
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photophysical characterization and fluorescence cell imaging applications of 4-N-substituted benzothiadiazoles†
Susanne Doloczki a,
Karl O. Holmbergb,
Ignacio Fdez. Galvána,
Fredrik J. Swartlingb and
Christine Dyrager*a
a,
Karl O. Holmbergb,
Ignacio Fdez. Galvána,
Fredrik J. Swartlingb and
Christine Dyrager*a
aDepartment of Chemistry – BMC, Uppsala University, Box 576, 75123 Uppsala, Sweden. E-mail: christine.dyrager@kemi.uu.se
bDepartment of Immunology, Genetics and Pathology, Rudbeck Laboratory, Uppsala University, 75185 Uppsala, Sweden
First published on 13th May 2022
Abstract
In this work, a series of fluorescent 2,1,3-benzothiadiazole derivatives with various N-substituents in the 4-position was synthesized and photophysically characterized in various solvents. Three compounds emerged as excellent fluorescent probes for imaging lipid droplets in cancer cells. A correlation between their high lipophilicity and lipid droplet specificity could be found, with log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P ≥ 4 being characteristic for lipid droplet accumulation.
P ≥ 4 being characteristic for lipid droplet accumulation.
Introduction
The 2,1,3-benzothiadiazole (BTD) unit is a key component of numerous highly tuneable fluorophores. BTD is electron-deficient, and its derivatives generally exhibit high photostability, large Stokes shifts and solvatochromic properties.1–4 These advantages have been broadly recognized in materials science for use as building blocks in conjugated polymers (e.g., in solar cell components).5–10 Recently, the BTD-motif has also shown its potential as a bioimaging scaffold for fluorescence cell microscopy.2,3 Our contribution to the latter has been the development of BTD-based dyes that specifically stain intracellular lipid droplets (LDs),11–13 lipid-rich organelles that are strongly associated with cancer progression and cancer cell survival.14–17 LDs serve as a biomarker for cancer diagnosis and prognosis and their relative quantity can be used as a measure for therapeutic response.14–17 Thus, LD-specific fluorophores are important molecular tools in the field of oncology. Several fluorescent probes for imaging LDs have been reported,18,19 including a few BTD derivatives described by other research groups.3,20–23 While many of these have shown advantages over the commonly used commercialized fluorescent probes (e.g., Nile Red and BODIPY™ 493/503), they often feature drawbacks, including unspecific staining, small Stokes shifts or not being easily accessible.18 Our previously reported LD-specific dye, LD-BTD1 (Fig. 1), overcomes each of these drawbacks.11 LD-BTD1 is readily synthesized in one step and displays large Stokes shifts, strong solvatochromism, and high fluorescence quantum yields in apolar solvents, as well as specific and bright staining of lipid droplets in fixed or live cancer cells.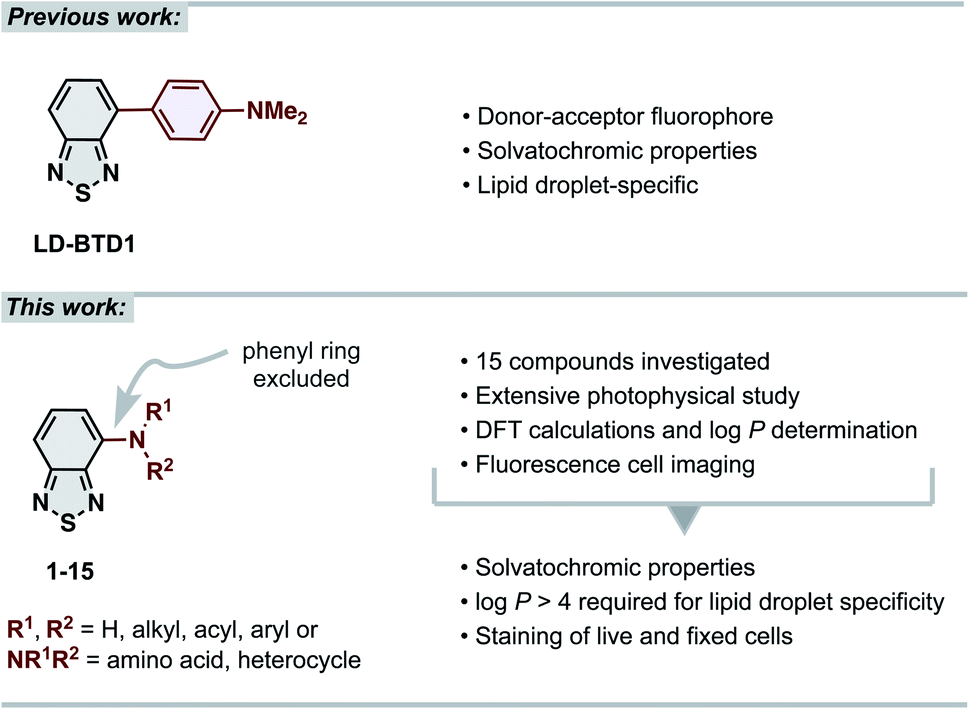 | ||
| Fig. 1 (Top) previous work: our previously reported lipid droplet dye, LD-BTD1.11 (Bottom) this work: extensive photophysical study of 4-N-substituted BTD derivatives and their utility as imaging agents for fluorescence cell microscopy. | ||
In this work we have explored the photophysical properties and cell imaging potential of 4-N-substituted BTDs – structures that lack the phenyl ring between the acceptor unit (BTD) and the amine donor, as present in LD-BTD1 (Fig. 1). We hypothesized that this approach could provide new solvatochromic fluorophores that retain selectivity for lipid droplets if they possess an appropriate hydrophobicity. Fourteen BTD derivatives with various N-substituents in the 4-position were synthesized and, together with 4-amino-BTD (BTD-NH2, 1, Scheme 1), subjected to an extensive photophysical study in solvents of different polarity. The octanol/water partition coefficient (logP) was calculated and, if practicable, experimentally determined. In addition, all compounds were investigated as potential fluorescent probes for cancer cell imaging (e.g., for staining LDs).
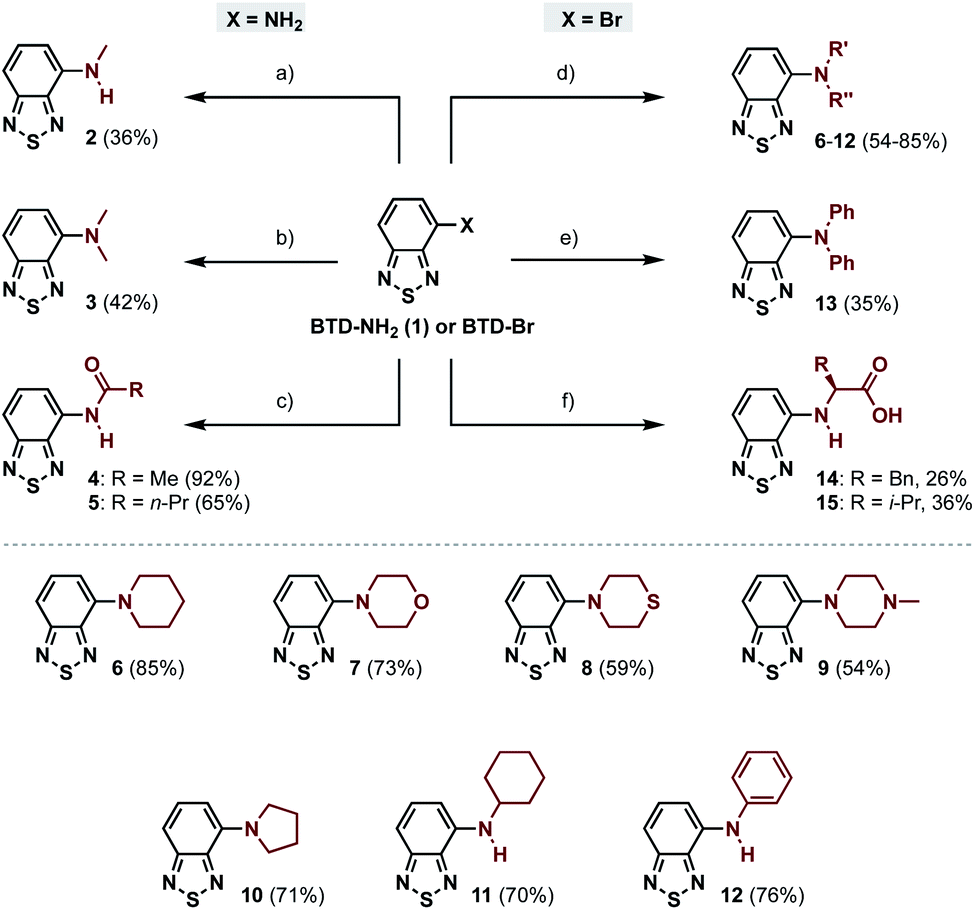 | ||
| Scheme 1 Synthesis of amino- and amido-BTD derivatives (2–15) starting from BTD-NH2 (2–5) or BTD-Br (6–15). (a) MeI, K2CO3, DMF, 50 °C, 20.5 h; (b) paraformaldehyde, NaBH3CN, AcOH, rt, 26 h; (c) acyl chloride, pyridine, CH2Cl2, 0 °C to rt, 2.5–3.5 h; (d) amine, PEPPSI-IPr, t-BuOK, toluene, 120 °C, 3.5–4.5 h; (e) HNPh2, Pd2(dba)3, RuPhos, Cs2CO3, toluene, 120 °C, 24 h; (f) L-amino acid, CuI, K2CO3, DMA, 90 °C, 5–6 d. | ||
Results and discussion
Synthesis
The 4-amino-BTDs (2, 3 and 6–15) were synthesized from BTD-NH2(1) or BTD-Br in one step, e.g., via metal-catalyzed cross-coupling reactions, whereas the 4-amido-BTDs (4 and 5) were obtained through nucleophilic addition (Scheme 1).Photophysical characterization
The photophysical properties of compounds 1–15 were investigated using UV-vis absorption and fluorescence spectroscopy in solvents of different polarity (Table 1). Measurements in aqueous solution were performed in water with 5% DMSO to facilitate solubilization.| Compd. | Solvent | λAmaxa | λEmaxa | Stokes shifta | εb | φF |
|---|---|---|---|---|---|---|
| a In nm and (cm−1).b In M−1 cm−1.c 5% DMSO in water.d Emission from precipitating particles.e Not determined due to poor solubility. n.e. = no emission detected. n.d. = not determined. | ||||||
| 1 | Hexane | 400 | 501 | 101 (5040) | 2000 | 0.37 |
| Toluene | 410 | 537 | 127 (5768) | 2700 | 0.24 | |
| THF | 425 | 568 | 143 (5924) | 2200 | 0.14 | |
| DMSO | 437 | 618 | 181 (6702) | 3000 | <0.01 | |
| MeOH | 421 | 628 | 207 (7829) | 2700 | <0.01 | |
| H2Oc | 403 | n.e. | — | 2400 | — | |
| 2 | Hexane | 420 | 513 | 93 (4316) | 3500 | 0.42 |
| Toluene | 430 | 553 | 123 (5173) | 3200 | 0.34 | |
| THF | 437 | 568 | 131 (5278) | 3400 | 0.22 | |
| DMSO | 446 | 615 | 169 (6161) | 2800 | 0.03 | |
| MeOH | 437 | 629 | 192 (6985) | 3300 | <0.01 | |
| H2Oc | 429 | n.e. | — | 2300 | — | |
| 3 | Hexane | 424 | 523 | 99 (4464) | 3600 | 0.41 |
| Toluene | 432 | 558 | 126 (5227) | 2800 | 0.34 | |
| THF | 434 | 576 | 142 (5680) | 2700 | 0.24 | |
| DMSO | 440 | 624 | 184 (6702) | 3200 | 0.06 | |
| MeOH | 432 | 625 | 193 (7148) | 2800 | <0.01 | |
| H2Oc | 394 | n.e. | — | 2800 | — | |
| 4 | Hexane | 372 | 436 | 64 (3946) | 3100 | 0.50 |
| 465 | 93 (5376) | |||||
| Toluene | 374 | 483 | 109 (6034) | 3400 | 0.79 | |
| THF | 374 | 488 | 114 (6246) | 3900 | 0.72 | |
| DMSO | 373 | 510 | 137 (7202) | 4200 | 0.72 | |
| MeOH | 362 | 516 | 154 (8244) | 3800 | 0.17 | |
| H2Oc | 352 | 536 | 184 (9752) | 3200 | 0.02 | |
| 5 | Hexane | 373 | 439 | 66 (4031) | 4800 | 0.58 |
| 467 | 94 (5396) | |||||
| Toluene | 375 | 485 | 110 (6048) | 3400 | 0.76 | |
| THF | 374 | 486 | 112 (6162) | 4000 | 0.69 | |
| DMSO | 373 | 509 | 136 (7163) | 4100 | 0.73 | |
| MeOH | 364 | 515 | 151 (8055) | 3700 | 0.16 | |
| H2Oc | 354 | 537 | 183 (9627) | 3100 | 0.02 | |
| 6 | Hexane | 413 | 533 | 120 (5451) | 3700 | 0.48 |
| Toluene | 420 | 569 | 149 (6235) | 3000 | 0.38 | |
| THF | 420 | 586 | 166 (6745) | 3100 | 0.28 | |
| DMSO | 423 | 627 | 204 (7692) | 2500 | 0.07 | |
| MeOH | 413 | 635 | 222 (8465) | 2700 | <0.01 | |
| H2Oc | 388 | 575d | 187 (8382) | n.d.e | n.d.d | |
| 7 | Hexane | 403 | 530 | 127 (5946) | 3300 | 0.59 |
| Toluene | 409 | 563 | 154 (6688) | 4000 | 0.50 | |
| THF | 408 | 578 | 170 (7209) | 3800 | 0.32 | |
| DMSO | 412 | 620 | 208 (8143) | 3000 | 0.08 | |
| MeOH | 402 | 629 | 227 (8977) | 2900 | <0.01 | |
| H2Oc | 380 | n.e. | — | 2600 | — | |
| 8 | Hexane | 405 | 526 | 121 (5680) | 3100 | 0.63 |
| Toluene | 414 | 557 | 143 (6201) | 3000 | 0.60 | |
| THF | 412 | 573 | 161 (6820) | 3200 | 0.36 | |
| DMSO | 417 | 614 | 197 (7694) | 2800 | 0.13 | |
| MeOH | 408 | 618 | 210 (8329) | 2700 | <0.01 | |
| H2Oc | 381 | n.e. | — | n.d.e | — | |
| 9 | Hexane | 409 | 532 | 123 (5653) | 3500 | 0.55 |
| Toluene | 414 | 565 | 151 (6455) | 2700 | 0.19 | |
| THF | 414 | 583 | 169 (7002) | 4000 | 0.05 | |
| DMSO | 416 | 620 | 204 (7909) | 3100 | <0.01 | |
| MeOH | 402 | 624 | 222 (8850) | 3100 | <0.01 | |
| H2Oc | 380 | n.e. | — | 2800 | — | |
| 10 | Hexane | 446 | 526 | 80 (3410) | 4900 | 0.53 |
| Toluene | 458 | 563 | 105 (4072) | 5000 | 0.44 | |
| THF | 457 | 575 | 118 (4491) | 4200 | 0.28 | |
| DMSO | 464 | 620 | 156 (5423) | 3100 | 0.08 | |
| MeOH | 459 | 633 | 174 (5989) | 4300 | <0.01 | |
| H2Oc | 460 | 579d | 119 (4468) | n.d.e | n.d.d | |
| 11 | Hexane | 431 | 523 | 92 (4081) | 3500 | 0.52 |
| Toluene | 438 | 554 | 116 (4781) | 3500 | 0.34 | |
| THF | 442 | 570 | 128 (5081) | 3400 | 0.23 | |
| DMSO | 451 | 619 | 168 (6018) | 3200 | 0.03 | |
| MeOH | 443 | 629 | 186 (6675) | 3100 | <0.01 | |
| H2Oc | 438 | n.e. | — | n.d.e | — | |
| 12 | Hexane | 432 | 511 | 79 (3579) | 6300 | 0.58 |
| Toluene | 439 | 544 | 105 (4397) | 5300 | 0.43 | |
| THF | 443 | 563 | 120 (4811) | 6000 | 0.25 | |
| DMSO | 450 | 615 | 165 (5962) | 5500 | <0.01 | |
| MeOH | 441 | n.e. | — | 5400 | — | |
| H2Oc | 441 | 562d | 121 (4882) | n.d.e | n.d.d | |
| 13 | Hexane | 448 | 531 | 83 (3489) | 4900 | 0.45 |
| Toluene | 453 | 573 | 120 (4623) | 4000 | 0.29 | |
| THF | 451 | 594 | 143 (5338) | 3100 | 0.13 | |
| DMSO | 451 | 638 | 187 (6499) | 3700 | 0.01 | |
| MeOH | 449 | n.e. | — | 3900 | — | |
| H2Oc | Insufficient solubility | |||||
| 14 | Hexane | 409 | 507 | 98 (4726) | n.d.e | 0.53 |
| Toluene | 414 | 535 | 121 (5463) | 2800 | 0.54 | |
| THF | 426 | 562 | 136 (5681) | 2900 | 0.34 | |
| DMSO | 434 | 598 | 164 (6319) | 3000 | 0.05 | |
| MeOH | 427 | 617 | 190 (7212) | 2800 | <0.01 | |
| H2Oc | 430 | n.e. | — | 2800 | — | |
| 15 | Hexane | 411 | 507 | 96 (4607) | 2200 | 0.52 |
| Toluene | 416 | 535 | 119 (5347) | 2800 | 0.47 | |
| THF | 427 | 553 | 126 (5336) | 2800 | 0.35 | |
| DMSO | 433 | 600 | 167 (6428) | 2700 | 0.07 | |
| MeOH | 428 | 615 | 187 (7104) | 3200 | <0.01 | |
| H2Oc | 434 | n.e. | — | 2200 | — | |
The lowest energy absorption maximum (λAmax) of most amines (1–3, 6–12, 14 and 15) displayed small bathochromic shifts (a few nm) with increasing solvent polarity (i.e., positive solvatochromism). However, this trend did not generally apply in polar protic solvents (MeOH and H2O). For tertiary amines (3, 6–9) even a hypsochromic shift of about 25 nm in water compared to hexane was observed. Blue-shifted λAmax of aromatic amines in polar protic solvents are commonly ascribed to hydrogen bonding interactions between the nitrogen lone pair and solvent protons.24–26 Primary and secondary amines are capable of acting as hydrogen bond donors (in addition to their hydrogen bond acceptor ability) which leads to an inverse effect (i.e., red-shift) on λAmax. Therefore, primary and secondary amines 1, 2, 11, 12, 14, and 15 displayed red-shifted λAmax, whereas tertiary amines 3 and 6–9 featured blue-shifted λAmax in water compared to hexane (for correlations with hydrogen bond energy see ESI, Fig. S27†). A few deviations from these trends were found among the studied compounds. For instance, the absorption wavelength of 13 was independent of solvent polarity, likely due to N lone pair conjugation with the three aromatic rings. The amides 4 and 5 were insensitive to aprotic solvents but exhibited hypsochromic shifts in polar protic solvents (e.g., ca. 20 nm in water compared to hexane). Furthermore, pyrrolidino-BTD 10 did not behave like the other tertiary amines as it featured red-shifted absorption in water (14 nm compared to hexane). DFT calculations of 6 and 10 indicated that the nitrogen atom is less pyramidalized in the latter case (Fig. 2A). A flattened geometry around the nitrogen atom facilitates conjugation of the nitrogen lone pair with the BTD π-system and makes 10 less susceptible to hydrogen bonding with the solvent (calculated hydrogen bond energies are 21.4 kJ mol−1 for 6 and 12.0 kJ mol−1 for 10).24 Further consequences of this were red-shifted λAmax (Fig. 2B) as well as higher molar extinction coefficients (ε) and brightness (ε × φF) of 10 in comparison with the larger cyclic amines (6–9).
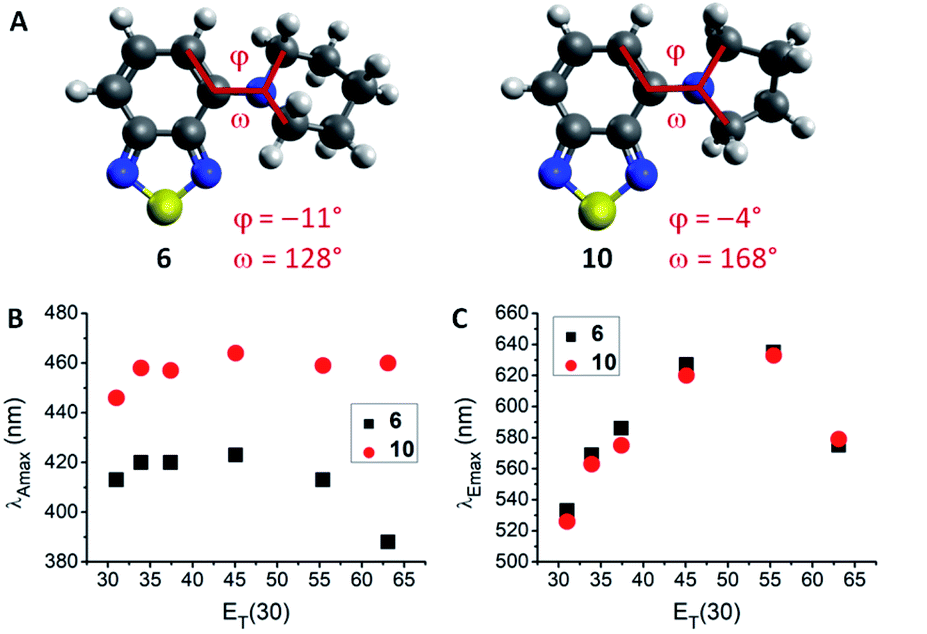 | ||
| Fig. 2 Comparison of 6 and 10. (A) Optimized ground state geometries in hexane. Dihedral angles (φ and ω) indicating the geometry around the nitrogen atoms are displayed, with 6 being more pyramidalized. (B) Absorption wavelength dependence on ET(30) solvent polarity parameter. (C) Emission wavelength dependence on ET(30) solvent polarity parameter. Emission in water could be ascribed to aggregates/precipitation. | ||
Regarding the maximum emission wavelength (λEmax), a clear trend was observed for all amino-BTDs, independent of their hydrogen bonding properties. For instance, compounds 6 and 10 showed the same behaviour in terms of λEmax (Fig. 2C). Furthermore, all studied compounds (1–15) showed positive solvatochromic fluorescence with a linear relationship between λEmax and Reichardt's ET(30) solvent polarity parameter27 in aprotic solvents (for amides also in protic solvents; ESI, Fig. S20†).
Emission wavelengths for the amines were in the range of 501–638 nm and blue-shifted to 436–537 nm for the amides. A linear correlation in aprotic solvents was also observed between fluorescence quantum yields (φF) and ET(30) (ESI, Fig. S20†). Thus, the fluorescence quantum yields of the amino-BTDs (1–3 and 6–15) decreased steadily upon increasing the solvent polarity, with complete quenching in polar protic solvents. The detected fluorescence of 6, 10 and 12 in water could be ascribed to aggregation and visible precipitation of the compounds, which are fluorescent in the solid state. Their blue-shifted emission was also indicative of the presence of aggregates.28 The fluorescence quantum yields for the amido-BTDs (4 and 5) remained high in aprotic solvents (φF ≈ 0.70) while MeOH (φF = 0.17 and 0.16, respectively) and water (φF = 0.02) caused partial quenching. The reason for less pronounced quenching in comparison with the amines might be decreased hydrogen bonding ability due to electron delocalization. Another unique feature within the series of compounds was observed for 4 and 5, as these showed dual emission in hexane (ESI, Fig. S4 and S5†). This feature is in line with findings from our recent study on substituted aryl-BTDs, which showed dual emission arising from the locally excited (LE) and intramolecular charge transfer (ICT) excited states – the latter of which was promoted by strongly electron-donating substituents and polar solvents.12 This ICT effect has also been discussed for other BTD-based fluorophores.29,30 It therefore seems reasonable to suggest that single emission profiles from amines in this work arise from the ICT state, whereas the weakened donor ability of the amides causes competing LE/ICT emission in hexane that is pushed to sole ICT emission in more polar solvents.
A few other notable differences in photophysical properties for some of the compound classes could be observed. For instance, the six-membered cyclic amines (6–9) had comparable photophysical behaviour independent of the integrated heteroatom. However, 9 experienced considerably stronger fluorescence quenching with increasing solvent polarity. Compounds 6–9 featured very large Stokes shifts (120–227 nm), with 7 representing the largest value of all our studied BTD derivatives. Large Stokes shifts were seen throughout the whole series (1–15) – a characteristic that is favourable for bioimaging applications.
Many of the compounds showed low molar extinction coefficients (ε), typically <4000 M−1 cm−1. However, addition of one phenyl substituent on the nitrogen (12) increased ε almost two-fold in comparison with the corresponding aliphatic derivative 11 (cf. 6300 vs. 3500 M−1 cm−1 in hexane). Consequently, 12 displayed the highest brightness among the studied amines 1–3 and 6–15 (notably, the amides were brighter in most solvents). Addition of a second phenyl substituent (13) did not have an amplifying effect. On the contrary, it generated lower ε and φF values. Nevertheless, compound 13 featured the most red-shifted fluorescence (e.g., 638 nm in DMSO) in the series of compounds.
Methylation of 1 to give 2 led to red-shifted absorption and emission (20 and 12 nm in hexane, respectively). The impact of a second methyl group (3), especially on λAmax, was smaller than that of the first methyl group (only 4 nm red-shift in hexane). The ε and φF values for 2 and 3 were comparable and in general higher than for 1. It should be noted that compound 3 in comparison to its π-extended equivalent LD-BTD1 (ref. 11) (Fig. 1) featured similar absorption and emission wavelengths. The fluorescence quantum yields, however, were generally lower for 3.
Plots of Stokes shifts vs. the ET(30) solvent polarity parameter showed a better linear correlation (R2 ≥ 0.892) than ordinary Lippert–Mataga plots31,32 (R2 ≥ 0.608), which are based on the solvent orientation polarizability parameter (ESI, Fig. S16–S19†). This linearity indicated that the observed spectral shifts are mainly caused by general solvent effects such as polarity. The relatively large positive slopes of the Lippert–Mataga plots indicated a significant increase in dipole moment upon excitation, which is consistent with the expected increase in charge separation in the ICT excited state.
The smallest compounds in this series, 1–4, were chosen for computational studies using hexane, THF and water as representative solvents of various polarity (ESI, Table S2†). DFT and TDDFT calculations using CAM-B3LYP/6-31G** level of theory resulted in absorption wavelengths that are in good agreement with the experimental data (maximal 30 nm deviation). Trends based on solvent (red-shift from hexane to THF and blue-shift in water) or molecular changes (red-shift with increasing donor strength) were largely well-described. Calculated emission wavelengths (based on optimized S1 excited state geometries) for 1–4 in hexane and for 4 in water and THF were in good agreement with the experimental data (1–3 were not emissive in water). A conformer and tautomer search did not reveal any likely candidates for the dual emission of 4, supporting the hypothesis of LE/ICT competition. The red-shifted emission with increasing solvent polarity was correctly represented in the calculations. However, it was notably overestimated, indicating an excessive stabilization of the excited state (for 1–3). Trends in λEmax based on molecular modifications were well-represented in the calculated data (except for 1 in THF). Good starting geometries with explicit solvent molecules hydrogen-bonded (as both, hydrogen bond donor and -acceptor) to the compound of interest were crucial for reliable computational results. Natural transition orbital analysis showed that the lowest transition corresponds to a HOMO–LUMO excitation, where the HOMO is mainly localized on the amine nitrogen and the benzo-moiety of the BTD, whereas the LUMO is BTD-centred (ESI, Table S3†). This indicated charge-transfer from the amine/amide substituent to the BTD acceptor for absorption into the first excited state. Comparison of optimized ground state (S0) and excited state (S1) structures of 1–4 in hexane further supported the putative ICT (ESI, Fig. S26†). Planarization of the amine moiety (coplanar to BTD) and shortening of the BTD–N bond in S1 indicated a strong sp2 character of the amine nitrogen after excitation. Furthermore, significant elongation of the BTD S–N bonds (ca. 4%) showed that the heterocycle acts as electron acceptor in the excited state. Similar effects on BTD-based fluorophores have previously been computed and discussed in literature.29 The length of the intramolecular hydrogen bond in compounds 1, 2 and 4 was significantly shortened (ca. 5%) in S1 as compared to their corresponding S0 structure, which is in line with the other observations.
Cell studies
Compounds 1–15 were investigated as imaging agents for fluorescence cell microscopy using two different cancer cell lines: SK-MEL-28 (melanoma) and MDA-MB-231 (breast cancer). Initial compound screening of live cells (30 μM, 24 h incubation) gave three potential imaging candidates (11, 12 and 13) that showed distinct punctate staining patterns in the green channel33 of the fluorescence microscope. These compounds were further submitted to immunocytochemical colocalization with markers for various cellular organelles (lipid droplets, mitochondria, endosomes, lysosomes, endoplasmic reticulum, and Golgi; ESI, Fig. S33–S37†). As exemplified by 13 (Fig. 3), the results showed colocalization with an ADFP antibody, which targets an enzyme that is prevalent on the surface on lipid droplets. The initial cell experiments were performed at 30 μM to compensate for the rather low brightness of the compounds. However, due to precipitation of 13, the concentration in all further experiments was reduced to 10 μM, which was proven to be sufficient for cell imaging. The lipid droplet distribution in cells treated with 13 seemed to be more located around the nucleus (Fig. 3), whereas LDs stained with 11 or 12 were distributed in a uniform manner over the entire cell (ESI, Fig. S31 and S32†).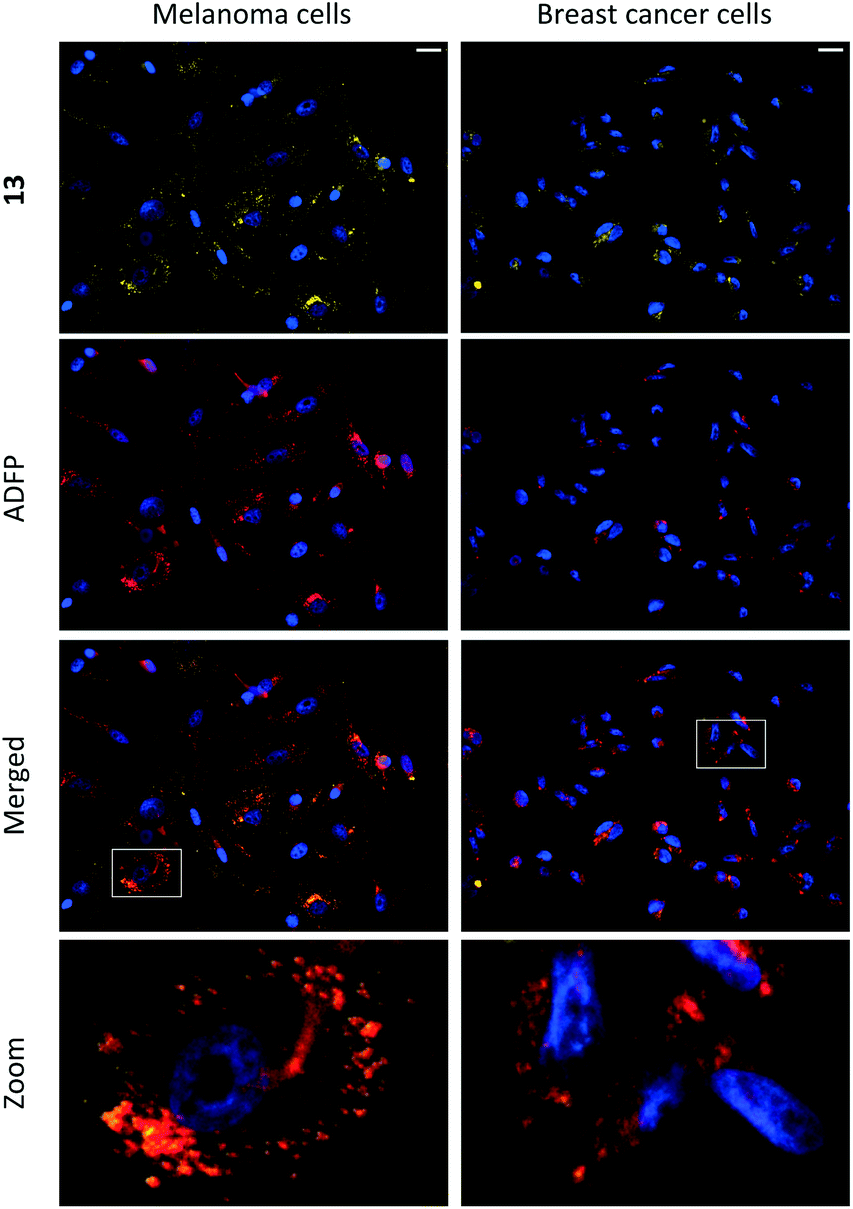 | ||
| Fig. 3 Fluorescence microscopy images of the colocalization of 13 (seen in yellow; ex. 460–500 nm; em. 512–542 nm) and anti-ADFP antibody (1:1000; seen in red; ex. 590–650 nm; em. 662–738 nm) in melanoma (SK-MEL-28) and breast cancer (MDA-MB-231) cells. Scale bar 20 μm. Staining was performed on live cells, which were fixed after incubation (10 μM, 24 h) and then subjected to immunocytochemistry. Cell nuclei were stained with DAPI (seen in blue; ex. 325–375 nm; em. 435–485 nm). | ||
Furthermore, the cell viability after treatment with 11–13 was investigated using the resazurin assay (ESI, Fig. S38 and S39†). Compounds 11 and 12 showed no cytotoxic effects in the tested cancer cell lines at 10 μM concentration after 24 h incubation. The viability measurements of compound 13 indicated some level of toxicity, although with high statistical uncertainty after performing multiple replicates. Due to this, the possibility of staining fixed cells was explored. Staining cells with 11, 12 and 13 (10 μM, 1 h) after fixation resulted in fluorescence images with high intensity and good signal-to-background ratios (e.g., 13, Fig. 4, left).
 | ||
| Fig. 4 Fluorescence microscopy images of melanoma (SK-MEL-28) cells stained with 13 (seen in green; ex. 460–500 nm; em. 512–542 nm) in the absence or presence of lipid supplementation using oleic acid (100 μM, 24 h). Scale bar 10 μm. Staining with 13 (10 μM, 1 h) was performed on fixed cells. Cell nuclei were stained with DAPI (seen in blue; ex. 325–375 nm; em. 435–485 nm). | ||
Addition of oleic acid to the cell culture medium is known to stimulate lipid accumulation in cells.34–36 Live cells were therefore supplemented with oleic acid (100 μM, 24 h) and fixed prior treatment with 11, 12 or 13 (10 μM, 1 h). The growth of lipid droplets could be visualized clearly in melanoma cells compared to cells without fatty acid supplementation (Fig. 4 and ESI, Fig. S28–S30†). The effects on the breast cancer cell line, which presumably contains lower amounts of LDs, were less distinct (ESI, Fig. S30†). These results further support that compounds 11–13 indeed are specific for staining lipid droplets in cancer cells. Moreover, we noticed that only the most lipophilic structures in the series were suitable for cell imaging (i.e., as LD-specific dyes). Therefore, the octanol/water partition coefficients (logP) were calculated37 and for the less hydrophobic compounds also experimentally determined (ESI, Table S1†). The calculated data largely matched the experimental logP values (maximal deviation of 0.6). The calculated values without an experimental counterpart were therefore considered as adequate approximations. As expected, most of the compounds were less lipophilic than LD-BTD1 (calculated logP = 3.9). Moreover, a logP ≥ 4, as determined for 11–13, seemed to be required for lipid droplet accumulation – referring to the studied compounds herein (i.e., 1–15).
Further photophysical studies of the lipid droplet-specific dyes 11–13
The LD-specific dyes 11–13 were further studied for their environmental sensitivity to lipophilic media using concentrations of sodium dodecyl sulfate (SDS), below and above the critical micelle concentration (ca. 10 mM)38 in a 5% DMSO/water mixture. Compounds 11 and 12 exhibited increased fluorescence intensity in the presence of SDS, with more intense emission at higher SDS concentrations (ESI, Fig. S22 and S23†) – an effect that has previously been reported for BTD derivatives that stain lipophilic structures in cells.21,29,39 In contrast, 13 initially aggregated in the presence of SDS (4 mM), which resulted in increased emission intensity (ESI, Fig. S24†). However, a significant gradual decrease of the intensity was observed at higher SDS concentrations (8 and 12 mM). To further probe the aggregation behaviour, additional experiments were performed in DMSO/water mixtures with increasing fractions of water (fw). Measurements of 13 showed diminished fluorescence intensity with increasing fw in the range of 0–80%, while fw values of 90% and 95% engendered aggregation-induced emission (AIE)40,41 (see ESI, Fig. S24†). This AIE effect has been described for similar BTD structures.12,20,39,42–44 Compounds 11 and 12 did not exhibit this behaviour; their fluorescence intensity only decreased with increasing fw (ESI, Fig. S22 and S23†). Based on these results, we concluded that SDS strongly affects the aggregation behaviour of 13. In addition, the photostability of compounds 11–13 was investigated in toluene using continuous irradiation for 30 min. Compound 13 showed excellent photostability while the emission intensity of 11 and 12 decreased to approximately 85% of its original value (ESI, Fig. S21†).Conclusions
We have described the synthesis, photophysical characterization and cell imaging utility of a series 4-N-substituted BTD derivatives. Measurements in solvents of different polarity revealed strong solvatochromism, large Stokes shifts and fluorescence quenching with increasing solvent polarity. Three compounds (11–13) were successfully applied as fluorescent probes for imaging lipid droplets in melanoma and breast cancer cells. These dyes displayed high signal-to-background ratios, in both live and fixed cells, and their lipid droplet specificity was found to correlate with high lipophilicity (logP ≥ 4).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Swedish Research Council (Dnr: 2018-03524). We are grateful to Professor Katarina Edwards and Associate Professor Victor Agmo Hernandez for help with the fluorescence spectrometer. This study made use of the NMR Uppsala infrastructure, which is funded by the Department of Chemistry – BMC and the Disciplinary Domain of Medicine and Pharmacy. Calculations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) partially funded by the Swedish Research Council through grant agreement no. 2018-05973. We also thank Dr Lukasz Pilarski for proof-reading the manuscript.Notes and references
- B. A. D. Neto, A. A. M. Lapis, E. N. da Silva Júnior and J. Dupont, Eur. J. Org. Chem., 2013, 2013, 228–255 CrossRef CAS.
- B. A. D. Neto, P. H. P. R. Carvalho and J. R. Correa, Acc. Chem. Res., 2015, 48, 1560–1569 CrossRef CAS PubMed.
- B. A. D. Neto, J. R. Correa and J. Spencer, Chem.–Eur. J., 2022, 28, e202103262 CrossRef CAS PubMed.
- J. Du, M. C. Biewer and M. C. Stefan, J. Mater. Chem. A, 2016, 4, 15771–15787 RSC.
- Y. Wang and T. Michinobu, J. Mater. Chem. C, 2016, 4, 6200–6214 RSC.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- Z. Li, J. Lu, S.-C. Tse, J. Zhou, X. Du, Y. Tao and J. Ding, J. Mater. Chem., 2011, 21, 3226–3233 RSC.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- X. Gong, G. Li, C. Li, J. Zhang and Z. Bo, J. Mater. Chem. A, 2015, 3, 20195–20200 RSC.
- N. Wang, Z. Chen, W. Wei and Z. Jiang, J. Am. Chem. Soc., 2013, 135, 17060–17068 CrossRef CAS PubMed.
- H. Appelqvist, K. Stranius, K. Börjesson, K. P. R. Nilsson and C. Dyrager, Bioconjugate Chem., 2017, 28, 1363–1370 CrossRef CAS PubMed.
- K. Colas, S. Doloczki, A. Kesidou, L. Sainero-Alcolado, A. Rodriguez-Garcia, M. Arsenian-Henriksson and C. Dyrager, ChemPhotoChem, 2021, 5, 632–643 CrossRef CAS.
- K. Colas, K. O. Holmberg, L. Chiang, S. Doloczki, F. J. Swartling and C. Dyrager, RSC Adv., 2021, 11, 23960–23967 RSC.
- A. L. S. Cruz, E. d. A. Barreto, N. P. B. Fazolini, J. P. B. Viola and P. T. Bozza, Cell Death Dis., 2020, 11, 105 CrossRef PubMed.
- Z. Li, H. Liu and X. Luo, Am. J. Cancer Res., 2020, 10, 4112–4122 CAS.
- J. A. Olzmann and P. Carvalho, Nat. Rev. Mol. Cell Biol., 2019, 20, 137–155 CrossRef CAS PubMed.
- T. Petan, E. Jarc and M. Jusović, Molecules, 2018, 23, 1941 CrossRef PubMed.
- T. K. Fam, A. S. Klymchenko and M. Collot, Materials, 2018, 11, 1768 CrossRef PubMed.
- H. Tian, A. C. Sedgwick, H.-H. Han, S. Sen, G.-R. Chen, Y. Zang, J. L. Sessler, T. D. James, J. Li and X.-P. He, Coord. Chem. Rev., 2021, 427, 213577 CrossRef CAS.
- Y. Yu, H. Xing, H. Park, R. Zhang, C. Peng, H. H. Y. Sung, I. D. Williams, C. Ma, K. S. Wong, S. Li, Q. Xiong, M.-H. Li, Z. Zhao and B. Z. Tang, ACS Mater. Lett., 2022, 4, 159–164 CrossRef CAS.
- S. I. Suarez, C. C. Warner, H. Brown-Harding, A. M. Thooft, B. VanVeller and J. C. Lukesh, Org. Biomol. Chem., 2020, 18, 495–499 RSC.
- G. A. Medeiros, J. R. Correa, L. P. de Andrade, T. O. Lopes, H. C. B. de Oliveira, A. B. Diniz, G. B. Menezes, M. O. Rodrigues and B. A. D. Neto, Sens. Actuators, B, 2021, 328 Search PubMed.
- A. A. R. Mota, P. H. P. R. Carvalho, B. C. Guido, H. C. B. de Oliveira, T. A. Soares, J. R. Corrêa and B. A. D. Neto, Chem. Sci., 2014, 5, 3995–4003 RSC.
- M. J. Kamlet, R. R. Minesinger, E. G. Kayser, M. H. Aldridge and J. W. Eastes, J. Org. Chem., 1971, 36, 3852–3856 CrossRef CAS.
- P. Suppan, J. Photochem. Photobiol., A, 1990, 50, 293–330 CrossRef CAS.
- M. J. Kamlet, R. R. Minesinger and E. G. Kayser, J. Org. Chem., 1971, 36, 1342–1345 CrossRef CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- Q. Wu, T. Zhang, Q. Peng, D. Wang and Z. Shuai, Phys. Chem. Chem. Phys., 2014, 16, 5545–5552 RSC.
- S. T. A. Passos, G. C. Souza, D. C. Brandão, D. F. S. Machado, C. K. Grisolia, J. R. Correa, W. A. da Silva and B. A. D. Neto, Dyes Pigm., 2021, 186, 109005 CrossRef CAS.
- A. A. Mota, J. R. Correa, P. H. Carvalho, N. M. de Sousa, H. C. de Oliveira, C. C. Gatto, D. A. da Silva Filho, A. L. de Oliveira and B. A. Neto, J. Org. Chem., 2016, 81, 2958–2965 CrossRef CAS PubMed.
- E. Lippert, Zeitschrift für Naturforschung A, 1955, 10, 541–545 CrossRef.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- Green channel: excitation 460–500 nm, emission 512–542 nm.
- A. Rohwedder, Q. Zhang, S. A. Rudge and M. J. O. Wakelam, J. Cell Sci., 2014, 127, 3104–3115 CAS.
- S. Nakajima, M. Gotoh, K. Fukasawa, K. Murakami-Murofushi and H. Kunugi, Brain Res., 2019, 1725, 146484 CrossRef CAS PubMed.
- Y. Fujimoto, J. Onoduka, K. J. Homma, S. Yamaguchi, M. Mori, Y. Higashi, M. Makita, T. Kinoshita, J.-i. Noda, H. Itabe and T. Takanoa, Biol. Pharm. Bull., 2006, 29, 2174–2180 CrossRef CAS PubMed.
- Molinspiration Cheminformatics free web services, https://molinspiration.com/, last accessed March 2021.
- B. Mandal, S. P. Moulik and S. Ghosh, Colloid Polym. Sci., 2014, 292, 2485–2495 CrossRef CAS.
- E. R. Sodre, B. C. Guido, P. E. N. de Souza, D. F. S. Machado, V. H. Carvalho-Silva, J. A. Chaker, C. C. Gatto, J. R. Correa, T. A. Fernandes and B. A. D. Neto, J. Org. Chem., 2020, 85, 12614–12634 CrossRef CAS PubMed.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- X. Cai and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 9868–9886 CrossRef CAS PubMed.
- W. Li, L. Wang, H. Tang and D. Cao, Chem. Commun., 2019, 55, 4491–4494 RSC.
- X. Shi, S. H. P. Sung, M. M. S. Lee, R. T. K. Kwok, H. H. Y. Sung, H. Liu, J. W. Y. Lam, I. D. Williams, B. Liu and B. Z. Tang, J. Mater. Chem. B, 2020, 8, 1516–1523 RSC.
- Y. Xu, H. Zhang, N. Zhang, R. Xu, Z. Wang, Y. Zhou, Q. Shen, D. Dang, L. Meng and B. Z. Tang, Mater. Chem. Front., 2021, 5, 1872–1883 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, photophysical and computational data, cell pictures and viability, and NMR spectra. See https://doi.org/10.1039/d2ra01404a |
| This journal is © The Royal Society of Chemistry 2022 |