents.
Results and discussion
Synthesis and solution studies
The silver(I) complexes [1–Ag–1]PF6 (1a) and [2–Ag–2]PF6 (2a) were synthesised quantitatively from the two sterically bulky pyridine-based ligands, 2-(diphenylmethyl)pyridine (1) and 2-(1,1-diphenylethyl)pyridine (2), respectively (Scheme 1). The synthesis of the iodine(I) analogues, [1–I–1]PF6 (1b) and [2–I–2]PF6 (2b), were performed by addition of an equivalent of elemental iodine. The reactions were all followed by 1H and 1H–15N correlated NMR spectroscopy. | ||
| Scheme 1 The synthesis of silver(I) (1a, 2a) and iodine(I) (1b, 2b) complexes of 2-(diphenylmethyl)pyridine (1) and 2-(1,1-diphenylethyl)pyridine (2). | ||
The 1H NMR spectra of the free ligand 1, to Ag+ complex 1a, and finally to I+ complex 1b revealed that all peaks demonstrate noticeable shifts for each transformation, with the ranges of 0.04–0.56 ppm (1 to 1a) and 0.01–0.84 ppm (1a to 1b), the most apparent being those for the downfield pyridyl and upfield methine resonances that are free from overlapping chemical shifts with the pendant phenyl rings (Fig. 1). Whilst the 1H NMR data did provide clear indications of clean reactions occurring, they could not themselves point toward the identity of the products. It should be noted that the pyridyl resonances in 1a (green) do not follow the trend of shifting toward downfield as observed for 1 to 1b, which is likely due to the increased electron density on the pyridyl rings due to retro-donation with the Ag+ metal centre.
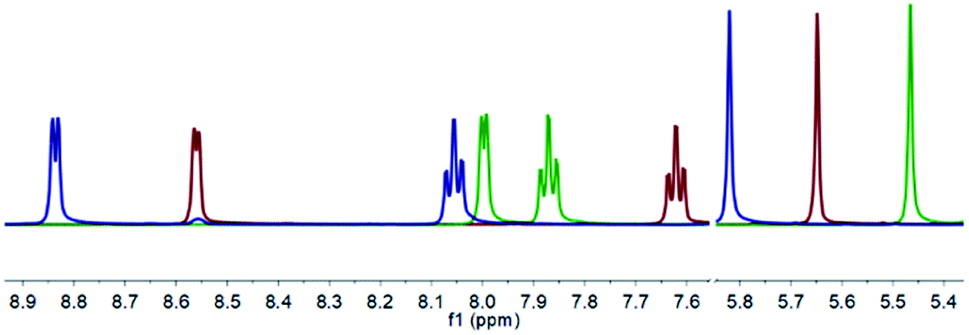 | ||
| Fig. 1 The superimposed 1H NMR spectra of 1 (red), 1a (green), and 1b (blue) of their non-overlapping pyridyl and methine resonances in CD2Cl2 (500 MHz, 298 K). | ||
In contrast, from the 1H–15N HMBC NMR experiments, the 15N NMR chemical shifts of −65.4 ppm (1), −118.6 ppm (1a), and −165.7 ppm (1b) are characteristic of the desired conversions having been achieved, resembling results observed for other iodine(I) complexes of 4-substituted pyridine analogues.12,13 Anticipating an increased likelihood of decomposition due to the steric hindrance of the substituents in the 2-positions of the pyridyl rings, the protonated (1c) and hydronium (1d) complexes were deliberately synthesised for comparison, which gave 15N NMR chemical shifts of −167.5 and −122.5 ppm, respectively. Given the reactivity of halogen(I) complexes, their propensity to decompose to protonated species, and the similarity of the 15N NMR chemical shifts of 1a to 1d (Δδ15N = 3.9 ppm) and 1b to 1c (Δδ15N = 1.8 ppm), caution must always be taken in characterising halogen(I) species based solely on NMR spectroscopy data. Nevertheless, the identity of 1a and 1b were definitely confirmed by single crystal X-ray diffraction studies.
The conversion of the free ligand 2 to the Ag+ complex 2a demonstrated similar changes in the 1H NMR chemical shifts as observed for 1 to 1a, with a range of 0.07–0.90 ppm, and 15N NMR chemical shifts of −65.1 ppm (2) and −111.4 ppm (2a). However, despite the precipitation of the AgI by-product of cation exchange upon addition of elemental iodine to effect the transformation of 2a to 2b, NMR studies suggested that the desired iodine(I) complex 2b had already begun to decompose within minutes of its inception. The 1H NMR spectrum showed that the pyridyl protons were significantly broadened, and the concomitant 1H–15N HMBC experiment gave a 15N NMR chemical shift of −121.5 ppm.‡ This 15N NMR chemical shift was far from the expected value of approximately −165 ppm for the desired iodine(I) complex 2b, though it did match well to the independently synthesised hydronium species [2–H–2]PF6 (2d), which had a 15N NMR chemical shift of −123.0 ppm.
Solid-state studies
The solid-state structure of 1a contained two half cations which self-completed by symmetry, and similarly 1b contained just one half, which in both instances ensured that all N–Ag–N and N–I–N angles were symmetry enforced, i.e., perfectly linear. The slightly elongated Ag–N bond lengths of 2.151(2) and 2.162(2) Å in 1a, in combination with the I–N bond length of 2.273(3) Å in 1b, were as expected and unremarkable when compared to those observed for [Ag(pyridine)2]PF6 (2.129(6) Å) and [I(pyridine)2]PF6 (2.268(2) Å),24,25 or even the more closely related 2-ethylpyridine (2-Etpy) derivatives [Ag(2-Etpy)2]PF6 (2.128(3)/2.130(3) Å) and [I(2-Etpy)2]PF6 (2.270(2) Å),13 with only the silver(I) comparisons being slightly beyond a 3σ tolerance and therefore crystallographically distinguishable from one another. It should be noted that the previously reported [I(2-Etpy)2]PF6 adopted a counterintuitive syn-configuration of the 2-ethyl substituents, with concomitant loss of co-planarity (an angle of 32.4° between the planes of the two pyridyl rings was found). However, in both 1a and 1b, the ligands were co-planar and, as expected, assumed anti-configurations (Fig. 2) due to steric considerations, with one of the two pendant phenyl rings pointing directly away from the Ag+ or I+ centres, respectively.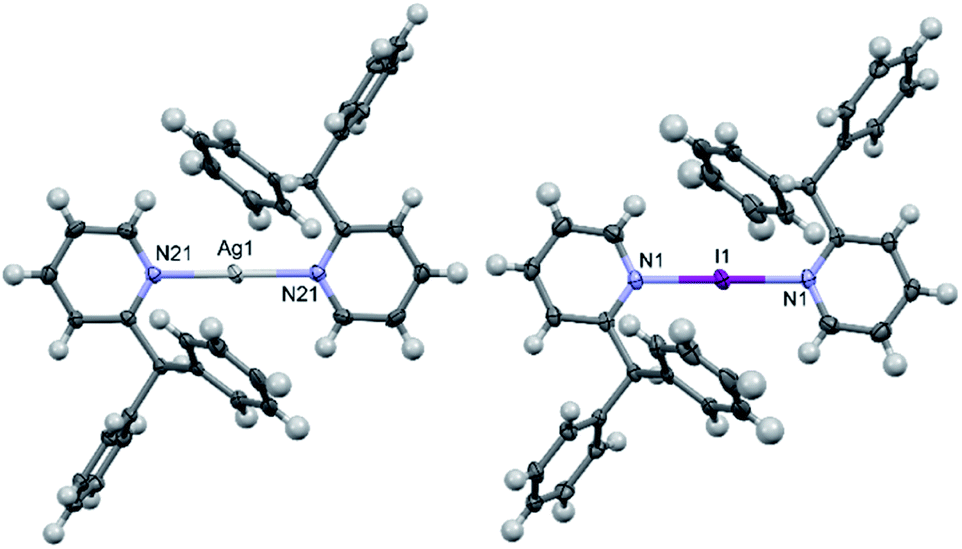 | ||
| Fig. 2 The X-ray crystal structures of the cations of [1–Ag–1]PF6 (1a; left) and [1–I–1]PF6 (1b; right) showing the anti-configuration of the ligands due to steric considerations (PF6 anions omitted for clarity; thermal ellipsoids at 50% probability). | ||
Interestingly, the I+ centre of 1b is noticeably exerting an increased repulsion on the 2-substituents, despite the ligands being further apart from one another and the I+ when compared to the Ag+ in 1a. This distortion is not readily apparent in the previously discussed bond lengths or angles, but can be quantified by comparison of the I+⋯pyridyl(C2)/I+⋯pyridyl(C6) through-space distances in 1b of 3.074(5)/3.252(4) Å, which are reminiscent of those observed for [I(2-Etpy)2]PF6 of 3.086(2)/3.244(2) Å, and are clearly deviating from the more alike pair of distances of Ag+⋯pyridyl(C2)/Ag+⋯pyridyl(C6) in 1a of 3.065(2)/3.070(2) Å and 3.050(3)/3.085(2) Å. Whilst a few examples of discrete silver(I) complexes have been reported with similarly sterically bulky 2-substituted pyridines,17,26 though all less sterically encumbered than 1a and 2a, no examples of such sterically endowed iodine(I) complexes currently exist in the literature.
The solid-state structure of 2a (Fig. 3) revealed significantly lengthened Ag–N bond lengths of 2.210(2) and 2.210(2) Å, which are, as best as can be determined,17 some of the longest known to date for a discrete, linear silver(I) complex incorporating 2-substituted pyridines, only rivalled by those of [Ag(4-(phenylethynyl)pyridine)2]+ (2.214(5) and 2.217(5) Å).27 Whilst longer Ag–N bond lengths are present in the literature, in those instances the elongation can be associated to the complexes exhibiting significantly distorted N–Ag–N angles due to partial coordination from another donor to the silver(I) centre, such as coordination from an anion,28,29 or from strain caused by the ligand system due to it being dimeric/polymeric in nature,30 or both.31 Unlike in 1a and 1b, the pyridyl ligands in 2a are not co-planar, nor do they assume an anti-configuration, with a N–Ag–N angle of 173.04(7)° and an angle of 78.4° between the planes of the two pyridyl rings. If the deviation from linearity of the N–I–N angle in 2b was similar to that observed for 2a, then this could contribute to the high reactivity of 2b, given that the largest observed deviation from linearity for an iodine(I) complex is for [I(2-Etpy)2]PF6 with a N–I–N angle of 173.62(10)°.13
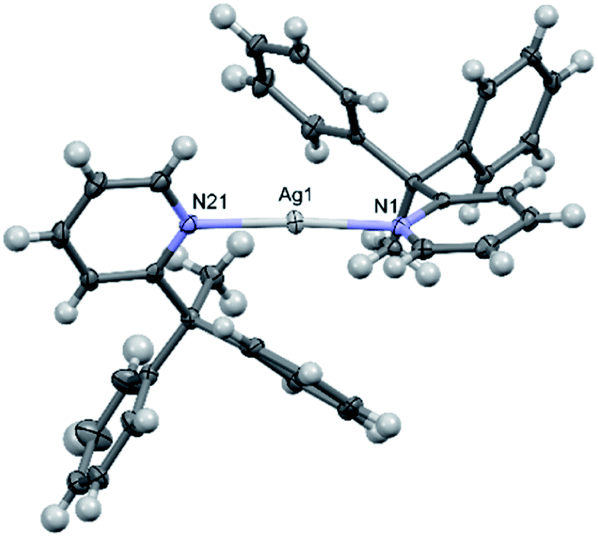 | ||
| Fig. 3 The X-ray crystal structure of the cation of [2–Ag–2]PF6 (2a) (PF6 anion omitted for clarity; thermal ellipsoids at 50% probability). | ||
Whilst an increase in reactivity for 2b was anticipated relative to 1b due to the slightly increased steric bulk of its substituent, the rapid decomposition of 2b was particularly striking in comparison to the only minimal decomposition that was observed after 8 days for a sample of 1b kept in solution for the duration. The persistence of 1b is drastically longer than for many other known iodine(I) complexes incorporating Lewis bases with sterically negligible substituents in the 2-positions, such as 2-ethylpyridine and 1-ethylpiperidine,14 both of which demonstrated behaviour resembling that of 2b.§.
Computational studies
DFT calculations (M06-2X/def2-TZVP level of theory, see ESI† and theoretical methods below for details) were performed to investigate the counterintuitive syn-configuration of 2a and also to characterise computationally the elusive 2b structure that could not be structurally characterised by X-ray diffraction methods. Fig. 4 shows the optimised structure of 2a and the hypothetical anti-configuration (denoted as 2a′), that exhibits a perfectly linear N–Ag–N angle and co-planar pyridyl rings. This configuration is 2.3 kcal mol−1 less stable than the syn-configuration, in line with the experimental observation.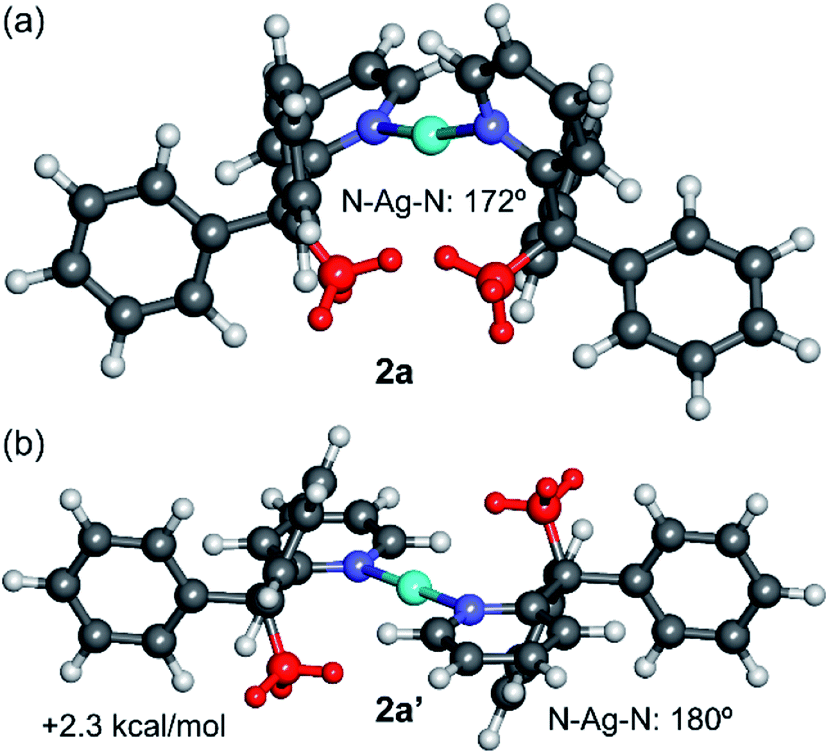 | ||
| Fig. 4 M06-2X/def2-TZVP optimised geometries of the syn (a) and anti (b) configurations of the cation of 2a, with indication of the relative energy. The methyl groups are represented in red. | ||
The DFT optimised structure exhibits a N–Ag–N angle of 172° and angle between the pyridyl rings of 81°, in good agreement with the experimental values (cf. 173° and 78°, respectively). Similar agreement, including distances, was observed for the rest of complexes (see ESI, Table S2†), thus giving reliability to this level of theory. The larger stability of the syn-configuration in 2a is most likely due to the contribution of van der Waals interactions between the methyl groups and the aromatic rings, as revealed by the noncovalent interaction plot analysis (NCIPlot index, see Fig. S43 in the ESI†).
The geometries of the syn- and anti-configurations of compound 2b were also calculated (Fig. 5). The calculations reveal that both isomers are practically isoenergetic (the syn-configuration is only 0.2 kcal mol−1 more stable). A likely explanation is that the stabilisation due to the co-planarity of the rings in the anti-configuration is more important in the iodine(I) complex 2b than in the silver(I) complex 2a. In fact, the anti-configuration facilitates the back-donation from the iodine atom (free lone pair) to the π-system, which is important in iodine(I) complexes.14 This effect compensates for the van der Waals interactions that are established in the syn-configuration, which are also less important in 2b, as revealed by the NCIPlot index (Fig. S43†). The geometries of the Ag+ and I+ compounds (2a and 2b) in the anti-configuration are very similar. In contrast, those in the syn-configuration (2a′ and 2b′) are more different, especially regarding the pyridyl ring plane angles that differ by 21° (cf. 60° in 2b′ and 81° in 2a′).
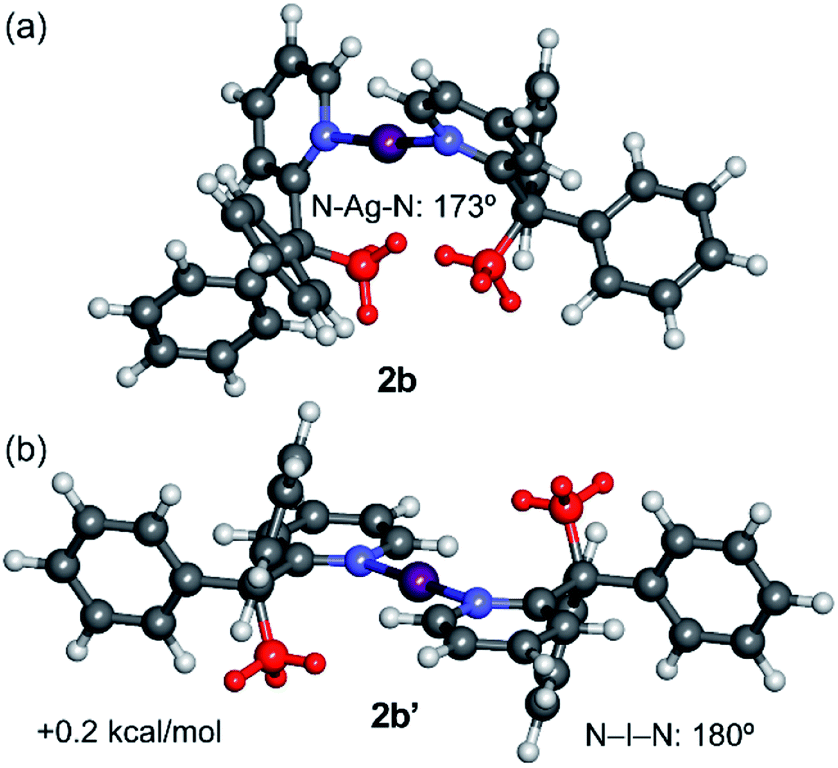 | ||
| Fig. 5 M06-2X/def2-TZVP optimised geometries of the syn (a) and anti (b) configurations of the cation of 2b, with indication of the relative energy. The methyl groups are represented in red. | ||
Finally, the dissociation energies (measured as [L–I–L]+ to I+ + 2L; Edis) and I–N distances of the iodine(I) complexes 1b and 2b were compared with previously reported examples instead incorporating the quinuclidine (quin) and dimethylaminopyridine (DMAP) ligands (Table 1).14 The results show the greatest stability for the [I(quin)2]+ and [I(DMAP)2]+ complexes followed by 1b and 2b in line with the I–N distances that are significantly longer in the latter complexes. The lowest dissociation energy corresponds to compound 2b, which agrees with its higher reactivity as observed in the NMR experiments.
| 1b | 2b(syn) | 2b (anti) | [I(quin)2]+ | [I(DMAP)2]+ | |
|---|---|---|---|---|---|
| Edis | 126.8 | 119.8 | 119.6 | 179.7 | 181.8 |
| d | 2.273 | 2.305 | 2.301 | 2.288 | 2.245 |
| α | 180 | 173.8 | 180 | 180 | 180 |
Anion and packing effects
In halogen(I) chemistry, the BF4 and PF6 anions are traditionally used owing to their weakly coordinating nature, which therefore do not complicate the well-established Ag+ to X+ (X = Br, I) cation exchange process used to synthesise halogen(I) complexes. In the solid state, both the silver(I) complexes 1a and 2a showed meaningful interactions with their respective PF6 anions, though with 2a as a discrete ion pair and 1a as a continuous 1D array of Ag+⋯F–PF4–F⋯Ag+⋯F–PF4–F contacts (Fig. 6). The closest Ag+⋯F distances of 2.918(2)/2.990(2) Å for the two independent molecules in 1a were below the combined van der Waals radii of for these atoms (Ag + F = 3.19 Å), though are comparable to other previously reported linear silver(I) complexes, such as [Ag(2-Etpy)2]PF6 and [Ag(DMAP)2]PF6.12,13 Similarly, 1a was able to successfully synthesise 1b via cation exchange, just as has been reported for the aforementioned literature complexes. However, the closest Ag+⋯F distance of 2.810(1) Å in 2a is significantly shorter than those observed in 1a, and is reminiscent of silver(I) complexes with more strongly coordinating anions, such as those bearing potential oxygen donors like the nitrate anion. The more strongly bound PF6 anion in 2a is likely an outcome of the sterically bulky ligands preventing optimal electronic stabilisation for the Ag+ centre, which was similarly indicated by the long Ag–N bond lengths observed in 2a (vide supra).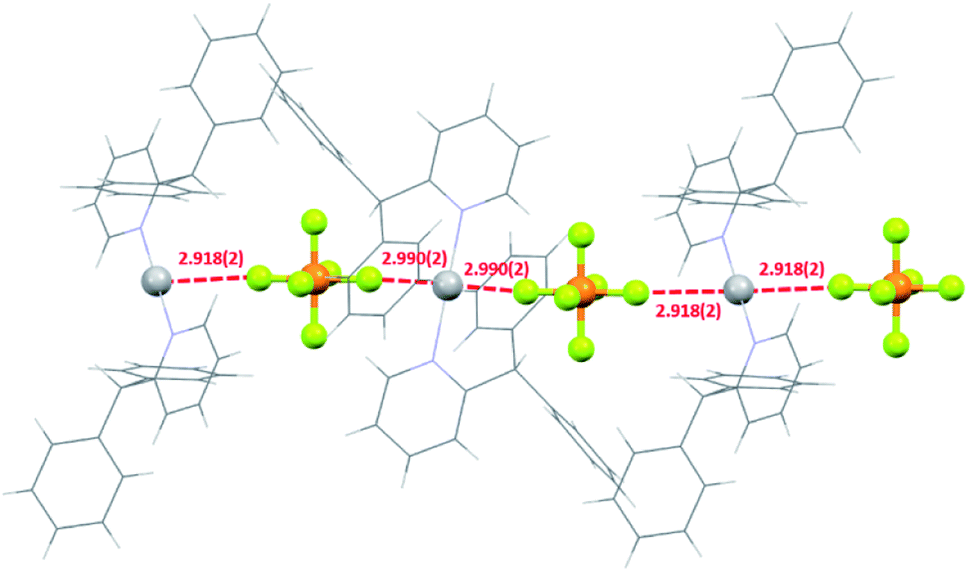 | ||
| Fig. 6 The packing of three molecules of 1a showing the 1D network of short Ag+–F intermolecular interactions (all distances in Å; ligands simplified for clarity). | ||
With respect to 1b, similar to 2a, it also existed as a discrete ion pair. Previous studies have confirmed that iodine(I) ions intrinsically impose a linear 2-coordination sphere, and unlike their silver(I) counterparts, are insensitive to the identity of the anion present.25 This is apparent in 1b with a pair of shortest I+⋯F distances of 3.618(5), which were reminiscent of those observed in [I(2-Etpy)2]PF6 (shortest I+⋯F distances = 3.693(2) Å),13 both of which were well over the combined van der Waals radii of these atoms (cf. van der Waals radii of I + F = 3.45 Å), indicating that neither were meaningful interactions.
Intrigued by the influence of the anions on the silver(I) precursors, the BF4, [1–Ag–1]BF4 (1e) and [2–Ag–2]BF4 (2e), and OTf, [1–Ag–1]OTf (1f; OTf = triflate) and [2–Ag–2]OTf (2f), anion analogues were also prepared and crystallised so comparisons could be made (Fig. 7). These anions were selected as they are commonly used in the preparation of halogen(I) complexes, so were more relevant than anions such as nitrate, and safer than others such as the perchlorate anion. Both 1e and 2e exist as discrete ion pairs with the BF4 anions, with the shortest Ag+⋯F distances of 2.828(5)/2.965(8) Å (the BF4 anion was found to equally occupy two positions in the solid state) and 2.743(2) Å, respectively. As expected, the OTf anion complexes 1f and 2f were observed as neutral T-shaped species, with the OTf anion bound to the Ag+ centres with Ag+–O bond lengths of 2.685(2) and 2.578(2) Å, respectively. These matched reported examples of OTf silver(I) complexes incorporating 2-substituted pyridines in the literature, [Ag(2-methoxypyridine)2]OTf and [Ag(2-methylsulfanylpyridine)2]OTf,32,33 which demonstrated the same T-shaped geometry and similar Ag+–O bond lengths of 2.679(3) and 2.673(3) Å (minor position (19%) of the disordered OTf anion ignored), respectively.
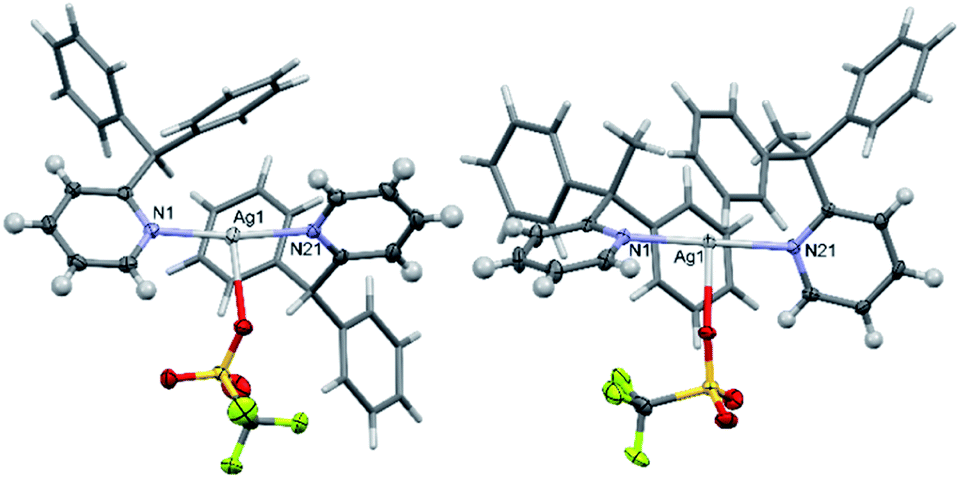 | ||
| Fig. 7 The X-ray crystal structures of [1–Ag–1]OTf (1f; left) and [2–Ag–2]OTf (2f; right) showing the bound OTf anions (pyridyl substituents simplified for clarity; thermal ellipsoids at 50% probability). | ||
These solid-state observations were reflected in the solution-state studies of the 1H–15N HMBC determined 15N NMR chemical shifts (Table 2). The values show that the comparisons of the BF4 (1e and 2e) and PF6 (1a and 2a) complexes only exhibit negligible differences for the same ligand (1a vs. 1e and 2a vs. 2e), whilst the OTf complexes showed small, but significant, differences of 4.1 (1a vs. 1f) and 1.4 (2a vs. 2f) ppm when compared to their PF6 analogues, possibly indicating that the less sterically encumbered complex 1f continues to interact with the OTf anion in solution, at least more so than 2f.
| Complex | δN | Complex | δN |
|---|---|---|---|
| [1–Ag–1]PF6 (1a) | –118.6 | [2–Ag–2]PF6 (2a) | –111.4 |
| [1–Ag–1]BF4 (1e) | –117.6 | [2–Ag–2]BF4 (2e) | –111.9 |
| [1–Ag–1]OTf (1f) | –114.5 | [2–Ag–2]OTf (2f) | –110.0 |
A common feature of all silver complexes is that the anion is close to the Ag(I) atom, establishing semi-coordination bonds, or coinage bonds (CiB) according to the nomenclature proposed by some authors.34,35 These contacts likely influence the Ag–N distance along with the bulkiness of the ligands. The CiBs in complexes 1a, e, f and 2a, e, f were analysed using the quantum theory of atoms-in-molecules (QTAIM).36 Fig. 8 shows the QTAIM representation of the six compounds showing in all cases a bond critical point (CP, represented as red sphere) and bond path (orange line) connecting one atom of the anion to the silver atom, thus confirming the existence of the interaction. In all cases both the Laplacian (∇2ρ) of the electron density and the total energy density (Hr) at the bond CP are positive, thus revealing that the Ag⋯F(O) contacts are noncovalent in nature (CiBs). This is corroborated by the small values or ρ (see Table 3), confirming the weak and closed shell nature of the CiBs. The strength of these CiBs was estimated by using the total energy density at the bond CP and the equation proposed by Espinosa et al.37 This method is convenient to evaluate the noncovalent interaction without the contribution of the pure coulombic attraction between the counterions. These values are indicated in Fig. 8 (annotated in red close to the CPs). The energies range from 2.3 kcal mol−1 in 1a to 9.5 kcal mol−1 in 2f, in line with the Ag⋯anion distances (see Table 3). It is interesting to highlight that for the complexes with the shortest Ag⋯anion distance of each series (complexes 1f and 2f) the density (ρ, see Table 3) at the bond CP is greater than that in the rest of complexes, thus suggesting a greater charge transfer from the anion to the Ag(I), thus causing the elongation and weakening of the N–Ag–N bonds, as evidenced by the longer Ag–N distances and smaller Wiberg bond indexes (see Table 3)38 in complexes 1f and 2f.
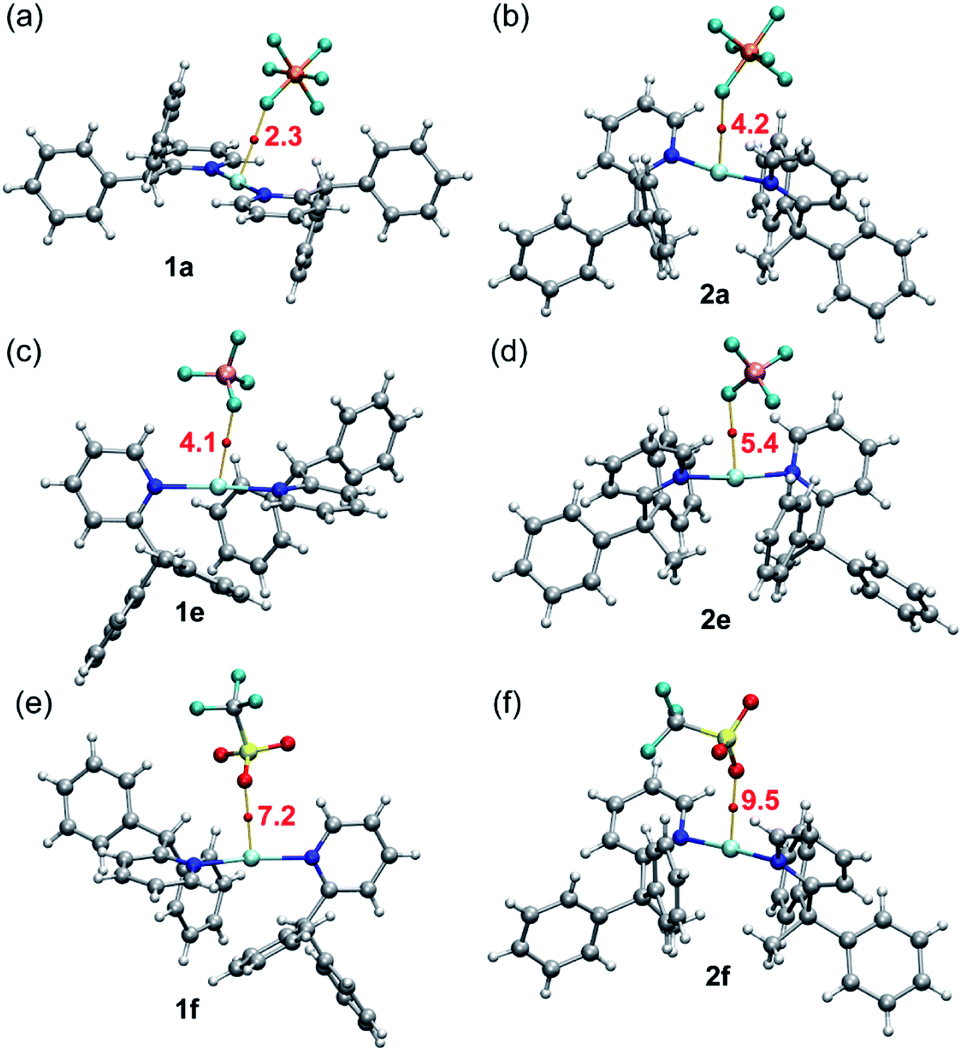 | ||
| Fig. 8 QTAIM analysis (only the Ag⋯anion contact is represented for clarity) of compounds 1a (a), 2a (b), 1e (c), 2e (d), 1f (e) and 2f (f). The dissociation energies are indicated in red adjacent to the bond CPs (red spheres). | ||
| Complex | Ag–N | Ag–X | ρ × 102 | WBI (Ag–N) |
|---|---|---|---|---|
| 1a | 2.151 | 2.990 | 0.97 | 0.164 & 0.159 |
| 1e | 2.150 & 2.155 | 2.828 | 1.46 | 0.144 & 0.150 |
| 1f | 2.170 & 2.175 | 2.685 | 2.23 | 0.138 & 0.133 |
| 2a | 2.210 | 2.818 | 1.45 | 0.092 & 0.095 |
| 2e | 2.215 & 2.219 | 2.743 | 1.75 | 0.081 & 0.091 |
| 2f | 2.224 & 2.223 | 2.578 | 2.67 | 0.080 & 0.082 |
Conclusions
In conclusion, silver(I) and iodine(I) complexes of sterically bulky 2-substituted pyridines were synthesised and spectroscopically characterised to investigate the steric limitations of iodine(I) ion formation. Three of these complexes, including two silver(I) and one iodine(I) complex, were also definitively confirmed by X-ray diffraction studies, thus demonstrating the possibility to form iodine(I) complexes with sterically bulky ligands in close proximity to the I+ centre. The effect of the anions on the sterically bulky silver(I) complexes was examined and further explored with a range of different anions, commonly used in halogen(I) chemistry, through extensive DFT studies. DFT calculations were utilised to explain the formation of the syn-isomer of 2a, as well as to study the decreased stability of 2b with respect to 1b and other iodine(I) complexes reported in the literature, highlighting the potential of steric control in future halogen(I) chemistry.Experimental
General considerations
All reagents and solvents were obtained from commercial suppliers and used without further purification, except for 2-(diphenylmethyl)pyridine (1) and 2-(1,1-diphenylethyl)pyridine (2) which were synthesised according to literature procedures.39,40 For structural NMR assignments, 1H NMR and 1H–15N NMR correlation spectra were recorded on a Bruker Avance III 500 MHz spectrometer at 25 °C in CD2Cl2. Chemical shifts are reported on the δ scale in ppm using the residual solvent signal as internal standard (CH2Cl2 in CD2Cl2: δH 5.32), or for 1H–15N NMR spectroscopy, to an external d3-MeNO2 standard. For the 1H NMR spectroscopy, each resonance was assigned according to the following conventions: chemical shift (δ) measured in ppm, observed multiplicity, observed coupling constant (J Hz), and number of hydrogen atoms. Multiplicities are denoted as: s (singlet), d (doublet), t (triplet), m (multiplet), and br (broad). For the 1H–15N HMBC spectroscopy, spectral windows of 4 ppm (1H) and 300 ppm (15N) were used, with 1024 points in the direct dimension and 512 increments used in the indirect dimension, with subsequent peak shape analysis being performed to give the reported 15N NMR resonances.The single crystal X-ray data for 1c was collected at 120 K using an Agilent SuperNova dual wavelength diffractometer with an Atlas detector using mirror-monochromated Cu-Kα (λ = 1.54184 Å) radiation. The single crystal X-ray data for 1a, 1b and 2a was collected at 120 K using an Agilent SuperNova diffractometer with an Eos detector using mirror-monochromated Mo-Kα (λ = 0.71073 Å) radiation The program CrysAlisPro41 was used for the data collection and reduction on the SuperNova diffractometer, and the intensities were absorption corrected using a Gaussian face index absorption correction method. All structures were solved by intrinsic phasing (SHELXT)42 and refined by full-matrix least squares on F2 using the OLEX2,43 utilizing the SHELXL-2015 module.44 Anisotropic displacement parameters were assigned to non-H atoms and isotropic displacement parameters for all H atoms were constrained to multiples of the equivalent displacement parameters of their parent atoms with Uiso(H) = 1.2 Ueq (aromatic) or Uiso(H) = 1.5 Ueq (alkyl) of their respective parent atoms. The X-ray single crystal data and CCDC numbers of all new structures are included below.
Synthesis and characterisation
All silver(I) and iodine(I) complexes were prepared using the same quantitative general methods, which are given below using [1–Ag–1]PF6 (1a) and [1–I–1]PF6 (1b) as examples.![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2), a = 8.5415(3) Å, b = 11.2632(6) Å, c = 16.8492(7) Å, α = 91.301(4)°, β = 97.022(3)°, γ = 101.834(4)°, V = 1572.71(12) Å3, Z = 2, Dcalc = 1.570 g cm−3, F(000) = 752, μ = 0.76 mm−1, T = 120.0(1) K, θmax = 29.2°, 7314 total reflections, 5852 with Io > 2σ(Io), Rint = 0.028, 7314 data, 461 parameters, 186 restraints, GooF = 1.06, 0.57 < dΔρ < −0.55 eÅ−3, R[F2 > 2σ(F2)] = 0.036, wR(F2) = 0.079.
(No. 2), a = 8.5415(3) Å, b = 11.2632(6) Å, c = 16.8492(7) Å, α = 91.301(4)°, β = 97.022(3)°, γ = 101.834(4)°, V = 1572.71(12) Å3, Z = 2, Dcalc = 1.570 g cm−3, F(000) = 752, μ = 0.76 mm−1, T = 120.0(1) K, θmax = 29.2°, 7314 total reflections, 5852 with Io > 2σ(Io), Rint = 0.028, 7314 data, 461 parameters, 186 restraints, GooF = 1.06, 0.57 < dΔρ < −0.55 eÅ−3, R[F2 > 2σ(F2)] = 0.036, wR(F2) = 0.079.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pentane (1:3) solution of 1b. Crystal data for 1b: CCDC-2144043, [C36H30IN2]PF6, M = 762.49, colourless plate, 0.04 × 0.10 × 0.23 mm3, monoclinic, space group C2/c, a = 20.8912(5) Å, b = 8.1839(2) Å, c = 18.9412(7) Å, β = 94.605(3)°, V = 3227.95(16) Å3, Z = 4, Dcalc = 1.569 g cm−3, F(000) = 1528, μ = 1.11 mm−1, T = 120.0(1) K, θmax = 28.0°, 3844 total reflections, 2994 with Io > 2σ(Io), Rint = 0.043, 3844 data, 230 parameters, 51 restraints, GooF = 1.09, 0.74 < dΔρ < −0.75 eÅ−3, R[F2 > 2σ(F2)] = 0.046, wR(F2) = 0.100. (No. 2), a = 11.3073(5) Å, b = 11.6452(4) Å, c = 15.0627(7) Å, α = 68.117(4)°, β = 85.174(4)°, γ = 71.906(4)°, V = 1748.34(14) Å3, Z = 2, Dcalc = 1.420 g cm−3, F(000) = 760, μ = 0.69 mm−1, T = 120.0(1) K, θmax = 29.2°, 8052 total reflections, 6904 with Io > 2σ(Io), Rint = 0.029, 8052 data, 424 parameters, no restraints, GooF = 1.05, 0.44 < dΔρ < −0.54 eÅ−3, R[F2 > 2σ(F2)] = 0.033, wR(F2) = 0.077.
:pentane (1:3) solution of 1b. Crystal data for 1b: CCDC-2144043, [C36H30IN2]PF6, M = 762.49, colourless plate, 0.04 × 0.10 × 0.23 mm3, monoclinic, space group C2/c, a = 20.8912(5) Å, b = 8.1839(2) Å, c = 18.9412(7) Å, β = 94.605(3)°, V = 3227.95(16) Å3, Z = 4, Dcalc = 1.569 g cm−3, F(000) = 1528, μ = 1.11 mm−1, T = 120.0(1) K, θmax = 28.0°, 3844 total reflections, 2994 with Io > 2σ(Io), Rint = 0.043, 3844 data, 230 parameters, 51 restraints, GooF = 1.09, 0.74 < dΔρ < −0.75 eÅ−3, R[F2 > 2σ(F2)] = 0.046, wR(F2) = 0.100. (No. 2), a = 11.3073(5) Å, b = 11.6452(4) Å, c = 15.0627(7) Å, α = 68.117(4)°, β = 85.174(4)°, γ = 71.906(4)°, V = 1748.34(14) Å3, Z = 2, Dcalc = 1.420 g cm−3, F(000) = 760, μ = 0.69 mm−1, T = 120.0(1) K, θmax = 29.2°, 8052 total reflections, 6904 with Io > 2σ(Io), Rint = 0.029, 8052 data, 424 parameters, no restraints, GooF = 1.05, 0.44 < dΔρ < −0.54 eÅ−3, R[F2 > 2σ(F2)] = 0.033, wR(F2) = 0.077.Theoretical methods
For the optimisations and single point calculations the M06-2X46/def2-TZVP47 level of theory and the Turbomole 7.2 program48 was used. This level of theory was previously used to study similar complexes.14–16 The def2-TZVP implementation used in this work employs for Ag the ECP-28 set and scalar relativistic effects.47 Frequency calculations were used to verify that the geometries correspond to true minima on the potential surface (no imaginary frequencies). No symmetry constraints were imposed for the calculations. Solvent calculations were considered using the conductor-like screening model (COSMO).49 QTAIM and NCIplot index, that is adequate to reveal noncovalent interactions in real space,50 were computed at the same level of theory by means of the MULTIWFN program51 and represented using the VMD software.52 The Wiberg bond index was computed using the NBO 7.0 program53 at the same level of theory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Magnus Ehrnrooth Foundation (J. S. W.), the MICIU/AEI of Spain (A. F. project PID2020-115637GB-I00, FEDER), the Academy of Finland (K. R. grant no. 317259), and the University of Jyvaskyla, Finland for financial support.Notes and references
- J. A. Creighton, I. Haque and J. L. Wood, Chem. Commun., 1966, 229 RSC.
- I. Haque and J. L. Wood, J. Mol. Struct., 1968, 2, 217–238 CrossRef CAS.
- J. Barluenga, J. M. González, M. A. Garcia-Martin, P. J. Campos and G. Asensio, J. Chem. Soc., Chem. Commun., 1992, 1016–1017 RSC.
- J. Ezquerra, C. Pedregal, C. Lamas, J. Barluenga, M. Pérez, M. A. García-Martín and J. M. González, J. Org. Chem., 1996, 61, 5804–5812 CrossRef CAS.
- G. Espuña, G. Arsequell, G. Valencia, J. Barluenga, M. Pérez and J. M. González, Chem. Commun., 2000, 1307–1308 RSC.
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033–14036 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Chem, 2017, 3, 861–869 CAS.
- A. Vanderkooy, A. K. Gupta, T. Földes, S. Lindblad, A. Orthaber, I. Pápai and M. Erdélyi, Angew. Chem., Int. Ed., 2019, 58, 9012–9016 CrossRef CAS PubMed.
- G. Gong, S. Lv, J. Han, F. Xie, Q. Li, N. Xia, W. Zeng, Y. Chen, L. Wang, J. Wang and S. Chen, Angew. Chem., Int. Ed., 2021, 60, 14831–14835 CrossRef CAS PubMed.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdélyi, J. Am. Chem. Soc., 2016, 138, 9853–9863 CrossRef CAS PubMed.
- J. S. Ward, G. Fiorini, A. Frontera and K. Rissanen, Chem. Commun., 2020, 56, 8428–8431 RSC.
- J. S. Ward, A. Frontera and K. Rissanen, Chem. Commun., 2021, 57, 5094–5097 RSC.
- J. S. Ward, A. Frontera and K. Rissanen, Dalton Trans., 2021, 50, 8297–8301 RSC.
- S. Yu, P. Kumar, J. S. Ward, A. Frontera and K. Rissanen, Chem, 2021, 7, 948–958 CAS.
- J. S. Ward, A. Frontera and K. Rissanen, Inorg. Chem., 2021, 60, 5383–5390 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- G. D. Brayer and M. N. G. James, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1982, 38, 654–657 CrossRef.
- A. A. Neverov, H. X. Feng, K. Hamilton and R. S. Brown, J. Org. Chem., 2003, 68, 3802–3810 CrossRef CAS PubMed.
- A. S. Batsanov, J. A. K. Howard, A. P. Lightfoot, S. J. R. Twiddle and A. Whiting, Eur. J. Org. Chem., 2005, 2005, 1876–1883 CrossRef.
- T. Okitsu, S. Yumitate, K. Sato, Y. In and A. Wada, Chem.–Eur. J., 2013, 19, 4992–4996 CrossRef CAS PubMed.
- L. C. F. Morgan, Y. Kim, J. N. Blandy, C. A. Murray, K. E. Christensen and A. L. Thompson, Chem. Commun., 2018, 54, 9849–9852 RSC.
- N. W. Alcock and G. B. Robertson, J. Chem. Soc., Dalton Trans., 1975, 2483–2486 RSC.
- C. Y. Chen, J. Y. Zeng and H. M. Lee, Inorg. Chim. Acta, 2007, 360, 21–30 CrossRef CAS.
- M. Bedin, A. Karim, M. Reitti, A.-C. C. Carlsson, F. Topić, M. Cetina, F. Pan, V. Havel, F. Al-Ameri, V. Sindelar, K. Rissanen, J. Gräfenstein and M. Erdélyi, Chem. Sci., 2015, 6, 3746–3756 RSC.
- C.-Q. Wan, Q.-S. Li, H.-B. Song, F.-B. Xu and Z.-Z. Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m1973–m1975 CrossRef CAS.
- J. V Knichal, W. J. Gee, C. A. Cameron, J. M. Skelton, K. J. Gagnon, S. J. Teat, C. C. Wilson, P. R. Raithby and A. D. Burrows, Eur. J. Inorg. Chem., 2017, 2017, 1855–1867 CrossRef.
- M. A. M. Abu-Youssef, R. Dey, Y. Gohar, A. A. Massoud, L. Öhrström and V. Langer, Inorg. Chem., 2007, 46, 5893–5903 CrossRef CAS PubMed.
- B. Kilduff, D. Pogozhev, S. A. Baudron and M. W. Hosseini, Inorg. Chem., 2010, 49, 11231–11239 CrossRef CAS PubMed.
- Z. Qin, L. Zhao, Z. Li, S. Tian, Q. Xiao, Y. Deng, J. Zhang, G. Li and C. Wan, Dalton Trans., 2019, 48, 6730–6737 RSC.
- X.-D. Chen and T. C. W. Mak, Chem. Commun., 2005, 3529–3531 RSC.
- C. Di Nicola, Effendy, F. Marchetti, C. Nervi, C. Pettinari, W. T. Robinson, A. N. Sobolev and A. H. White, Dalton Trans., 2010, 39, 908–922 RSC.
- M. I. Rogovoy, T. S. Frolova, D. G. Samsonenko, A. S. Berezin, I. Y. Bagryanskaya, N. A. Nedolya, O. A. Tarasova, V. P. Fedin and A. V Artem’ev, Eur. J. Inorg. Chem., 2020, 2020, 1635–1644 CrossRef CAS.
- A. C. Legon and N. R. Walker, Phys. Chem. Chem. Phys., 2018, 20, 19332–19338 RSC.
- A. Daolio, A. Pizzi, G. Terraneo, M. Ursini, A. Frontera and G. Resnati, Angew. Chem., Int. Ed., 2021, 60, 14385–14389 CrossRef CAS PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- D. A. Klumpp, M. Garza, G. V. Sanchez, S. Lau and S. de Leon, J. Org. Chem., 2000, 65, 8997–9000 CrossRef CAS PubMed.
- X. Ji, T. Huang, W. Wu, F. Liang and S. Cao, Org. Lett., 2015, 17, 5096–5099 CrossRef CAS PubMed.
- CrysAlis Pro, Agilent Technologies Ltd, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- U. Pieper and D. Stalke, Organometallics, 1993, 12, 1201–1206 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- E. D. Glendening, K. Badenhoop, J. A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, 2018 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, NMR, computational details, and X-ray. CCDC 2144042–2144045, 2150094–2150097. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ra01390h |
| ‡ The 1H NMR spectrum was collected within 5 minutes of I2 addition, but a satisfactory 1H–15N HMBC experiment took several hours to complete to give the chemical shift stated. |
| § Despite decomposition of [I(2-Etpy)2]PF6 being observed within minutes of its synthesis by NMR studies, it was robust enough for its solid-state structure to be obtained.13 |
| This journal is © The Royal Society of Chemistry 2022 |