
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmerging application of biochar as a renewable and superior filler in polymer composites
Tengku Arisyah Tengku Yasim-Anuarae,
Lawrence Ng Yee-Foong b,
Abubakar Abdullahi Lawalcd,
Mohammed Abdillah Ahmad Farida,
Mohd Zulkhairi Mohd Yusufa,
Mohd Ali Hassanac and
Hidayah Ariffin*ab
b,
Abubakar Abdullahi Lawalcd,
Mohammed Abdillah Ahmad Farida,
Mohd Zulkhairi Mohd Yusufa,
Mohd Ali Hassanac and
Hidayah Ariffin*ab
aDepartment of Bioprocess Technology, Faculty of Biotechnology and Biomolecular Sciences, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia. E-mail: hidayah@upm.edu.my
bLaboratory of Biopolymer and Derivatives, Institute of Tropical Forestry and Forest Products, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
cDepartment of Process and Food Engineering, Faculty of Engineering, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
dDepartment of Agricultural and Environmental Resources Engineering, Faculty of Engineering, University of Maiduguri, Maiduguri, Borno State, Nigeria
eNextgreen Pulp & Paper Sdn. Bhd., Green Technology Park, Paloh Inai, 26600 Pekan, Pahang, Malaysia
First published on 11th May 2022
Abstract
Biochar is conventionally and widely used for soil amendment or as an adsorbent for water treatment. Nevertheless, the need for transition to renewable materials has resulted in an expansion of biochar for use as a filler for polymer composites. The aim is to enhance the physical, chemical, mechanical and rheological properties of the polymer composite. The reinforcement of biochar into a polymer matrix however is still new, and limited reports are focusing on the effects of biochar towards polymer composite properties. Hence, this review highlights the unique properties of biochar and its effect on the crystallization, thermal, flammability, electrical conductivity, and mechanical properties of polymer composites. This review does not solely summarize recent studies on biochar–polymer-based composites, but also offers insights into a new direction of biochar as a renewable and superior polymer filler in the future.
1. Introduction
Given the high demand for smart materials, most materials such as metals, glass and ceramics have been replaced by polymer composites mainly due to their light weight, easy processing and low production cost.1 Polymer composites have been used for various purposes ranging from low-cost household products to high-performance industrial products. According to the Plastics Market Size Share Industry Analysis Report released by Grand View Research in February 2020, the plastics market growth has been increased from USD 72.7 billion in 2016 to USD 568.7 billion by 2019 due to the high demand from various industrial sectors including the wind energy, aerospace, packaging, construction and automotive industries. This was highly attributed to the versatility of polymers as they are light in weight, have good strength, are corrosion-resistant, cheap and easy to produce and can be reused multiple times. Despite their diverse properties, polymers have some drawbacks associated with their components such as low thermal stability, low conductivity and low flame retardancy. Research has been carried out to unfold such issues and one of the most effective solutions is by adding a filler to improve and modify the composite properties.2–7 Since there is great interest in low-cost, sustainable and environmentally friendly materials, biochar has received great interest for use as a filler, as an alternative to the other non-environmentally and non-economically viable carbon fillers such as carbon black, carbon nanotubes and graphene.In contrast to the other carbon fillers which require complex synthetic production methods, biochar which is a porous carbonaceous solid residue can be produced by slowly pyrolysing the biomass at a high temperature ranging from 500 to 700 °C.8 Similar to other carbon fillers, biochar is porous, thermally stable, has large specific surface area, and consists of several functional groups: hydroxyl, carboxyl, carbonyl and others.5,9,10 The use of biomass for biochar production to be used as filler in polymer composites will not only solving the waste management issue, but it also promotes the potential of biomass for the production of high-value-added products.
Over the recent years, there has been an influx of research done to assess the potential of biochar to act as a cost-efficient, sustainable and renewable filler for polymers. The major aim of reinforcing biochar is to enhance the mechanical, thermal and electrical conductivity properties of polymer composites.11 Based on the previous research, biochar has been proven as a superior filler compared to other fillers, especially natural fibers.12–16 In contrast to the natural fibers, the properties of biochar can be altered by modifying the pyrolysis condition to achieve high hydrophobicity properties, which can help in enhancing the compatibility with the polymer matrix. In fact, the thermal stability of biochar composites was found to be higher than the composites reinforced with natural fibers, thus diversifying their usage for various purposes. For instance, Khan et al. (2017)14 reported the use of biochar as a filler in epoxy composites and compared with carbon nanotubes (CNT). It was revealed that the ultimate tensile strength of epoxy composites reinforced with 2 wt% biochar was higher than that of epoxy composites reinforced with 2 wt% CNT. In the same report, tensile toughness was superior in the case of biochar as compared to CNT. In fact, the microwave permittivity and conductivity among epoxy/biochar and epoxy/CNT composites were comparable. This shows the potential of biochar to replace the expensive carbon nanotubes as a filler for polymer composites.
In general, biochar reinforced polymer composites have potential applications in various industries. Biochar reinforced polymer composites could be beneficial in the packaging industry and for the manufacturing of interior components for cars or aeroplanes, as it is light-weight and has fire-resistant property.17 Nevertheless, there is still limited research published on biochar-polymer-based composites. This might be due to the lacking of understanding on the potential effect of biochar on the polymers. Hence, this review intends to highlight the unique properties of biochar, to explain its effect on the polymer matrix. Comparison on the properties, processing methods, and interaction with the polymers between biochar and other carbon fillers are also discussed, to provide the new insights of biochar as a potential renewable filler to enhance the properties of polymer composites.
2. Production and characterization of biochar
2.1. Biochar production
Thermochemical decomposition of biomass has been the most common and convenient conversion technique for biochar production. The thermochemical conversion refers to the controlled thermal treatment to activate and sustain decomposition and/or oxidation of biomass to produce energy carriers (biochar, bio-oil and syngas) or heat.18 Several thermochemical schemes have been developed to produce biochar from biomass feedstock. Among the various thermochemical conversion technologies, pyrolysis and hydrothermal carbonization are the preferred methods for high carbon yield.19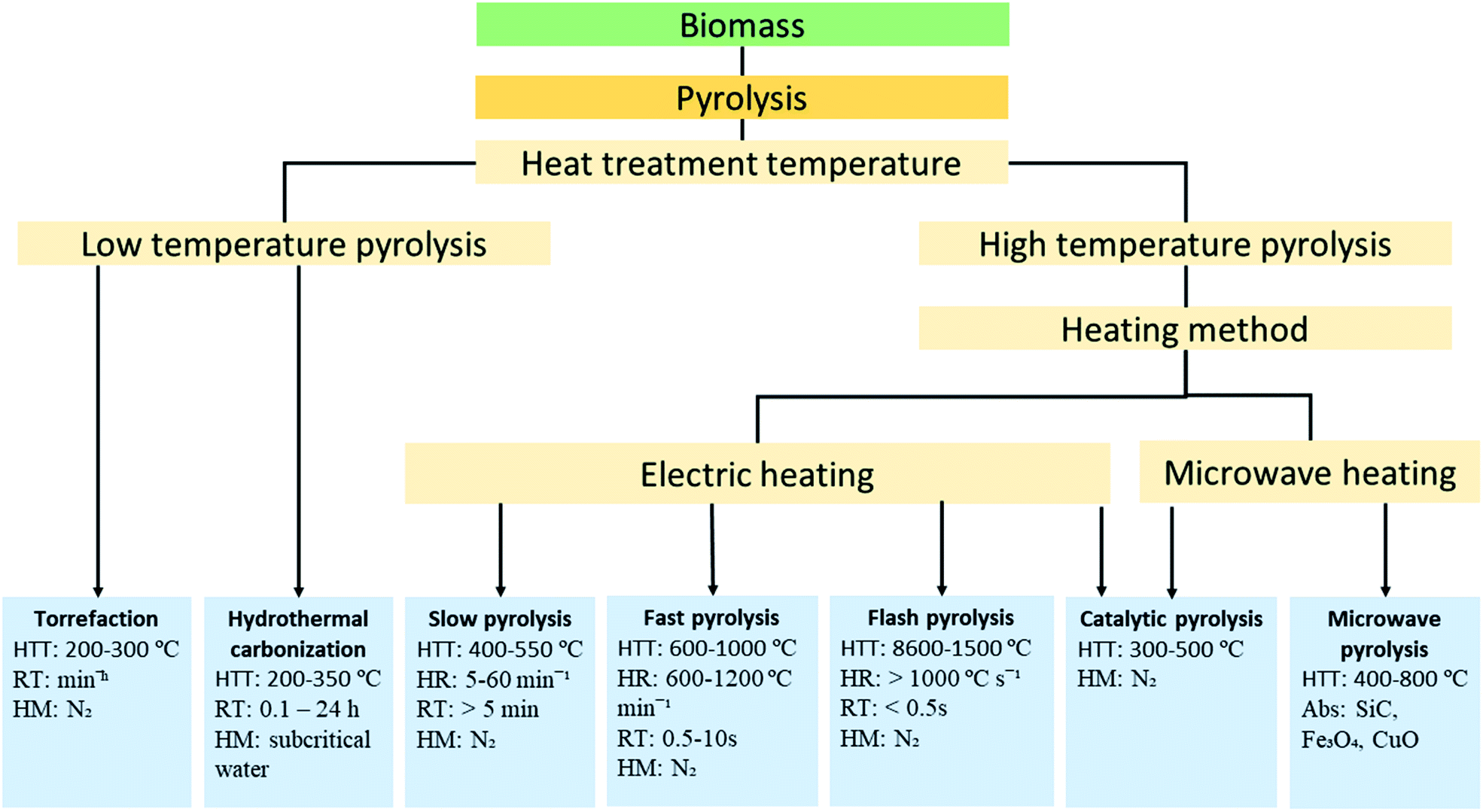 | ||
| Fig. 1 Schematic of classification pyrolysis of biomass feedstock. HTT (heat treatment temperature), HR (heating rate), RT (residence time), HM (heating medium), and Abs (absorber). | ||
The other modes of high-temperature pyrolysis are microwave and catalytic pyrolysis. In microwave pyrolysis, the components of biomass are exposed to radiation respond by a sort of movement (dipole rotation or ion migration) and generates heat due to the friction between the molecules.24 This in situ heating of the biomass components drastically limits the mass transfer of condensable volatile components within the biomass particle and encourages continuous reaction to form biochar. Biochar yield from microwave pyrolysis is comparable to slow pyrolysis. Depending on the type of catalyst used, catalytic pyrolysis can either favour the production of biochar or bio-oil. The research trend on catalytic pyrolysis is toward bio-oil production. Torrefaction and hydrothermal carbonization are types of biomass pyrolysis processes carried out at low-temperature (<350 °C) aiming to produce biochar as the main product.25 For hydrothermal carbonization, yields of gas and liquid by-products are substantially low.26 The degree of biochar carbonization is low compared to products of high-temperature pyrolysis and contains a high degree of oxygen functional groups. Autoclave reactors (pressure vessels) are used for hydrothermal carbonization of biomass. Autoclave reactors are designed to operate at elevated pressures and temperatures typical of the hydrothermal carbonization process and can efficiently convert wet biomass feedstock without extra energy input.
2.2 Factors influencing biochar properties
The properties of biochar are known to depend on the characteristics of its feedstock and production methods, and they were summarised in Table 1. Biomass feedstocks are mostly composed of different proportions of cellulose, hemicellulose, lignin, extractives and inorganic compounds. These components decompose differently during thermal treatment and thus have a great influence on the properties of the biochar produced. Meanwhile, the dependence of biochar properties on lignin and ash contents could be related to their low volatility.| Factors | Descriptions |
|---|---|
| Proportion of biomass feedstocks composition | Influence of lignin: high yield of biochar and fixed carbon content,32 low surface area,33 high surface pH and functional groups,34 high ion exchange capacity25 |
| Influence of inorganic constituent:35 high biochar yield through bond dissociation energy alteration between organic and inorganic carbon, loss of volatile matter, high electrical conductivity of biochar correlated with potassium and sodium fractions of the total ash | |
| Production condition | Influence of high temperature: low yield of biochar,37 high fixed carbon and low volatile matter contents,42 high specific surface area of biochar,43,44 high total pore volume biochar,40 high crushing and impact strengths of biochar,45 high pH and electrical conductivity46 |
| Influence of low temperature: better cation exchange capacity25 |
The properties of biochar are also significantly influenced by the production conditions such as temperature, residence time and heating rate. The peak temperature is an important and dominant process parameter in determining the stage of biomass degradation, which invariably has a significant effect on the yield and quality of biochar. Biochar prepared from biomass show the common trend of yield reduction with the rise in temperature. Antal and Grønli (2003)36 and Weber and Quicker (2018)25 have succinctly summarized the biochar properties with different carbonization temperatures. The drastic yield reduction at the initial stage of thermal treatment (<500 °C) followed by a slow decrease thereafter has been observed for lignocellulosic biomass feedstocks.37–41 These trends of reduction in biochar yield reflect the devolatilization of organic polymers of the feedstock and the slow carbonization of the solid residue. Quality properties such as fixed carbon and volatile matter contents increase and decrease with temperature, respectively. A rise in carbonization temperature results in variations in the element composition and atomic ratios of the biomass by releasing oxygen and hydrogen groups resulting in carbon-rich and hydrophobic products.42
Changes in textural properties of biochar have been identified to correlate with carbonization temperature. Biomass pyrolysed at milder peak temperatures typically <400 °C does not produce biochar with a relatively high specific surface area compared to its original biomass. However, a significant increase in the specific surface area between the production temperature range of 450 and 700 °C was observed.43,44 Removal of volatile matter through destruction of aliphatic alkyl and ester groups, exposure of aromatic lignin core, and restructuring (re-polymerization and aromatization) of fixed carbons through higher production temperature could be responsible for the higher surface area.20 As production temperature further increases from 700 to 1000 °C, the specific surface area starts to decline possibly due to softening and sintering of high molecular weight volatile matter which leads to shrinkage of biochar.40 The pore volume is another textural property that is relevant in many biochar applications. The total pore volume increases with temperature which corresponds to a decrease in biochar particle density. Again, as the volatile matter is released from the biomass and solid carbons are re-polymerized and aromatized due to temperature rise, pores are gradually developed within the biochar matrix.
Crushing and impact strengths are relevant in the design of material handling and transportation systems. Both crushing and impact strengths of biochar exhibit a similar trend to the surface area with temperature rise. M. Kumar et al. (1999)45 reported that both crushing and impact strengths of biochar produced from acacia and eucalyptus wood biochars increased with temperature and further decreased after the production temperature of 600 °C. Clear trends were also observed between temperature and changes in electrical conductivity and ion exchange capacities of biochar. Low-temperature biochar gives rise to a better cation exchange capacity due to the presence of a sufficient amount of negatively charged functional groups.25 Biochar pH and electrical conductivity were reported to increase with production temperature due to loss of hydrogen![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :carbon ratio and volatile matter resulting in high ash content production.46
:carbon ratio and volatile matter resulting in high ash content production.46
2.3 General applications of biochar
Biochar now becomes a more appealing carbon material owing to its features of being a sustainable, green, easy-produced and cost-effective alternative to other forms of carbon materials. The high surface area, porosity and large amounts of surface functional groups are key properties influencing the suitability of biochar for various applications. Initially, soil improvement, waste management, climate change mitigation and energy production motivated the application of biochar on environmental management.47 Currently, advancement in production, modification and characterization of biochar is motivating its application beyond environmental management services to emerging high-end applications. Biochar is identified as an adsorbent for the removal of contaminants and pollutants in liquid and gas phases.48–50 Today, biochar is acknowledged to perform efficiently by accelerating the decomposition of substrates and improving compost quality,51 having good specific capacitance and current density,52 increasing catalytic removal of nitrogen oxides,53 improving the stiffness and flame retardancy of glass-fiber reinforced composites,54 and improving the mechanical properties of polypropylene composites.123. Effects of biochar addition on the characteristics of polymers composites
The porous structure of biochar is extremely advantageous for the purpose of filtration and soil remediation. Recently, researchers have found the potential of biochar to be used as a filler for polymer composites, owing to its porous structure, excellent thermal stability, low-cost production and minimal negative impact on the environment.55 Theoretically, different kinds of fillers will act differently on polymer matrix, so does biochar. This section discusses the effect of biochar on the crystallization, thermal, flammability, electrical conductivity and mechanical properties of polymer composites.3.1 Crystallization behavior
Crystallization behaviour is important due to the role they play in the mechanical properties of the polymer composites. Knowing the crystallization kinetics of a polymer or composite is also vital when it comes to assessing the processability of these plastics during downstream manufacturing. The crystallization kinetics of materials can be observed through a combination of differential scanning calorimetry (DSC) and X-ray diffraction (XRD). The effect of biochar on the crystallinity of the different polymers is mixed. There are multiple explanations for the fluctuations in crystallinity of the polymers due to the addition of biochar. Poulose et al., (2018)13 and Zhang et al., (2018)56 explained that the decrease in crystallinity and the slower crystallization rates were due to the hindrance of mobility of the polymer chains caused by biochar. However, although the crystallinity of the composite was reduced, the crystallization onset temperature (Tc) of the composites was slightly higher than the neat polypropylene (PP).13 This suggested that biochar was able to act as a nucleating agent triggering nucleation at a higher temperature as compared to neat PP. This is in agreement with the report by Li et al., (2018),57 which stated that well distribution of biochar in the polymer matrix has contributed to the functionality of the biochar as nucleating agent to initiate the crystallization of the polymer. This was supported by their findings on the decrease of crystallinity at higher loadings of biochar, which caused mobility restrictions of the molecular chains. In another study involving polyethylene terephthalate (PET)/biochar composites, the addition of biochar caused a decrease in the cold-crystallization temperature and a significant increase in the crystallization rate as well as the crystallinity of the composite when compared to the neat PET.58 Zhang et al., (2019),59 experienced similar effect where the addition of poplar biochar caused the crystallization of high-density polyethylene (HDPE) to occur earlier. They explained that the poplar biochar acted as a nucleating agent to decrease the amount of energy required for the crystallization of the composites.3.2 Thermal properties and thermal stability
The thermal properties of polymers are typically assessed with the help of DSC. The DSC curves will provide information on the melting temperatures, glass transition temperatures as well as the various enthalpies that are required to induce the phase transitions in the polymers or composites. According to Poulose et al., (2018),13 the melting temperature (Tm) of PP was not affected by the addition of biochar. Additionally, Das et al., (2016a)12 discovered that while the addition of biochar did not give an effect on the Tm of PP, the melting enthalpy (ΔHm), however, was increased and attributed. Also in agreement with the previous two studies, Zhang et al., (2019)59 revealed that the addition of poplar biochar to the HDPE had no effect on the composite's Tm but also no change in ΔHm.Thermal stability is essentially the resistance of a certain molecule to high levels of heat. The thermal stability of polymers is usually determined using thermogravimetric analysis (TGA). Depending on the raw material, biochar by itself has extremely high thermal stability as proven by numerous studies by Das et al., (2015a),62 Nan et al., (2015)60 and Jeon et al., (2019).61 This was especially true for biochar that has undergone pyrolysis at very high temperatures as it was found that biochar seems to be thermally stable below their pyrolysis temperatures. Therefore, the higher the temperature at which the biochar is produced, the more thermally stable it will be due to a higher degree of carbonization.63 The addition of biochar seems to consistently improve the thermal stability of various polymers. Nan et al., (2015)60 reported that both thermal degradation and weight loss of polyvinyl alcohol (PVA) were delayed due to the addition of biochar into the matrix and this was highly attributed to the superior thermal stability of biochar particles. Li et al., (2018)57 also reported similar results where the increasing content of biochar in the ultrahigh molecular weight polyethylene (UHMWPE)/linear low-density polyethylene (LLDPE) matrix delayed the onset of thermal degradation even further. The authors attributed this to the faster heat transfer from the polymer matrix to the filler due to higher biochar content. However, they also noted that the maximum degradation temperatures of the composites were below that of the neat polymer blend. This was due to the higher thermal conductivity of biochar.
Another study done on biochar/PET composites suggested that the improvement in thermal stability could be due to the biochar acting as a barrier to volatiles and that the oxygen permeation into the polymer matrix could have been affected by the presence of biochar.58 They found that the addition of biochar to recycled PET increased the onset degradation temperature from 381.8 °C of neat PET to 387.2 °C with the addition of 5 wt% of biochar. Das et al., (2015a)12 conducted their research on the addition of biochar to conventional PP. Yet again, biochar has proved to be significantly advantageous to the thermal properties of polymers. In this case, the authors recorded a clear increase of residual mass after thermal degradation as the biochar content in the PP/biochar composites increased.
3.3 Flammability and flame retardancy
Flammability or combustibility, unlike thermal stability, is the ability of a substance or chemical to combust or ignite, causing a fire. Flame retardancy, on the other hand, is the ability of a substance to prevent combustion. Combustion of materials can only occur in the presence of fuel and sufficient heat and oxygen. The absence of one of these requirements will prevent a fire from starting. Logically, most applications of polymers would benefit from a material with low flammability and high flame retardancy. Zhang et al., (2017),64 did a study where they tested the effects of biochar on the flammability properties of Al(OH)3/HDPE/wood composites where Al(OH)3 is used as a flame retardant. They found that the addition of biochar to the composite yielded a limiting oxygen index (LOI) of up to 30% with flame retardant content of 40 wt%. This is an improvement to the LOI of the composite without biochar at about 27.1%. It was explained that the addition of biochar helped to delay the heat degradation velocity which in turn, boosted the flame retardancy of the overall material.64 Another study tested the flammability properties of PP/biochar.12 From their research, they stated that while the time to ignition (TTI) of the composites decreased when compared to neat PP, the peak heat release rate (PHRR) decreased significantly with the addition of biochar to PP. In another study by the same principal researcher, they found that the LOI of PP was increased from 18% to 22.08% with the addition of 25 wt% biochar.163.4 Electrical conductivity
Poulose et al., 2018,13 measured the surface resistivity of PP upon the addition of date palm biochar. They tested the effects of biochar produced at two different pyrolysis temperatures (700 and 900 °C) on the properties of PP. They found that as the biochar content increased, the surface resistivity of the composites decreased. A decrease in surface resistivity means that the electrical conductivity of the composites increased by up to 4 orders of magnitude with the incorporation of 15% w/w biochar. It was also found that biochar pyrolyzed at a higher temperature (900 °C) lead to a more drastic decrease in surface resistivity. However, it is noted that the improvement to the electrical conductivity of PP with the addition of biochar is not to the level of other carbonaceous materials such as carbon black.65 This could be due to the agglomeration of biochar or the presence of impurities and high ash content which could lead to the disruption of the conductive carbon network.13Another study by Nan et al., (2015)60 on the electrical conductivity of PVA/biochar composites revealed that PVA by itself has no electrical conductivity properties and the addition of small amounts of biochar to PVA (2% w/w) did little to improve the electrical conductivity of PVA. This could be explained by the same reasoning used by Poulose et al., (2018),13 where there was not enough biochar to form a conductive network. However, upon higher loadings of biochar (6% and 10% w/w), the conductivity of the composite seemed to increase together with the biochar load. This is because an increase in biochar content provided enough biochar to reduce the insulated space in the matrix, forming a conductive network of carbon. The results from this study are in agreement with a study by Li et al., (2018)57 who combined UHMWPE/LLDPE with biochar to increase the electrical conductivity of the polymer. They also found that the electrical conductivity of a polymer would increase with higher loadings of biochar. It was also noted that at 80% w/w of biochar, they achieved one of the highest electrical conductivity (107.6 S m−1) of a composite fabricated via melt processing. Interestingly, to further prove the electrical conductivity properties of the UHMWPE/LLDPE/biochar composite, they successfully lit a blue LED bulb using the composite as part of the circuit.57 In short, this proves that the addition of biochar in a polymer matrix is beneficial for the production of high electrical conductivity composites including electromagnetic interference shielding.57
3.5 Mechanical properties
Out of all the properties of materials, the mechanical properties of materials are perhaps the ones that have garnered the most interest from researchers. Depending on the application, a material has to be strong and stiff or even flexible and stretchable. Researchers are always on the hunt for additives that could improve the mechanical properties of polymers. Biochar, among other green, environmentally friendly materials, has been the center of focus among material scientists to be used as a reinforcing material for various polymers.The tensile strength of a material is the ability of the material to resist breaking under tension. A material of high tensile strength would be able to undergo a higher degree of tensile stress without breaking as compared to a material with weaker tensile strength. Poulose et al., (2018)13 found that the addition of biochar to PP seemed to decrease its tensile strength. It was implied that the decrease in tensile properties could be due to poor interaction between the biochar particles and the PP matrix. Das et al., (2015a)62 who experienced a similar decrease in tensile strength with the addition of biochar to PP/wood, suggested that the presence of voids, which are empty spaces within the blend caused by biochar, could have contributed to the lower tensile strength of the polymer. Nevertheless, in another paper by the same author, they found that the addition of biochar to PP without the presence of wood did not cause any significant negative effects on the tensile strength of PP, so it could be that the reduction in tensile strength was caused by the presence of wood particles instead of biochar.12
Nan et al., (2015)60 added that a drop in tensile strength is theorized to be due to the particle size and dispersion into the matrix. It is said that the addition of biochar could have interfered with the cross-linking of the polymer network.60 Upon higher loadings of biochar, the drop in mechanical properties can be attributed to aggregations formed by the biochar particles. Therefore, biochar of a smaller scale could have the potential to form proper interfacial bonding with the polymer matrix and in turn, improve the tensile strength of the resulting composite. Interestingly, while biochar may have negative effects on the tensile strength of some polymers, they have been found to be beneficial to the tensile strength of other polymers. Idrees et al., (2018)58 found that PET with biochar content up to 5% w/w had higher tensile strength when compared to the neat PET. This was attributed to the good interfacial bonding between the two components due to the porous structure of biochar which allows for mechanical interlocking of the matrix to the biochar additive. However, it was also noted that lower loadings of biochar (0.5% w/w) lead to a higher tensile strength as compared to higher loadings of biochar (1, 3, 5% w/w). This is because the lower content of biochar allowed for a more well dispersed composite mixture with less agglomeration. The addition of biochar to UHMWPE/LLDPE also seemed to increase the tensile strength of the polymer mixture.57 They found that there was good homogenous dispersion of the additive even with biochar content of up to 60% w/w.
The elongation at break of a material is the ratio of the difference between the length of the sample at break and the initial length of the sample before tensile stress is applied. The elongation at break is a useful parameter to assess the ductility of a material. Poulose et al., (2018)13 reported a general decrease in elongation at break as a function of increased biochar load. This is caused by an increase in the toughness due to the incorporation of biochar with PP which led to the composite being more resistant to deformation. Nan et al., (2015)60 also found a decrease in ductility of PVA upon the addition of biochar. However, they found an increase in the tensile modulus which is a measure of polymer toughness, due to the biochar's naturally higher rigidity as compared to PVA. Das et al., (2015b)66 also discovered that the PP/wood composites had a higher elongation at break as compared to PP/wood/biochar composites.
Another important aspect of the mechanical properties of a material is its flexural strength. In other words, flexural strength is the ability of the material to be bent without fracturing. Zhang et al., (2018)56 compared the flexural strength of HDPE/rice husk (RH) and HDPE risk husk biochar (RHB). They found that although both RH and RHB had positive effects on the flexural strength of HDPE, at higher loadings, RHB continued to strengthen the flexural properties of the polymer whereas, RH started to cause a decline in tensile strength. With a maximum flexural strength of 53.7% at 70% w/w RHB, they attributed this to the fact that RHB limited the mobility of the polymer chains leading to less deformation of the matrix in its elastic zone.56
A similar study combined poplar biochar with HDPE and found that the addition of biochar up to 50% w/w significantly improved the flexural strength of HDPE.59 However, further loading of biochar at 70% w/w caused the flexural strength of the composite to drop below that of neat HDPE. This was because, at 70% w/w biochar, there was not enough polymer matrix to properly bond with the biochar particles. Additionally, higher biochar content would lead to a higher degree of agglomeration which could be detrimental to the mechanical properties of the composites.59 Das et al., (2016b)67 also had positive results in terms of flexural properties with the addition of biochar to PP. They found that the flexural strength of neat PP increased from 51.08 MPa to 58.26 MPa with the addition of 35% w/w biochar.
The ability to withstand a sudden force applied to the surface of a material is called impact strength. The impact strength is also one of the key properties of materials when referring to mechanical strength. The effect of biochar on the impact strength of polymers varies. In one study, it was found that the addition of biochar to HDPE had a negative effect on the impact strength of the composite.56 The decline in impact strength increased with further addition of biochar content. It was explained that the high rigidity of the biochar fillers resulted in increased brittleness and decreased toughness in the composite. However, the authors also compared the results to those of wood/HDPE and found that biochar/HDPE had significantly higher impact strength than wood/HDPE, especially at higher filler loadings. This is attributed to the presence of pores in the biochar which allows a more uniform dispersion of filler due to HDPE being embedded into the pores.56 In a similar study, Zhang et al., (2019)59 also found that biochar addition worsened the impact strength of the composite. While the presence of pores within the biochar structure may prove beneficial for filler–polymer dispersion and interaction, it also limits the mobility of the polymer chains resulting in a weakened ability to absorb the energy of an impact force. Nagarajan et al., (2016)3 stated that the size of the biochar particles had a profound effect on the impact strength of the composite. The authors found that by decreasing the size of the biochar particles, they were able to slightly increase the impact strength of poly(trimethylene terephthalate) (PTT) and PLA blends.
4. Comparison of biochar with other carbon materials as a reinforcement material in polymer composites
Besides biochar, other carbon materials especially carbon nanotubes (CNT) and graphene have been of great interest to be used as a reinforcement material as they can effectively enhance the properties of polymer matrices as well as their utilities to a large extent.68 In this sub-topic, the effect of reinforcing CNTs and graphene for composite production will be discussed and compared with biochar.4.1 Carbon nanotubes
Carbon nanotubes (CNTs) are cylindrical molecules that consist of graphite sheets, with a diameter size of about 100 nm, and their length can reach several micrometers.69 In general, there are two types of CNTs which are single-walled (SWCNT) and multi-walled (MWCNT).70 The major difference between these two types is their graphene layer. SWCNT can be described as a single hexagonal graphene layer with an approximate diameter between 0.4–3 nm. While MWCNT consists of a number of rolled-up hexagonal graphene sheets, and the diameter was around 1–3 nm.71Unlike biochar which can be produced by a simple carbonization process, CNTs require a complicated process to be produced. Arc discharge, laser ablation, and chemical vapor deposition methods are among the common methods used to produce CNT from graphite. Nevertheless, these methods are costly and only able to produce a limited amount of CNTs. In fact, those produced CNTs need to be further purified prior to being used as a reinforcement material due to the presence of impurities associated with the catalyst that is needed during the production process as well as due to the presence of amorphous carbon and non-tubular fullerene structures in them.71
Despite their costly and complex production process, CNTs have been proven as a superior nanomaterial that can enhance the electrical and thermo-mechanical properties of matrices owing to their unique structure, outstanding thermal conductivity, and high aspect ratio.68,72 Findings by Thi et al. (2020)68 revealed that by incorporating only 1–3 wt% SWCNT in polycarbonate (PC) is enough in enhancing both electrical and mechanical properties of polymer matric. Similarly, Liu et al. (2019)72 also discovered that the incorporation of CNTs in microcapsules with dodecanol core and melamine-formaldehyde (MF) resin shell was able to enhance the thermal conductivity of the microcapsules by 35.2%.
On the other hand, the increase of CNTs loading in the polymer matrix may lead to a heterogeneous dispersion by forming clusters and agglomeration within the polymer matrix, thus reducing the mechanical properties. Hence, a high shear force is needed to disperse high CNTs loading into the polymer matrix.73 In this particular scenario, pre-compounding is necessary to de-agglomerate CNTs by sonication or/and mechanical stirring, where the shearing force is higher than a typical screw-extruder used during compounding. However, too high of shearing severity may likely cause CNTs degradation.
4.2 Graphene
Graphene is a single atomic sheet composed of sp2 carbon atoms arranged in a honeycomb pattern.74 It has high mechanical strength and modulus of elasticity, has unparalleled electronic properties, and a large surface area that can be chemically functionalized.75 In general, graphene has two derivatives, namely graphene oxide (GO) and reduced graphene oxide (rGO).76 Like any other carbon filler, the underlying reinforcement mechanisms of graphene in polymer composites are dependent on filler concentration, aspect ratio (interfacial area), homogeneous dispersion, and interfacial adhesion.73Nevertheless, graphene is often chemically modified to either GO or rGO before being incorporated into the matrix. This might be because of their larger surface area, layered structure, and oxygen-containing functional groups compared to graphene. For example, epoxy composites with GO inclusion demonstrate improvement in tensile strength, Young's modulus, and fracture resistance over pure epoxy composites. The covalent bonds formation along with GO interface within polymer matrix attributes to effective load transfer.77 Also, with a strong interfacial interaction it provides, the behavioral polymer chains mobility at a high temperature can be impeded, hence increasing the resulting composites' glass transition temperature (Tg).78
The addition of graphene also shows an improvement in electrical conductivity and this was proven by Wang et al. (2012).73 The author revealed that graphene is capable of providing electrical conductivity to the insulating polymer matrix. It has high intrinsic conductivity which leads to a lower electrical percolation threshold. With the addition of graphene, it promotes the formation of a percolating network within the matrix which reduces the electron conduction resistance. The conductivity output of nanocomposites is influenced by filler dispersion and alignment, as well as the intrinsic characteristics of the filler such as aspect ratio, morphology (defects), and inter-spacing between graphene sheets.73
A highly disperse graphene, however, has not necessarily promoted the onset of electrical percolation as it barely formed a continuous graphene–graphene conductive network within the matrix so that the electron can be percolated from one's end to another end of the nanocomposites.79 The 2D geometry of graphene also offers a lower interfacial thermal resistance (high thermal conductivity) in the nanocomposites.80 Similar to CNTs, high loading of graphene may also lead to the reduction of mechanical properties as graphene tends to agglomerate. In order to overcome this issue, a high shear force is needed to break the agglomeration. Nevertheless, this method is unsuitable for GO and rGO. Byrne and Guin'Ko (2010)81 reported that the poor thermal resilience and the small mass density of chemically modified graphene make it unsuitable for this method. The use of high shear force during mixing may result in graphene sheets shortening, thus reducing its aspect ratio. Besides that, this technique often leads to unintended oxidative depolymerization at high temperatures.
4.3 Comparison of biochar, carbon nanotubes and graphene-based composites
Overall, Table 2 summarises the comparison of composites reinforced with biochar, CNTs and graphene.| Biochar-based composites | CNTs-based composites | Graphene-based composites | |
|---|---|---|---|
| Raw materials and their production method | Biochar can be produced by pyrolysed any biomass at a very high temperature (>500 °C)82 | CNTs is produced by separating it from graphite by either:83–85 catalytic chemical vapor disposition (CCVD), laser ablation, arc discharge | Graphene is produced by separating it from graphite by either: micromechanical exfoliation, liquid exfoliation, chemical vapor deposition, flame synthesis, pulsed laser deposition |
| Advantages of filler as a reinforcement material | Biochar is a renewable material and the use of it to replace non-renewable carbon materials would reduce the ubiquitous dependency on fossil fuels, minimise wastes and promote sustainability. The addition of biochar in the composites may improve their thermal stability. E.g. the addition of 20 and 30 wt% biochar to epoxy composites have increased the Td10% by 7–20%. Overall, the thermal degradation of the epoxy/biochar composites was delayed.86 Biochar exhibits aliphatic functional groups on its surface, which make it hydrophobic. Due to this, when biochar is incorporated in wood and polymer composites, it may lower the resulting moisture absorption of the entire composite.66 | CNTs possess excellent adsorption ability, owing to its ability in creating a strong interaction with other molecules. E.g. CNTs has been used as an adsorbent for various heavy metal ions such as copper, nickel, cobalt, vanadium, silver, cadmium and other earth elements.87 | Graphene recorded the highest thermal conductivity than biochar and CNTs which is about 5300 W mK−1.72 E.g. the incorporation of rGO into n-eicosane/silica microcapsules increased the thermal conductivity by 83–193% and decreased the latent heat by 6–15%.88 |
| Disadvantages of filler as a reinforcement material | The properties of biochar are mainly dependent on the properties of biomass (raw material) and thermal conditions during pyrolysis.9 Modification is needed to increase the properties of biochar, i.e. by increasing the specific surface area and pore fraction, forming functional groups, etc.89 The different feedstock used to produce biochar will produce biochar with different properties. E.g. the percentage of biochar loading at which best mechanical properties obtained was inconsistent for epoxy composites with three different biochars; plastic waste char, wood shavings char, and pine cone char.86 | Homogenous dispersion of CNTs especially at high loading is difficult to achieve. Modification is needed to reduce the aggregation and improve the dispersion of CNTs caused by the inactive surface of CNTs.70 The high cost of CNTs does not compensate for the enhancement of properties on numerous occasions, unless for premium end-products.87 | Graphene may cause a reduction in the mechanical strength of composites, mainly due to poor interface with matrices. Hence, it needs to be modified to GO or rGO. E.g. the mechanical strength and deformation at break of epoxy/graphene composites are much lower than the neat epoxy resin due to the poor interface between the nanofiller and matrix.90 Similar to CNTs, graphene is expensive and considering the cost, it can only be used for premium end-products.87 The cohesive energy between graphene layers is around 2 eV nm−2 which is considered very high, and this causes graphene to irreversibly agglomerate or restack when compounded with molten polymer or when solvents evaporate from graphene dispersion.91 |
5. Challenges in the preparation of biochar-based polymer composites
The incorporation of biochar in polymers leads to the creation of new polymer composites with improved structural and functional properties due to the synergistic effects of the biochar and polymers. Nevertheless, there are challenges in utilizing biochar in the polymers such as the possibility of ash formation during polymer composite melt-compounding, and the appropriate methods to maintain the safe and environmentally friendly production.In the context of polymer composites preparation, three common processes namely compounding, blending, and mixing are usually applied. Compounding is a process in which polymers are softened, melted, and intermingled with fillers to produce a composite. Blending is referring to a process in which two or more materials are physically intermingled without causing any physical changes to the materials. While mixing describes both compounding and blending processing, which involves the intermingling of polymers with fillers or other additional materials without any specific restrictions, to produce composite.92
These three processes play big roles in ensuring polymers, fillers, and other materials are homogeneously mixed to produce a desirable composite with superior properties. In the case of biochar-based composites, the major challenge comes from the varied properties of biochar itself. According to Bartoli et al., (2019),93 biochar from different biomass acts differently when being used as a filler for epoxy resin. In his findings, wheat straw (WS) and oilseed rape (OSR) derived biochars were able to increase tensile strength and Young's modulus of epoxy resin by approximately 35–50%, mainly due to the presence of pores that can act as anchoring sites for the resin to create high tensioned structures. Miscanthus straw (MS) and mixed softwoods (SW) derived biochars, however, showed a different behavior, as they were only able to increase elongation of the epoxy resin while reducing Young's modulus. These differences could be attributed to the chemical composition of the biomass itself, especially cellulose.
According to Das et al., (2018),94 biomass that contains a high amount of cellulose such as wood resulted in better mechanical properties. As the main constituent of biomass, the strong and stiff cellulose remains aligned with the longitudinal axis of the fibers which is responsible for the resistance towards stress and indirectly helps to increase the composite's mechanical properties after being reinforced into the polymer matrix. Besides the properties of biomass or feedstock, the amount of biochar to be reinforced into the polymer matrix need to be identified as well. Ho et al. (2015)95 revealed that biochar composition gave significant improvement to both tensile and flexural modulus. This was highly attributed to the reduction of intra-particle distances by the incorporation of a higher amount of biochar. Nevertheless, an excess amount of incorporated biochar may as well lead to the reduction of mechanical properties, mainly due to aggregation. The aggregated biochar may enhance the brittleness of composites, thus reducing the elongation at break and this limits the composite's usage.
As a biomaterial produced by pyrolysis process which involves extremely high temperature, biochar consists of high carbon content which may cause health deterioration if being inhaled. Direct exposure to carbon especially during polymer composite compounding may lead to respiratory problems and skin inflammation, while chronic inhalation exposure may raise the risk of cancer and permanent damage to the lungs. Hence, protective equipment which may offer protection to the wearer as stipulated under the Occupational Safety and Health (Use and Standard of Exposure to Chemical Hazardous to Health) Regulations 2000 need to be worn throughout the processing. The aim is to eliminate, minimize and prevent hazards from being in contact with the worker.
6. Future recommendations and summary
A wide range of biomass have been explored as the potential feedstock for biochar production, and their effectiveness as filler for various polymer matrices were also reported. Despite of using various feedstock, all of them are facing similar issues after being reinforced into the polymer matrices, aggregation and heterogeneous dispersion of biochar. In regards to this matter, biochar properties need to be modified to ensure it can be used commercially as a polymer filler.Reducing its size from macro/microsize into nanosize particles could be an interesting approach to further understand its potential as a filler. Nanosized biochar can be obtained by subjecting the biochar to physical treatment such as ball milling as illustrated in Fig. 2.96 Biochar in its microsize has been found to improve the mechanical properties of polymer composites as mentioned earlier. It is expected that nanosized biochar would provide a better mechanical property due to the fact that the interfacial area of nanomaterial is higher as compared to the microsize material. The stress transfer from the polymer matrix to the nanomaterials is greater at a higher interfacial area and this leads to the improved mechanical properties when nanomaterial is used as a filler.97 Nevertheless, agglomeration tends to occur for nanomaterial at high concentrations, it is, therefore, important to optimize the nanosized biochar loading into the polymer matrix, which could be the study of interest in the future.
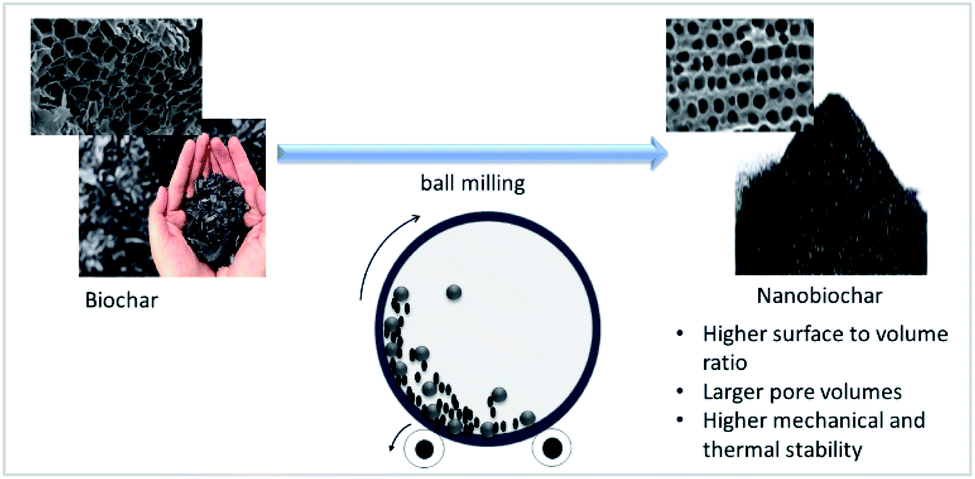 | ||
| Fig. 2 Schematic diagram of nanobiochar production (modified from Naghdi et al., (2017)96). | ||
The interaction of biochar and polymer matrices may also be enhanced by the addition of compatibilizers. Compatibilizers can help in modifying the interfacial properties of a blend of immiscible materials including biochar–polymer blends, making them bind tightly to each other. This will indirectly enhance the interfacial adhesion between biochar–polymer, thus increase the mechanical properties of the composite afterward. Besides physical modification by using a compatibilizer, biochar can also be modified using a chemical treatment. The use of chemicals such as HNO3 and NaOH for surface modification of biochar can improve the biochar/polymer composite properties due to the introduction of amino and carbonyl/carboxyl groups on biochar surface.98
Despite some pointed difficulties that have to be overcome, the utilization of biochar as a renewable filler is seen as one of the alternatives to replace non-renewable filler. In fact, the use of biochar as a filler for biocomposite production fulfilled several principles of green chemistry, which are the 1st principle: prevention, 3rd principle: less hazardous chemical syntheses, 7th principle: use of renewable feedstocks and 10th principle: design for degradation.99 The rationale behind the fulfillment of the 1st and 7th principles is since biochar is mostly produced from agricultural biomass, its usage as an alternative filler is seen to be able to reduce and prevent biomass accumulation, which if not efficiently managed, might cause environmental issues. This indirectly maximizes the utilization of biomass for high-value-added products. In terms of the risk of chemical synthesis, the overall biochar and biocomposite process do not generate toxic substances that may be harmful to humans and the environment, and biochar will also be degraded on its own over time, thus fulfilling the requirement of the 3rd and 10th principles.
7. Conclusion
Biochar from biomass is indeed potentially viable in terms of its performance and commercial value to be utilized as a renewable and superior filler for polymer composite. Further studies will be needed to determine the toxicity, rheology, and stability of the biochar/polymer composites in order to evaluate the potential applications of this material.Conflicts of interest
The authors have no conflict of interest to declare.Acknowledgements
The authors would like to thank the Ministry of Higher Education (MOHE), Malaysia for financial support through Fundamental Research Grant Scheme, FRGS/1/2019/TK05/UPM/02/1 (vot no. 5540321).References
- G. Mittal, K. Y. Rhee, V. Mišković-Stanković and D. Hui, Composites, Part B, 2018, 138, 122–139 CrossRef CAS.
- H. Essabir, M. O. Bensalah, D. Rodrigue, R. Bouhfid and A. Qaiss, Mech. Mater., 2016, 93, 134–144 CrossRef.
- V. Nagarajan, A. K. Mohanty and M. Misra, ACS Omega, 2016, 1, 636–647 CrossRef CAS PubMed.
- T. A. T. Yasim-Anuar, H. Ariffin, M. N. F. Norrrahim, M. A. Hassan, T. Tsukegi and H. Nishida, J. Cleaner Prod., 2019, 207, 590–599 CrossRef CAS.
- M. Giorcelli, A. Khan, N. M. Pugno, C. Rosso and A. Tagliaferro, Biomass Bioenergy, 2019, 120, 219–223 CrossRef CAS.
- S. Manafi and S. R. Kiahosseini, Iran. Polym. J., 2020, 29, 25–35 CrossRef CAS.
- J. Wang, H. Li, R. Liu, L. Li, Y. H. Lin and C. W. Nan, Compos. Sci. Technol., 2018, 157, 1–9 CrossRef CAS.
- B. Wang, B. Gao and J. Fang, Crit. Rev. Environ. Sci. Technol., 2017, 47, 2158–2207 CrossRef CAS.
- A. A. Lawal, M. A. Hassan, M. A. A. Farid, T. A. T. Yasim-Anuar, M. Z. M. Yusoff, M. R. Zakaria, A. M. Roslan, M. N. Mokhtar and Y. Shirai, Environ. Technol. Innovation, 2020, 18, 1–12 Search PubMed.
- T. Väisänen, A. Haapala, R. Lappalainen and L. Tomppo, Waste Manag., 2016, 54, 62–73 CrossRef PubMed.
- S. Vivekanandhan, Biochar as Sustainable Reinforcement for Polymer Composites, Elsevier Ltd, 2019 Search PubMed.
- O. Das, D. Bhattacharyya, D. Hui and K. T. Lau, Composites, Part B, 2016, 106, 120–128 CrossRef CAS.
- A. M. Poulose, A. Y. Elnour, A. Anis, H. Shaikh, S. M. Al-Zahrani, J. George, M. I. Al-Wabel, A. R. Usman, Y. S. Ok, D. C. W. Tsang and A. K. Sarmah, Sci. Total Environ., 2018, 619–620, 311–318 CrossRef CAS PubMed.
- A. Khan, P. Savi, S. Quaranta, M. Rovere, M. Giorcelli, A. Tagliaferro, C. Rosso and C. Q. Jia, Polymers, 2017, 642(9), 1–14 Search PubMed.
- J. George, L. B. Azad, A. M. Poulose, Y. An and A. K. Sarmah, Composites, Part A, 2019, 124, 1–9 CrossRef.
- O. Das, N. K. Kim, A. K. Sarmah and D. Bhattacharyya, J. Cleaner Prod., 2017, 144, 78–89 CrossRef.
- P. Savi, S. Member and S. P. Josè, in IEEE MTT-S International Microwave Workshop Series on Advanced Materials and Processes, IMWS-AMP, 2017 Search PubMed.
- P. Tanger, J. L. Field, C. E. Jahn, M. W. DeFoort and J. E. Leach, Front. Plant Sci., 2013, 218(4), 1–20 Search PubMed.
- X. Tan, Y. Liu, G. Zeng, X. Wang, X. Hu, Y. Gu and Z. Yang, Chemosphere, 2015, 125, 70–85 CrossRef CAS PubMed.
- M. Ahmad, A. Upamali, J. Eun, M. Zhang, N. Bolan, D. Mohan, M. Vithanage, S. Soo and Y. Sik, Chemosphere, 2014, 99, 19–33 CrossRef CAS PubMed.
- M.-K. Bahng, C. Mukarakate, D. J. Robichaud and M. R. Nimlos, Anal. Chim. Acta, 2009, 651, 117–138 CrossRef CAS PubMed.
- A. W. Palumbo, C. J. Bartel, J. C. Sorli and A. W. Weimer, Chem. Eng. Sci., 2019, 196, 527–537 CrossRef CAS.
- Q.-V. Bach and W.-H. Chen, Bioresour. Technol., 2017, 246, 88–100 CrossRef CAS PubMed.
- Y.-F. Huang, P.-T. Chiueh, W.-H. Kuan and S.-L. Lo, Bioresour. Technol., 2013, 142, 620–624 CrossRef CAS PubMed.
- K. Weber and P. Quicker, Fuel, 2018, 217, 240–261 CrossRef CAS.
- A. Jain, R. Balasubramanian and M. P. Srinivasan, Chem. Eng. J., 2016, 283, 789–805 CrossRef CAS.
- D. Mohan, A. Sarswat, Y. S. Ok and C. U. Pittman, Bioresour. Technol., 2014, 160, 191–202 CrossRef CAS PubMed.
- B. Cheng, R. J. Zeng and H. Jiang, Bioresour. Technol., 2017, 246, 224–233 CrossRef CAS PubMed.
- C. E. Brewer, K. Schmidt-Rohr, J. A. Satrio and R. C. Brown, Environ. Prog. Sustainable Energy, 2009, 28, 386–396 CrossRef CAS.
- S. You, Y. S. Ok, S. S. Chen, D. C. W. Tsang, E. E. Kwon, J. Lee and C.-H. Wang, Bioresour. Technol., 2017, 246, 242–253 CrossRef CAS PubMed.
- J. Idris, Y. Shirai, Y. Andou, A. A. Mohd Ali, M. R. Othman, I. Ibrahim and M. A. Hassan, J. Cleaner Prod., 2015, 89, 257–261 CrossRef CAS.
- X. Sun, R. Shan, X. Li, J. Pan, X. Liu, R. Deng and J. Song, GCB Bioenergy, 2017, 9, 1423–1435 CrossRef CAS.
- J. Li, Y. Li, Y. Wu and M. Zheng, J. Hazard. Mater., 2014, 280, 450–457 CrossRef CAS PubMed.
- Y. Huang, F. Li, J. Meng and W. Chen, BioResources, 2018, 13, 5153–5163 CAS.
- K. B. Cantrell, P. G. Hunt, M. Uchimiya, J. M. Novak and K. S. Ro, Bioresour. Technol., 2012, 107, 419–428 CrossRef CAS PubMed.
- M. J. Antal and M. Grønli, Ind. Eng. Chem. Res., 2003, 42, 1619–1640 CrossRef CAS.
- N. Abdullah, F. Sulaiman, A. A. Safana and I. I. Abdullahi, in AIP Conf. Proc., 2016, pp. 020041–0200046 Search PubMed.
- K. H. Khor, K. O. Lim and Z. A. Z. Alimuddin, Energy Sources, Part A, 2010, 32, 518–531 CrossRef CAS.
- K. K. Hooi, Z. A. Zainal Alauddin and L. K. Ong, J. Oil Palm Res., 2009, 21, 577–587 CAS.
- A. C. Lua, F. Y. Lau and J. Guo, J. Anal. Appl. Pyrolysis, 2006, 76, 96–102 CrossRef CAS.
- K. H. Khor and K. O. Lim, Int. Energy J., 2008, 9, 181–188 Search PubMed.
- M. Keiluweit, P. S. Nico, M. G. Johnson and M. Kleber, Environ. Sci. Technol., 2010, 44, 1247–1253 CrossRef CAS PubMed.
- N. Claoston, A. Samsuri, M. Ahmad Husni and M. Mohd Amran, Waste Manage. Res., 2014, 32, 331–339 CrossRef CAS PubMed.
- S. Yavari, A. Malakahmad and N. Sapari, Appl. Mech. Mater., 2014, 567, 150–154 CAS.
- M. Kumar, B. B. Verma and R. C. Gupta, Energy Sources, 1999, 21, 675–685 CrossRef.
- Z. Liu, W. Niu, H. Chu, T. Zhou and Z. Niu, BioResources, 2018, 13, 3429–3446 CAS.
- J. Lehmann and S. Joseph, in Biochar for Environmental Management Science and Technology, ed. J. Lehmann and S. Joseph, Earthscan, London, First edn, 2009, p. 416 Search PubMed.
- S.-H. Liu and Y.-Y. Huang, J. Cleaner Prod., 2018, 175, 354–360 CrossRef CAS.
- S.-H. Ho, S. Zhu and J.-S. Chang, Bioresour. Technol., 2017, 246, 123–134 CrossRef CAS PubMed.
- M. J. Ahmed and B. H. Hameed, J. Cleaner Prod., 2018, 195, 1162–1169 CrossRef CAS.
- R. Xiao, M. K. Awasthi, R. Li, J. Park, S. M. Pensky, Q. Wang, J. J. Wang and Z. Zhang, Bioresour. Technol., 2017, 246, 203–213 CrossRef CAS PubMed.
- B.-H. Cheng, K. Tian, R. J. Zeng and H. Jiang, Sustainable Energy Fuels, 2017, 1, 891–898 RSC.
- X. Xiong, I. K. M. Yu, L. Cao, D. C. W. Tsang, S. Zhang and Y. Sik, Bioresour. Technol., 2017, 246, 254–270 CrossRef CAS PubMed.
- R. K. Dahal, B. Acharya, G. Saha, R. Bissessur, A. Dutta and A. Farooque, Composites, Part B, 2019, 175, 107169 CrossRef CAS.
- S. Kim, S. Wi, J. H. Park, Y. S. Ok, S. W. Yang and J. Jeon, Environ. Res., 2019, 172, 637–648 CrossRef PubMed.
- Q. Zhang, W. Yi, Z. Li, L. Wang and H. Cai, Polymers, 2018, 10, 1–10 CrossRef PubMed.
- S. Li, A. Huang, Y. J. Chen, D. Li and L. S. Turng, Composites, Part B, 2018, 153, 277–284 CrossRef CAS.
- M. Idrees, S. Jeelani and V. Rangari, ACS Sustainable Chem. Eng., 2018, 6, 13940–13948 CrossRef CAS.
- Q. Zhang, M. U. Khan, X. Lin, H. Cai and H. Lei, Composites, Part B, 2019, 175, 107151 CrossRef CAS.
- N. Nan, D. DeVallance, X. Xie and J. Wang, J. Compos. Mater., 2015, 50, 1161–1168 CrossRef.
- J. Jeon, J. H. Park, S. Wi, S. Yang, Y. S. Ok and S. Kim, Chemosphere, 2019, 236, 124269 CrossRef CAS PubMed.
- O. Das, A. K. Sarmah and D. Bhattacharyya, Waste Manag., 2015, 38, 132–140 CrossRef CAS PubMed.
- S. X. Zhao, N. Ta and X. D. Wang, Energies, 2017, 10, 1–15 CrossRef.
- Q. Zhang, H. Cai, K. Yang and W. Yi, Results Phys., 2017, 7, 2391–2395 CrossRef.
- A. Manjaly Poulose, A. Anis, H. Shaikh, J. George and S. M. Al-Zahrani, Polym. Compos., 2015, 38, 2472–2479 CrossRef.
- O. Das, A. K. Sarmah and D. Bhattacharyya, Sci. Total Environ., 2015, 512–513, 326–336 CrossRef CAS PubMed.
- O. Das, D. Bhattacharyya and A. K. Sarmah, J. Cleaner Prod., 2016, 129, 159–168 CrossRef.
- T. B. N. Thi, S. Ata, T. Morimoto, T. Yamada, T. Okazaki and K. Hata, Mater. Today: Proc., 2020, 1–4 Search PubMed.
- N. Anzar, R. Hasan, M. Tyagi, N. Yadav and J. Narang, Sensors Int, 2020, 1, 100003 CrossRef.
- K. Babu, G. Rendén, R. A. Mensah, N. K. Kim, L. Jiang, Q. Xu, Á. Restás, R. E. Neisiany, M. S. Hedenqvist, M. Försth, A. Byström and O. Das, Polymers, 2020, 12, 1–19 CrossRef PubMed.
- B. V. Basheer, J. J. George, S. Siengchin and J. Parameswaranpillai, Nano-Struct. Nano-Objects, 2020, 22, 100429 CrossRef CAS.
- Z. Liu, Z. Chen and F. Yu, Sol. Energy Mater. Sol. Cells, 2019, 192, 72–80 CrossRef CAS.
- M. Wang, C. Yan and L. Ma, in Intech, 2012, p. 38.
- M. Bhattacharya, Materials, 2016, 9, 1–35 CrossRef PubMed.
- F. Mindivan and M. Göktaş, Mater. Today: Proc., 2020, 27, 3119–3123 CAS.
- H. Xie, T. Cao, F. J. Rodríguez-Lozano, E. K. Luong-Van and V. Rosa, Dent. Mater., 2017, 33, 765–774 CrossRef CAS PubMed.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098–7105 RSC.
- R. D. Priestley, C. J. Ellison, L. J. Broadbelt and J. M. Torkelson, Science, 2005, 309, 456–459 CrossRef CAS PubMed.
- P. M. Ajayan, L. S. Schadler and P. V. Braun, Nanocomposite Science and Technology, WILEY-VCH Verlag GmbH & Co., Weinheim, Germany, 2003 Search PubMed.
- L. Monica Veca, M. J. Meziani, W. Wang, X. Wang, F. Lu, P. Zhang, Y. Lin, R. Fee, J. W. Connell and Y. P. Sun, Adv. Mater., 2009, 21, 2088–2092 CrossRef.
- M. T. Byrne and Y. K. Guin'Ko, Adv. Mater., 2010, 22, 1672–1688 CrossRef CAS PubMed.
- A. A. Lawal, M. A. Hassan, M. A. A. Farid, T. A. T. Yasim-Anuar, M. Z. M. Yusoff, M. R. Zakaria, A. M. Roslan, M. N. Mokhtar and Y. Shirai, J. Cleaner Prod., 2020, 265, 121643 CrossRef CAS.
- F. Su and M. Miao, Synth. Met., 2014, 191, 99–103 CrossRef CAS.
- J. M. Wernik, B. J. Cornwell-Mott and S. A. Meguid, Int. J. Solids Struct., 2012, 49, 1852–1863 CrossRef CAS.
- J. Chen, X. Gao and W. Song, Results Phys., 2019, 15, 102771 CrossRef.
- G. Ahmetli, S. Kocaman, I. Ozaytekin and P. Bozkurt, Polym. Compos., 2013, 34, 500–509 CrossRef CAS.
- J. Chawla, R. Kumar and I. Kaur, J. Water Supply: Res. Technol.–AQUA, 2015, 64, 641–659 CrossRef.
- W. Wang, C. Wang, T. Wang, W. Li, L. Chen, R. Zou, J. Zheng and X. Li, Mater. Chem. Phys., 2014, 147, 701–706 CrossRef CAS.
- B. H. Cheng, R. J. Zeng and H. Jiang, Bioresour. Technol., 2017, 246, 224–233 CrossRef CAS PubMed.
- S. G. Prolongo, R. Moriche, A. Jiménez-Suárez, M. Sánchez and A. Ureña, Eur. Polym. J., 2014, 61, 206–214 CrossRef CAS.
- S. J. Han, H. Il Lee, H. M. Jeong, B. K. Kim, A. V. Raghu and K. R. Reddy, J. Macromol. Sci., Part B: Phys., 2014, 53, 1193–1204 CrossRef CAS.
- A. V. Shenoy, Rheology of Filled Polymer Systems, Springer Science & Business Media, 1999 Search PubMed.
- M. Bartoli, M. Giorcelli, C. Rosso, M. Rovere, P. Jagdale and A. Tagliaferro, Appl. Sci., 2019, 9, 3109 CrossRef CAS.
- O. Das, N. K. Kim, M. S. Hedenqvist, R. J. T. Lin, A. K. Sarmah and D. Bhattacharyya, Environ. Manage., 2018, 62, 403–413 CrossRef PubMed.
- M. P. Ho, K. T. Lau, H. Wang and D. Hui, Composites, Part B, 2015, 81, 14–25 CrossRef CAS.
- M. Naghdi, M. Taheran, S. K. Brar, T. Rouissi, M. Verma, R. Y. Surampalli and J. R. Valero, J. Cleaner Prod., 2017, 164, 1394–1405 CrossRef CAS.
- M. A. Ashraf, W. Peng, Y. Zare and K. Y. Rhee, Nanoscale Res. Lett., 2018, 13, 1–7 CrossRef CAS PubMed.
- T. Sizmur, T. Fresno, G. Akgül, H. Frost and E. Moreno-jiménez, Bioresour. Technol., 2017, 246, 34–47 CrossRef CAS PubMed.
- L. M. Gilbertson, J. B. Zimmerman, D. L. Plata, J. E. Hutchison and P. T. Anastas, Chem. Soc. Rev., 2015, 44, 5758–5777 RSC.
| This journal is © The Royal Society of Chemistry 2022 |