
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColloidal silver-based lateral flow immunoassay for detection of profenofos pesticide residue in vegetables
Kuo-Hui Wu *a,
Wen-Chien Huanga,
Shu-Chen Changb and
Rong-Hwa Shyuc
*a,
Wen-Chien Huanga,
Shu-Chen Changb and
Rong-Hwa Shyuc
aDepartment of Chemical and Materials Engineering, Chung Cheng Institute of Technology, National Defense University, Taoyuan, 33551, Taiwan. E-mail: ccitkhwu@gmail.com
bApplied Zoology Division, Taiwan Agricultural Research Institute, Taichung 41362, Taiwan
cInstitute of Preventive Medicine, National Defense Medical Center, 90048, Taipei, Taiwan
First published on 29th April 2022
Abstract
A colloidal silver nanoparticle (AgNP)-based lateral flow immunoassay (LFIA) was evaluated in terms of the rapid detection of profenofos (PEO) pesticide residue in vegetables. Colloidal AgNPs, of a diameter of approximately 20 nm, were surface-modified with trisodium citrate dehydrate (TSC) in order to improve their stability and dispersion. An anti-profenofos polyclonal antibody (pAb) was successfully immobilized on the surface of the AgNPs by ionic interaction and characterized using UV-vis, SEM, TEM, FTIR and XPS analyses. Surface modification of Ag–pAb conjugates of varying pH, pAb content and cross-reactivity was employed to design and prepare labels for use in an LFIA to examine whether these factors affect the performance of the assay. The visible detection limit and optical detection limit of the PEO test strip were 0.20 and 0.01 ppm, respectively, in PEO standard solution. This assay showed no cross-reaction with omethoate, methamidophos or pyraclofos. Finally, the PEO test strip was effectively applied for the detection of PEO in liquid vegetables A and B, with optical detection limits of 0.09 and 0.075 ppm, respectively.
1. Introduction
The lateral flow immunoassay assay (LFIA) is a solid-phase immunoassay combining the principles of thin-layer chromatography and immune recognition reaction. The LFIA is a powerful tool for rapid and low-cost on-site detection through the naked eye; as such, this technique does not require any instrumentation, and only utilizes reagents stored in dry form. Various LFIA products are commercially-available for the detection of biomarkers, proteins, drugs, and hormones in biomedicine, as well as chemical contaminants, bio-toxins, and pathogens in food safety and environmental monitoring.1–5 Novel nanoparticle (NP) labels have been developed to improve the analytical performance of traditional AuNP-based LFIAs. Currently, the analytical sensitivity and/or limit of detection (LOD) of LFIAs can be enhanced using three types of NP as alternative labels: colored NPs (e.g., AuNPs, carbon NPs and colloidal magnetic NPs), luminescent NPs [e.g., quantum dots (QDs) and dye-doped NPs] and magnetic NPs (MNPs).6–11Metal nanoparticles have been developed for a variety of applications, such as biosensors, anti-bacterial agents, drug-delivery vehicles, contrast agents, and so on. Silver nanoparticles (AgNPs) have attracted much attention due to their ease of synthesis, chemical stability, good conductivity and antimicrobial properties.12 A widely-used method for AgNP synthesis is the reduction of Ag ions in aqueous solution in the presence of a capping agent, such as citrate molecules, which impart negative surface charges that prevent nanoparticle aggregation through repulsion forces.13 AgNPs have a large number of surface atoms, which translates into a significant increase in the surface energy. A tendency to reduce their large surface energy by interacting with surrounding components that contain donating or accepting sites has been observed.14 Data reported in the literature showed that nanoparticles introduced in a biological environment tend to interact with proteins and form a protein corona shell.15
Organophosphorous pesticides (OPPs) vary in toxicity, but most are poisonous and can cause acute or chronic damage to humans and livestock. In recent years, owing to overuse of OPPs, pesticide residues in agricultural products have greatly exceeded the maximum residue limits, polluting the environment. For example, OPPs in environmental water originate primarily from agriculture and industry, and are difficult to degrade. Profenofos is a kind of moderately-toxic and non-systemic broad-spectrum organophosphate insecticide. With action on contact and stomach-poisoning activity, profenofos has a good control effect on pests that affect rice, cotton, fruit trees, vegetables and other crops.16,17 Conventional pesticide residue detection methods include gas chromatography, liquid chromatography, gas chromatography-mass spectrometry, and liquid chromatography-mass spectrometry. These detection methods can measure various components simultaneously with high accuracy, but require expensive instruments, long and complicated preprocessing procedures, and professional operators.18 Therefore, establishing a reliable, sensitive, rapid, and simple analytic procedure for the detection of profenofos residue is essential in controlling pesticide residues and preserving the ecological environment and health.
Many papers report the AuNPs-based LFIA, herein, we present a study focused on AgNPs-based LFIA. Accordingly, it is important to develop accurate and convenient methods for effective monitoring of the profenofos contents of various products. Profenofos detection devices currently available for on-site and in-home testing are a one-step, enzyme-linked immunosorbent assay,19 a gold-based colorimetric aptasensor20 and an aptamer-based fluorescence assay.17 Considering the drawbacks of current methods of profenofos detection, the aims of this study were to employ colloidal AgNPs for binding to an antibody against profenofos (pAb) and prepare a lateral flow test strip based on a colloidal AgNP immunoassay. The preparation of AgNPs is based on citrate reduction and stabilization; we present a study of the conformation of citrate molecules adsorbed on AgNPs using Fourier transform infrared spectrometry (FTIR) and X-ray photoelectron spectroscopy (XPS). We also present studies the effects of varying pAb content and solution pH on Ag–pAb size combining the spectroscopy results with structure-based modeling in addition to observations described in the literature. Based on the modeling result, we design and prepare labels for use in a LFIA and applied to detect of PEO. The test strip was prepared simply, and proved convenient, cost-effective, rapid and specific for sensitive profenofos detection in vegetables. Finally, ImageJ software was employed to analyze the optical densities of the test line (T) and control lines (C), which was inversely proportional to the amount of profenofos.
2. Experimental
2.1 Materials and apparatus
Silver nitrate (AgNO3) and trisodium citrate dehydrate (TSC) were purchased from J.T. Baker Chemical Company. Sodium borohydride (NaBH4) was purchased from Sigma-Aldrich. Donkey anti-rabbit immunoglobulin G (IgG) was purchased from Rainbow Biotechnology Co., Ltd. Nitrocellulose membrane (NC membrane), sample pads, conjugate pads and absorbent pads were purchased from Advance Merck Millipore. Profenofos (PEO), anti-PEO polyclonal antibody (pAb) and OVA-PEO conjugate were obtained from Taiwan Agricultural Research Institute. Pyraclofos, omethoate and methamidophos were obtained from Chem Service Inc. Bovine serum albumin (BSA) was purchased from Jackson ImmunoResearch Inc. Trehalose was purchased from Alfa Aesar. Polyoxyethylene sorbitan monolaurate (Tween-20) was purchased from Bio Basic Canada Inc. All chemicals were of analytical grade and were used without further purification. In addition, the following buffers were used: (1) 0.1 M phosphate-buffered saline (PBS), at pH 7.4, containing 8.0 g NaCl, 0.2 g KCl, 1.42 g Na2HPO4 and 0.27 g KH2PO4; (2) PBST solution (0.1% Tween-20 in 10 mM PBS); (3) conjugate pad solution (0.1% BSA, 10% trehalose and 0.05% Tween-20 in 10 mM PBS).The formation of an Ag–pAb conjugate was confirmed by UV-vis (UV3101PC, Shimadzu, Kyoto, Japan) spectrophotometry and an energy-dispersive X-ray (EDX) microanalysis system (JSM-6330F). The morphologic details of the AgNPs and Ag–pAb were studied by transmission electron microscopy (TEM, Hitachi H-7100) and scanning electron microscopy (SEM, JSM-6330F). The chemical compositions of the products were obtained using Fourier transform infrared (FTIR) spectrometry (PerkinElmer Spectrum 100) and X-ray photo electron spectroscopy (XPS, VG Scientific ESCALAB 250, UK). Optical images of the test strips were acquired using a smart phone (ASUS) and then processed using ImageJ software for analysis of the optical densities of the test line (T) and control line (C).
2.2 Preparation of AgNPs and Ag–pAb conjugate
AgNPs were synthesized by a simple chemical reduction method using sodium borohydride as the reducing agent.21 30 mL of 0.04 M aqueous NaBH4 solution were placed in an Erlenmeyer flask, and 10 mL of 0.01 M AgNO3 solution were added at approximately 1 drop per second under vigorous stirring. The reaction was allowed to continue until the solution turned from light yellow to brighter yellow. The AgNO3 reduction reaction can be written as:| 2AgNO3 + 2NaBH4 → 2Ag + H2 + B2H6 + 2NaNO3 |
Finally, 1 mL of 0.02 M TSC solution was added to stabilize the colloidal AgNPs. TSC stabilized the colloidal AgNPs in a colloidal state owing to the repulsive force that existed among the particles, and this was maintained by a net negative charge on their surface. These charged particles were very sensitive to changes in the solution dielectric. Therefore, binding of anti-PEO pAb to the colloidal AgNP surface would maintain a suspended state by blocking the salt-induced precipitation of colloidal AgNPs.
In addition to titration to judge the minimum amount of pAb, it was also necessary to determine the correct pH for conjugation. A 1.0 mL aliquot of colloidal AgNPs was added to 7 of 1.5 mL Eppendorf tubes and the pH adjusted from 7.0 to 10.0 by 0.1 M K2CO3. Then, 6.0 μg (0.1 mg mL−1) of pAb solution were added to the 7 tubes of adjusted colloidal AgNP solution and incubated at 37 °C for 30 min. 100 μL of 10 wt% NaCl were added, followed by agitation for 3 h. Another 1.0 mL aliquot of colloidal AgNPs was added to 6 of 1.5 mL Eppendorf tubes, and the pH was adjusted to 9.0 by 0.1 M K2CO3. Then, pAb was added to each tube in a series of amounts from 0 μg to 10 μg. The 6 tubes of adjusted colloidal AgNP solution were incubated at 37 °C for 30 min. 100 μL of 10 wt% NaCl were then added to each tube, followed by agitation for 3 hours. The concentration of pAb at which the colloidal silver surface became saturated and no aggregation occurred was determined by observing the color change.
The colloidal AgNP solution (10 mL) was adjusted to pH 9.0 with 0.1 M K2CO3, and then 6.0 μg of pAb were added dropwise. After the mixture had been gently agitated at 37 °C for 1 h, pAb was conjugated to the colloidal AgNP surface by monodentate or bidentate bonding. 2.5 mL of 5 wt% BSA solution were added to the mixture to block the non-coated colloidal AgNP surfaces for 15 min, and the mixture was then centrifuged at 6000 rpm for 20 min. Colloidal AgNP-conjugated pAb (Ag–pAb) in soft pellet form was collected after being washed twice by centrifugation at 6000 rpm for 20 min. Finally, Ag–pAb was re-suspended in 50 μL of dilution buffer and stored at 4 °C before use (Fig. 1a).
 | ||
| Fig. 1 (a) Schematic illustration of the preparation of Ag–pAb conjugate, (b) Ag–pAb based LFIA for detection of PEO. | ||
2.3 Assembly of PEO test strips
The PEO test strip consisted of three pads (sample, conjugate and absorbent pad), one nitrocellulose membrane (NC membrane) with a test line and a control line, and one polyvinyl chloride (PVC) baseplate. OVA-PEO conjugate (7.14 mg mL−1) was applied to the NC membrane and used as the test line, while anti-rabbit IgG (0.1 mg mL−1) was applied to the NC membrane and used as the control line. The conjugate pad was prepared by immobilizing the Ag–pAb conjugate on the pad. First, the conjugate pad was submerged in 10 mM phosphate-buffered saline, pH 7.4, containing 0.1% (w/v) BSA, 10% (w/v) trehalose and 0.05% (w/v) Tween 20, and dried at 37 °C for 2 h. Then, 2 μL of Ag–pAb conjugate solution were spotted onto the conjugate pad. The resultant conjugate pad was incubated at 37 °C for 1 h until fully dried. The NC membrane was pasted onto the PVC baseplate, and then the Ag–pAb conjugate pad was pasted on the plate, overlapping the NC membrane by 5 mm. The absorbent pad and sample pad were pasted to the lower and upper portions of the baseplate, respectively, with a 1–2 mm overlap of the NC membrane on both sides (Fig. 1b).2.4 Sensitivity and cross-reactivity of the LFIA strip
Sensitivity testing of the LFIA strip was performed as follows: in brief, 100 μL of a solution of standard PEO at a series of concentrations (0, 0.1, 0.2, 0.3, 0.4 and 0.5 ppm) in 0.01 M PBS buffer (pH 7.4) were applied to the sample pad. When PEO aqueous solution was applied to the sample pad, the Ag–pAb conjugate was rehydrated and consequently released into the migrating liquid; it then migrated across both the T-line and the C-line driven by capillary force. As both PEO in solution and the PEO residue immobilized on the T-line could specifically bind with the Ag–pAb conjugate, as shown in Fig. 1, they will be in competition to bind to the limited binding sites on the Ag–pAb conjugate. Consequently, the color of the T-line, as determined by the amount of colored conjugate, inversely reflected the amount of PEO. In contrast, all particles that migrated across the C-line were captured by the secondary antibody immobilized on the C-line, independent of the combination between PEO and the colored conjugate. Therefore, the color of the C-line reflected the effective release of Ag–pAb conjugate from the conjugate pad, and was used to test the validity of the run.In the present study, the value of the detection limit was defined as the lowest PEO concentration that inhibited apparent color development of the test line. If the PEO concentration was lower than the value of the detection limit (sample considered negative), both the T-line and C-line were colored. In contrast, only a colored C-line appeared if the PEO concentration in the sample was higher than the value of the detection limit (sample considered positive). Finally, the obtained color signals were analyzed using ImageJ software to calculate the optical density of the T-line. Samples containing 5.0 ppm of profenofos, omethoate, methamidophos and pyraclofos were prepared in phosphate buffer (pH 7.4) and analyzed using an LFIA strip to evaluate cross-reactivity.
2.5 Analysis of the simulated samples
For the purpose of assessing the practicability of using the LFIA strip under natural conditions, two different vegetables were used as chemical-poison diluents to mimic actual samples. The various types of sample to be analyzed were mimicked with profenofos standard solution (100 ppm, prepared in 0.01 M PBS, pH 7.4). The vegetable samples needed to be diluted, because the particles would have blocked the pores of the membrane. 0.5 g of vegetable was finely ground, then added to 1.25 mL PBS, and standard PEO (100 ppm) of 0, 1.25, 2.50, 3.75, 5.00 and 6.25 μL was added, respectively. The final PEO concentrations were 0, 0.1, 0.2, 0.3, 0.4 and 0.5 ppm in the vegetable samples. Subsequently, 100 μL of each sample were introduced onto the strip sample region in a sensitivity assay. Four replicates of each sample were tested using LFIA strips. The simulated samples were analyzed in order to calculate the detection limit and repeatability.3. Results and discussion
3.1 Structure characterization and morphology
UV-vis, SEM, TEM and EDX provided evidence of the formation of AgNPs and the Ag–pAb conjugate. UV-vis spectroscopy is extensively used for studies of metal nanoparticles, and shows the surface plasmon resonance phenomenon in the metals. AgNPs exhibit a plasmon band in the range of 350 nm to 550 nm.22 In this study, the citrate-capped AgNPs presented surface plasmon resonance bands (SPR) at 382 and 427 nm, while pAb had a λmax centered at 280 nm (Fig. 2a and c). The peak at 382 nm was attributed to the SPR band of the AgNPs, while the peak at 427 nm was attributed to the larger AgNPs. Therefore, the synthesis adopted here may be heterogeneous. The λmax value of 382 nm corresponded to the average size of 10–20 nm, which has already been established in the literature.23 The citrate-terminated AgNPs form the Ag–pAb conjugate by a ligand exchange reaction of pAb.24 The SPRs of the Ag–pAb conjugate shifted to higher wavelengths, with a decrease in the absorbance value. This result indicated that the residual vacancies on the surface of the citrate-labelled AgNPs were occupied by pAb, leading to further aggregation of the AgNPs.25 It was observed that the color of the AgNPs and Ag–pAb solution was light yellow, as shown in the Fig. 2 insert. This indicated that, after reacting with pAb, no agglomeration of AgNPs occurred. This was supported by TEM measurements. The sizes and morphologies of the AgNPs and Ag–pAb conjugate were observed using SEM, TEM and EDX, as shown in Fig. 3. Most of the colloidal AgNPs and Ag–pAb displayed well-distributed spherical nanocrystals, with diameters below 25 nm. The EDX spectrum revealed a strong signal in the Ag and biomolecules region, confirming the formation of colloidal AgNPs and the Ag–pAb conjugate. The elemental Ag signal was confirmed in the EDX spectrum, and the carbon and oxygen indicated that citrate was observed on the surface of the AgNPs. In addition, the nitrogen element was observed in the EDX spectrum of Ag–pAb, indicating the presence of pAb on the AgNP surface. | ||
| Fig. 2 UV-vis absorption spectra of (a) AgNPs, (b) Ag–pAb and (c) pAb, inset: photograph of (a) AgNPs and (b) Ag–pAb. | ||
 | ||
| Fig. 3 (a) TEM, (b) SEM and (c) EDX images of AgNPs; (d) TEM, (e) SEM and (f) EDX images of Ag–pAb. | ||
Fig. 4 illustrates the UV absorbance and particle size of Ag–pAb at varying pAb contents and solution pHs. Depending on the UV absorbance and particle size of the reaction system, different possible interactions occurred. Fig. 4a shows the effect of pH on the maximum absorption and particle size of Ag–pAb. It was clear that as the pH value increased, the particle size decreased. The optimal pH for binding of antibodies to colloidal AgNPs was determined to be 9.0. It was evident that an optically-transparent yellow solution was obtained when the appropriate pH was applied. In pH below 8.5, the presence of cations in the salt solution negated this charge repulsion and caused the particles to agglomerate and finally precipitate. In acidic pH the particle size of NPs was larger than that in basic pH.27 Fig. 4b shows the effects of pAb content on the maximum absorption and particle size of Ag–pAb. With an increasing antibody content, the color of the solution changed from colorless to yellow. The variation in color could be attributed to the surface plasmon resonance, as the strong interaction between AgNPs and light resulted in collective oscillations of conduction electrons on the surface when excited by light at specific wavelengths.26 As the particles decreased in size, their absorption intensity drastically increased. In contrast, at sufficient or excess amounts of antibodies, colloidal Ag–pAb remained in a stable state with regards to UV absorbance and particle size. Spectral change in the maximum adsorption of the surface plasmon band determined the optimal amount of antibodies to bind to colloidal AgNPs. The antibody-stabilized colloidal AgNPs remained as clusters of particles without aggregation. As shown in Fig. 4b, at 6.0 μg of anti-PEO pAb per 1 mL of colloidal AgNP solution, the anti-PEO pAb was adequately bound to the colloidal AgNPs.
 | ||
| Fig. 4 The effects of (a) pH and (b) anti-PEO pAb content on absorption and particle size of Ag–pAb (insets: photographic images of corresponding solutions). | ||
3.2 Mechanism of the formation of AgNPs and Ag–pAb
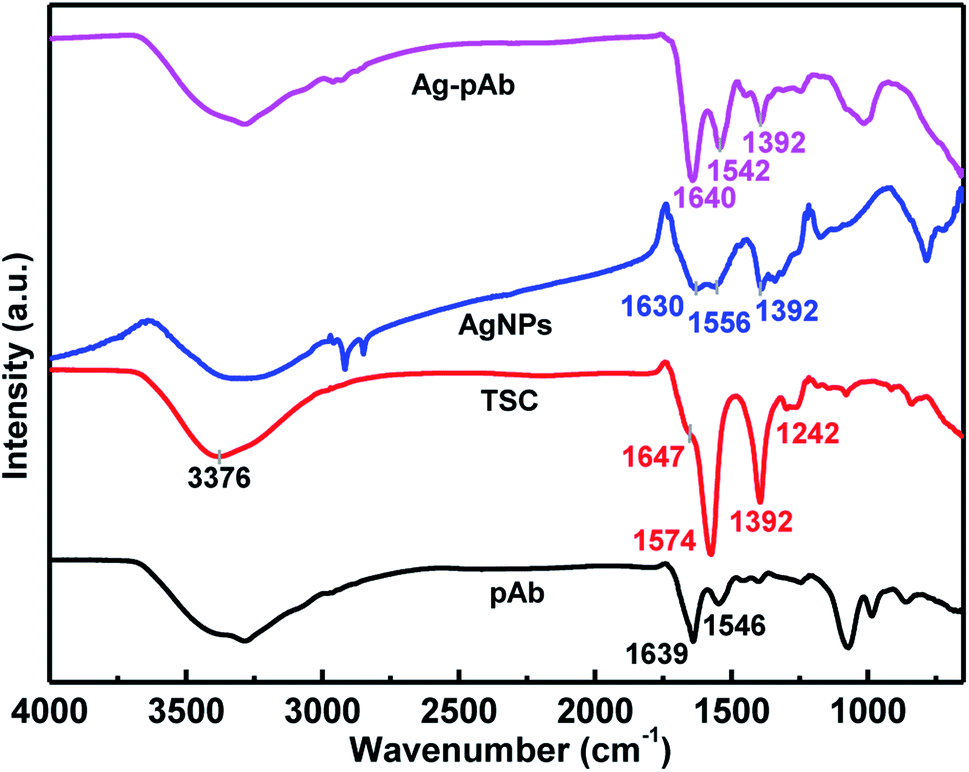
We used FTIR spectroscopy and XPS to characterize the interactions of citrate and pAb molecules on the surface of the AgNPs. Fig. 5 depicts the FTIR spectra of the Ag–pAb conjugate, AgNPs, TSC and pAb. In the IR spectrum of citrate (TSC), the peaks at 1647 and 1574 cm−1 are due to asymmetric stretching of COO−, and the peak at 1392 cm−1 is due to symmetric stretching of COO−. The broad 3376 cm−1 peak is due to the OH stretching mode. In the AgNP spectrum, the absorption bands at 1630/1556 and 1392 cm−1 are assigned to the asymmetric and symmetric stretching of COO− from the citrate molecule present on the surface of the AgNPs.27 In the pAb spectrum, the characteristic adsorption peaks at 1639 and 1546 cm−1 are attributed to amide I and amide II bands, respectively.28 The amide I band is due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the peptide bonds, and the amide II band is due to C–N stretching vibration in combination with N–H bending. These absorption bands were also present in the Ag–pAb conjugate spectrum at 1640 and 1542 cm−1, rendering the IR spectra of pAb and Ag–pAb almost identical. The characteristic absorption bands of Ag–pAb at 1640/1542 and 1392 cm−1 were also attributed to the asymmetric and symmetric stretching vibrations of COO− from the citrate and pAb molecules present on the surface of the AgNPs.
O stretching vibration of the peptide bonds, and the amide II band is due to C–N stretching vibration in combination with N–H bending. These absorption bands were also present in the Ag–pAb conjugate spectrum at 1640 and 1542 cm−1, rendering the IR spectra of pAb and Ag–pAb almost identical. The characteristic absorption bands of Ag–pAb at 1640/1542 and 1392 cm−1 were also attributed to the asymmetric and symmetric stretching vibrations of COO− from the citrate and pAb molecules present on the surface of the AgNPs.
 | ||
| Fig. 5 FTIR spectra of Ag–pAb conjugate. | ||
A carboxylate (COO−) group can coordinate metals in different forms: (1) monodentate, (2) bridging bidentate, and (3) chelating bidentate, which can be distinguished from the extent of the Δν = νas(COO−) − νs(COO−) shift. The value of Δν weakens in the order of monodentate, bridging bidentate, chelating bidentate form. The values of Δν with a high probability in monodentate complexes are expected to be larger than 200 cm−1; complexes with 200 > Δν > 140 cm−1 have bridging bidentate carboxylate groups; and complexes with Δν < 110 cm−1 have chelating bidentate carboxylate groups.29 Furthermore, the Δν(νas − νs) values of AgNPs are 238 (νas(1630) − νs(1392)) and 164 (νas(1556) − νs(1392)) cm−1, respectively, with two bonding structures of the monodentate and bridging bidentate type. Similarly, the Δν(νas − νs) values of the Ag–pAb were 248 (νas(1640) − νs(1392)) and 150 (νas(1542) − νs(1392)) cm−1, respectively, with two bonding structures of the monodentate and bridging bidentate type. Thus, for the Ag–pAb sample, the increase of the 1640 cm−1 band attributed to pAb confirmed the successful monodentate bonding of AgNPs with the pAb conjugate. These results demonstrated that citrate (TSC) coordinated AgNPs in the monodentate and bridging bidentate type, but pAb coordinated AgNPs in the monodentate type.
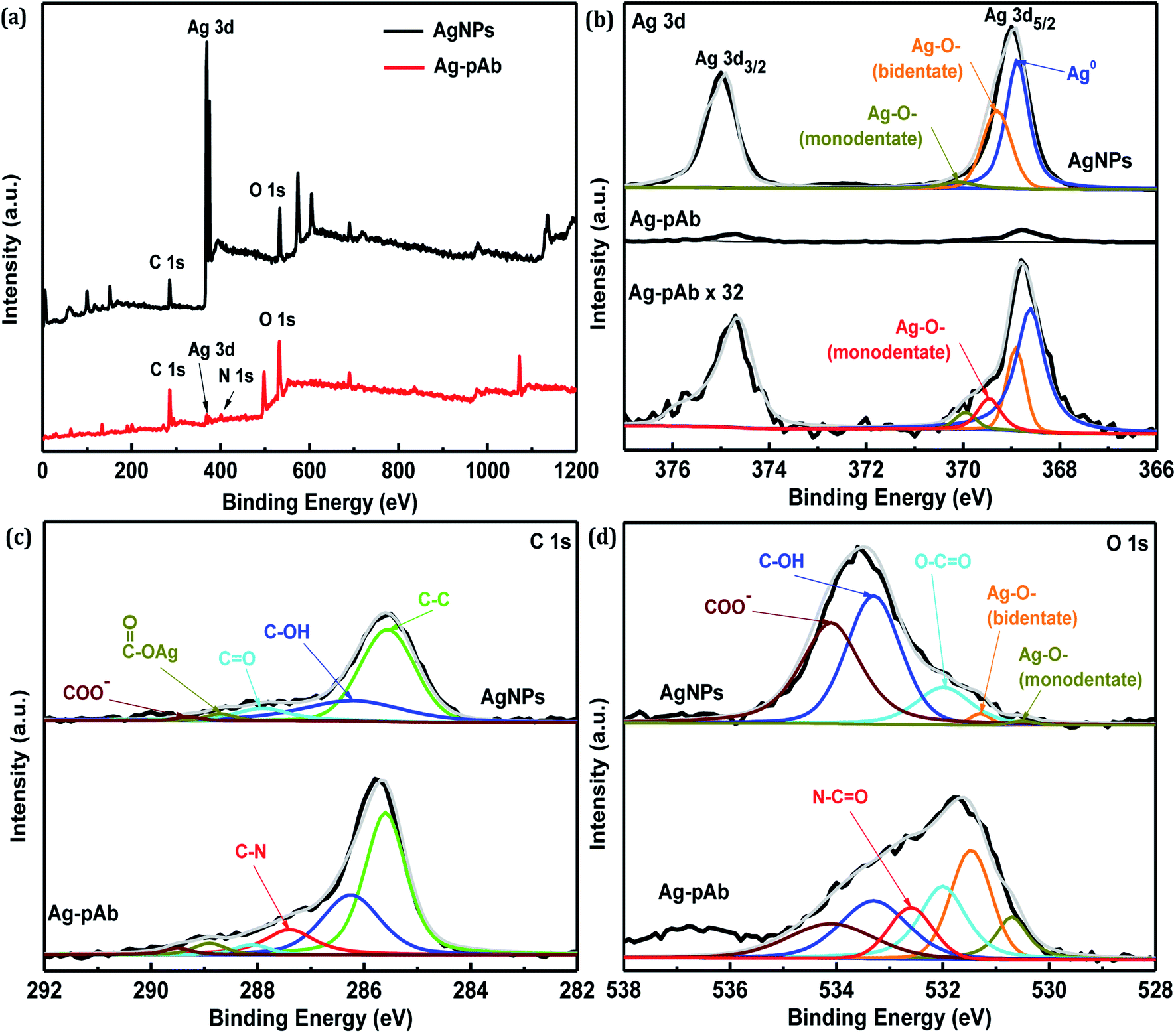
The full-range XPS spectra of AgNPs and Ag–pAb are depicted in Fig. 6a, which illustrate the binding energies of all elements, including Ag, C, O and N. In the AgNP spectrum, we observed the characteristic Ag 3d, C 1s and O 1s peaks, which were compositionally consistent with the elemental compositional content of AgNPs. The full-range XPS spectrum of Ag–pAb showed strong signals for C and O, along with weak Ag and N peaks, which were for pAb and citrate bound to the surface of the AgNPs, indicating the presence of pAb. It was observed from Fig. 6b that the Ag 3d3/2 and Ag 3d5/2 peaks appeared at binding energies of 374.7 and 368.8 eV, respectively. According to curve-fitting for Ag 3d5/2, colloidal AgNPs have three Ag 3d5/2 peaks; one at 368.7 eV, which may be due to Ag0, and two at 369.2 (bidentate) and 370.0 (monodentate) eV, which may be due to the Ag–O bond of the carboxylates in citrate bonding to Ag atoms.30 However, the Ag 3d5/2 spectra of Ag–pAb exhibited a new Ag–O bond (monodentate) at 369.5 eV, which arose mainly from the carboxyl (COO−) oxygen in pAb bonding to the AgNP surface. As shown in Fig. 6c, AgNPs have five C 1s peaks at 289.3, 288.7, 287.9, 286.2 and 285.5 eV, which are attributed to the COO−, COO–Ag, CO, C–OH and C–C groups, respectively. Moreover, Ag–pAb has six C 1s peaks at 289.5, 288.9, 288.0, 287.4, 286.3 and 285.5 eV. A new peak at 287.4 eV was attributed to C–N in pAb.
 | ||
| Fig. 6 XPS spectra of AgNPs and Ag–pAb in (a) full range, (b) Ag 3d, (b) C 1s, (d) O 1s regions. | ||
As shown in Fig. 6d, AgNPs have five O 1s peaks at 534.2, 533.3, 531.9, 531.2 and 530.4 eV, which are attributed to COO−, C–OH, O–CO, COO–Ag (bidentate) and COO–Ag (monodentate), respectively. Moreover, Ag–pAb has six O 1s peaks at 534.1, 533.3, 532.6, 532.0, 531.4 and 530.6 eV. A new peak at 532.6 eV was attributed to the amide oxygen (N–CO) in pAb. There were remarkable contributions of O 1s from N–CO, O–CO, COO–Ag (bidentate) and COO–Ag (monodentate) in the Ag–pAb sample, contributing to the peak intensity increase. This may indicate that citrate and pAb were bound to the surface of the AgNPs. It is known from previous investigations that when carboxylic acids are adsorbed from solution to a metal surface, there may exist two different types of bonding of carboxylate groups to the metal, either a bidentate bond through two equivalent oxygen atoms or a monodentate bond with inequivalent oxygen atoms.31,32 According to the results of FTIR spectroscopy and XPS, we were able to draw up a schematic representation of the ligand exchange in the citrate-stabilized AgNPs (Fig. 7). In the first stage, Ag+ ions were reduced to metallic Ag0 with the aid of NaBH4. In the second stage, the carboxylate groups of citrate protected the AgNPs by charge stabilization. Citrate adsorbed on the surface of the AgNPs provided steric repulsion against aggregation. The steric effect of citrate kept each nanoparticle apart, which resulted in the formation of stable colloids at a high ionic strength. In the third stage, pAb was added to the citrate-stabilized AgNP solution, leading to ligand exchange. Because some of the outer-sphere Ag atoms had an Ag+ oxidation state, the carbonyl groups of pAb were coordinated with outer-sphere silver atoms during Ag–pAb formation.33
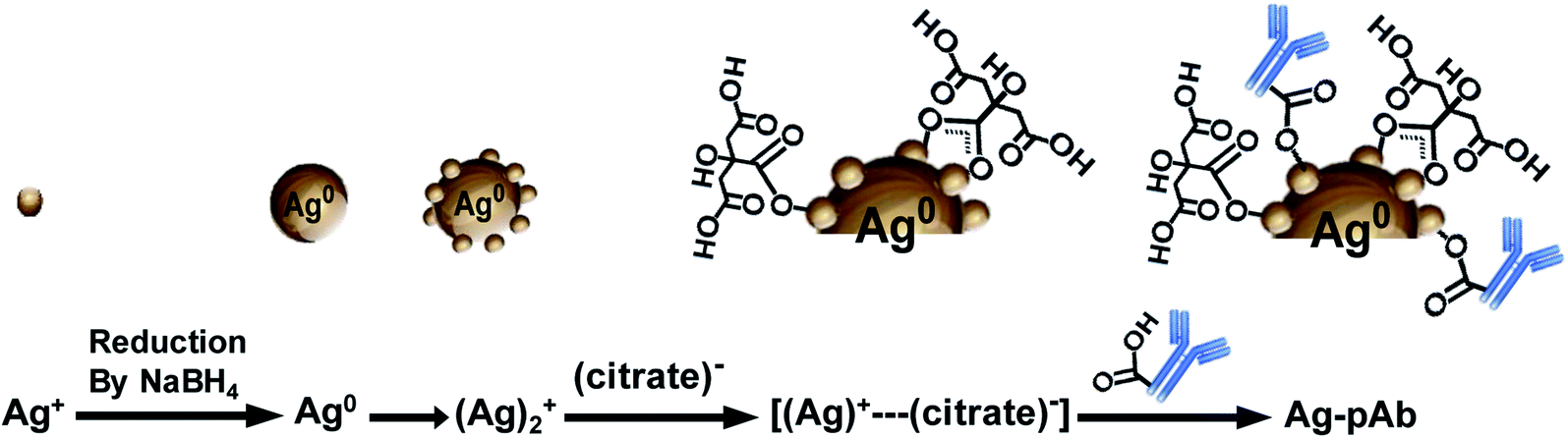 | ||
| Fig. 7 Schematic illustration of citrate and pAb conformation on the surface of AgNPs. | ||
3.3 Sensitivity and specificity of PEO test strips
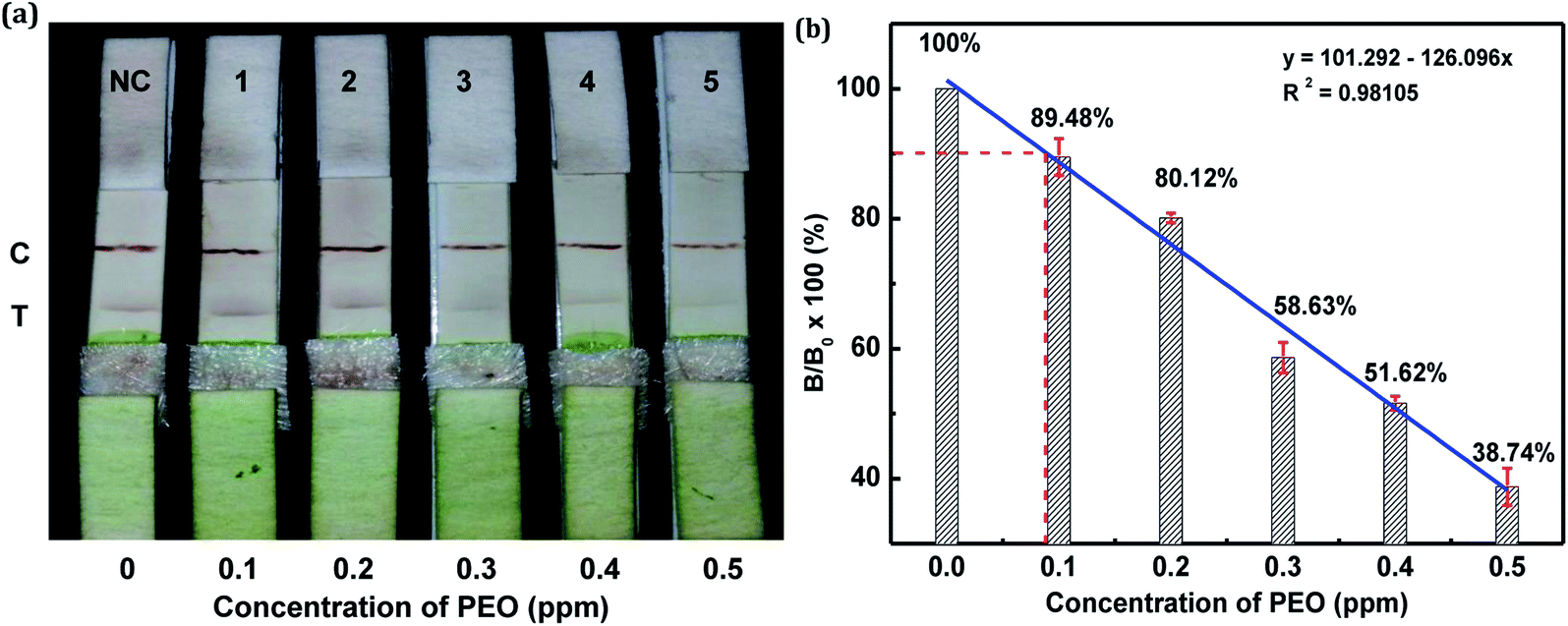
The sensitivity of the LFIA strip was determined by testing of PEO standard samples at concentrations ranging from 0 to 0.5 ppm. The visual inspection limit was defined as the minimum analyte concentration required for no apparent color to be present on the T-line to the naked eye.34 According to this definition, a PEO concentration of 0.4 ppm caused a slight but visually-distinguishable difference in the test line intensity as compared with the negative control, as shown in Fig. 8a. The detection procedure was repeated three times. All the results were highly-reproducible throughout the assay. In order to quantitatively study the detection performance of the LFIA strip, the optical density of the T-line was measured to test the sensitivity. The signals on the T-line averaged from five parallel runs were digitized to the optical density using ImageJ software and expressed as the integral area of the cross-section of the T-line (area T) within a fixed peak width. The relative optical density (ROD), defined as area T (sample)/area T (negative control), was used in signal analysis.35 Fig. 8b shows the ROD profiles of the T-line at different PEO concentrations. Generally, the ROD decreased gradually with increasing concentration of PEO. Defining the detection limit as the minimum concentration of analyte required to induce a 10% relative optical signal decrease,35 0.01 ppm of PEO was considered to be the limit of detection (LOD) for the LFIA strip. To determine the specificity of the developed strip test for PEO, a study of cross-reactivity was carried out under optimized conditions. Cross-reactants tested included omethoate, methamidophos and pyraclofos. Standard solutions of each compound at concentrations of 5.0 ppm were applied as the reactant mixture on the LFIA test strip instead of PEO. As shown in Fig. 9, red colors appeared obviously on the T-line for all three test strips, showing no detection performance for the referenced cross-reactants. The LFIA strip displayed good resistance and a high selectivity for PEO.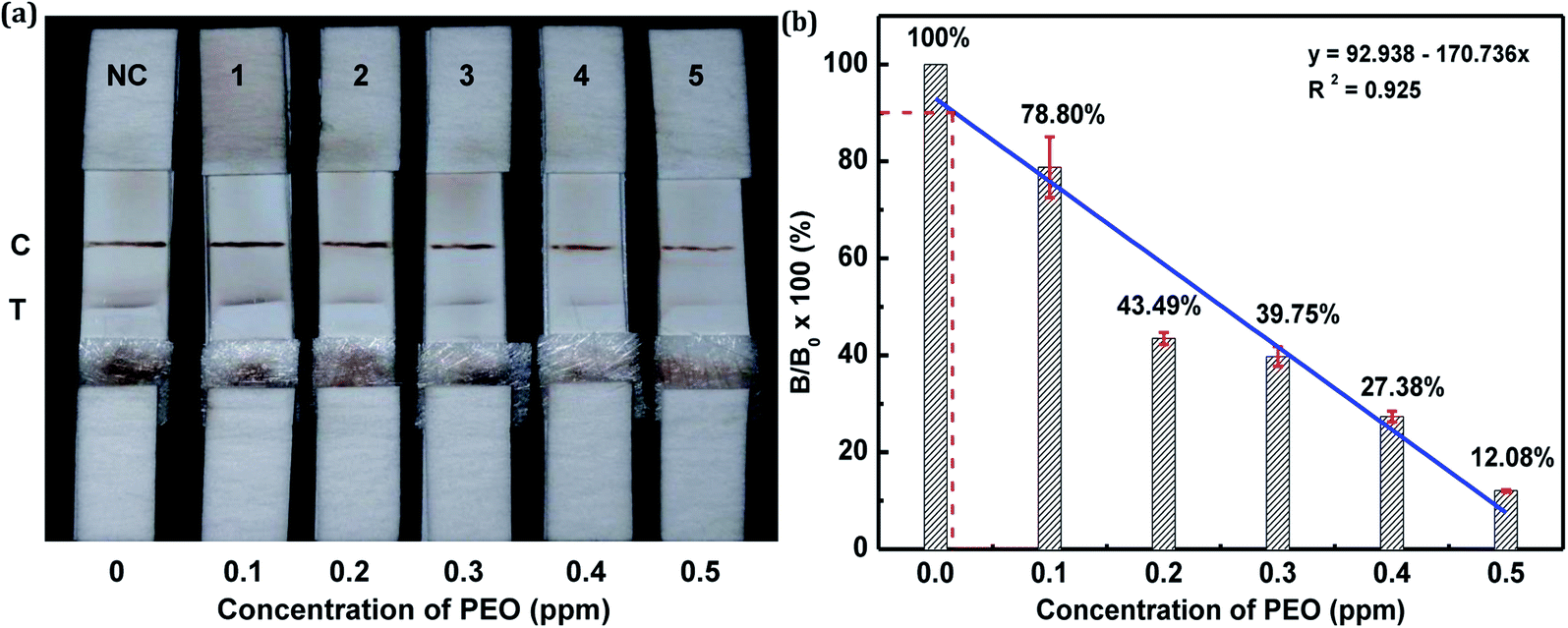 | ||
| Fig. 8 Sensitivity of PEO test strips. (a) PEO in PBS (0.1–0.5 ppm) was tested by the PEO test strips. (b) Mean intensity ratio (T-line/C-line) versus PEO concentration of test strip (error bar: standard deviation, n = 3). | ||
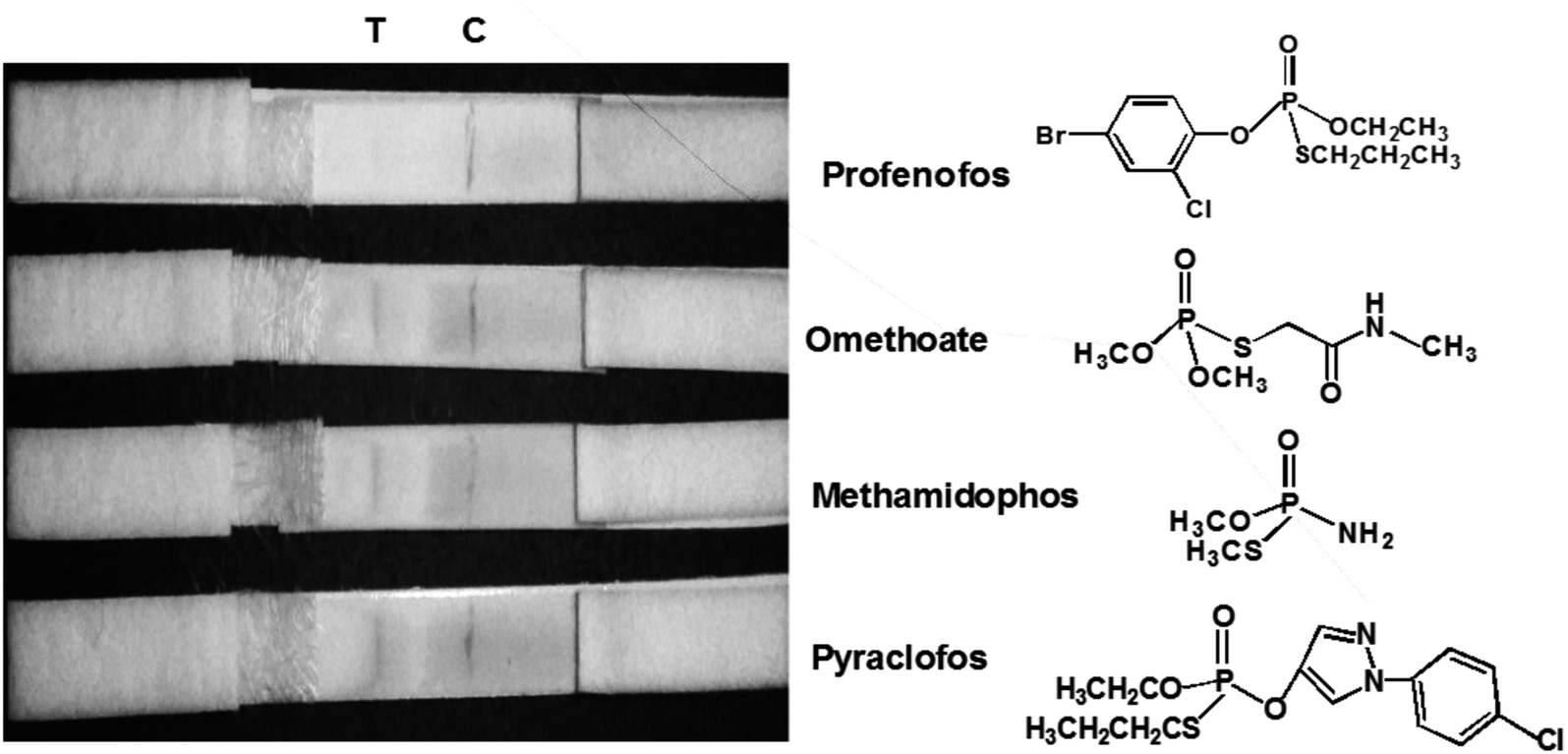 | ||
| Fig. 9 Specificity of PEO test strips. Samples containing 5.0 ppm of various substances (omethoate, methamidophos and pyraclofos) were applied. | ||
In this study, the matrix effect was determined for vegetables A and B. The sample pretreatment process prior to conducting the strip test was very simple and rapid. Liquid vegetables were spiked with different concentrations of standard PEO to determine the detection limit of the strip. Finally, the visual LODs of PEO in vegetables A and B were determined to be 0.3 ppm (Fig. 10a) and 0.2 ppm (Fig. 11a), respectively. The optical-density LODs of PEO in vegetables A and B were determined to be 0.09 ppm (Fig. 10b) and 0.075 ppm (Fig. 11b), respectively. The same detection process was conducted three times for both of the samples, and similar detection limits were obtained, which indicated good reproducibility. The results demonstrated that the developed method was suitable for on-site determination of PEO residue in vegetables.
 | ||
| Fig. 10 (a) PEO diluted in PBS and commercially available vegetable A at different concentration (0.1–0.5 ppm) was tested by the PEO test strips. (b) Optical density profiles of the T-line with different PEO concentration. NC: uncontaminated Qingjiang cuisine (error bar: standard deviation, n = 3). | ||
 | ||
| Fig. 11 (a) PEO diluted in PBS and commercially available vegetable B at different concentration (0.1–0.5 ppm) was tested by the PEO test strips. (b) Optical density profiles of the T-line with different PEO concentration. NC: uncontaminated Chinese cabbage (error bar: standard deviation, n = 3). | ||
4. Conclusions
In the present study, we primarily aimed to establish a colloidal Ag-based lateral flow immunoassay for the detection of PEO in vegetable samples. Ag-based LFIAs for PEO detection have rarely been reported, and few investigations of the properties of Ag–pAb conjugates have been conducted; for example, their behavior under changes in variables such as the amount of antibody and the pH, which generate different interactions and particle sizes of the Ag–pAb conjugate, has not been analyzed. The results demonstrated that the optimal pH value was 9.0, and the optimum amount of pAb was 6.0 μg mL−1 for preparation of the Ag–pAb conjugate. In addition, we demonstrated the binding mode and reaction mechanics between colloidal AgNPs and citrate/pAb using FTIR spectroscopy and XPS. The strip was easy to use, and the results were obtained within 15 min, sensitively and without cross-reaction with homologues. The optical-density LODs of PEO in liquid vegetables A and B were determined to be 0.09 and 0.075 ppm, respectively. Therefore, the Ag-based LFIA strip can be used as a simple, semiquantitative, quantitative and sensitive screening tool for routine monitoring of residue of PEO in large numbers of vegetable and fruit products.Author contributions
Kuo-Hui Wu: (1) the conception and design of the study, (2) drafting the article, (3) final approval of the version to be submitted. Wen-Chien Huang: acquisition of data, analysis and interpretation of data. Shu-Chen Chang and Rong-Hwa Shyu: revising the article critically for important intellectual content.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors thank the Ministry Science and Technology for supporting this work (MOST 108-2623-E-606-006-D).Notes and references
- A. E. Urusov, A. V. Zherdev and B. B. Dzantiev, Biosensors, 2019, 9, 89 CrossRef CAS PubMed.
- M. Yin, Y. W. Nie, H. Liu, L. Liu, L. Tang, Y. Dong, C. M. Hu and H. Y. Wang, BMC Nephrol., 2022, 23, 30 CrossRef CAS PubMed.
- K. Luo, H. Y. Kim, M. H. Oh and Y. R. Kim, Crit. Rev. Food Sci. Nutr., 2020, 60, 157–170 CrossRef CAS PubMed.
- G. Liu, Y. She, S. Hong, J. Wang and D. Xu, Appl. Sci., 2018, 8, 560 CrossRef.
- A. Raysyan and R. J. Schneider, Biosensors, 2021, 11, 231 CrossRef CAS PubMed.
- T. Bu, X. Yao, L. Huang, L. Dou, B. Zhao, B. Yang, T. Li, J. L. Wang and D. H. Zhang, Talanta, 2020, 206, 120204 CrossRef CAS PubMed.
- Y. Li, L. Liu, S. Song, H. Kuang and C. Xu, Nanomaterials, 2018, 8, 144 CrossRef.
- N. Wiriyachaiporn, H. Sirikett, W. Maneeprakorn and T. Dharakul, Microchim. Acta, 2017, 184, 1827–1835 CrossRef CAS.
- Y. K. Jung, Y. J. Heo, J. J. Lee, A. Deering and E. Bae, J. Microbiol. Methods, 2020, 168, 105800 CrossRef CAS PubMed.
- D. D. Lou, L. Fan, T. Jiang and Y. Zhang, VIEW, 2022, 3, 20200125 CrossRef.
- C. H. Chou, T. H. Huang, P. C. Hsieh, N. Y. J. Ho, C. A. Chen, K. Wu and T. T. Tsai, Anal. Chim. Acta, 2022, 1196, 339544 CrossRef CAS.
- Y. Huang, T. Xu, W. Wang, Y. Q. Wen, K. Li, L. S. Qian, X. Zhang and G. D. Liu, Microchim. Acta, 2020, 187, 70 CrossRef CAS.
- C. T. Matea, T. Mocan, F. Zaharie, C. Iancu and L. Mocan, Chem. Cent. J., 2015, 9, 55–62 CrossRef PubMed.
- H. N. Abdelhamid and H. F. Wu, Trends Anal. Chem., 2015, 65, 30–46 CrossRef CAS.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- F. T. Rodrigues, E. Marchioni, S. Lordel-Madeleine, F. Kuntz, A. L. C. H. Villavicencio and D. Julien-David, Radiat. Phys. Chem., 2020, 166, 108441 CrossRef.
- L. Wang, H. Ye, H. Q. Sang and D. D. Wang, Chin. J. Anal. Chem., 2016, 44, 799–803 CAS.
- V. Cutillas, C. Ferrer and A. R. Fernández-Alba, Anal. Bioanal. Chem., 2021, 413, 5849–5857 CrossRef CAS PubMed.
- Y. Wang, X. X. Liu, C. Chen, Y. D. Chen, Y. Li, H. Ye, B. Wang, H. Chen, J. H. Guo and X. Ma, ACS Nano, 2022, 16, 180–191 CrossRef CAS PubMed.
- W. Bai, C. Zhu, J. Liu, M. Yan, S. Yang and A. Chen, Environ. Toxicol. Chem., 2015, 34, 2244–2249 CrossRef CAS PubMed.
- M. U. Rashid, M. K. H. Bhuiyan and M. E. Quayum, Dhaka Univ. J. Pharm. Sci., 2013, 12, 29–33 CrossRef.
- N. Hooshmand and M. A. El-Sayed, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19299–19304 CrossRef CAS.
- K. Mavani and M. Shah, Int. J. Eng. Res. Technol., 2013, 2, 1–5 Search PubMed.
- S. Szunerits, J. Spadavecchia and R. Boukherroub, Rev. Anal. Chem., 2014, 33, 153–164 CAS.
- A. Gebregeorgis, C. Bhan, O. Wilson and D. Raghavan, J. Colloid Interface Sci., 2013, 389, 31–41 CrossRef CAS.
- M. K. Alqadi, O. A. Abo Noqtah, F. Y. Alzoubi, J. Alzouby and K. Aljarrah, Materials, 2014, 32, 107–111 CAS.
- J. W. Park and J. S. Shumaker-Parry, J. Am. Chem. Soc., 2014, 136, 1907–1921 CrossRef CAS.
- C. T. Matea, T. Mocan, F. Zaharie, C. Iancu and L. Mocan, Chem. Cent. J., 2015, 9, 55–62 CrossRef.
- P. Uznanski, J. Zakrzewska, F. Favier, S. Kazmierski and E. Bryszewska, J. Nanopart. Res., 2017, 19, 121 CrossRef PubMed.
- M. S. Bootharaju and T. Pradeep, J. Phys. Chem. C, 2010, 114, 8328–8336 CrossRef CAS.
- S. Jeong, S. H. Lee, Y. Jo, S. S. Lee, Y. H. Seo, B. W. Ahn, G. Kim, G. E. Jang, J. U. Park, B. H. Ryu and Y. Choi, J. Mater. Chem. C, 2013, 1, 2704–2710 RSC.
- M. I. Azócar, G. Gómez, P. Levín, M. Paez, H. Muñoz and N. Dinamarca, J. Coord. Chem., 2014, 67, 3840–3853 CrossRef.
- S. Govindaraju, S. R. Ankireddy, B. Viswanath, J. Kim and K. Yun, Sci. Rep., 2017, 7, 40298 CrossRef CAS.
- J. Wang, L. Zhang, Y. Huang, A. Dandapat, L. Dai, G. Zhang, X. Lu, J. Zhang, W. Lai and T. Chen, Sci. Rep., 2017, 7, 41419 CrossRef CAS PubMed.
- C. Liu, Q. Jia, C. Yang, R. Qiao, L. Jing, L. Wang, C. Xu and M. Gao, Anal. Chem., 2011, 83, 6778–6784 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |