
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of calcium carbonate nanoparticles through the assembling effect of glucose and the influence on the properties of PDMS
Dengkui Shanga,
Nifan Zhoua,
Zhengguan Daib,
Nengyu Songb,
Zongrong Wang *a and
Piyi Dua
*a and
Piyi Dua
aState Key Lab of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou, Zhejiang province 310027, China. E-mail: zrw@zju.edu.cn
bAL Mine Co., Ltd, Jiande, Zhejiang province 311600, China
First published on 5th May 2022
Abstract
In order to prepare calcium carbonate nanoparticles in a green and environmentally friendly way, the concept of bio-mineralization has been proposed. Glucose, as a common small molecular organic substance found in organisms, participates in the mineralization process in cells. By adding glucose as a chemical additive, long chains of calcium carbonate form at the initial stage and then break granularly via over-carbonation. The average size of the calcium carbonate nanoparticles is about 40 nm based on the statistical analyses of three hundred particles. The growth mechanism of calcium carbonate under the influence of glucose is obtained. After the calcium carbonate nanoparticles are modified by sodium stearate, they are introduced to the PDMS matrix to achieve the composite material. Compared with pure PDMS, the composite with additional 3% calcium carbonate has its elongation at break and tensile strength increased by 23.96% and 48.15%, respectively.
1. Introduction
Calcium carbonate is widely used because of its excellent properties at the nanometer scale as a building filler1–4 and a drug carrier.5–9 In recent years, an increasing number of researchers have adopted bio-mineralization methods to synthesize calcium carbonate.10,11 This process mainly relies on various organic molecules in the organisms used.12,13 Carbonation is the main method for the large-scale industrial production of calcium carbonate,14,15 and organic molecules can also be used as additives to control the synthesis of calcium carbonate.16 In order to analyze and study the effect of additives on the growth of calcium carbonate, we chose a common and simple molecule, glucose, as a research subject. The oxygen in glucose can interact with calcium to influence the growth of calcium carbonate.17,18 PDMS has excellent properties such as high transparency, air permeability, and low toxicity.19,20 However, its mechanical properties are poor, and fillers need to be added for reinforcement.21In this study, glucose was adopted as an additive and the reaction mechanism of the calcium carbonate formation process was also investigated in detail. The calcium carbonate nanoparticles (CCNPs) were modified with sodium stearate, and the modified calcium carbonate was added to the PDMS matrix to effectively improve the elongation at break, tensile strength and stability of the composite.
2. Experiment
2.1 Raw material
Calcined lime (91 wt%, purchased from Zhejiang AL Mine Co., Ltd), carbon dioxide (99.995 vol%, purchased from Hangzhou Jingong Special Gas Co., Ltd), polydimethylsiloxane (PDMS, SYLGARD 184, purchased from Dow Corning), glucose (AR, purchased from Shanghai Hutri Laboratory Equipment Co., Ltd) and sodium stearate (AR, purchased from Shanghai Hutri Laboratory Equipment Co., Ltd) were used.2.2 Synthetic procedure of calcium carbonate nanoparticles and composite
Firstly, calcium oxide was added to deionized water for 3 hours in a 70 °C water bath. By fully slaking the calcium hydroxide, the undissolved particles were made uniform and small, which helped dissolve the excess calcium hydroxide immediately after the subsequent reaction was completed. Secondly, after the solution cooled, glucose was added to the calcium hydroxide slurry. Then, the solution was put in a 25 °C water bath and carbon dioxide was introduced into the solution with porous quartz stone. When the pH of the solution was close to 6, the ventilation was stopped. Sodium stearate was added to the solution as a modifier, and reacted in a water bath at 70 °C for 1 hour. Then, the solution was filtered and washed to obtain the calcium carbonate nanoparticles. The modified CCNPs (1–25% by weight) were added to the monomer, and after mixing uniformly, one-tenth of the weight of curing agent was added. After mixing uniformly again, the composite material samples were obtained by curing in an oven at 80 °C for 3 hours.2.3 Instrumentation
X-Ray powder diffraction (XRD) (Cu Kα, 40 kV, 20 mA) was conducted to identify the crystalline phases of the precipitates. The morphology and microstructure were examined using scanning electron microscopy (SEM) and transmission electron microscopy (TEM), respectively. The shift of the functional group peak position was measured by Fourier transform infrared spectroscopy (FTIR). A universal testing machine was used to test the tensile properties of the samples.3. Result and discussion
Fig. 1 shows SEM images of the calcium carbonate product without chemical additives and with different reaction times after adding glucose. This was used to determine whether the addition of glucose resulted in great changes in the morphology of calcium carbonate. | ||
| Fig. 1 SEM images of calcium carbonate from different stage of preparation: (a) no additives, reaction for (b) 5 min; (c) 10 min; (d) 15 min; (e) 20 min; (f) 25 min; (g) 30 min; (h) 45 min; (i) 60 min; and (j) 75 min after adding glucose. | ||
As shown in Fig. 1(a), without chemical additives, the reaction products were spindle-shaped particles with irregular morphology. In Fig. 1(b), it can be seen that at the beginning of the reaction, long-chain calcium carbonate appeared. From Fig. 1(c)–(j), the length of calcium carbonate decreases, finally affording circular calcium carbonate. After 75 minutes of reaction, 300 calcium carbonate particles at random positions were counted and divided into different groups. The proportion of each group is shown in Fig. 2. According to the statistics, particles with a size of 40 nm accounted for the highest proportion.
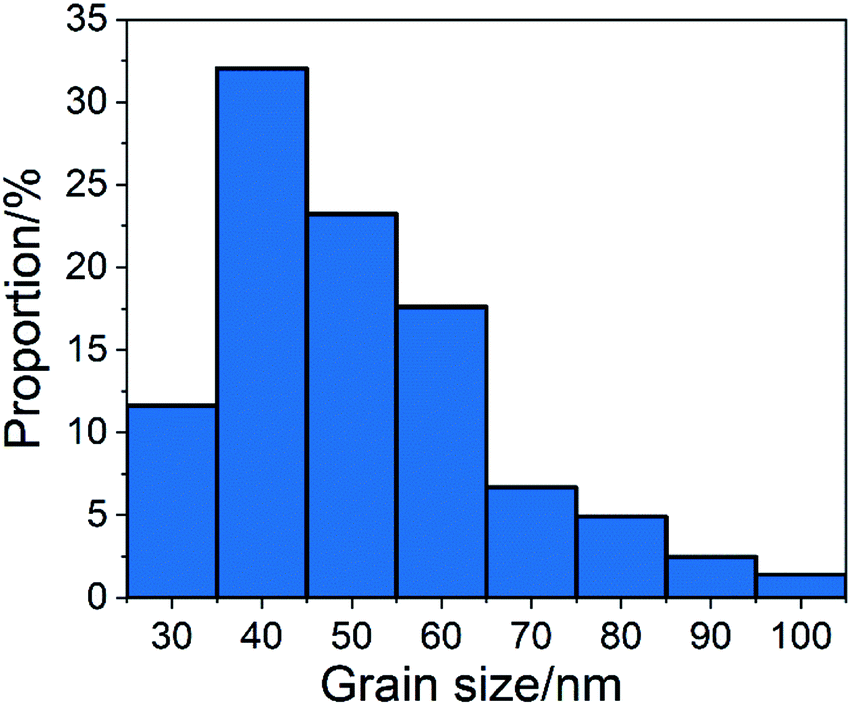 | ||
| Fig. 2 Grain size distribution of CCNPs after 75 minutes of reaction after adding glucose. | ||
Fig. 3 shows the change of pH and zeta potential over time during the preparation of calcium carbonate. As shown in Fig. 3, it can be found that after adding glucose to the system, the zeta potential is reduced. The zeta potential is an indicator that reflects the stability of the system.22–24 The decrease in zeta potential indicates that the stability of the system is destroyed, that is, the positively charged calcium ions are adsorbed by glucose molecules and agglomerate. From 0 to 15 minutes, the zeta potential increased. Due to the introduction of carbon dioxide, hydroxide ions were consumed and a large amount of carbonate ions were produced. The concentration of hydroxide ions and calcium ions in the system dropped rapidly, and the excess calcium hydroxide solid began to dissolve. The calcium hydroxide solids in the system were consumed, and calcium carbonate solids were generated instead. In the presence of large quantities of calcium hydroxide solids in solution, the potential-determining ions were calcium and hydroxide ions. At the beginning of the reaction process, the charges of the two were equal, so the zeta potential of the system was neutral. When a large amount of calcium carbonate was formed, the potential-determining ions were converted into calcium ions and carbonate ions. At this time, the carbonate ions in the solution were consumed immediately, and the calcium ions were continuously dissolved and replenished by the calcium hydroxide solid. Therefore, the amount of calcium ions was greater than that of the carbonate ions, and the zeta potential of the system was biased towards positive. In the subsequent reaction process, the amount of calcium carbonate increased, but the proportion of the total was not changed, so the zeta potential of the system increased slowly. After glucose was removed by washing and filtering, the calcium carbonate had a higher zeta potential, which caused the absorptivity of glucose on the calcium carbonate. The experiment did not stop when the pH was 7 because, as can be seen from Fig. 1(h), there was a large amount of chain-like calcium carbonate. At 75 minutes, the pH was close to 6. At this time, the granular morphology of the calcium carbonate product accounted for the majority.
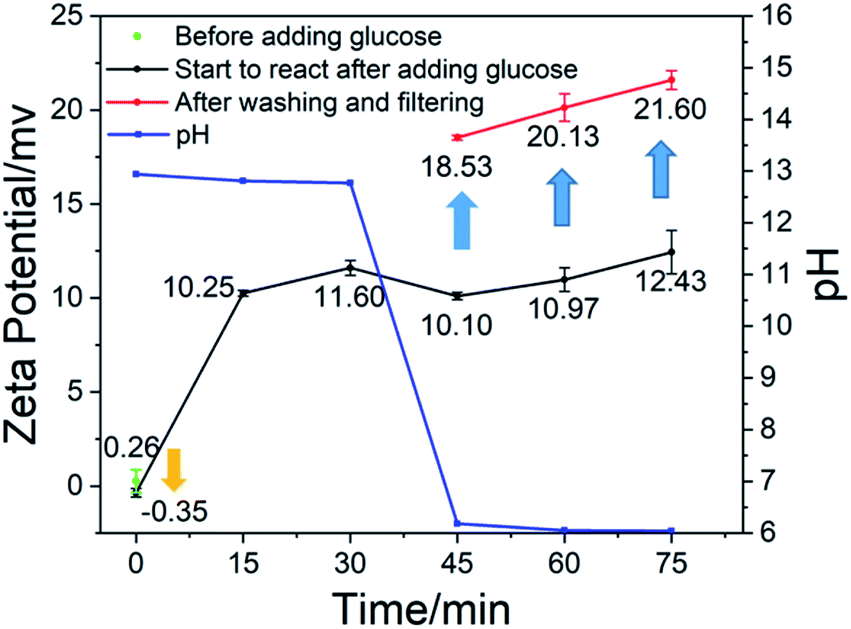 | ||
| Fig. 3 Graph of pH and zeta potential over time. | ||
Fig. 4 shows the XRD patterns of calcium carbonate at different times. It can be seen from Fig. 4 that the characteristic peak of calcium hydroxide disappeared at 15 minutes, indicating that there was no excessive undissolved calcium hydroxide solid at this time. As shown in Fig. 3, when the zeta potential of the system was detected, almost all the solid components in the solution were calcium carbonate. The characteristic peaks of calcium carbonate in Fig. 4 become increasingly obvious, and its crystal phase was calcite. Fig. 5 shows the TEM image of calcium carbonate. The distribution of calcium at the position indicated by the arrow in a2 was obviously sparse, which proved the absence of calcium. This showed that calcium carbonate was missing here. However, the distribution of oxygen in a3 is uniform, and oxygen can only come from glucose, indicating that this position was occupied by glucose. From the TEM image in Fig. 5(b2), it can be known that the interplanar spacing was 0.31 nm in the (104) crystal plane, and the crystal grew along the axial direction.25 There were some non-stripe regions on the side of the stripes. These regions may be occupied by glucose, destroying the orderly arrangement of the crystals, causing the calcium carbonate here to fail at forming stable lattice stripes.
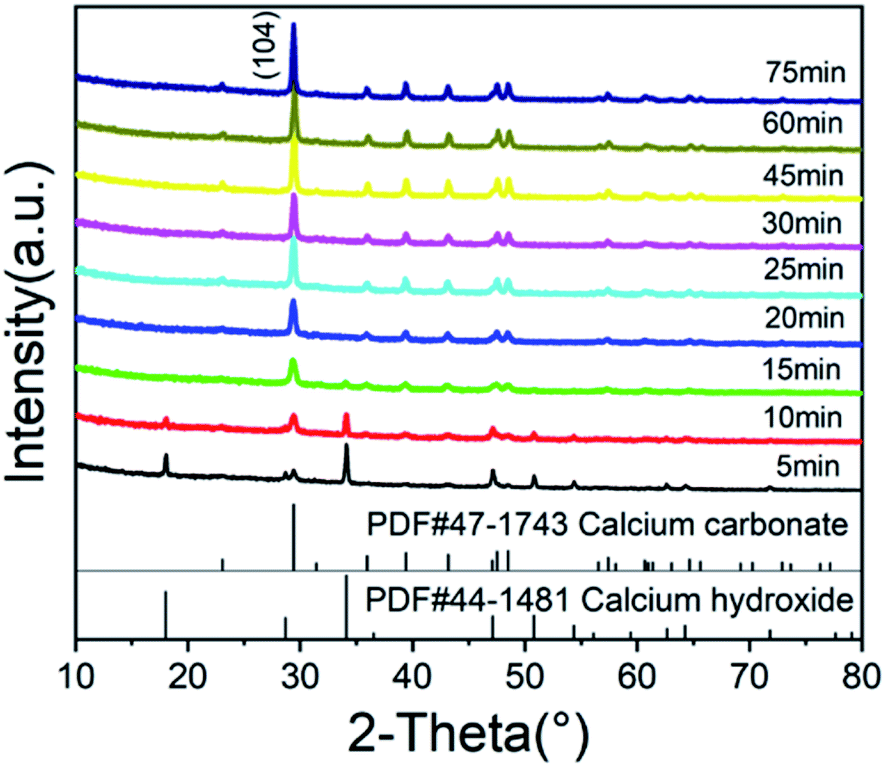 | ||
| Fig. 4 XRD patterns of the CCNPs at different times. | ||
 | ||
| Fig. 5 (a) Elemental analysis by mapping: (a1) morphology; (a2) calcium distribution; (a3) oxygen distribution. (b) (b1) TEM morphology and (b2) high-resolution images; (b3) FFT. | ||
Fig. 6 shows that the peak of calcium hydroxide was at 3642 cm−1, and there was no obvious movement. After 15 minutes, the characteristic peak of calcium hydroxide was significantly weakened. At this time, calcium hydroxide was consumed. The vibration peak at 1796 cm−1 was the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration peak of carbonate. At the beginning of the reaction, there was a strong absorption peak near 1415 cm−1, which was the characteristic absorption peak of v3 of calcium carbonate, and represented the asymmetric stretching vibration of the C–O bond. However, by the end of the reaction, this peak moved to 1456 cm−1. This may be due to the influence of glucose. Most of the calcium ions in calcium carbonate formed at the initial stage of the reaction were derived from free calcium ions. At the end of the reaction, they were derived from the calcium ions of the complex formed by glucose, which resulted in a red shift of the peak due to the influence of glucose. The absorption peaks at 874 cm−1 and 713 cm−1 were the v2 and v4 absorption peaks in calcite crystals, respectively. They were related to the bending vibration of the C–O bond. Normally, a peak should appear at 3404 cm−1 due to hydrogen bonding on the second and third carbons of glucose. However, the weak peak actually appeared in the range of 3421–3455 cm−1, and a significant transition to a high frequency and broad peak occurred. This may be due to the rearrangement of the hydrogen bond system caused by the 1
O vibration peak of carbonate. At the beginning of the reaction, there was a strong absorption peak near 1415 cm−1, which was the characteristic absorption peak of v3 of calcium carbonate, and represented the asymmetric stretching vibration of the C–O bond. However, by the end of the reaction, this peak moved to 1456 cm−1. This may be due to the influence of glucose. Most of the calcium ions in calcium carbonate formed at the initial stage of the reaction were derived from free calcium ions. At the end of the reaction, they were derived from the calcium ions of the complex formed by glucose, which resulted in a red shift of the peak due to the influence of glucose. The absorption peaks at 874 cm−1 and 713 cm−1 were the v2 and v4 absorption peaks in calcite crystals, respectively. They were related to the bending vibration of the C–O bond. Normally, a peak should appear at 3404 cm−1 due to hydrogen bonding on the second and third carbons of glucose. However, the weak peak actually appeared in the range of 3421–3455 cm−1, and a significant transition to a high frequency and broad peak occurred. This may be due to the rearrangement of the hydrogen bond system caused by the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 coordination of calcium and glucose.26 This weak peak may be due to the low amount of glucose. The calcium and oxygen of the hydroxyl group on the first and second carbons of the two glucoses form an eight-coordination complex with four water molecules. The native hydrated calcium ions formed a six-coordination complex.27 In the preparation of this experiment, glucose had a 2 mmol concentration, the calcium hydroxide slurry was supersaturated, and up to 4 mmol of Ca(OH)2 was dissolved in 200 ml of the solution. Therefore, the ratio of calcium ions coordinated with glucose to hydrated calcium ions was 1:3. The smaller size and higher quantity of hydrated calcium ions formed a stable crystal lattice on the inside, while the larger size and lower quantity of coordinated calcium ions formed an unstable structure on the outside. After passing in excess carbon dioxide, it was easier to dissolve and break.
:2 coordination of calcium and glucose.26 This weak peak may be due to the low amount of glucose. The calcium and oxygen of the hydroxyl group on the first and second carbons of the two glucoses form an eight-coordination complex with four water molecules. The native hydrated calcium ions formed a six-coordination complex.27 In the preparation of this experiment, glucose had a 2 mmol concentration, the calcium hydroxide slurry was supersaturated, and up to 4 mmol of Ca(OH)2 was dissolved in 200 ml of the solution. Therefore, the ratio of calcium ions coordinated with glucose to hydrated calcium ions was 1:3. The smaller size and higher quantity of hydrated calcium ions formed a stable crystal lattice on the inside, while the larger size and lower quantity of coordinated calcium ions formed an unstable structure on the outside. After passing in excess carbon dioxide, it was easier to dissolve and break.
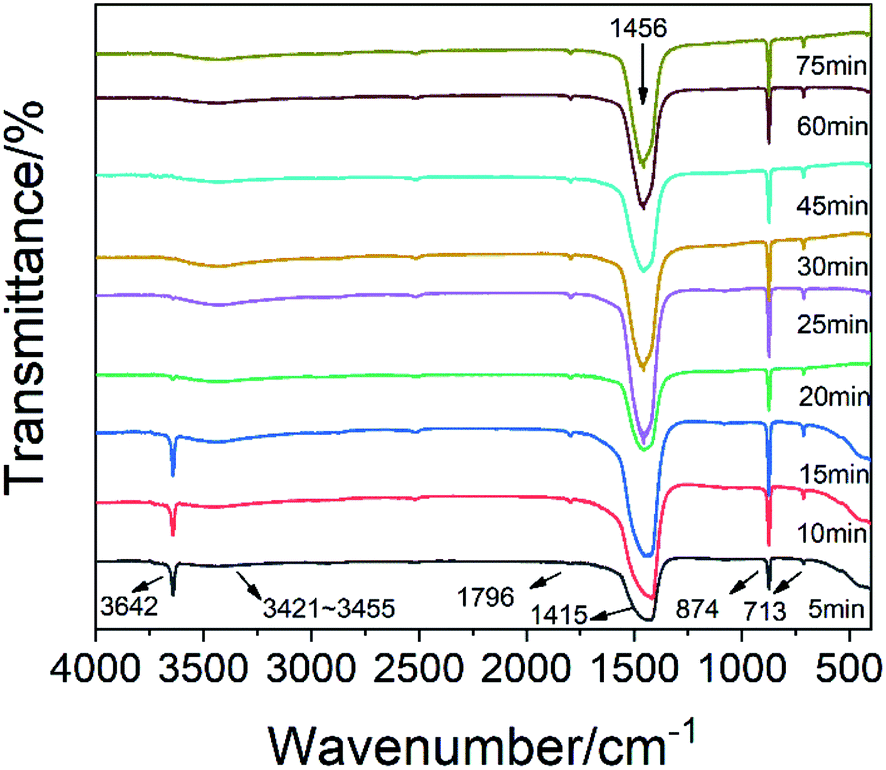 | ||
| Fig. 6 FTIR spectra of the CCNPs at different times. | ||
Fig. 7 shows the TGA result of the CCNPs. The characteristic decomposition temperature data are shown in Table 1. As shown in Fig. 7, there was no significant weight loss at 200–400 °C. Glucose decomposes in this temperature range.28 This showed that there was no glucose residue in the samples. The residual weight of the final sample was 56.0% of the initial weight, which was consistent with the process of calcium carbonate (Mw = 100.09) decomposition to form calcium oxide (Mw = 56.08).
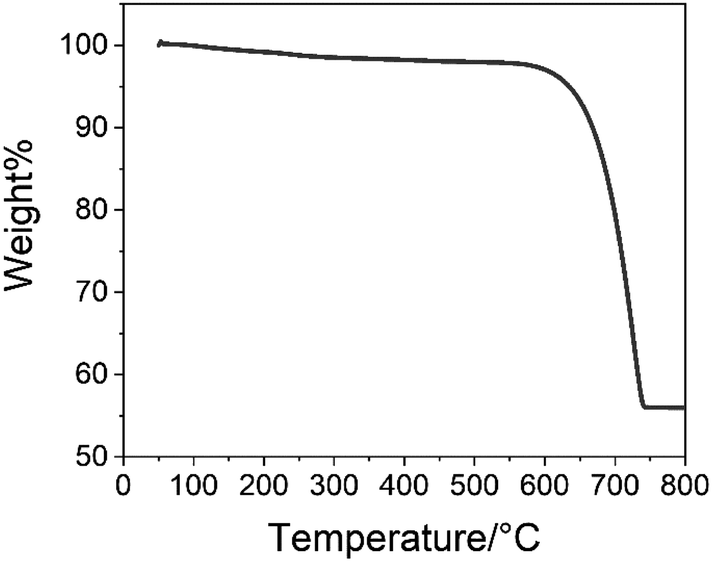 | ||
| Fig. 7 TGA of the CCNPs. | ||
| Sample | Temperature for 5% weight loss (°C) | Temperature for 20% weight loss (°C) | Residue at 800 °C (%) |
|---|---|---|---|
| CCNPs | 635 | 699 | 56.0 |
According to the above results, the growth mechanism of calcium carbonate is shown in Fig. 8. In the first step, glucose was added to convert a part of the calcium ions from hydrated calcium ions to calcium ions coordinated with glucose. In the second step, carbon dioxide gas was introduced to the calcium hydroxide solution to generate carbonate ions, which will combine with both the calcium ions coordinated with glucose and the hydrated calcium ions to form calcium carbonate molecules adsorbed on glucose (coordinated-CCs) and free calcium carbonate molecules (free-CCs). In the third step, the free-CCs are likely to combine with the coordinated-CCs to form the cluster structure of two kinds of molecules with one kind connecting with another.29 Inside this cluster, the zone with free-CCs could pack more closely to form stable and regular calcium carbonate lattices with a preferential orientation of (104), whereas coordinated-CCs are unstable due to the influence of glucose. In the fourth step, after dehydration and crystallization,30,31 a stable lattice is formed inside. The region near the coordinated-CCs is destabilized by glucose. Therefore, unstable links are formed between stable lattices. In the fifth step, since the links are occupied by coordinated-CCs, excess carbon dioxide is inclined to react with calcium carbonate at the link to generate calcium bicarbonate, which breaks the link to obtain the nanoparticles.
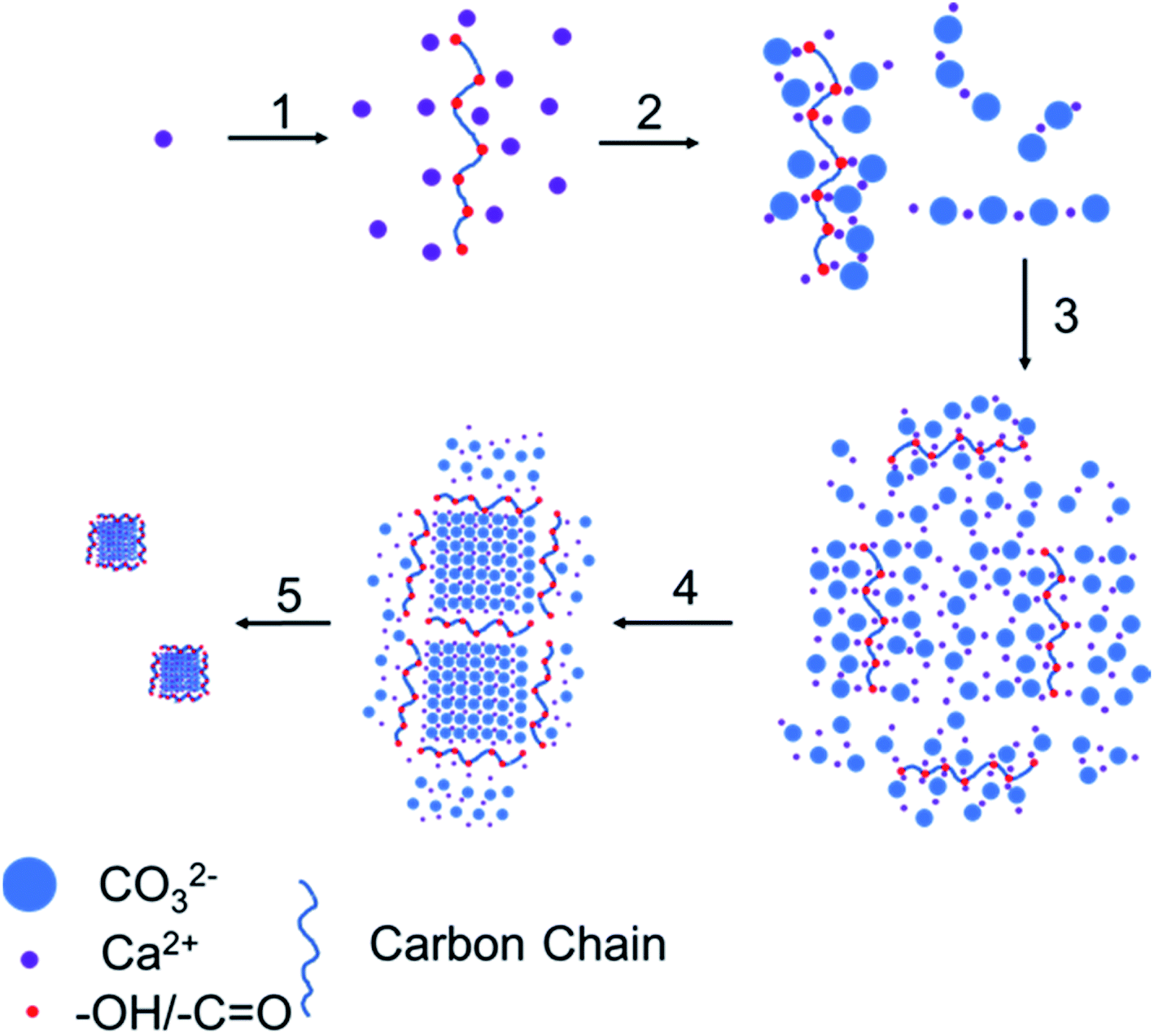 | ||
| Fig. 8 Growth process of the CCNPs: (1) add glucose; (2) introduce carbon dioxide; (3) cluster aggregation; (4) dehydration crystallization; (5) break at the chain link. | ||
In order to better combine the CCNPs with the matrix, we modified the CCNPs with sodium stearate. As seen in the FTIR spectrum of sodium stearate (Fig. 9), the characteristic peaks of sodium stearate appeared around 2917 cm−1 and 2849 cm−1, which were ascribed to the antisymmetric stretching vibration and the symmetric stretching vibration of the methylene group, respectively.32 The characteristic peaks at 1558 cm−1 and 1422 cm−1 were the antisymmetric stretching vibration and the symmetrical stretching vibration of the carboxylate group, respectively,33 and the characteristic peak at 1471 cm−1 was the vibration of the C–O group. In the FTIR spectrum of the modified CCNPs, the characteristic peaks of sodium stearate appeared at 2918 cm−1 and 2850 cm−1, which proved the formation of coordinated-CCs. The CCNPs before modification had a sharp peak at 1466 cm−1, while the modified CCNPs had a broad peak at 1459 cm−1. The shape broadening and position shift of the peaks may be due to the influence of the carboxyl groups of sodium stearate combined with calcium. Sodium stearate was bound to calcium carbonate via ionic bonds with the carboxylate.34
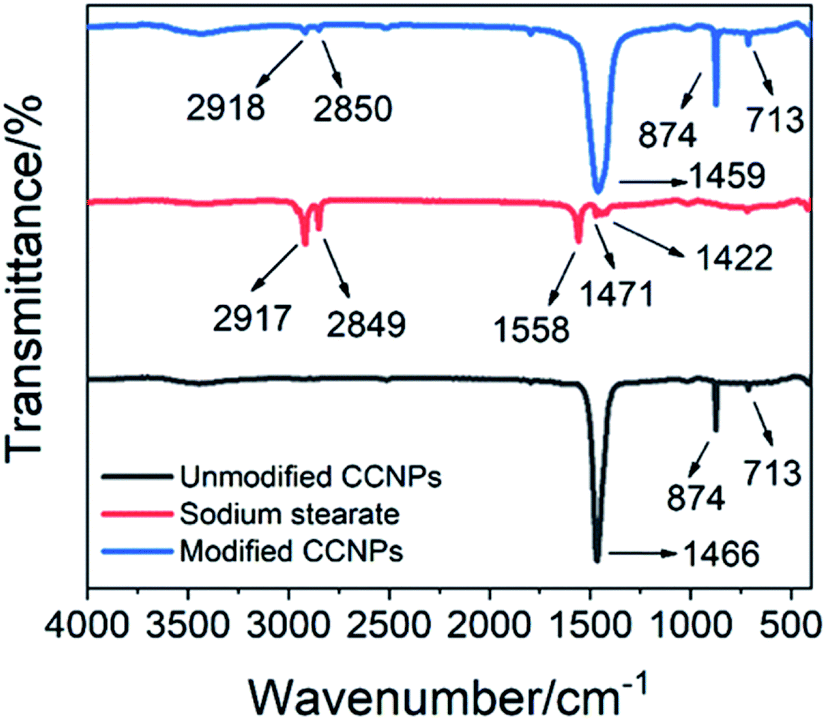 | ||
| Fig. 9 FTIR spectra of the CCNPs before and after modification and of sodium stearate. | ||
The CCNPs before and after modification were added to the matrix to make a composite, and the tensile properties were tested, as shown in Fig. 10. The tensile properties of the CCNPs after modification were obviously better than those before modification. The addition of the CCNPs to PDMS before modification even led to worse tensile properties of the composite, inferior to that of pure PDMS. Since the CCNPs are hydrophilic, they had poor compatibility with the matrix. Therefore, the modification process was necessary for the CCNPs to be added to PDMS to synthesize composites.
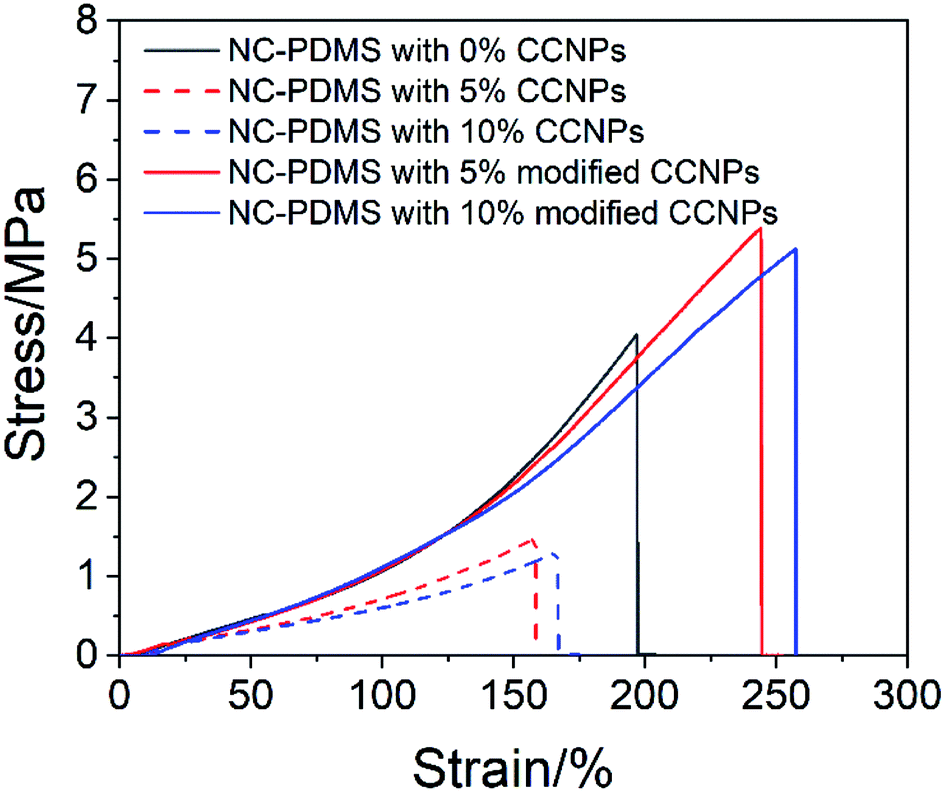 | ||
| Fig. 10 Tensile properties of the composite with the CCNPs before and after modification. | ||
Next, the effect of the addition amount of modified CCNPs on the tensile properties of the composites was investigated. As shown in Fig. 11, after the addition of the CCNPs to PDMS, the elongation at break increased, but the tensile strength increased first and then decreased. With the addition of calcium carbonate from 0% to 25%, the elongation of the sample increased from 197.16% to 308.56%: an increase of 56.50%. With the addition from 0% to 3%, the tensile strength increased from 4.05 MPa to 6.00 MPa, which is an increase of 48.15%. After adding more than 20% CaCO3, the tensile strength was lower than that of the pure PDMS sample.
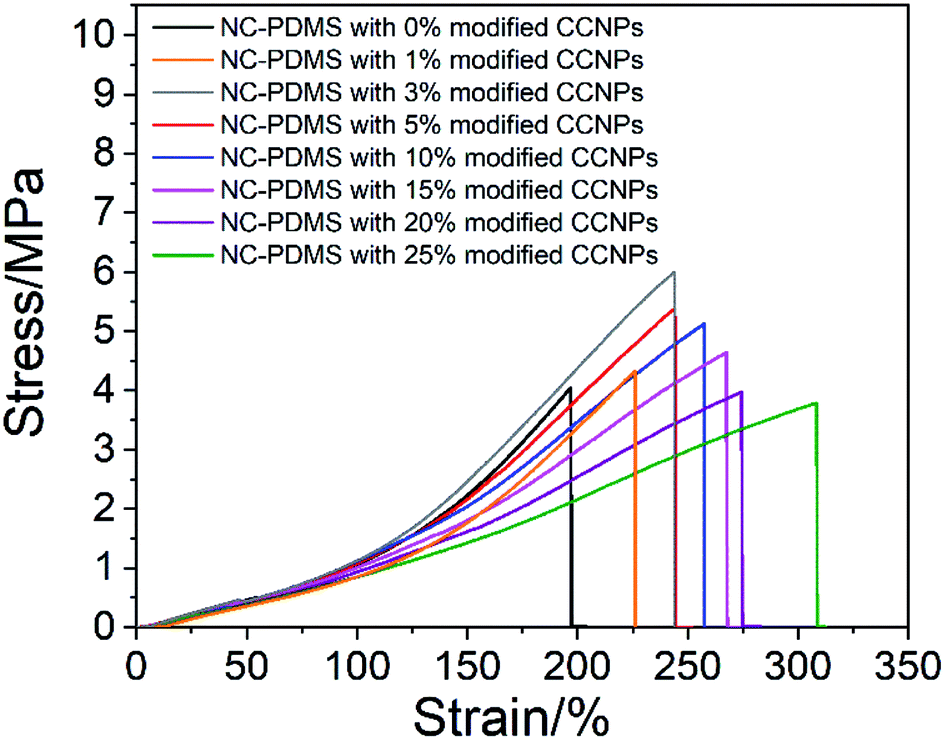 | ||
| Fig. 11 Tensile properties of the composite with different amount-modified CCNPs. | ||
After the modified CCNPs were added to the matrix as a filler for the composite, the long carbon chains of sodium stearate complexed on the particle surface combined with the rubber chains of the matrix in a physically entangled manner,35 thereby restricting the movement of the matrix.36 Since the modulus of the CCNPs was much higher than that of the matrix, the microscopic deformation of the composites was mainly contributed by the matrix between the particles. The stress was concentrated at the interface of the nanoparticle and the matrix, leading to debonding and resulting in the cavitation mechanism.37 The voids caused by debonding altered the stress state in the host matrix polymer surrounding the voids, which was a typical shear yielding mechanism, and a lot of energy could be absorbed.38 The deformation of the composite through the process of debonding effectively improved the elongation at break. With the addition of the filler, the area of debonding increased, resulting in an increase in the elongation at break of the composite. However, the tensile strength decreased after the filler content exceeded a certain amount. This was due to the excessive concentration of stress caused by the increase of filler particles, resulting in a decrease in the limit of stress that can be tolerated.38
Fig. 12 shows the cross section of the test sample after fracture. As shown in Fig. 12, with 3% CCNPs, the particle distribution was uniform, and there was no large-particle agglomerate. However, with 25% CCNPs, the size of the agglomerate was large, which may be the reason for the decreased tensile strength of the composite. As shown in Fig. 12(e), after 3 months, the composite with 3% CCNPs still did not show obvious agglomeration and had excellent stability.
 | ||
| Fig. 12 SEM images of the cross section of the composite: (a) 0% CCNPs; (b) 3% CCNPs; (c) 25% CCNPs; and the same sample after 3 months at room temperature: (d) 0% CCNPs; (e) 3% CCNPs; (f) 25% CCNPs. | ||
Adding nanoparticles as filler in the matrix can effectively improve the thermal stability of the composite. The test results of TGA are shown in Fig. 13. The important characteristic degradation data of all samples are listed in Table 2. Although the initial characteristic temperature at the 5% weight loss of pure PDMS was higher, the decomposition rate was the fastest in the subsequent heating process. The residue was the least at 650 °C, with only 53.6% left. For the composite with 3% CCNPs, although the initial characteristic temperature at 5% weight loss was low, the subsequent decomposition rate was slow, and 59.3% residue remained. The composite with 25% CCNPs had the lowest temperature at the initial 5% weight loss, but the residue was the most at 650 °C, with 65.0% remaining. It showed that the CCNPs as fillers interact with the matrix, and can effectively improve the thermal stability of the composites. The presence of nanoparticles in the composite enhanced the formation of the carbon layer, which hindered the diffusion of volatile decomposition products in the matrix material,39 thereby improving the thermal stability of the composite.
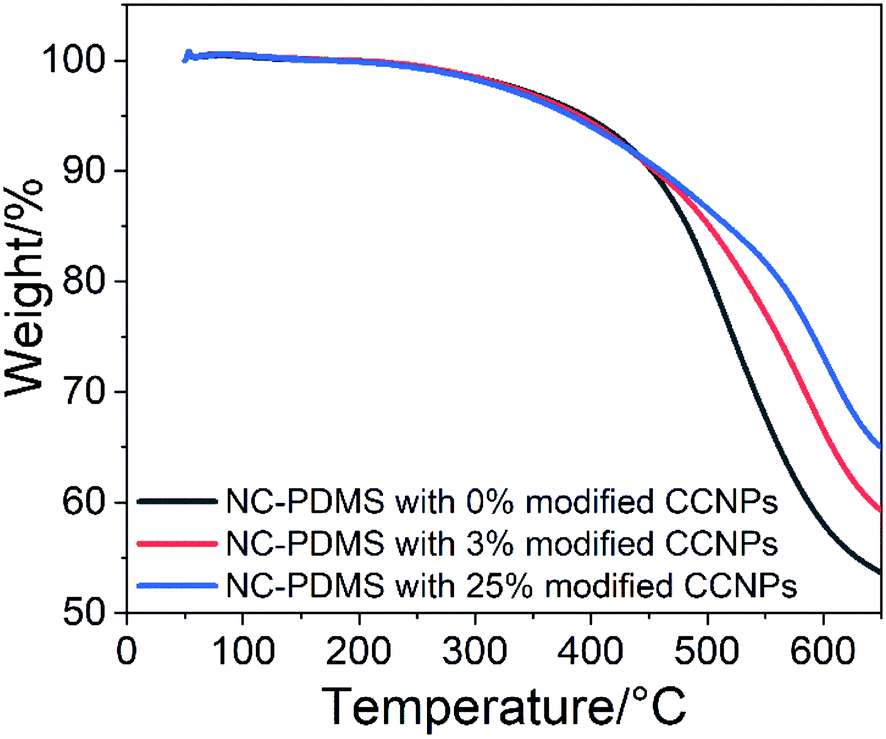 | ||
| Fig. 13 TGA of the composites with different amount of CCNPs. | ||
| Sample no. | Temperature for 5% weight loss (°C) | Temperature for 20% weight loss (°C) | Residue at 650 °C (%) |
|---|---|---|---|
| 0% CCNPs | 394 | 504 | 53.6 |
| 3% CCNPs | 389 | 534 | 59.3 |
| 25% CCNPs | 383 | 563 | 65.0 |
4. Conclusion
This work mainly studied the growth process of CCNPs with glucose as a chemical additive. It was found that calcium carbonate formed a long chain-like structure during the early stage of growth, which then gradually broke due to the introduction of excess carbon dioxide in the later stage, affording the CCNPs. Modification of the CCNPs with sodium stearate allowed them to bond with the matrix components more strongly. By adding 3% CCNPs, the elongation at break and the tensile strength increased by 23.96% and 48.15%, respectively. The addition of CCNPs to PDMS can dissipate a large amount of energy through the cavitation mechanism via debonding between the filler and the matrix. The thermal stability of the composites was positively correlated with the amount of nanoparticle filler, and the presence of nanoparticles hindered the decomposition of the matrix and improved the thermal stability of the composites. Exploration of the CCNP growth process and composite application will help to better develop and utilize this material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant number 51702285) and Natural Science Foundation of Zhejiang Province (Grant number LY17F040003).References
- M. A. Khalaf, C. B. Cheah, M. Ramli, N. M. Ahmed, A. M. A. Al-Asady, A. M. A. Ali, A. Al-Shwaiter and W. Tangchirapat, Constr. Build. Mater., 2021, 267, 120878 CrossRef CAS.
- J. Feng, F. Yang and S. Qian, Constr. Build. Mater., 2021, 269, 121249 CrossRef CAS.
- G. Momen and M. Farzaneh, Rev. Adv. Mater. Sci., 2011, 27, 1–13 CrossRef CAS.
- Y. Zhou, S. Wang, Y. Zhang, Y. Zhang, X. Jiang and D. Yi, J. Appl. Polym. Sci., 2006, 101, 3395–3401 CrossRef CAS.
- O. Długosz, K. Lis and M. Banach, Nanotechnology, 2021, 32, 025715 CrossRef PubMed.
- B. Dong, H. Li, J. Sun, Y. Li, G. M. Mari, X. Yu, W. Yu, K. Wen, J. Shen and Z. Wang, J. Hazard. Mater., 2021, 402, 123942 CrossRef CAS PubMed.
- M. Gautam, D. Santhiya and N. Dey, Mater. Today Commun., 2020, 25, 101394 CrossRef CAS.
- X. Pan, S. Chen, D. Li, W. Rao, Y. Zheng, Z. Yang, L. Li, X. Guan and Z. Chen, Front. Chem., 2018, 5, 130 CrossRef PubMed.
- A. M. Ferreira, A. S. Vikulina and D. Volodkin, J. Controlled Release, 2020, 328, 470–489 CrossRef CAS PubMed.
- Y. Chen, X. Ji, G. Zhao and X. Wang, Powder Technol., 2010, 200, 144–148 CrossRef CAS.
- A. Sergeeva, A. S. Vikulina and D. Volodkin, Micromachines, 2019, 10, 357 CrossRef PubMed.
- A. Xu, Y. Ma and H. Colfen, J. Mater. Chem., 2007, 17, 415–449 RSC.
- C. Yao, A. Xie, Y. Shen, J. Zhu and T. Li, J. Chil. Chem. Soc., 2013, 58, 2235–2238 CrossRef CAS.
- Y. Boyjoo, V. K. Pareek and J. Liu, J. Mater. Chem. A, 2014, 2, 14270–14288 RSC.
- W. Chuajiw, K. Takatori, T. Igarashi, H. Hara and Y. Fukushima, J. Cryst. Growth, 2014, 386, 119–127 CrossRef CAS.
- C. Wang, P. Xiao, J. Zhao, X. Zhao, Y. Liu and Z. Wang, Powder Technol., 2006, 170, 31–35 CrossRef CAS.
- D. Konopacka-Łyskawa, N. Czaplicka, B. Koscielska, M. Łapinski and J. Gebicki, Crystals, 2019, 9, 117 CrossRef.
- A. García-Hernández, H. Arzate, I. Gil-Chavarría, R. Rojo and L. Moreno-Fierros, Bone, 2012, 50, 276–288 CrossRef PubMed.
- J. E. Mark, Acc. Chem. Res., 2004, 37, 946–953 CrossRef CAS PubMed.
- E. J. Park, J. K. Sim, M.-G. Jeong, H. O. Seo and Y. D. Kim, RSC Adv., 2013, 3, 12571–12576 RSC.
- Q. Xu, M. Pang, L. Zhu, Y. Zhang and S. Feng, Mater. Des., 2010, 31, 4083–4087 CrossRef CAS.
- H. Collini, S. Li, M. D. Jackson, N. Agenet, B. Rashid and J. Couves, Fuel, 2020, 266, 116927 CrossRef CAS.
- P. Moulin and H. Roques, J. Colloid Interface Sci., 2003, 261, 115–126 CrossRef CAS PubMed.
- E. Chibowski, L. Hotysz and A. Szczes, Colloids Surf., A, 2003, 222, 41–54 CrossRef CAS.
- M. Takasaki, Y. Kimura, T. Yamazaki, Y. Oaki and H. Imai, RSC Adv., 2016, 6, 61346–61350 RSC.
- H.-A. Tajmir-Riam, Carbohydr. Res., 1988, 183, 35–46 CrossRef.
- L. M. Hamm, A. F. Wallace and P. M. Dove, J. Phys. Chem. B, 2010, 114, 10488–10495 CrossRef CAS PubMed.
- L. Chunbo, S. Qinpeng, Y. Guangyu, S. Xiaoxi, H. Pei, W. Kunmiao, Z. Hongyu, L. Zhihua and M. Mingming, Phys. Test. Chem. Anal., Part B, 2014, 50, 1342–1347 Search PubMed.
- R. A. Metzler, G. A. Tribello, M. Parrinello and P. U. P. A. Gilbert, J. Am. Chem. Soc., 2010, 132, 11585–11591 CrossRef CAS PubMed.
- D. Gebauer, A. Völkel and H. Cölfen, science, 2008, 322, 1819–1822 CrossRef CAS PubMed.
- G. A. Tribello, F. Bruneval, C. Liew and M. Parrinello, J. Phys. Chem. B, 2009, 113, 11680–11687 CrossRef CAS PubMed.
- S. Wang, X. Zuo, H. Cheng, Y. Yang and Q. Liu, J. Braz. Chem. Soc., 2016, 27, 1311–1318 CAS.
- C. Liu, Q. Zhao, Y. Wang, P. Shi and M. Jiang, Appl. Surf. Sci., 2016, 360, 263–269 CrossRef CAS.
- B. Shentu, J. Li and Z. Weng, Chin. J. Chem. Eng., 2006, 14, 814–818 CrossRef CAS.
- P. J. Flory, J. Macromol. Sci., Part B: Phys., 1976, 12, 1–11 CrossRef.
- S. Mishra, N. G. Shimpi and A. D. Mali, J. Polym. Res., 2011, 18, 1715–1724 CrossRef CAS.
- A. F. Yee and R. A. Pearson, J. Mater. Sci., 1986, 21, 2462–2474 CrossRef CAS.
- W. C. J. Zuiderduin, C. Westzaan, J. Huetink and R. J. Gaymans, Polymer, 2003, 44, 261–275 CrossRef CAS.
- S. Mishra, U. D. Patil and N. G. Shimpi, Polym.-Plast. Technol. Eng., 2009, 48, 1078–1083 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |