
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of a novel ene reductase from Pichia angusta with potential application in (R)-levodione production†
Baoqi Zhang ,
Jiale Sun,
Yanqiu Zheng,
Xinlei Mao,
Jinping Lin* and
Dongzhi Wei
,
Jiale Sun,
Yanqiu Zheng,
Xinlei Mao,
Jinping Lin* and
Dongzhi Wei
State Key Laboratory of Bioreactor Engineering, New World Institute of Biotechnology, East China University of Science and Technology, Shanghai 200237, People's Republic of China. E-mail: jplin@ecust.edu.cn
First published on 10th May 2022
Abstract
Asymmetric reduction of electronically activated alkenes by ene reductases (ERs) is an attractive approach for the production of enantiopure chiral products. Herein, a novel FMN-binding ene reductase (PaER) from Pichia angusta was heterologously expressed in Escherichia coli BL21(DE3), and the recombinant enzyme was characterized for its biocatalytic properties. PaER displayed optimal activity at 40 °C and pH 7.5, respectively. The purified enzyme was quite stable below 30 °C over a broad pH range of 5.0–10.0. PaER was identified to have a good ability to reduce the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of various α,β-unsaturated compounds in the presence of NADPH. In addition, PaER exhibited a high reduction rate (kcat = 3.57 s−1) and an excellent stereoselectivity (>99%) for ketoisophorone. Engineered E. coli cells harboring PaER and glucose dehydrogenase (for cofactor regeneration) were employed as biocatalysts for the asymmetric reduction of ketoisophorone. As a result, up to 1000 mM ketoisophorone was completely and enantioselectively converted to (R)-levodione with a >99% ee value in a space–time yield of 460.7 g L−1 d−1. This study provides a great potential biocatalyst for practical synthesis of (R)-levodione.
C bond of various α,β-unsaturated compounds in the presence of NADPH. In addition, PaER exhibited a high reduction rate (kcat = 3.57 s−1) and an excellent stereoselectivity (>99%) for ketoisophorone. Engineered E. coli cells harboring PaER and glucose dehydrogenase (for cofactor regeneration) were employed as biocatalysts for the asymmetric reduction of ketoisophorone. As a result, up to 1000 mM ketoisophorone was completely and enantioselectively converted to (R)-levodione with a >99% ee value in a space–time yield of 460.7 g L−1 d−1. This study provides a great potential biocatalyst for practical synthesis of (R)-levodione.
1 Introduction
CC bond reduction is a valuable reaction in organic synthesis that can generate up to two stereogenic centers simultaneously. In the past, chemical catalysts (such as organocatalysts and metal catalysts) were widely used in catalyzing the stereoselective reduction of conjugated CC bonds of α,β-unsaturated compounds.1 As an attractive tool in organic chemistry, several FMN-dependent ene reductases (ERs) have been identified to catalyze the trans-hydrogenation of activated alkenes in recent years.
ERs have attracted interest due to their ability to reduce CC bonds of α,β-unsaturated compounds with inherent advantages such as mild reaction conditions, excellent chemo-selectivities, improved stereoselectivity and environmental friendliness.2 Thus, ERs have been employed on industrial-scale preparative applications, such as in the synthesis of (2R,5R)-dihydrocarvone and pregabalin.3–12 Furthermore, levodione serves as an intermediate in the synthesis of zeaxanthin, cryptoxanthin and xanthoxin, which can be synthesized through enzymatic reduction of the activated CC double bond in ketoisophorone by the action of ene reductase.8,9
However, wider large-scale implementation of ERs has proved challenging. Major limitations of most ERs lie in their low substrate loading tolerance, poor stability and unsatisfied enantioselectivity.10 Therefore, access to novel and robust ERs is vital to biotechnological applications. To data, novel ERs are universally distributed in bacteria, fungi and plants, and generally operate under mild reaction conditions.10,11,13,14 Actually, as an important consideration of enzyme employed in industrial biocatalysis, the excellent thermostable ability has attracted more attention for ERs. Consequently, a handful of thermostable ERs was identified from extremophile organisms in the past few years.14–17 Meanwhile, the ene reductase in yeast, which as the important sources of OYEs, has not been fully explored. Aiming at increasing the protein diversity of yeast OYEs, we commenced investigating the OYE homologues from Pichia angusta, which is a heat-resistant yeast.18
In this study, a new ene reductase (named PaER) was identified from P. angusta by data mining based on sequence homology. PaER was heterologously expressed and its biochemical properties, including the enzymatic properties, activity and selectivity for α,β-unsaturated compounds, were investigated in detail. Furthermore, the capability of PaER catalyzing ketoisophorone to produce (R)-levodione has been assessed. Finally, by using the whole cells of the engineering E. coli which co-expressed PaER and BmGDH, an efficient and desirable biocatalytic process for optically pure (R)-levodione production was evaluated and developed.
2 Materials and methods
2.1 Materials
All chemicals and solvents were of analytical grade and purchased from commercial sources. NADH, NADPH, NAD+ and NADP+ were purchased from Yeasen Biotechnology (Shanghai, China). Regents for PCR, FastPfu DNA polymerase, Genomic DNA extraction kit, and Plasmid MiniPrep kit were purchased from Transgen (Beijing, China). The vector pET28a(+), pACYC184 and pETDuet-1 were purchased from Novagen (San Diego, CA, USA). Oligonucleotide primers were synthesized by Personal Biotechnology (Shanghai, China). DNA sequencing was performed by Tsingke Biological Technology (Beijing, China). E. coli BL21(DE3) was purchased from Transgen (Beijing, China).2.2 Phylogenetic analysis
The PaER phylogenetically analyzed was other reported OYEs with known functions. The OYEs homologue sequences were retrieved from the NCBI database and used to generate the distance neighbor-joining tree using the MEGA 7.0 software (Temple University, Philadelphia, PA, USA). The CLUSTAL W algorithm was used for alignment.2.3 Clone, expression, and purification of PaER
The PaER gene was amplified via PCR and ligated into the vector pET28a(+). The resulting plasmid pET28a-PaER was sequenced to confirm the fidelity and then transformed into host E. coli BL21(DE3). Pre-cultures were performed in 5 mL LB medium containing 50 mg L−1 kanamycin at 37 °C and 200 rpm. After shaking overnight, preparative cultures were performed in 500 mL LB medium at 37 °C and 200 rpm until optical density OD600 = 0.8 was reached. Afterward expression was induced with 0.2 mM IPTG, and incubation was continued at 20 °C and 150 rpm for 14 h. Cells were harvested by centrifugation (6000 rpm, 10 min, and 4 °C) and washed twice with 0.9% NaCl solution. The pellets were resuspended in the PBS (20 mM, pH 7.5), and then sonicated on ice water bath. The supernatant was collected by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 30 min, and 4 °C). Afterwards, PaER was purified by nickel affinity chromatography according to previous research.3 Protein concentration was determined using the Bradford Protein Assay Kit (Beyotime, Shanghai, China), and the purity of the target protein was analyzed by SDS-PAGE.
000 rpm, 30 min, and 4 °C). Afterwards, PaER was purified by nickel affinity chromatography according to previous research.3 Protein concentration was determined using the Bradford Protein Assay Kit (Beyotime, Shanghai, China), and the purity of the target protein was analyzed by SDS-PAGE.
2.4 Enzyme analysis
The native molecular weight was determined by size-exclusion chromatography using a TSK-GEL G3000SW column (30 cm × 7.5 mm, 10 μm, TOSPH, King of Prussia, PA, USA) equilibrated with sodium phosphate buffer (pH 7.0) and 100 mM Na2SO4 at a flow rate of 0.4 mL min−1. Bovine erythrocytes carbonic anhydrase (29 kDa), yeast alcohol dehydrogenase (150 kDa), sweet potato β-amylase (200 kDa) and horse spleen apoferritin (443 kDa) were used as molecular weight standards.The specific activity of PaER was determined by monitoring the consumption of NADPH at 340 nm (ε = 6.22 mM−1 cm−1). In the case of ketoisophorone, the assay was performed at 365 nm (ε = 3.51 mM−1 cm−1) as described previously.15 The standard assay mixture contained 100 mM PBS (pH 7.5), 5 mM substrate, 0.2 mM NADPH and an appropriate amount of purified enzyme and was carried out at 30 °C. All experiments were conducted in triplicates.
For the determination of the optimum pH, the specific activity was determined by standard activity assay at different pH using sodium citrate buffer (pH 4.0–5.5), sodium phosphate buffer (pH 5.5–8.5) and Gly/NaOH buffer (pH 8.5–10.5). The optimum temperature was determined under standard conditions at different temperature (20–50 °C).
For pH stability measurements, purified PaER was incubated in different pH buffers (20 mM, 5.0–10.0) at 30 °C. Furthermore, the thermal stability of PaER was examined by incubating the purified PaER in PBS buffer (20 mM, pH 7.5) at temperatures between 4 and 40 °C. Samples were withdrawn at regular intervals and assayed for residual activity using the standard assay. The pseudo-first-order rate constants for enzyme inactivation (kin) were obtained from the natural logarithm residual activity values plotted against the time axis. The half-lives were calculated according to the following equation:
| t1/2 = ln(2)/kin. |
For the determination of the kinetic parameters (Km and kcat) on several compounds, 0.01–10 mM of substrates, ketoisophorone (3a), α-methylcinnamaldehyde (10a), and citral (12a), were applied in the standard assay. For determination of the Km values for NADPH, 0.005–1 mM of NADPH was applied in the standard assay with a fixed α-methylcinnamaldehyde concentration of 5 mM. Data were fitted using the Michaelis–Menten equation by the Origin 2020b software (OriginLab, Northampton, MA, USA).
To determine the flavin content of PaER, the purified PaER was incubated at 100 °C to release flavin. Subsequently, the denatured protein was removed by centrifugation. The flavin content of PaER was assayed by RP-HPLC using a Extend-C18 column (250 mm × 4.6 mm, Agilent, Santa Clara, CA, USA) as described previously.11
The effect of metal ions on the activity of PaER was determined by adding 1 mM Na2-EDTA, NaCl, KCl, LiCl, CaCl2, CuCl2, MnCl2, BaCl2, NiCl2, ZnCl2, MgCl2, FeSO4, or Fe2(SO4)3 into the standard assay mixture, then the residual activity was assayed under standard conditions.
2.5 General procedure for enzymatic reduction of α,β-unsaturated compounds
The enzymatic reduction of prochiral compounds were performed in 100 mM PBS buffer (pH 7.5) containing 10 mM substrate, 0.5 mg mL−1 purified PaER, 2.5 U mL−1 purified GDH, 0.5 mM NADP+, and 20 mM glucose. The reaction mixture was shaken at 30 °C and 200 rpm for 2 h. After that, the reactions were stopped by the addition of ethyl acetate. The organic phases were dried over Na2SO4 and the resulting samples were used for GC analysis.2.6 Homology modeling and molecular docking
The structure mode of PaER was undertaken using the SWISS-MODEL web server based on the known structure of OYE2.6 from S. stipitis CBS 6054 (PDB ID: 3TJL, 1.50 Å, 45.67% sequence identity to PaER).19 The model quality was evaluated using Verify-3D and Ramachandran plot, results presented in Fig. S4.† Substrate–enzyme docking was performed using the AutoDock 4.2 program. The substrate ketoisophorone was set as “flexible” and amino acid residues were treated as “rigid” except for His185, His188 and Tyr190. Ultimately, ketoisophorone was docked into the active center pocket of PaER. Structural analysis was performed using the PyMOL-edu program.2.7 Analytical methods
GC analyses were performed on a 7890A GC system (Agilent), equipped with an FID detector. HPLC analyses were carried on a 1100A HPLC system (Agilent), equipped with an VWD detector. The product yield and ee value of (R)-levodione were determined by GC using a CP-ChiraSil-DEX CB column (25 m × 0.25 mm × 0.25 μm, Agilent) as described previously.3,11 The product yield and de value of dihydrocarvone were determined by GC using a DB-5 column (30 m × 0.32 mm × 0.25 μm, Agilent) as described previously.3 The yield and ee value of 2-methyl-hydrocinnamaldehyde (12b) was determined by HPLC using a Chiralcel OJ-H column (250 mm × 4.6 mm, Daicel, Shanghai, China) as described previously.11 The conversion of citral (12a) was performed on GC using a HP-5 column (30 m × 0.32 mm × 0.25 μm, Agilent) as described previously.3 The ee value of citronellal was determined by GC using a Beta DEX 225 column (30 m × 0.25 mm × 0.25 μm, Supelco, Bellafonte, PA, USA) as described previously.32.8 Construction of recombinant cells co-expressing PaER and GDH
The open reading frame (ORF) of GDH from pET28a-GDH was cloned into the backbone of pET28a-PaER behind PaER to create pET28a-PaER–GDH. Then pET28a-PaER–GDH was transformed into E. coli BL21(DE3) to have a co-expression strain (Strain 1). The PaER gene was amplified via PCR and ligated into the MCS1 of vector pETDuet-1 to create pETDuet-1-PaER. Subsequently, the glucose dehydrogenase (GDH) gene was inserted into the MCS2 of vector pETDuet-1-PaER to create pETDuet-1-PaER–GDH. Then pETDuet-1-PaER–GDH was transformed into E. coli BL21(DE3) to acquire a co-expression strain (Strain 2). The gene of GDH was cloned in vector pACYC184 (pACYC184-GDH) as described previously.20 Then pET28a-PaER and pACYC184-GDH were co-transformed into E. coli BL21(DE3) to gain a co-expression strain (Strain 3). The fusion expression of PaER and GDH was constructed in pET28a(+) using the SOE-PCR, and a 10 nm rigid α-helical ER/K linker was selected as the linker peptide as described previously.21 Then the resulting plasmid (pET28a-GDH–ERK–PaER) was transformed into E. coli BL21(DE3) to have a fusion expression strain (Strain 4). The primers were listed in the Table S1.†2.9 Asymmetric synthesis of (R)-levodione
The reaction system for synthesis of (R)-levodione from ketoisophorone contained 1 M ketoisophorone, 1.05 M glucose, NADP+ (0.05–0.2 mM), 10 g L−1 lyophilized E. coli cell (Strain 3), and PBS buffer (100 mM, pH 7.5). The final volume of the reaction system was 20 mL. The reaction temperature was kept at 40 °C, and the pH was automatically adjusted to 7.5 by titrating with Na2CO3. The reaction mixture was withdrawn periodically and extracted with ethyl acetate (EtAc), and then analyzed by gas chromatography (GC). After the reaction was completed, the reaction mixture was extracted with EtAc. Then the combined organic phase was filtered and evaporated under vacuum by a rotary evaporator.3 Results and discussion
3.1 Identification and sequence homology analysis of PaER
To obtain novel ERs to access α,β-unsaturated compounds, OYE1 (Q02899.3) was used as a probe for candidate sequence searching in Pichia angusta by BLASTP. Based on sequence identity and conserved domains, PaER which showed 39.9% sequence identity with OYE1 was selected from P. angusta. In PaER, highly conserved catalytic residue (Try190) and substrate binding residues (His185 and His188) in old yellow enzyme family were found according to multiple sequence alignments (Fig. S1†). A phylogenetic analysis was carried out including PaER and 27 ene reductases from old yellow enzyme families (Fig. 1). In particular, PaER shares 48.8%, 48.2%, and 45.8% sequence identity with ERs isolated from the Candida albicans (EBP1), Clavispora lusitaniae (ClER), Meyerozyma guilliermondii (MgER), respectively. Both the results of phylogenetic analysis and similarities observed in the alignment (Fig. S1†) proved that the PaER belongs to the cluster of class II OYEs.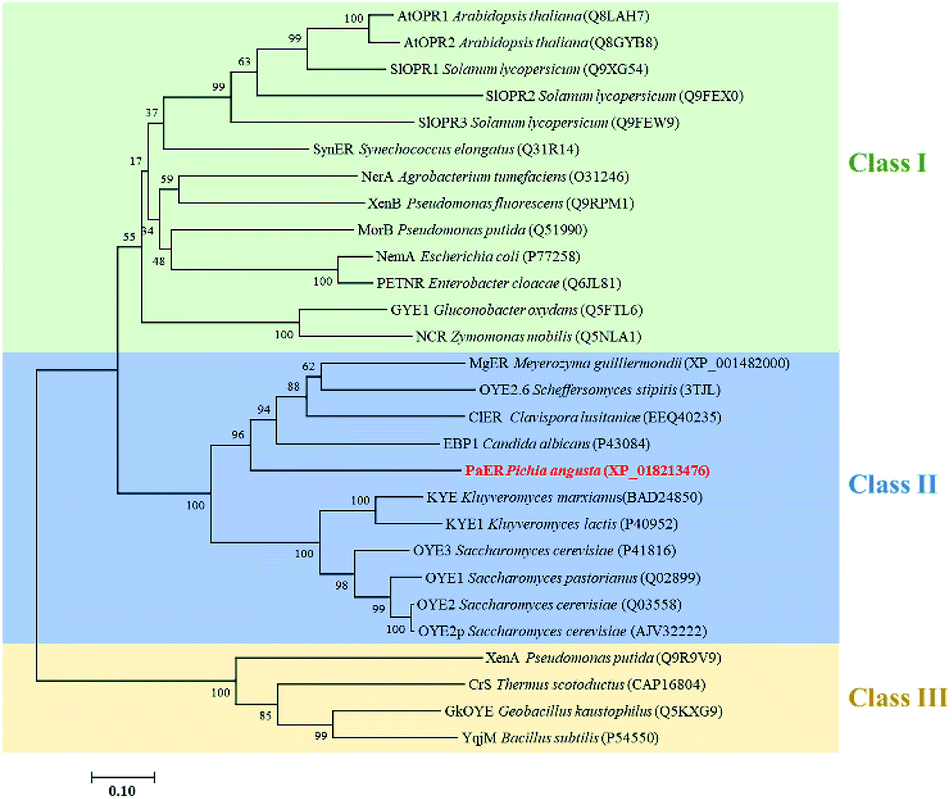 | ||
| Fig. 1 Phylogenetic relationship of amino acid sequences of PaER to other OYEs with known function. Bootstrap values at the nodes were expressed as percentages of 1000 replications. The accession number of OYEs is shown in brackets. | ||
3.2 Heterologous expression and purification of PaER
The gene fragment of PaER obtained by PCR was ligated to pET28a(+) vector and then introduced into E. coli BL21(DE3) for heterologous expression of PaER. The PaER was further purified by nickel-affinity chromatography. SDS-PAGE analysis showed that PaER was mainly present in soluble cell fraction and the apparent molecular weight of the purified protein was about 46 kDa, which was consistent with the predicted protein molecular mass (Fig. 2). The purified PaER displaying a yellow color as expected for FMN-containing protein and was also confirmed by HPLC (Fig. S2†). Moreover, the native molecular weight of PaER was determined to be 91 kDa by size-exclusion chromatography, indicating that PaER occurs as dimers in solution (Fig. S3†).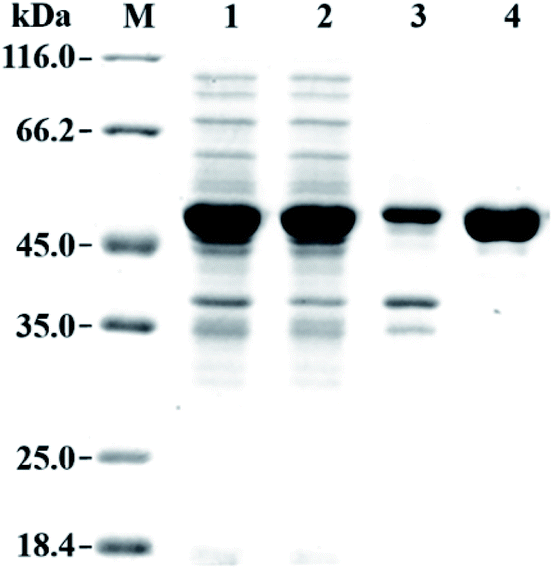 | ||
| Fig. 2 SDS-PAGE analysis of the expression and purification of PaER. Lane M, protein molecular weight marker; Lane 1, whole-cell of E. coli expressing PaER; Lane 2, crude extract of E. coli expressing PaER; Lane 3, precipitate of E. coli expressing PaER; Lane 4, purified PaER. | ||
3.3 Cofactor preference
In order to verify the cofactor preference of PaER, ketoisophorone was used as the substrate to test the oxidation of NADH or NADPH under standard conditions. When NADH or NADPH was used, the specific activity was 0.16 U mg−1 or 5.1 U mg−1, indicating NADPH as the preferred cofactor.3.4 Dependence of PaER activity on pH, temperature and metal ions
The effects of pH and temperature on the activity of PaER were performed at a pH range of 4.0–10.5, and a range of 20–50 °C. The activity-pH profile showed that the highest activity was observed at pH 7.5 in a PBS buffer (Fig. 3A). When the environment pH was lower than 6.0 or higher than 8.5, the enzyme activity was decreased significantly. The optimal temperature of PaER was 40 °C and it maintained high activity in the range of 25–45 °C (residual activity was more than 50%) (Fig. 3B).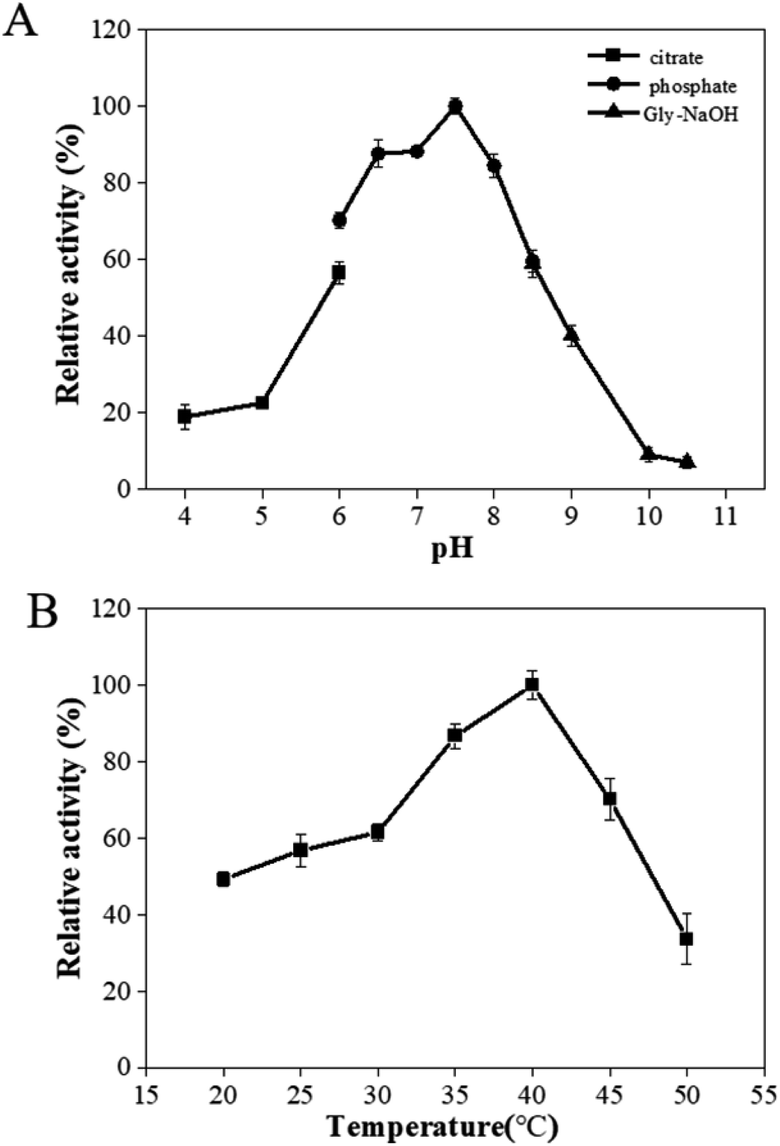 | ||
| Fig. 3 Effect of pH (A) and temperature (B) on enzyme activity of PaER. | ||
The pH stability and thermostability of PaER were also investigated. The enzyme was quite stable at a broad pH range of 5.0–10.0 at 30 °C, giving half-lives of 106 h, 118 h, 170 h, 102 h and 71 h, respectively (Fig. 4A). Moreover, the purified PaER exhibited an incredible stability from 4 °C to 40 °C (Fig. 4B). The half-lives of thermal inactivation of PaER amounted to 179 h at 30 °C and 89 h at 40 °C, much higher than those of known yeast OYEs reported so far (Table S2†). These results indicated that the PaER had relatively high pH stability and thermostability, which are favorable for good operational stability at industrial scale.
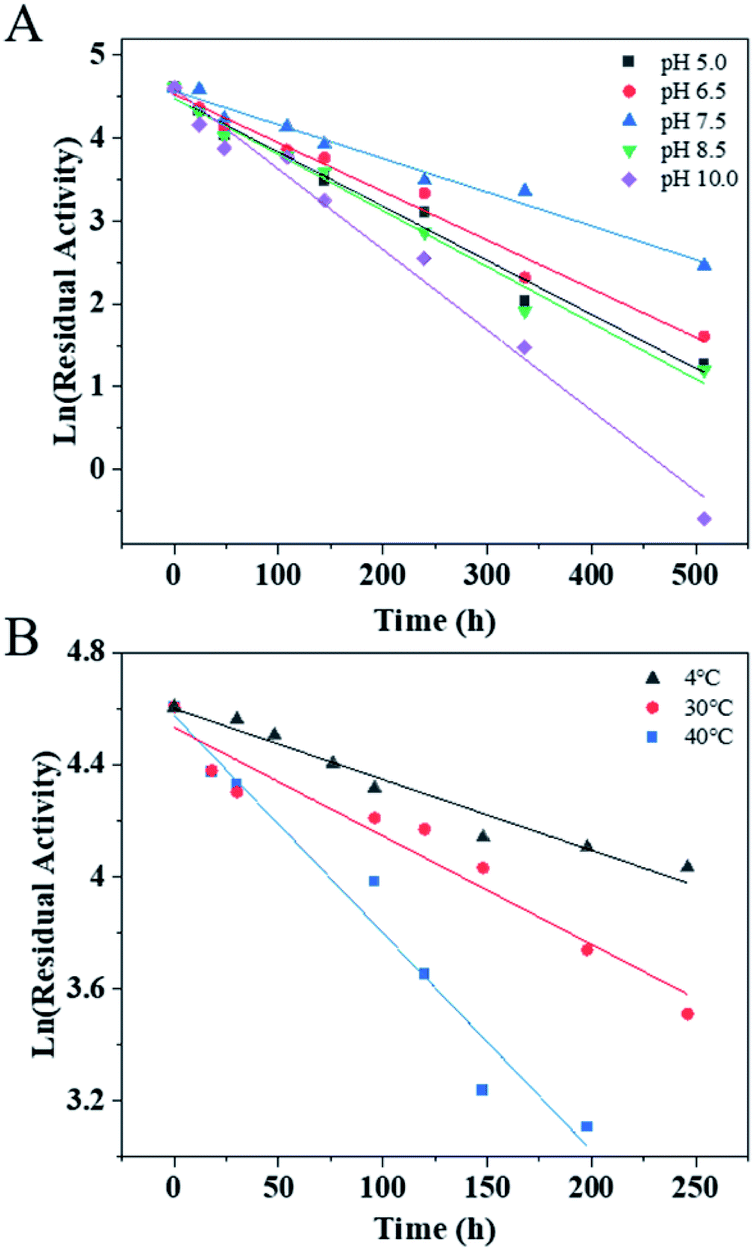 | ||
| Fig. 4 (A) The pH stability of PaER in various pH (5.0–10.0) conditions. (B) The thermostability of PaER at various temperatures (4, 30 and 40 °C). | ||
Metal ions dependence of PaER was also determined by incubating the purified PaER with different metal ions at the final concentration of 1 mM (Table 1). In the presence of EDTA, PaER still remained 85.46% of its normal activity, which indicated the metal ions are not necessary for PaER. Most of the tested metal ions had little effect on the activity of PaER, except Cu2+, Zn2+ and Fe2+, which partially inhibited the activity of PaER. In the presence of 1 mM K+ or Na+, the activity of PaER was slightly increased to 108.78% or 106.07%. All above indicated that PaER was not metal-dependent.
| Io | Relative activity (%) |
|---|---|
| Control | 100.00 ± 2.65 |
| Na2-EDTA | 94.87 ± 7.66 |
| Na+ | 106.07 ± 5.11 |
| K+ | 108.78 ± 5.74 |
| Li+ | 100.88 ± 3.45 |
| Ca2+ | 91.42 ± 4.72 |
| Cu2+ | 78.70 ± 4.62 |
| Mn2+ | 96.06 ± 7.65 |
| Co2+ | 96.25 ± 6.61 |
| Ba2+ | 97.73 ± 4.70 |
| Ni2+ | 93.98 ± 4.51 |
| Zn2+ | 83.63 ± 7.51 |
| Mg2+ | 101.13 ± 6.07 |
| Fe2+ | 88.26 ± 6.40 |
| Fe3+ | 101.64 ± 5.23 |
3.5 Substrate specificity and kinetic properties
Substrate specificity analysis could provide useful guidance on the application potential of biocatalysts. 14 α,β-unsaturated compounds bearing a ketone (1a–4a), imide (5a, 6a), ester (7a), aldehyde (9a–12a), and carboxylic acid (13a, 14a) as electron-withdrawing group were applied to study the specific activities of PaER. As shown in Fig. 5, PaER exhibited superior activity for substrates with two electron-withdrawing groups, such as ketoisophorone (3a), p-benzoquinone (4a), maleimide (5a), N-phenylmaleimide (6a) and dimethyl maleate (7a). The highest specific activity was observed toward maleimide (5a) (8.59 U mg−1). For 6a that had phenyl substituent at the N atom of 5a, the specific activity of PaER towards 6a was lower than that of 5a, indicating that the bulky substituent could reduce enzyme activity. The low catalytic activity of PaER towards 8a (0.16 U mg−1) indicated that the activity of PaER was decreased when the substrate contains a hydroxyl substituent. In addition, PaER performed considerable activity to α,β-unsaturated aldehyde (9a–12a), achieving 0.96–3.78 U mg−1. In the case of the substrate of 10a, which has a β-substituted methyl compared with 9a, PaER exhibited lower activity (2.69 U mg−1), which revealed that the methyl substituent at the CC bond was unfavorable to the reaction.
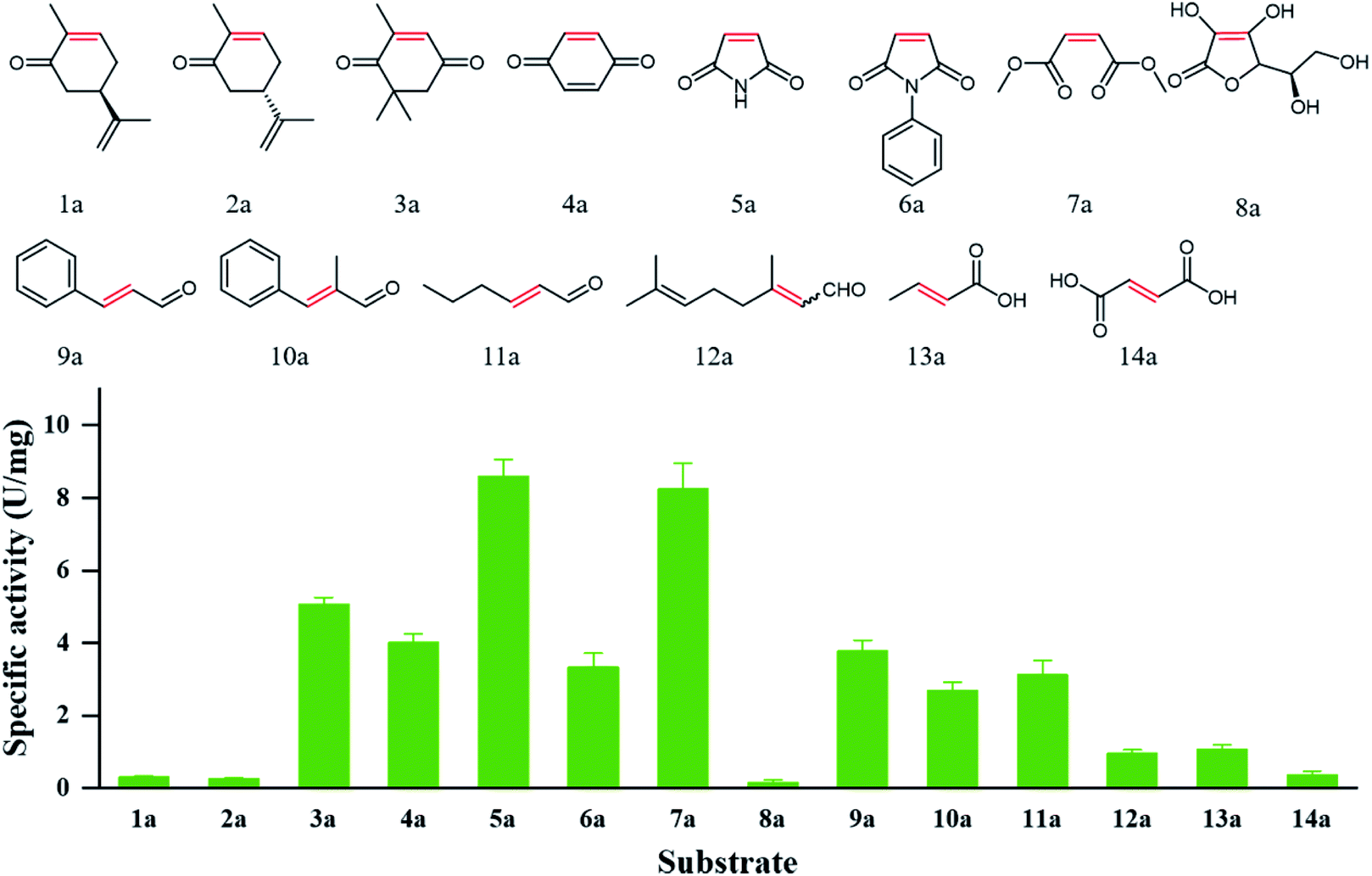 | ||
| Fig. 5 Catalytic profile of PaER and the measured specific activities toward 1a–14a of the substrate library. | ||
The stereoselectivities of PaER toward the prochiral α,β-unsaturated compounds (1–3a, 10a, 12a) were also determined. As shown in Table 2, (R)-carvone and (S)-carvone was reduced to (2R,5R)-dihydrocarvone and (2R,5S)-dihydrocarvone with unsatisfied conversion (64% and 59%) and low stereoselectivity (68% and 72%). Although PaER exhibited poor stereoselectivities toward α-methylcinnamaldehyde (10a) and citral (12a), albeit relatively high catalytic activities. Ketoisophorone (3a), which is recognized as a good substrate for OYEs, was reduced to (R)-levodione by PaER with excellent stereoselectivity (ee > 99%) and high catalytic constant (kcat = 3.57 s−1) (Table S3†). Taken together, these results suggest that PaER is feasible to manufacture important pharmaceutical intermediates such as (R)-levodione.
| Substrate | Conversion/% | ee/% | |
|---|---|---|---|
| a The reaction system (1 mL) contained 10 mM substrate, 0.5 mg purified PaER, 2.5 U purified BmGDH, 0.5 mM NADP+, 20 mM glucose, 100 mM PBS (pH 7.5). | |||
| 1a | 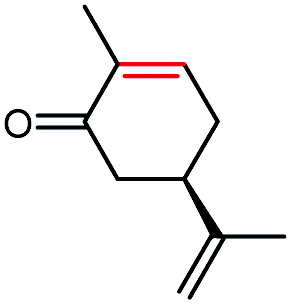 |
64 | 68 de (2R,5R) |
| 2a | 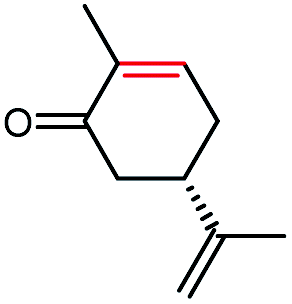 |
59 | 72 de (2R,5S) |
| 3a |  |
>99 | >99 (R) |
| 10a | 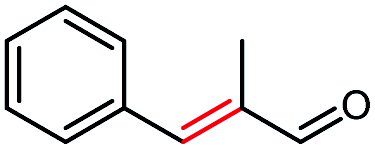 |
>99 | 29 (S) |
| 12a | 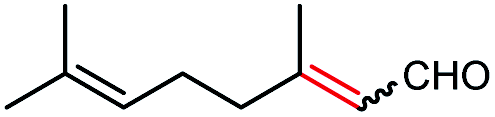 |
>99 | 25 (R) |
To further our understanding of the interactions between the high activity and stereoselectivity of PaER and ketoisophorone. The model of PaER was created from the OYE2.6 structure (PDB ID: 3TJL) using SWISS-MODEL and molecular docking was performed using the AutoDock 4.2 program. The Ramachandran plot of the resulting model showed that 99.4% of the residues fell into the allowed region (Fig. S4A†). The Verify-3D analysis results pointed out that the 100% of the amino acid for the model had 3D-1D score larger than 0 (Fig. S4B†), demonstrating the reliability of the model. The resulting model of PaER was further utilized for molecular docking analysis.
Mechanistically, the catalytic residue Tyr190 and FMNH2 donated their hydrogen atoms to the α-C and the β-C of the substrate ketoisophorone, respectively, resulting in the trans-hydrogenation of CC bond (Fig. 6A). The docking analysis (Fig. 6B) showed that the substrate ketoisophorone could enter the active pocket well and formed two hydrogen bonds between ketoisophorone-O1 and His185/His188, which produced a classical binding mode (R-selective) for ketoisophorone. Furthermore, the distance from Tyr190-OH to substrate α-C and the distance from FMN-N5 to substrate β-C were 3.7 Å and 3.4 Å, respectively, which was beneficial for the nucleophilic attack on the CC bond of ketoisophorone. This may be an important reason of why ketoisophorone can be reduced by PaER with high activity. Through homology modeling and molecular docking, the catalytic activity and stereoselective mechanism of PaER were studied and these results provided a basis for rational design in the future.
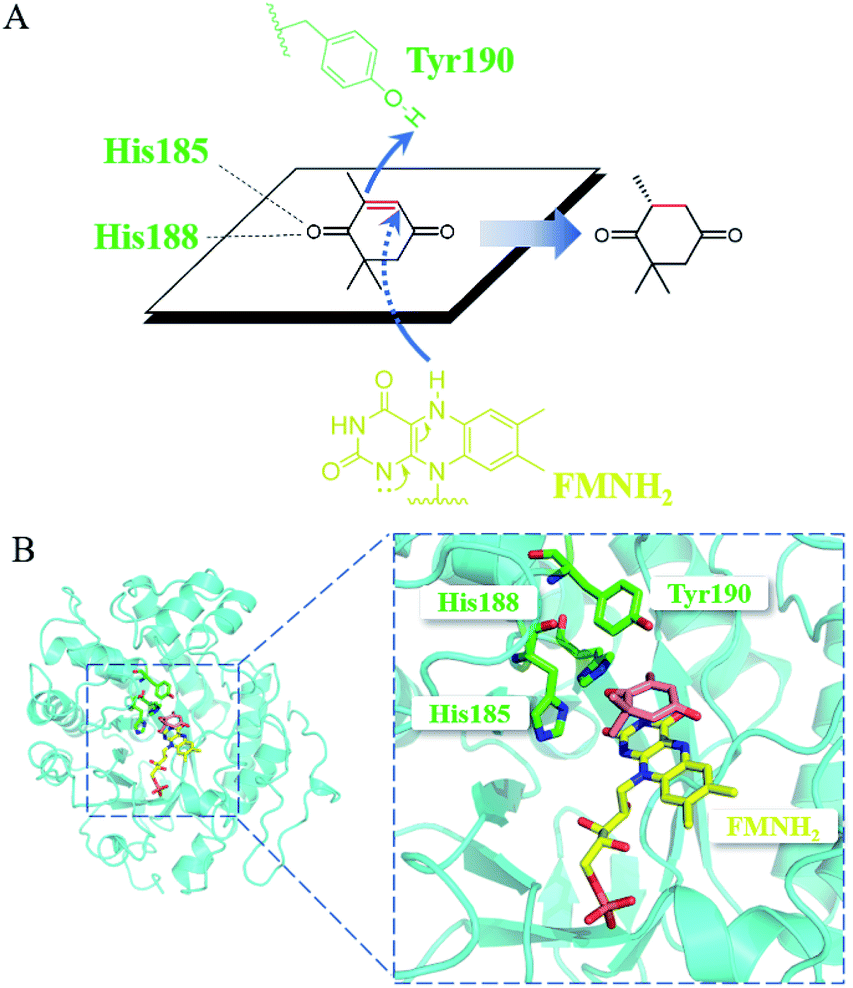 | ||
| Fig. 6 (A) Proposed catalytic mechanism of PaER for the reduction of ketoisophorone. (B) 3D structure generated by docking of ketoisophorone into the active pocket of PaER. | ||
3.6 Asymmetric synthesis of (R)-levodione using PaER
As an industrially applied building block in the synthesis of zeaxanthin and xanthoxin, (R)-levodione can be synthesized by stereo-selective CC bond bio-reduction of ketoisophorone (3a) (Fig. 7A). Compared with the catalysis of free enzyme, whole-cell catalysis is more conducive to the stability of enzyme and reduces the production cost, which makes it ideal to produce chiral compounds. Therefore, we attempted to develop an E. coli whole-cell biocatalyst for the bioreduction of ketoisophorone to (R)-levodione. Considering that PaER was NAD(P)H-dependent and GDH-glucose is the most commonly NAD(P)H regeneration system, BmGDH was used for NAD(P)H regeneration to promote the reduction of ketoisophorone by PaER. Therefore, PaER and BmGDH were co-expressed in E. coli BL21(DE3) cells in tandem using various types of vectors (named Strain 1–3, Fig. 7B and S5†). In addition, the long rigid linker peptide ER/K was used to fuse PaER and BmGDH following linker attachment to the N-terminal of PaER (named Strain 4, Fig. 6B and S4†). Four engineered E. coli strains were constructed and conducted to evaluate the industrial applicability of PaER. As shown in Fig. 7C, all the four resultant cells successfully produced (R)-levodione within 1 h. Among these recombinant strains, Strain 3 achieved the highest production of (R)-levodione (15.84 mM) as compared to other strains (0.16–14.78 mM).
 | ||
| Fig. 7 Asymmetric bioreduction of ketoisophorone. (A) PaER-mediated reduction reaction for preparation of (R)-levodione. (B) Construction of engineered E. coli strains. (C) Comparison of the (R)-levodione production in each constructed E. coli strain. The reaction system (1 mL) contained 20 mM substrate (3a), 1 g L−1 lyophilized E. coli cells, 0.2 mM NADP+, 40 mM glucose, and 100 mM PBS (pH 7.5). | ||
Finally, to demonstrate the potentiality of PaER for practical applications, the bioreduction of ketoisophorone was performed on a preparative scale using the whole cells of Strain 3 with addition of 0.2 mM NADP+. As shown in Fig. 8, as much as 1000 mM of ketoisophorone was completely converted to (R)-levodione in 8 h with ee of >99%, product titer of 153.6 g L−1 and a high space-time yield of 460.7 g L−1 d−1. Moreover, the substrate was almost totally converted in 20 h at the same substrate when the amount of external addition of NADP+ was decreased to 0.1 mM much lower than those of ERs-mediated reduction of ketoisophorone described in the literature (Table S4†). Most notably, addition of external cofactor is an important consideration for an efficient biocatalyst with feasibility in chemical manufacturing. When the NADP+ loading was decreased to 0.05 mM, the resultant concentration of (R)-levodione was only 647 mM in 24 h with 65.0% conversion (Fig. 8). This result can be attributed to the high Km value of PaER towards NADPH and the degradation of NADP(H).22
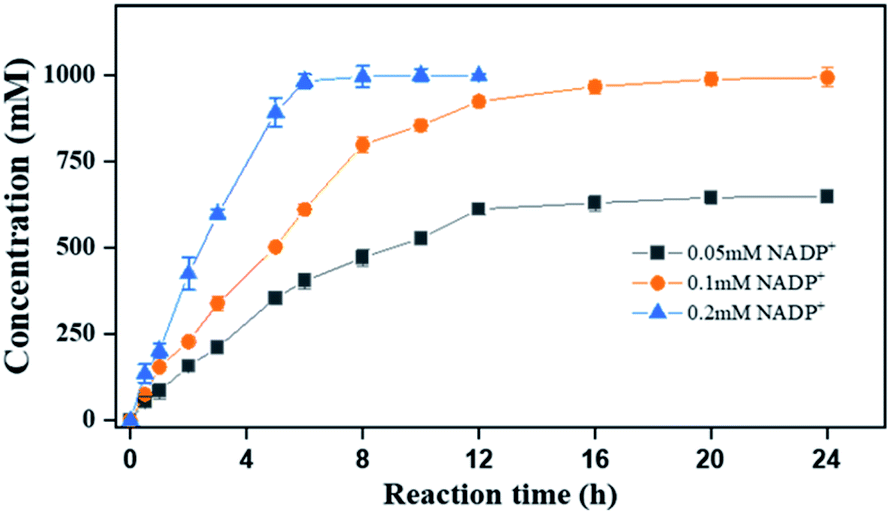 | ||
| Fig. 8 Reaction progress curves of ketoisophorone by Strain 3 on a preparative scale. Reaction mixtures (20 mL): ketoisophorone (1000 mM), glucose (1050 mM), NADP+ (0.05–0.2 mM), 10 g L−1 lyophilized E. coli cell (Strain 3), and PBS buffer (100 mM, pH 7.5). | ||
To date, numerous OYE homologues have been reported to catalyze the ene-reduction of ketoisophorone, while the substrate loads were quite low in most cases (Table S4†). As a representative case, Ni et al. reported that the crude enzyme of ClER could completely convert 500 mM ketoisophorone into (R)-levodione with >98% ee value.23 By contrast, PaER-expressing Strain 3 mediated ketoisophorone reduction in present study exhibited the highest product titer (153.6 g L−1) and excellent optical purity (>99% ee).
4 Conclusions
In the present study, a novel ene reductase (PaER) from P. angusta was identified as a promising biocatalyst for the asymmetric bioreduction of α,β-unsaturated compounds. PaER exhibited a broad substrate spectrum towards α,β-unsaturated compounds, as well as a high catalytic activity and stereoselectivity of ketoisophorone. Thus, PaER could be applied to produce (R)-levodione from ketoisophorone. The engineered E. coli BL21(DE3) cells co-expressing PaER and BmGDH could convert 1000 mM ketoisophorone into (R)-levodione with a >99% ee value and a space-time yield of 460.7 g L−1 d−1, indicating its potential applications as a biocatalyst in preparation of (R)-levodione. Further studies to change the coenzyme preference by protein engineering are now in progress for establishing economic bioprocess without assistance of external cofactor and make PaER a more efficient biocatalyst for industrial production.Author contributions
BZ and JL designed the experiments. BZ and JS performed the experiments. BZ and JL drafted the manuscript. JS and YZ provided advice in the experiments design and data analysis. JL and DW conceived the research. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2021YFC2102100), the Natural Science Foundation of Shanghai (No. 19ZR1412700), the Fundamental Research Funds for the Central Universities (No. 22221818014), and partially supported by the Open Funding Project of the State Key Laboratory of Bioreactor Engineering.References
- A. Zuliani, C. M. Cova, R. Manno, V. Sebastian, A. A. Romero and R. Luque, Green Chem., 2020, 22, 379–387 RSC.
- A. Scholtissek, D. Tischler, A. Westphal, W. van Berkel and C. Paul, Catalysts, 2017, 7, 130 CrossRef.
- L. Zheng, J. Lin, B. Zhang, Y. Kuang and D. Wei, Bioresour. Bioprocess., 2018, 5, 9 CrossRef.
- M. Hall, B. Hauer, R. Stuermer, W. Kroutil and K. Faber, Tetrahedron: Asymmetry, 2006, 17, 3058–3062 CrossRef CAS.
- C. Mahler, C. Burger, F. Kratzl, D. Weuster-Botz and K. Castiglione, Molecules, 2019, 24, 2550 CrossRef PubMed.
- N. Nett, S. Duewel, L. Schmermund, G. E. Benary, K. Ranaghan, A. Mulholland, D. J. Opperman and S. Hoebenreich, Mol. Catal., 2021, 502, 111404 CrossRef CAS.
- M. Kataoka, A. Kotaka, A. Hasegawa, M. Wada, A. Yoshizumi, S. Nakamori and S. Shimizu, Biosci., Biotechnol., Biochem., 2002, 66, 2651–2657 CrossRef CAS PubMed.
- M. Kataoka, A. Kotaka, R. Thiwthong, M. Wada, S. Nakamori and S. Shimizu, J. Biotechnol., 2004, 114, 1–9 CrossRef CAS PubMed.
- W. B. Black, L. Zhang, W. S. Mak, S. Maxel, Y. Cui, E. King, B. Fong, A. Sanchez Martinez, J. B. Siegel and H. Li, Nat. Chem. Biol., 2020, 16, 87–94 CrossRef CAS PubMed.
- D. Dobrijevic, L. Benhamou, A. E. Aliev, D. Méndez-Sánchez, N. Dawson, D. Baud, N. Tappertzhofen, T. S. Moody, C. A. Orengo, H. C. Hailes and J. M. Ward, RSC Adv., 2019, 9, 36608–36614 RSC.
- B. Zhang, L. Zheng, J. Lin and D. Wei, Biotechnol. Lett., 2016, 38, 1527–1534 CrossRef CAS PubMed.
- C. K. Winkler, D. Clay, S. Davies, P. O'Neill, P. McDaid, S. Debarge, J. Steflik, M. Karmilowicz, J. W. Wong and K. Faber, J. Org. Chem., 2013, 78, 1525–1533 CrossRef CAS PubMed.
- C. Magallanes-Noguera, F. M. Cecati, M. L. Mascotti, G. F. Reta, E. Agostini, A. A. Orden and M. Kurina-Sanz, J. Biotechnol., 2017, 251, 14–20 CrossRef CAS PubMed.
- A. Scholtissek, S. R. Ullrich, M. Muhling, M. Schlomann, C. E. Paul and D. Tischler, Appl. Microbiol. Biotechnol., 2017, 101, 609–619 CrossRef CAS PubMed.
- M. S. Robescu, M. Niero, M. Hall, L. Cendron and E. Bergantino, Appl. Microbiol. Biotechnol., 2020, 104, 2051–2066 CrossRef CAS PubMed.
- D. Tischler, E. Gadke, D. Eggerichs, A. Gomez Baraibar, C. Mugge, A. Scholtissek and C. E. Paul, ChemBioChem, 2020, 21, 1217–1225 CrossRef CAS PubMed.
- D. J. Opperman, B. T. Sewell, D. Litthauer, M. N. Isupov, J. A. Littlechild and E. van Heerden, Biochem. Biophys. Res. Commun., 2010, 393, 426–431 CrossRef CAS PubMed.
- D. Biswas, M. Datt, K. Ganesan and A. K. Mondal, Appl. Microbiol. Biotechnol., 2010, 88, 1311–1320 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS PubMed.
- B. Zhang, H. Du, Y. Zheng, J. Sun, Y. Shen, J. Lin and D. Wei, Microb. Biotechnol., 2021, 15, 1486–1498 CrossRef PubMed.
- Y. Liu, X. Mao, B. Zhang, J. Lin and D. Wei, Biotechnol. Lett., 2021, 43, 1–10 CrossRef PubMed.
- N. Xu, J. Zhu, Y.-Q. Wu, Y. Zhang, J.-Y. Xia, Q. Zhao, G.-Q. Lin, H.-L. Yu and J.-H. Xu, Org. Process Res. Dev., 2020, 24, 1124–1130 CrossRef CAS.
- Y. Ni, H. L. Yu, G. Q. Lin and J. H. Xu, Enzyme Microb. Technol., 2014, 56, 40–45 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01716d |
| This journal is © The Royal Society of Chemistry 2022 |