
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtonation-induced charge transfer and polaron formation in organic semiconductors doped by Lewis acids
Fabian Bauch*a,
Chuan-Ding Dong *a and
Stefan Schumacherab
*a and
Stefan Schumacherab
aDepartment of Physics and Center for Optoelectronics and Photonics Paderborn (CeOPP), Paderborn University, Warburger Strasse 100, 33098 Paderborn, Germany. E-mail: bauchfab@mail.uni-paderborn.de; cddong@mail.uni-paderborn.de
bWyant College of Optical Sciences, University of Arizona, Tucson, AZ 85721, USA
First published on 10th May 2022
Abstract
Lewis-acid doping of organic semiconductors (OSCs) opens up new ways of p-type doping and has recently become of significant interest. As for the mechanistic understanding, it was recently proposed that upon protonation of the OSC backbone, electron transfer occurs between the protonated polymer chain and a neutral chain nearby, inducing a positive charge carrier in the latter [B. Yurash, D. X. Cao, V. Brus et al., Nat. Mater., 2019, 18, 1327–1334]. To further clarify the underlying microscopic processes on a molecular level, in the present work, we theoretically analyze the influence of protons on the electronic properties of the widely used PCPDT-BT copolymer as a typical example. While we find that single protonation leads to formation of a localized polaron, double protonation leads to the release of a more delocalized polaron via an intrachain electron transfer. We also demonstrate the possibility of an interchain electron transfer. The vertical excitation spectra simulated for an ensemble of protonated polymers with different amounts of protons enable a detailed interpretation of the experimental observations and contribute to a molecular-level interpretation of the Lewis-acid doping process.
1 Introduction
Increasing and controlling the charge carrier density is fundamental for optimizing the performance of optoelectronic devices, such as light emitting diodes and field effect transistors.1,2 As a method to increase carrier density, doping is highly efficient and well understood in inorganic semiconductors. In contrast, molecular doping of organic semiconductors (OSCs) remains the subject of intense research activities and is not fully understood yet. The most common approach of molecular doping is to introduce dopant molecules that accept electrons from or donate electrons to the OSC, formally known as p-type and n-type doping leaving positive or negative charge carriers (polarons) in the OSC material, respectively.3–8 In recent years, tremendous experimental progress has been made in improving and understanding the molecular p-type doping, but their performance is limited due to limitations in solubility,9 doping efficiency, and hole mobility6 of the commonly used molecular dopants such as for example F4TCNQ (tetracyano-2,3,5,6-tetrafluoroquinodimethane). Lewis acid doping was proposed as an alternative that partially overcomes these limitations in P3HT (poly(3-hexylthiophene)).10 As recently reported by Yurash et al.,11 the doping of the polymer PCPDT-BT (poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta [2,1-b;3,4-b′]dithiophene)-alt-4,7(2,1,3-benzothiadiazole)]) with the strong Lewis acid BCF (tris(pentafluorophenyl)borane) exceeds the doping achieved using traditional molecular p-type doping with F4TCNQ. As for the origin of doping via BCF a multi-step mechanism was proposed. First, a Brønsted-type acid BCF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O complex is formed in solution which is then capable of protonating the polymers backbone and leaves behind a BCF:OH− counter anion. In a second step, an intermolecular electron transfer occurs in which a neutral polymer chain donates an electron to the protonated polymer chain, leaving a (positive) polaron on the un-protonated polymer chain. The formation of polaronic states is confirmed by UV-vis-NIR spectroscopic measurements and EPR and ENDOR measurements reveal the presence of unpaired electrons. Recent measurements on a related polymer, PCPDT-BT–SO3K, which can be protonated by H2O, show similar results.12,13 Detailed insights into the effect of Brønsted-type acid complex formation of BCF on the doping of P3HT were obtained identifying the complex formation as the driving factor of polaron formation, confirming the proposed mechanism.14 Theoretical investigations on the protonation pathway propose that a BCF:HO− counter anion energetically disfavors the protonation of PCPDT-BT, but formation of a [BCF:HO:BCF]− or [BCF:HO:H2O:BCF]− counter anion open an exergonic pathway and thus favors the protonation.15
:H2O complex is formed in solution which is then capable of protonating the polymers backbone and leaves behind a BCF:OH− counter anion. In a second step, an intermolecular electron transfer occurs in which a neutral polymer chain donates an electron to the protonated polymer chain, leaving a (positive) polaron on the un-protonated polymer chain. The formation of polaronic states is confirmed by UV-vis-NIR spectroscopic measurements and EPR and ENDOR measurements reveal the presence of unpaired electrons. Recent measurements on a related polymer, PCPDT-BT–SO3K, which can be protonated by H2O, show similar results.12,13 Detailed insights into the effect of Brønsted-type acid complex formation of BCF on the doping of P3HT were obtained identifying the complex formation as the driving factor of polaron formation, confirming the proposed mechanism.14 Theoretical investigations on the protonation pathway propose that a BCF:HO− counter anion energetically disfavors the protonation of PCPDT-BT, but formation of a [BCF:HO:BCF]− or [BCF:HO:H2O:BCF]− counter anion open an exergonic pathway and thus favors the protonation.15
In the present work we set out to further clarify the microscopic processes underlying Lewis acid doping on a molecular level. To this end, we theoretically analyze the influence of protons on the electronic properties of the widely used copolymer PCPDT-BT, with a particular focus on the protonation-induced charge transfer and polaron formation that may occur. This includes studies on the role of the counter ion in the doping process as well as intra- and inter-polymer charge transfer processes. We calculate vibrational infrared spectra showing clear polaron signatures in the form of particularly intense vibrational modes (IVMs)16 and we calculate vertical excitation spectra to support the interpretation of the experimentally measured spectra. Our results explicitly show that both single and double protonation of polymer chains may well contribute to the Lewis acid doping signatures observed experimentally, with the double doping inducing less localized and potentially more mobile charge carriers.
2 Computational details
To investigate the charge transfer in protonated PCPDT-BT, our method of choice is spin-unrestricted density functional theory (DFT) allowing for broken-spin symmetry (BS), which was previously used successfully in predicting charge densities as well as structure and spectroscopic properties also of doped conjugated polymers.17–24 The spin-unrestricted BS method allows the α- and β-orbitals to be treated separately, providing more freedom in optimizing the electronic ground state and potentially breaking the symmetric spatial distribution of the α-and β-orbitals. This allows for unequal charge density on different subregions of the polymer, as expected from a charge transfer process within the polymer backbone. This approach mimics parts of a multi-configurational method that yields similar electronic properties such as charge distribution and charge transfer in the electronic ground state at comparatively low computational cost. It should be noted that the spin-unrestricted BS method usually results in incorrect spin density due to spin contamination, but this is not of interest in the present study. For a more detailed discussion on the spin-unrestricted BS method we refer the reader to one of our earlier papers.25–28 In the present work, electronic states are described by a 6-31G** basis set with the range-separated cam-B3LYP functional. Molecular van der Waals interaction is covered at the level of Grimme's D3 dispersion correction with Becke–Johnson damping. As shown in one of our previous papers, the choice of the van der Waals method does not have a large effect on the qualitative results, but minor changes in the absolute orbital energies do occur, but these are of less interest in the present work.25 Time-dependent DFT (TD-DFT) is employed to calculate vertical excitation energies considering the first 25 excited states in the calculations. Solutions are modelled by the conductor-like polarizable continuum model (C-PCM) with chlorobenzene as solvent. All calculations are done with GAUSSIAN16.29 The Multiwfn software30 is used to analyze the density of states by the Hirshfeld method and the atomic dipole moment corrected Hirshfeld charge.31 The latter is used to analyze the partial charges of the individual donor and acceptor fragments of the polymer, providing insight into the spatial charge distribution and possible interpretation as charge transfer within the polymer backbone after protonation.The protonation is realized by explicit addition of hydrogen atoms bonded to the carbon-backbone of the PCPDT-BT conjugated polymer as illustrated in Fig. 1 with a +1e net positive charge per additional hydrogen atom. The resulting bonds are initiated with a typical carbon–hydrogen bond length of about 1.09 Å. All atoms including the additional hydrogen atoms are then freely relaxed in a geometry optimization procedure. This model approach allows to study the direct influence of the additional proton on the electronic properties of the protonated polymer, without considering the complexity of the chemical process that triggers the protonation. As in our previous work on PCPDT-BT,8,25,26 alkyl side chains are assumed to be of little influence to the frontier orbitals of the backbone and are replaced by methyl groups to reduce computational cost. The PCPDT-BT polymer is modelled by a rather long hexamer as depicted in Fig. 1. All structures are optimized in their singlet state allowing broken-spin symmetry, and in the triplet ground state, respectively, revealing the singlet ground state always to be energetically favorable. Thus, the results presented below address the spin-symmetry-broken singlet ground state.
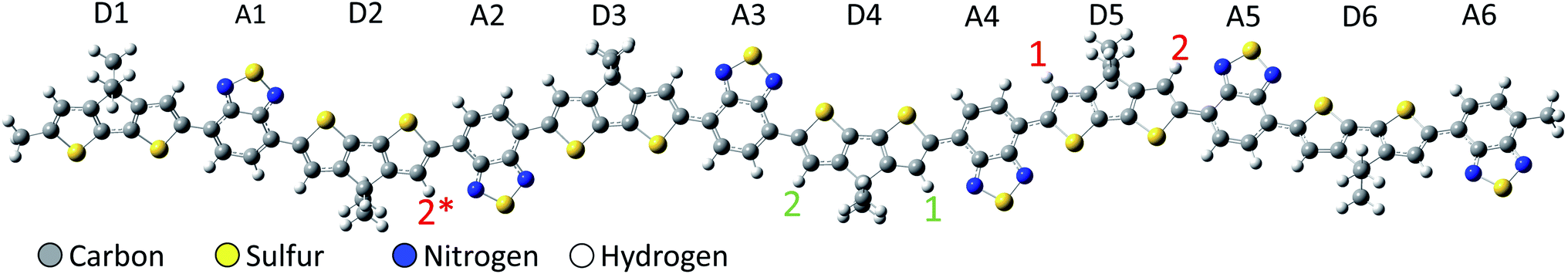 | ||
| Fig. 1 Optimized neutral PCPDT-BT with proton positions indicated. The red numbers 1 and 2 indicate the proton positions for the single protonated PCPDT-BT structure. The red 2* (between D2 and A2) denotes a proton position equivalent to the proton position 2 (between D5 and A5), but facing towards the middle of the polymer chain. The green numbers indicate the additional proton positions for the single protonated structure proton-2, resulting in double protonated chains. The labels D1, A1, D2, etc. label each donor (D) and acceptor (A) fragment used to visualize the fragment charges. | ||
3 Results and discussion
3.1 Single protonation and polaron formation
First, we focus on protonation of PCPDT-BT with a single proton at a time with proton positions as indicated in Fig. 1 (marked by red numbers). For simplicity, we refer to the protonated chain with the proton at position 1 and 2 as “proton-1” and “proton-2”, respectively. Proton-1 features a proton on the left side of the donor unit D5, facing the middle of the polymer. The resulting structure remains nearly planar as in a neutral PCPDT-BT without protonation. As shown in Fig. 2a, it forms a spin-symmetry-broken density of states (DOS) in its ground state in gas-phase. We note that the absolute energy values of the single-particle molecular orbitals (MOs) significantly depend on the specific method used, however, for the MOs and DOS discussed here we are mostly interested in trends observed and in their relative ordering. The lowest single unoccupied molecular orbital (SUMO) in the β-channel is depicted in Fig. 2b. It features as a bonding π-delocalized character, is situated on the left of the proton position at donor unit D5, and is delocalized from donor unit D3 to donor unit D5. This is accompanied by a high amount of positive charge on the donor units D3, D4, and D5 (a total of +0.88e), as shown in Fig. 2e (blue bars). Moreover, the calculated vibrational spectrum of proton-1 features a band of IVMs at wavenumbers of about 1100 cm−1 in good agreement with experimental reports on IVMs of PCPDT-BT doped with F4TCNQ.32 These results suggest that the proton on position 1 induces a polaron in an electronic state that is reflected by the β-SUMO. We note that the proton-induced polaron is spatially less delocalized than found for an individual singly charged PCPDT-BT and, consequently, the IVMs are shifted to higher wavenumbers compared to the IVMs of the singly charged PCPDT-BT (cf. Fig. 2d). We attribute the less delocalized nature of the proton induced polaron to a strong coupling between the proton and the polaron formed. For comparison we also show results where the proton is attached in a different position, in position 2 in Fig. 1 on the right side of donor unit D5 in a qualitatively different position with respect to the adjacent acceptor unit and facing the polymer edge (denoted as structure proton-2). The electronic structure for this proton position in gas-phase turns out to be spin-symmetric as shown in Fig. 2a with a lowest unoccupied molecular orbital (LUMO) localized on units D5 to A6 near the oligomer edge as illustrated in Fig. 2c. This orbital shows a dominant antibonding character near the proton. Moreover, large amounts of positive charge are found in the spatial range of this LUMO orbital (Fig. 2e) and, in contrast to proton-1, the acceptor units are also more charged. Together with the absence of an IVM (Fig. 2d), we conclude that the protonation in the structure proton-2 merely induces a slightly modified electronic behavior of a neutral polymer and does not induce a polaron.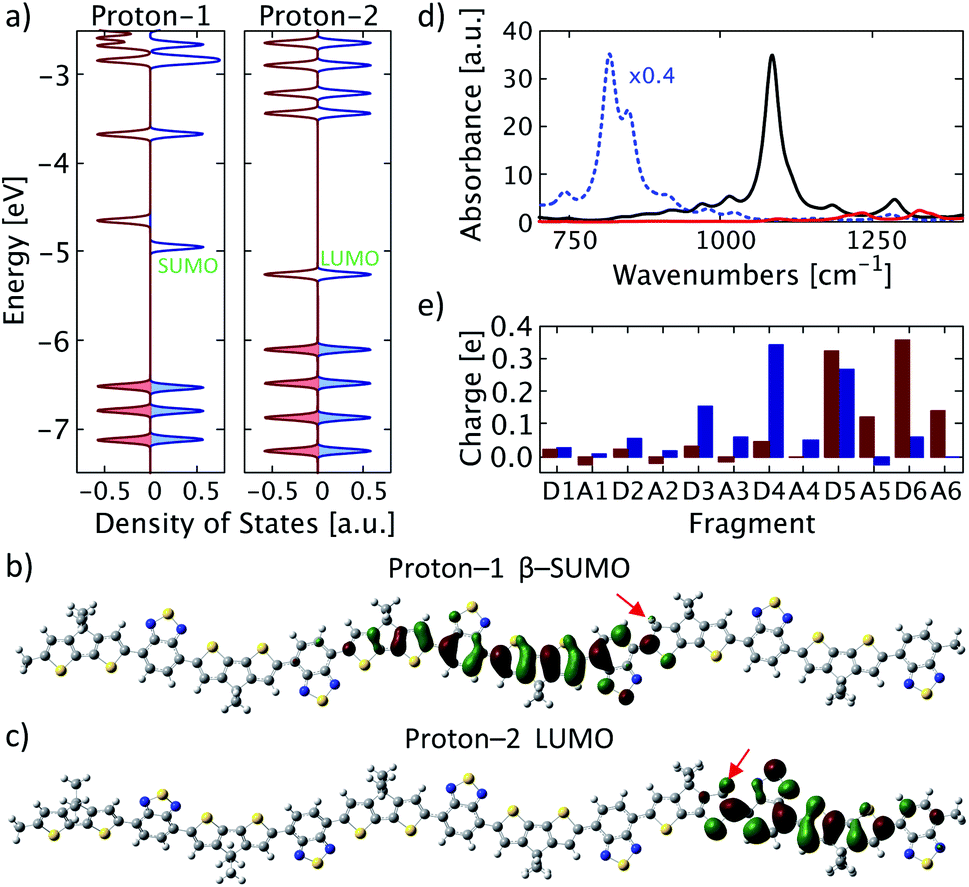 | ||
| Fig. 2 Single protonation. (a) Density of states for systems proton-1 and proton-2. Alpha states are shown in red and beta states in blue. Color-shaded states are occupied states. (b) and (c) Selected molecular orbitals with (b) proton-1 and (c) proton-2. The proton positions are indicated by red arrows. (d) Calculated IR spectra for proton-1 (solid black), proton-2 (solid red), and for a singly positively charged PCPDT-BT (dotted blue) rescaled by 0.4 for clarity. (e) Atomic dipole moment corrected Hirshfeld charge for each fragment of proton-1 (blue) and proton-2 (red) as labelled in Fig. 1. | ||
However, comparing with results where the proton is attached in position 2*, which is equivalent to position 2 but is further away from the edge of the oligomer modelled, we find very similar electronic behavior as in proton-1 with a positive polaron induced in the center of the PCPDT-BT conjugated chain. Therefore, we conclude that polaron formation in proton-2 is mostly hindered by the nearby edge of the oligomer modelled, which would be a very rare case to occur in experiments with long polymer chains. The precise positioning of the proton in the donor unit and relative positioning to the adjacent acceptor unit appears to have only little influence on the polaron formation.
In the following, we further investigate the robustness of the polaron induced in proton-1 configuration. First, we introduce chlorobenzene (ε = 5.6968) as a solvent which leads to formation of spin-symmetric electronic states for all proton positions. The corresponding LUMO of proton-1 is shown in Fig. 3a. The absence of spin-symmetry breaking and the formation of a localized LUMO just like for proton-2 without solvent (cf. Fig. 2c) indicates that the polaron formation is hindered by screening from the solvent. Next we investigate the influence of a nearby BCF:OH− counter anion. Placing a pre-optimized BCF:OH− counter anion above proton-1 and optimizing the complex with 0 net charge in gas-phase leads to a slightly curved structure of the polymer. The calculated electronic structure shows +0.78e net charge on the protonated polymer, and −0.78e on the BCF:OH− counter anion as shown in Fig. 3b, demonstrating that the counter anion and the proton attached to the polymer do not neutralize each other by recombining to a BCF:OH− complex. However, for this complex, spin-symmetric electronic states are formed with a LUMO very much resembling the LUMO of proton-1 in chlorobenzene (cf. Fig. 3a and c), indicating that the polaron formation on the polymer is also hindered by the presence of the counter anion. In addition to this geometry, where the OH group is facing away from the polymer, we have also considered a different position of BCF:OH− in which the OH group faces the polymer. The optimized geometry of the complex shows stronger bending of the polymer backbone with a charge of +0.818e and −0.818e for the polymer and BCF:OH− complex, respectively. Apart from the slightly stronger bending, which we attribute to a smaller distance between the anionic and cationic parts of the two systems, notably no qualitatively different behaviour in orbital formation is observed. Thus, we conclude that the relative position of the counter anion has no pronounced influence on the polaron behaviour. After removal of the anion of the shown complex and calculation of the electronic density without further geometry optimization with +1e net charge, a symmetry-broken electronic state is recovered with a polaron formed in the β-SUMO. These results indicate that the interaction between the counter anion and proton-1 hinders polaron formation, whereas the geometry distortion plays only a minor role.
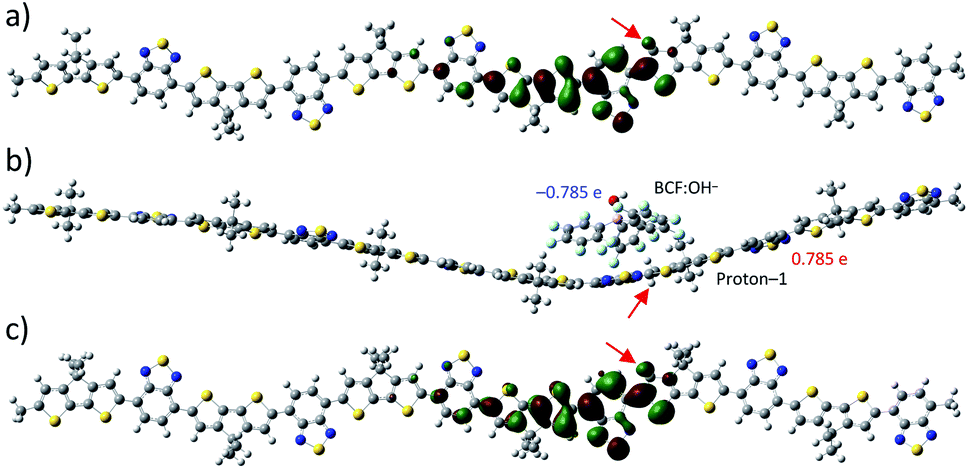 | ||
| Fig. 3 Proton-1 in solution and with counter anion. (a) LUMO of proton-1 in chlorobenzene. (b) Optimized structure of proton-1 with counter anion BCF:OH− in gas-phase and (c) corresponding LUMO (the anion is removed for clarity). The red arrows denotes the proton positions. The numbers in blue and red give the atomic dipole moment corrected Hirshfeld charge of the anion and proton-1, respectively. | ||
We note that the proton positions considered are the second most energetically favorable proton positions according to Marqués et al. calculated proton affinities,15 but the difference in calculated proton affinity is only 4.2 kcal mol−1 compared to their most favorable proton position with the highest proton affinity. Modeled protonated systems with the most favorable proton position result in a 130° twisted backbone, which is a major barrier to the formation of the electronic ground state in experimental environments. Nevertheless, we find that the electronic ground state for these proton positions has similar features as for proton-2, i.e., non-polaronic ground states are formed. We attribute this feature to the twist of the backbone, which leads to a splitting of the backbone into two separate, smaller polymer backbones, with the proton positions very close to the newly formed edges, hindering the formation of polarons similar to proton-2.
3.2 Double protonation and intrachain electron transfer
Next, a second proton is attached to the structure proton-2 on donor unit D4. This process may occur with increasing probability in systems with increasing acid concentrations. For a more complete picture, two possible positions of the second proton are considered: once on the right side of the donor unit (green number 1 in Fig. 1), designated double-1, and once on the left side of the of the donor unit (green number 2 in Fig. 1), designated double-2. A clear broken-spin symmetry is formed for double-1 in its optimized structure, as shown in Fig. 4a. The α-SUMO and β-SUMO are very close to each other in energy and the orbital profiles show a close resemblance (not shown). Both are located near the right edge of the chain with an orbital nature similar to that of the LUMO in proton-2 and do not represent polaron states. Instead, the proton-induced polaron state is found to be the α-SUMO+1 state, which is energetically very close to the α-SUMO (significantly overlapping in the DOS plot) but spatially well separated and of entirely different electronic nature. It is located on the left side of the conjugated chain as shown in Fig. 4b and the polaronic nature is reflected in the bonding state appearance and delocalization. In addition, the polaronic α-SUMO+1 state is accompanied by a high total amount of positive charge of about +0.96e on the donor units D1 to D3 (Fig. 4e, red bars) and leads to IVMs for frequencies below 1050 cm−1, as shown in Fig. 4d. A careful analysis of electronic orbitals reveals that a pronounced intrachain electron transfer occurs in double-1. The polaron turns out to be located on the left side of the chain as shown in Fig. 4b which indicates a substantial reorganization of electronic states within the double-protonated polymer. As the result, an originally unoccupied orbital with an antibonding appearance similar to the LUMO of proton-2, becomes the occupied α-SOMO as is shown in Fig. 4c. The electron transferred and polaron formed correspondingly roughly carry a charge of 1e, such that we term the electron reorganization an intrachain electron transfer process, resulting in the polaronic α-SUMO+1 and occupied α-SOMO, accompanied by the formation of non-polaronic α- and β-SUMO. This picture appears to be in agreement with the mechanism proposed by Yurash et al.11 This interpretation is further supported by the calculated total change in electron density induced by the addition of the second proton. In the calculation, the second proton (green number 1 in Fig. 1 and red arrow in Fig. 5) is removed from the optimized double-1 structure and a single-point calculation is performed, including the first proton as indicated by the green arrow in Fig. 5. Then, the total electron density of the optimized double-1 structure is subtracted from the total electron density resulting from this single-point calculation. The resulting change in electronic density is shown in Fig. 5 which illustrates directly how the total electron density changes upon addition of the second proton. A reduction of the total electron density in the double bonds where the polaron is located is observed, which is the typical consequence of polaron formation in conjugated systems.8 This is accompanied by a localized, strong increase in the total electron density at the proton position indicated by the red arrow in Fig. 5. Note that the electron density reduction and increase spatially coincide with the α-SUMO+1 (Fig. 4b) and α-SOMO (Fig. 4c), respectively. A calculation for an extended double-1 structure with two additional polymer repeat units added on the left edge shows that the polaron induced by the intrachain electron transfer has the same spatial distribution as for double-1, but is shifted even further away from the proton positions to the left side of the elongated chain. This indicates that the polaron delocalization is finite but the polaron itself is rather decoupled from the proton positions and thus might act as a free positive charge. To test the influence of the position of the first proton near the edge (proton-2), we tested another double proton position with similar spacing and charge overlap as for double-1, namely red number 1 (proton-1) and green number 2 in Fig. 1. We expect similar behavior to double-1, i.e., electron transfer within the chain. Indeed, we find qualitatively similar orbital formation and DOS. We conclude that the proximity of the first proton to the edge does not qualitatively affect the intrachain electron transfer that occurs when the charges of both protons may initially overlap.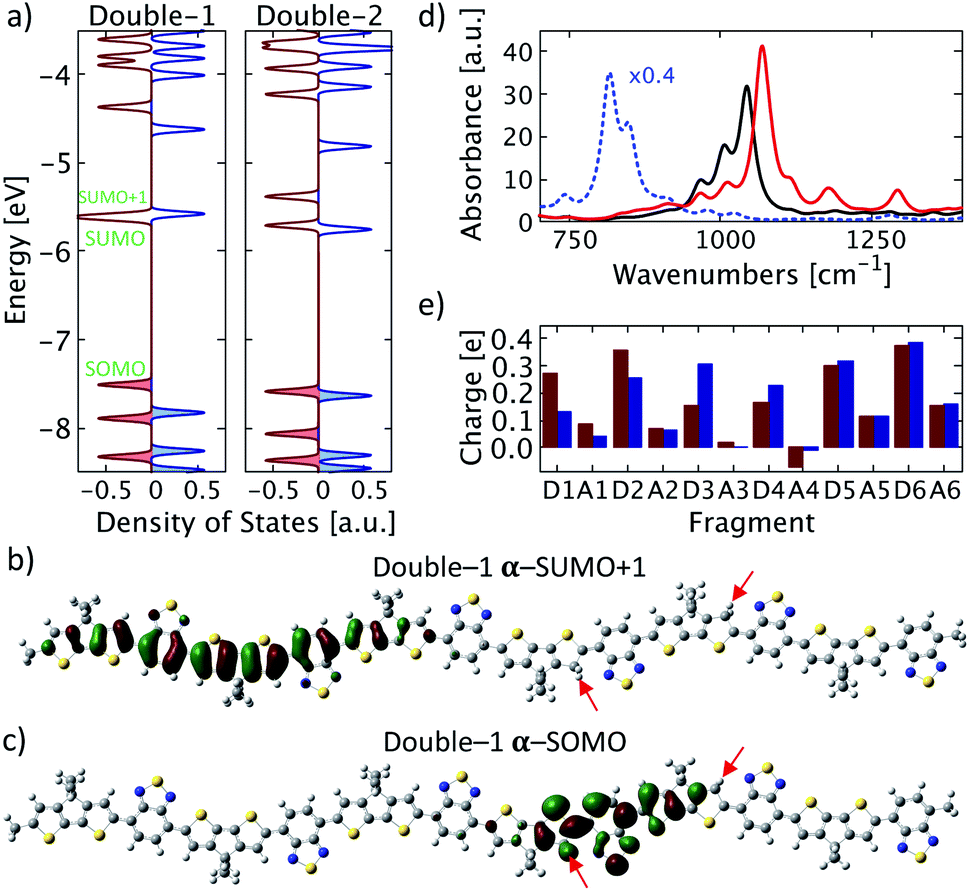 | ||
| Fig. 4 Double protonation. (a) Density of states for double-1 and double-2. Alpha states are shown in red and beta states in blue, color-shaded states are occupied states. α-SUMO and α-SUMO+1 in double-1 are almost energetically degenerate. (b) and (c) Selected molecular orbitals of double-1 with (b) α-SUMO+1 and (c) α-SOMO. The proton positions are indicated by red arrows. (d) Calculated IR spectra for double-1 (solid red), double-2 (solid black), and for a singly charged PCPDT-BT (dotted blue) rescaled by 0.4 for clarity. (e) Atomic dipole moment corrected Hirshfeld charge for each fragment of double-1 (red) and double-2 (blue) as labelled in Fig. 1. | ||
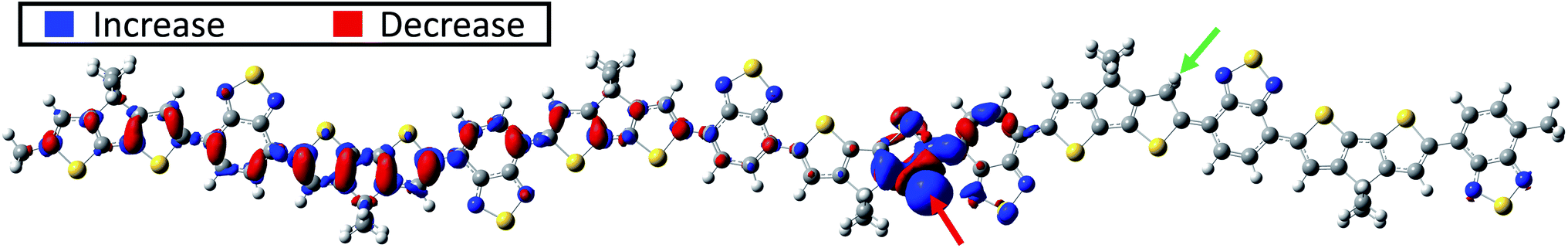 | ||
| Fig. 5 Intrachain electron transfer after double protonation. Shown is the change in total electron density induced by adding the second proton at the position marked by the red arrow in double-1. The green arrow marks the position of the first proton. The decrease in total electron density occurs in the double bonds on the left side of the conjugated chain, cf. α-SUMO+1 in Fig. 4b. This is accompanied by long-range electron transfer to the position of the second proton as marked by the red arrow. Isovalue is set to 0.0015. | ||
This intrachain electron transfer is not present in the other double protonated structure, namely double-2. Due to the different positioning of the second proton with respect to the middle of donor unit D4 (green number 2 in Fig. 1), a potential overlap of induced charges is avoided. The DOS of the optimized double-2 still features a broken-spin symmetry, but the orbitals induced by the protons are similar to the orbitals of the single protonated structures proton-1 and proton-2, i.e. the additional proton induces a polaron close to its position situated on the left side of donor unit D4. Moreover, the charge distribution is identical to the charge distribution expected from the results for single protonated structures (Fig. 4e, blue bars) and the IVM induced by the polaron has similar frequency as for proton-1 (cf. Fig. 4d and 2d).
3.3 Interchain electron transfer
In addition to the intrachain electron transfer demonstrated above, a more common interpretation of Brønsted acid doping is the occurrence of an interchain electron transfer.11 To investigate this mechanism, a neutral PCPDT-BT tetramer (four repeat units) and a double protonated PCPDT-BT tetramer are brought in proximity to each other and the structure of the entire complex is optimized. The resulting optimized structure is shown in Fig. 6b. In this structure an interchain electron transfer occurs that is manifested in a partial charge of each chain of about +1e and a broken-spin symmetry with singly unoccupied molecular orbitals on the un-protonated chain, which resemble the signatures of a positive polaron as illustrated in Fig. 6. Similar to the intrachain electron transfer, a SOMO is found on the double protonated chain featuring localized antibonding character, as shown in Fig. 6b. The occurrence of both intrachain and interchain electron transfer are in line with the pictures proposed in the literature.11 True interchain electron transfer is only observed for certain proton positions in our calculations and further depends on inter-molecular positioning. For other cases only charge transfer complex formation is found but no integer-charge transfer as reported in Fig. 6. These findings are also in agreement with previous reports on the coexistence of ICT and CTC in OSC materials doped with molecular dopants.33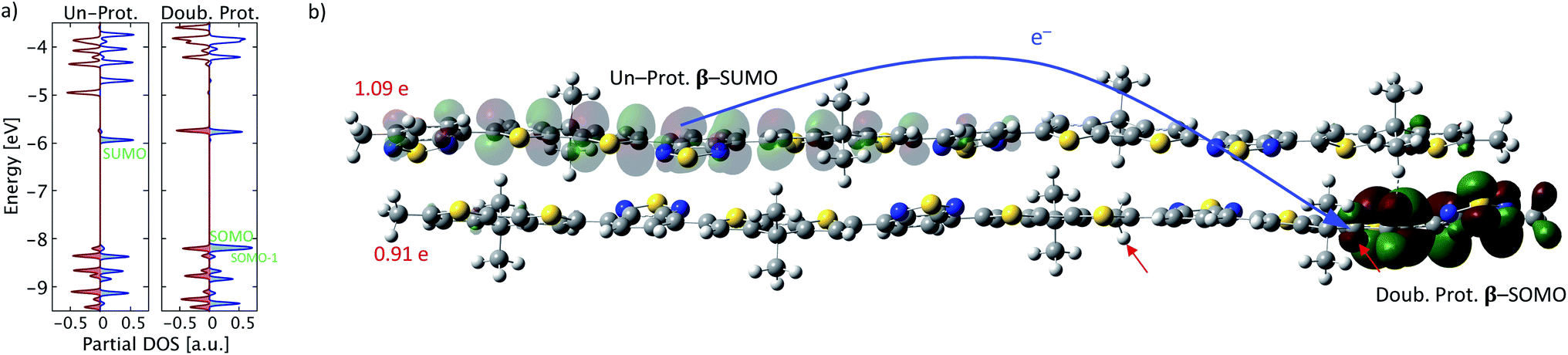 | ||
| Fig. 6 Interchain electron transfer between un-protonated and double protonated PCPDT-BT. (a) Partial density of states for both conjugated chains, un-protonated (Un-Prot.) and double protonated (Doub. Prot.), respectively. Alpha states are shown in red and beta states in blue. Color-shaded states are occupied. (b) Molecular orbitals involved in the electron transfer between chains as indicated by the blue arrow. The electron is transferred from the β-SUMO (semi-transparent) of the un-protonated PCPDT-BT, leaving behind a positively charged polaron, to the β-SOMO (solid) of the double protonated PCPDT-BT. The red arrows mark the proton positions. The numbers in red give the partial charge of each chain. | ||
3.4 Optical absorption spectra
Optical absorption measurements are widely used to identify the doping nature of OSC materials, with typical polaronic absorption features observed in the near infrared spectral range.34 Yurash et al.11 reported an increase of broad polaronic absorption features in the range of 1100 nm to 3000 nm in thin films of PCPDT-BT doped with increasing amounts of the Lewis acid BCF. Furthermore, measurements in solution (dry chlorobenzene) gave a more detailed insight into the absorption features below 1600 nm, revealing a doping induced absorption feature at about 800 nm (read value) quite close to the main absorption feature of pristine PCPDT-BT at 720 nm.35 The main absorption peak of pristine PCPDT-BT gets reduced accordingly in its amplitude as a result of the doping by BCF. In Fig. 7a and b we show the vertical absorption spectra calculated in gas-phase for each single and double protonated chain discussed in the present work. We also compare with the absorption spectra for pristine (neutral) PCPDT-BT and a singly positively charged (polaron) PCPDT-BT chain. Distinct differences are found for the singly protonated systems with (proton-1) and without (proton-2) polaron formation. As can be seen in Fig. 7a (blue), the single proton position that does not lead to the formation of a polaron (proton-2) still leads to a main peak reduced in intensity at 617 nm and an additional absorption feature at 795 nm. Both features together appear as a broadening of the main absorption peak compared to the pristine PCPDT-BT (neutral). In contrast, the polaron induced for the other proton position in proton-1 does lead to completely different absorption features (Fig. 7b, green). Comparing with the spectrum for the singly positively charged PCPDT-BT in Fig. 7a (polaron), in proton-1 we observe typical polaronic absorption features at 1114 nm and 1981 nm. The positions of the polaronic absorption peaks are slightly shifted compared to the polaron of PCPDT-BT (Fig. 7a, red) which is a result of the slightly reduced spatial extend of the polaron induced by the protonation. The double protonated structures appear to show the general features of both singly protonated structures as shown in Fig. 7b (orange and purple). We note that the spectral positions of absorption bands in the simulated spectra typically do not match the experimental values and depend on the method used. However, the appearance of doping induced features and relative trends and changes closely resemble the experimental observations as discussed further in the following. We also note that due to the increased computational cost, we do not explicitly study the vertical excitation of complexes with interchain electron transfer as shown in Fig. 6 here.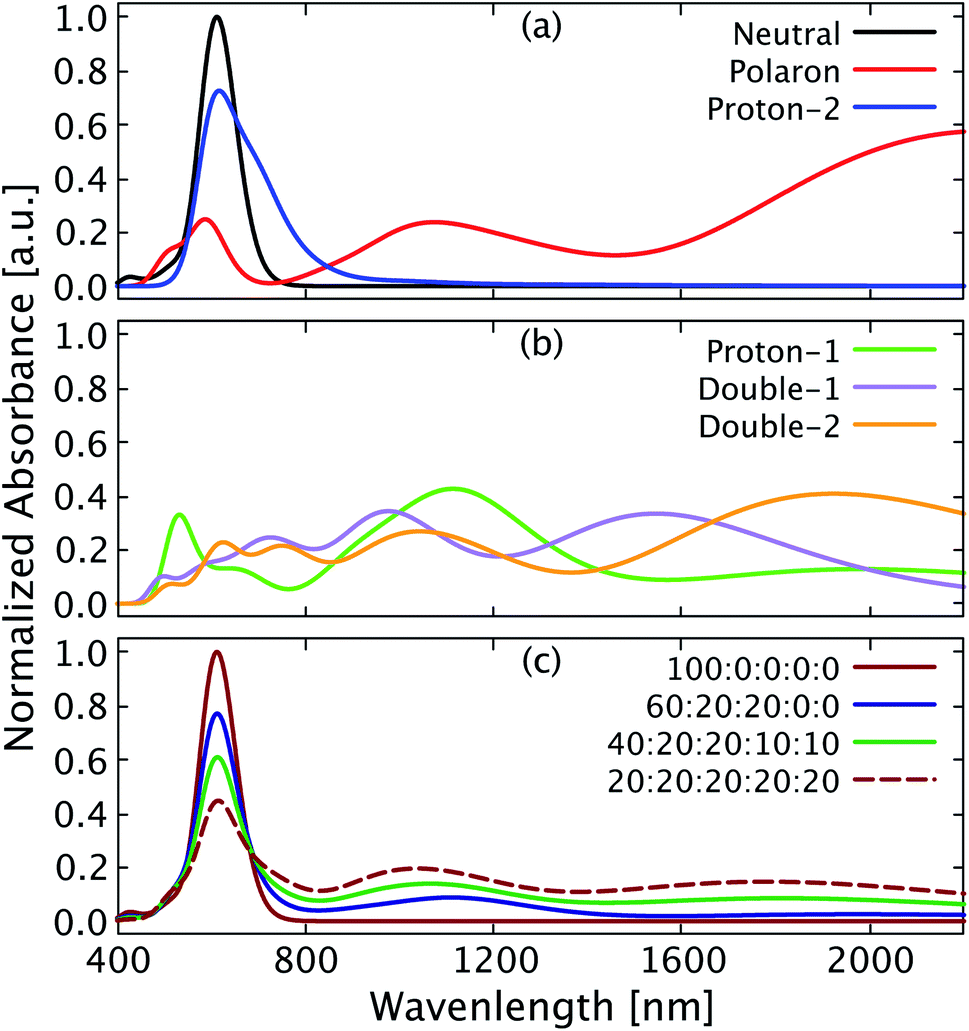 | ||
| Fig. 7 Optical absorption spectra for various species of PCPDT-BT. (a) Absorption of neutral, singly positively charged (polaron), and proton-2 PCPDT-BT. (b) Absorption of proton-1, double-1, and double-2. (c) Superposition of absorption of several PCPDT-BT species with ratios labelled as neutral:proton-1:proton-2:double-1:double-2. All spectra are normalized to the maximum in the neutral PCPDT-BT spectrum. The full width at half maximum is 0.3 eV. | ||
It appears reasonable to assume that the broad polaronic absorption observed in experiment is a combination of several (multi-)protonated structures, giving rise to a whole range of different polaronic absorption features. The doping induced absorption feature close to the main peak of PCPDT-BT also observed in the experiment appears to have its origin in protonation at a position for which polaron formation is suppressed (e.g., our case proton-2). Here we simulate a scenario with different contributions as a superposition of the absorption spectra found in the calculations for different structures as shown in Fig. 7c. With increasing concentration of BCF in the experiment, the proportion of double protonated species over single protonated ones will increase, while the proportion of the neutral PCPDT-BT will decrease, which is represented by the different ratios considered for the calculated spectra shown in Fig. 7c. As can be seen in Fig. 7c (dark blue), a ratio of 20% of each single protonated structure, corresponding to a low BCF concentration, mostly only leads to a reduction of the main peak intensity, with a slight broadening. If the amount of protonation is increased by introducing an additional 10% of each doubly protonated structure (Fig. 7c, dark green), the amplitude of the main peak decreases further and the broad polaronic absorption increases. Note that the amplitude of the polaronic absorption appears to be small, but compared to the reduction of the main peak it is in reasonable agreement with the experimental data.11 Finally, we address a 20% ratio of each single and double protonated structure (Fig. 7c, dashed dark red), corresponding to a high BCF concentration in the experiment.
4 Conclusion
In this work, we have theoretically investigated protonation induced polaron formation, a mechanism intensely discussed in the context of doping OSC materials with Lewis acids, in the widely used OSC PCPDT-BT as a representative example. On a microscopic level, we find that protonation of a conjugated chain with only one proton can induce formation of a typical polaronic state for which the induced positive charge is tied to the spatial position of the proton. We find that this polaron formation is hindered by the interaction with the BCF:OH− counter anion if it is still nearby. Also, with chlorobenzene as a solvent, the formation of such proton-induced polaron state appears more difficult.
Interestingly, with the attachment of a second proton to a given PCPDT-BT chain, we find that for certain spatial arrangement of the two protons, a polaron state is induced in the conjugated chain that is largely spatially decoupled from the proton sites. Similar behavior is found if the electron transfer process leaving the positive polaron behind occurs on an individual chain as an intrachain electron transfer process or between a protonated and an un-protonated chain as an interchain transfer process. In a film environment, we reckon that the polaron induced in this fashion would be able to move and act quite freely as a positive charge carrier, not tied to its site of origin. To further relate our theoretical results to the experimental observations, we also calculate the optical absorption spectra of doped PCPDT-BT. Taking into account species with different amounts of protonation, our results help to explain the experimentally observed doping induced spectral features in detail. We believe that the results reported in the present work will help to further clarify the microscopic mechanisms discussed in the literature to explain the potentially high efficiency achieved by doping OSC materials with Lewis acids.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the Deutsche Forschungsgemeinschaft through project SCHU 1980/13 and through the Heisenberg program (No. 270619725). A grant for computing time at the Paderborn Center for Parallel Computing (PC2) is gratefully acknowledged.References
- P. Beyer, D. Pham, C. Peter, N. Koch, E. Meister, W. Brütting, L. Grubert, S. Hecht, D. Nabok, C. Cocchi, C. Draxl and A. Opitz, Chem. Mater., 2019, 31, 1237–1249 CrossRef CAS.
- A. I. Hofmann, R. Kroon, S. Zokaei, E. Järsvall, C. Malacrida, S. Ludwigs, T. Biskup and C. Müller, Adv. Electron. Mater., 2020, 6, 2000249 CrossRef CAS.
- K.-H. Yim, G. L. Whiting, C. E. Murphy, J. J. M. Halls, J. H. Burroughes, R. H. Friend and J.-S. Kim, Adv. Mater., 2008, 20, 3319–3324 CrossRef CAS.
- E. E. Aziz, A. Vollmer, S. Eisebitt, W. Eberhardt, P. Pingel, D. Neher and N. Koch, Adv. Mater., 2007, 19, 3257–3260 CrossRef CAS.
- B. Lüssem, M. Riede and K. Leo, Phys. Status Solidi A, 2013, 210, 9–43 CrossRef.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS PubMed.
- D. T. Duong, C. Wang, E. Antono, M. F. Toney and A. Salleo, Org. Electron., 2013, 14, 1330–1336 CrossRef CAS.
- D. Di Nuzzo, C. Fontanesi, R. Jones, S. Allard, I. Dumsch, U. Scherf, E. von Hauff, S. Schumacher and E. Da Como, Nat. Commun., 2015, 6, 6460 CrossRef PubMed.
- J. Li, G. Zhang, D. M. Holm, I. E. Jacobs, B. Yin, P. Stroeve, M. Mascal and A. J. Moulé, Chem. Mater., 2015, 27, 5765–5774 CrossRef CAS.
- P. Pingel, M. Arvind, L. Kölln, R. Steyrleuthner, F. Kraffert, J. Behrends, S. Janietz and D. Neher, Adv. Electron. Mater., 2016, 2, 1600204 CrossRef.
- B. Yurash, D. X. Cao and V. Brus, et al., Nat. Mater., 2019, 18, 1327–1334 CrossRef CAS PubMed.
- D. X. Cao, D. Leifer and V. V. Brus, et al., Mater. Chem. Front., 2020, 4, 3556–3566 RSC.
- S. Kee, M. A. Haque and Y. Lee, et al., ACS Appl. Energy Mater., 2020, 3, 8667–8675 CrossRef CAS.
- E. H. Suh, J. G. Oh, J. Jung, S. H. Noh, T. S. Lee and J. Jang, Adv. Energy Mater., 2020, 10, 2002521 CrossRef CAS.
- P. S. Marqués, G. Londi, B. Yurash, T.-Q. Nguyen, S. Barlow, S. R. Marder and D. Beljonne, Chem. Sci., 2021, 12, 7012–7022 RSC.
- M. Zamadar, S. Asaoka, D. C. Grills and J. R. Miller, Nat. Commun., 2013, 4, 2818 CrossRef.
- M. Witte, U. Gerstmann, A. Neuba, G. Henkel and W. G. Schmidt, J. Comput. Chem., 2016, 37, 1005–1018 CrossRef CAS PubMed.
- F. Neese, Coord. Chem. Rev., 2009, 253, 526–563 CrossRef CAS.
- C. Blanchet-Boiteux and J. Mouesca, J. Am. Chem. Soc., 2000, 122, 861–869 CrossRef CAS.
- E. Ruiz, A. Rodríguez-Fortea, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 2003, 24, 982–989 CrossRef CAS PubMed.
- S. Herres-Pawlis, P. Verma, R. Haase, P. Kang, C. T. Lyons, E. C. Wasinger, U. Flörke, G. Henkel and T. D. P. Stack, J. Am. Chem. Soc., 2009, 131, 1154–1169 CrossRef CAS PubMed.
- C. J. Calzado, J. Cabrero, J. P. Malrieu and R. Caballol, J. Chem. Phys., 2002, 116, 3985–4000 CrossRef CAS.
- M. Zbiri, S. Saha, C. Adhikary, S. Chaudhuri, C. Daul and S. Koner, Inorg. Chim. Acta, 2006, 359, 1193–1196 CrossRef CAS.
- D. Fazzi, S. Fabiano, T.-P. Ruoko, K. Meerholza and F. Negri, J. Mater. Chem. C, 2019, 7, 12876–12885 RSC.
- C.-D. Dong and S. Schumacher, J. Phys. Chem. C, 2019, 123, 30863–30870 CrossRef CAS.
- C.-D. Dong and S. Schumacher, J. Mater. Chem. C, 2020, 8, 11929–11935 RSC.
- C.-D. Dong and S. Schumacher, J. Phys. Chem. C, 2021, 125, 21824–21830 CrossRef CAS.
- T. Bathe, C.-D. Dong and S. Schumacher, Microscopic Study of Molecular Double Doping, J. Phys. Chem. A, 2022, 126, 2075–2081 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian ∼ 16 Revision C.01, Gaussian Inc, Wallingford CT, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Theor. Comput. Chem., 2012, 11, 163–183 CrossRef CAS.
- M. Anderson, C. Ramanan, C. Fontanesi, A. Frick, S. Surana, D. Cheyns, M. Furno, T. Keller, S. Allard, U. Scherf, D. Beljonne, G. D'Avino, E. von Hauff and E. Da Como, Phys. Rev. Mater., 2017, 1, 055604 CrossRef.
- B. Neelamraju, K. E. Watts, J. E. Pemberton and E. L. Ratcliff, J. Phys. Chem. Lett., 2018, 9, 6871–6877 CrossRef CAS PubMed.
- C. Wiebeler, R. Tautz, J. Feldmann, E. von Hauff, E. Da Como and S. Schumacher, J. Phys. Chem. B, 2013, 117, 4454–4460 CrossRef CAS PubMed.
- F. S. U. Fischer, K. Tremel, A.-K. Saur, S. Link, N. Kayunkid, M. Brinkmann, D. Herrero-Carvajal, J. T. López Navarrete, M. C. Ruiz Delgado and S. Ludwigs, Macromolecules, 2013, 46, 4924–4931 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |