DOI:
10.1039/D2RA02049A
(Paper)
RSC Adv., 2022,
12, 13975-13984
Transition metal decorated phthalocyanine as a potential host material for lithium polysulfides: a first-principles study†
Received
30th March 2022
, Accepted 29th April 2022
First published on 11th May 2022
Abstract
The shuttle effect caused by the soluble long-chain lithium polysulfides greatly hinders the practical application of lithium–sulfur (Li–S) batteries. Therefore, the introduction of suitable anchoring materials is more effective to mitigate this problem. Transition metal phthalocyanines (TMPc) are regarded as a new class of sulfur host materials. Here, 4d transition metal (Y, Zr, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Cd) decorated phthalocyanines are designed and systematically researched for the performance analysis of anchoring S8/LiPSs by first-principles calculations. The results reveal that the bonding strength of LiPSs can be well adjusted by introducing suitable 4d transition metals into the phthalocyanine structure. The electronic structure analysis indicates the formation of TM–S bonds between the TMPc substrate materials and the LiPSs, which is essential to weaken the Li–S bonds and hence slow down the shuttle effect of LiPSs. ZrPc and NbPc both exhibit excellent potential and thermal stability for facilitating the conversion of LiPSs, as well as a better promoting effect for the sulfur reduction reactions (SRR) with a reduced Gibbs free energy in the rate-determining step (*Li2S2 → *Li2S) during the discharge reaction process. These findings in our work may encourage further experimental and theoretical research for anchoring LiPSs with TMPc as a host material.
1. Introduction
Diverse electrical energy storage systems are closely related to human society, such as rechargeable batteries with competitive costs, high specific energy power and long cycling life.1 These batteries play a leading role in the fields of portable electronic devices, electric vehicles, smart power grids, etc. Among the various batteries, lithium–sulfur (Li–S) batteries have attracted intense attention due to their exceptionally high theoretical capacity (1675 mA h g−1), energy density (2600 W h kg−1) and environmental friendliness.2,3 Compared with Li-ion batteries, Li–S batteries are expected to become one of the most promising alternatives for next-generation energy storage systems. Despite their great promise, the development of Li–S batteries is still hindered by some problems such as low active materials utilization, poor electrode stability, poor cycle life, and low Coulomb efficiency. The root cause of these matters lies in the low ion/electronic conductivity of elemental sulfur and discharge products Li2S/Li2S2 in the charge/discharge process, as well as the soluble long-chain polysulfides (Li2S4, Li2S6 and Li2S8) dissolved in the electrolyte solvent from the positive electrode (known as the “shuttle effect”).4–6 Therefore, solving these issues is particularly important for the design and the commercialization of Li–S batteries.
In the early days of Li–S batteries exploration, various two-dimensional (2D) materials with large special surface area and multiple adsorption sites, such as carbon nanotubes (CNTs),7 graphene sheets8 have been extensively used as the possible sulfur hosts to improve the conductivity of cathode and physically confine the shuttle effect of LiPSs. Among them, carbon-based materials can not only serve as a porous framework for adsorbing LiPSs, but also greatly improve the electronic conductivity of electrode materials. Therefore, the electrochemical performance of Li–S batteries has indeed been improved to a certain extent.9 However, until now, researchers have generally believed that the weak interaction between carbon-based materials and LiPSs is not sufficiently enough to prevent LiPSs from being transported into the electrolyte under high concentration gradients and internal electric fields.10 This phenomenon reveals that carbon-based materials alone cannot be used as a perfect matrix for promoting the overall performance of Li–S batteries. As an alternative, it has been proposed to anchor the LiPSs through relatively strong chemical interactions to enhance the capture capability11 and strengthen the binding ability between the substrate materials and LiPSs species.12 This method can effectively inhibit the dissolution of LiPSs into the electrolyte and is essential for improving the electrochemical performance of Li–S batteries. In the past few years, researchers have discovered that anchoring a single transition metal on a substrate material is an effective solution pattern, which can achieve a strong anchoring and catalytic effect on the basis of the Li anode and the S cathode.13 Moreover, it has been found that the interaction between substrate materials and adsorbents is mainly due to the transfer of charge between Li/S atoms in LiPSs and the substrate materials. For instance, Wu et al.14 investigated the InP3 monolayer as a viable substrate material for anchoring LiPSs. Their research showed that during the adsorption process of LiPSs, electron accumulation mainly occurs between Li and P atoms, and electron depletion mainly occurs around S atoms. Zheng et al.15 discussed the use of non-metallic monolayer materials as anchoring materials in Li–S batteries for the adsorption of LiPSs. The remarkable performance of C3N4/C2N is attributed to the bonding of Li–N/C–S and charge transfer. It can be seen that substrate materials with good physical and chemical properties are extremely important for the research of Li–S batteries.
Recently, materials based on phthalocyanine structure have received widespread discussion and research in the field of energy storage. Phthalocyanine (Pc) is a macrocyclic aromatic compound with a chemical formula of C32H16N8.16 It has four isoindole subunits connected by a methine or azamethine bridge. The characteristics of this structure make it having excellent physical and chemical properties, flexible structure adjustability. Benefitting from the central cavity of tetranitrogen coordination, it can provide a perfect position to anchor a single transition metal atom.17 Therefore, it can form transition metal phthalocyanine (TMPc) structures with the doping of various transition metal ions, making them natural single-atom catalysts, which are widely used in various catalytic reactions, such as oxygen reduction reaction (ORR),18 nitrogen reduction reaction (NRR)19 and carbon dioxide reduction reaction (CO2RR).20 At present, some investigations have successfully synthesized TMPc materials with excellent conductivity, which can be directly applied to electrochemical catalysis reactions and can meet the needs of the reaction by flexibly adjusting the coordination of transition metal atoms. Cheviri et al.21 researched the adsorption of LiPSs on the OH-functionalized CoPc. Attributed to the existence of the Li–O bonds and the Li–N bonds, the adsorption of LiPSs was significantly stronger than that of carbon-based materials, indicating that CoPc is a good substrate material. Kim et al.22 adopted cobalt(II)-centered fluorinated phthalocyanine (F–Co(II)Pc) as the positive electrode of a Li–S battery, which effectively promoted the conversion reaction between LiPSs due to the existence of catalytic site on Co(II) active center. In addition, during the charging process, the strong combination of Co(II) and S atoms in Li2S weakens the Li–S bonds, resulting in the lowest dissociation activation energy of Li2S on Co(II)Pc. By means of coupling FePc and rGO, Ma et al.23 designed a bi-functional anchored conversion composite material. This material can effectively capture the dissolved LiPSs, and the interconnected rGO substrate can also facilitate the transfer of ions and electrons during the reaction, thus speeding up the redox reaction of the lithium–sulfur battery. Yu et al.24 investigated the catalytic properties of different 3d transition metals anchored on the phthalocyanine structure and forecasted the potential application of TMPc as a battery cathode material in Li–S batteries. Besides, it is reported that the cavity of H2Pc can be occupied by about 70 different metal ions with excellent chemical stability.25,26 In view of the large family of TMPc members and the transition metal atoms, a systematical investigation of the promising transition metal phthalocyanine materials is necessary for the development and application of TMPc-based host materials for LiPSs.
Inspired by the work of predecessors,27–29 herein, we systematically investigated the anchoring capability and catalytic performance of all 4d transition metal (TM = Y, Zr, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Cd) phthalocyanines (TMPc) on a series of polysulfides S8/LiPSs. By first-principles calculations, a comprehensive picture of the structures and electronic properties of TMPc-S8/LiPSs complexes were provided. Overall, this work indicates that the binding ability of S8/LiPSs can be well regulated by the appropriate 4d TMPc structures, and Zr/NbPc exhibits the excellent potential (the binding energy of ZrPc and NbPc is 5.18 eV and 4.63 eV) for inhibiting the shuttle effect in Li–S batteries. In addition, the results of ab initio molecular dynamics (AIMD) simulations show that most of the TMPc structures provide a theoretical basis for the feasibility of the experiment. Our simulations not only are helpful for the designation of TMPc-based hosting materials for LiPSs, but also can attract further experimental research.
2. Computational methods
2.1 Computational details
All spin-polarized density functional theory (DFT) calculations for all TMPc structures were performed by the Vienna Ab initio Simulation Package (VASP) code.30,31 The projector augmented wave (PAW) method was employed for describing the ions–electron interaction and the exchange correlation functional within the generalized gradient approximation (GGA) parameterized by Perdew, Burke, and Enzerof (PBE) was used in all DFT calculations.32,33 In addition, the DFT-D3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 correction with Grimme scheme was applied to describe van der Waals interaction between the adsorbates and adsorbents. The cutoff energy was set to 520 eV for the plane-wave basis. The convergence standards for the energy and the residual force were set to 10−5 eV and 0.02 eV Å−1, respectively. The Brillouin zones were sampled with a 2 × 2 × 1 Monkhorst-Pack k-point grid for the H2Pc and all the TMPc structures. The crystal orbital Hamilton populations (COHP)35,36 implemented in LOBSTER37 was applied to describe the bonding/anti-bonding state between atoms.
34 correction with Grimme scheme was applied to describe van der Waals interaction between the adsorbates and adsorbents. The cutoff energy was set to 520 eV for the plane-wave basis. The convergence standards for the energy and the residual force were set to 10−5 eV and 0.02 eV Å−1, respectively. The Brillouin zones were sampled with a 2 × 2 × 1 Monkhorst-Pack k-point grid for the H2Pc and all the TMPc structures. The crystal orbital Hamilton populations (COHP)35,36 implemented in LOBSTER37 was applied to describe the bonding/anti-bonding state between atoms.
2.2 Binding energy
The binding strength between the S8/LiPSs and the H2PC/TMPc substrates was evaluated by the calculation of the binding energy (Eb) according to the following equation:38,39| | |
Eb = (Esubstrate + Eadsorbate) − Etotal
| (1) |
where Etotal, Esubstrate, and Eadsorbate are the total energies of H2Pc/TMPc–S8/LiPSs complexes, H2Pc/TMPc, and S8/Li2Sx (x = 1, 2, 4, 6, 8), respectively. The more positive the value of Eb, the stronger the interaction between the corresponding substrates and the adsorbates.
2.3 Charge density difference
The charge density difference was defined as follows:40| | |
Δρ = ρtotal − ρsubstrate − ρadsorbate
| (2) |
wherein ρtotal, ρsubstrate and ρadsorbate stand for the charge densities of H2Pc/TMPc–S8/LiPSs complexes, H2Pc/TMPc, and S8/LiPSs, respectively.
2.4 Free energy change of elementary reaction
The following eqn (3)–(8) described the overall discharge reactions or sulfur reduction reactions (SRRs) process in detail, where S8/LiPSs molecules with “*” marked indicate that they were adsorbed on the TMPc/H2Pc substrate materials.24 The energy of a single Li ion and an electron (Li+ + e−) pair was treated as the energy of a crystalline Li atom.| | |
S8 + 16Li+ → S8* + 16Li+ (ΔG1)
| (3) |
| | |
S8* + 16Li+ + 2e− → Li2S8* + 14Li+ (ΔG2)
| (4) |
| | |
Li2S8* + 14Li+ + 2e− → Li2S6* + Li2S2 + 12Li+ (ΔG3)
| (5) |
| | |
Li2S6* + Li2S2 + 12Li+ + 2e− → Li2S4* + 2Li2S2 + 10Li+ (ΔG4)
| (6) |
| | |
Li2S4* + 2Li2S2 + 10Li+ + 2e− → Li2S2* + 3Li2S2 + 8Li+ (ΔG5)
| (7) |
| | |
Li2S2* + 3Li2S2 + 8Li+ + 8e− → Li2S* + 7Li2S (ΔG6)
| (8) |
The Gibbs free energy (ΔG) of the above SRRs process was calculated as follows:41
| | |
ΔG = ΔEDFT + ΔEZPE − TΔS − neU + ΔGpH
| (9) |
Here ΔEDFT is the electronic energy difference before and after adsorption of reaction intermediates, ΔEZPE is the zero-point energy difference between the LiPSs and the adsorbed state, T is the room temperature (298.15 K), ΔS is the entropy change, n and U are the number of electrons transferred and the effect of an applied bias. ΔGpH is the correction of pH, which can be obtained via ΔGpH = 2.303 × kBT × pH, where kB is the Boltzmann constant and the calculations presented here are for pH = 0. For the battery systems, the ΔEZPE and TΔS can be ignored in the calculation, therefore, when U = 0 V, ΔG ≈ ΔEDFT. In order to gain further insight into the catalytic performance of the charging reaction (reverse reaction of SRRs), we carried out the transition state (TS) calculations for some structures. The climbing image nudged elastic band (CI-NEB) method42 is used to calculate the dissociation energy barrier of Li2S, and the cut-off energy, SCF convergence and max force are set to 520 eV, 1.0 × e−7 eV per atom and 0.03 eV Å−1, respectively.
2.5 Ab initio molecular dynamic (AIMD) simulation
Since the structure optimization process of the VASP calculation program is an optimization method using interatomic statics under standard 0 K conditions, it is not sufficient to understand the chemical reaction stability of the 4d TMPc structures in Li–S batteries. While density functional (DFT) ab initio molecular dynamics (AIMD) can reliably describe the time-evolution of solid-state systems directly embedded in real-world environments,43 its computational strategy for simulated annealing is an upgrade from the standard 0 K DFT basic computation. Therefore, in order to check the thermodynamic stability of all TMPc structures, calculations of AIMD simulations were implemented. The optimized TMPc structures were annealed with 1000 steps to increase the temperature to the target temperature of 400 K (time step is 1 fs, temperature from 100 K to 400 K), and then 4 ps equilibrium simulation calculation was performed at 400 K with a time step of 1 fs. Temperature control is achieved through the Nosé thermostat model.
3. Results and discussions
3.1 Structural stability analysis
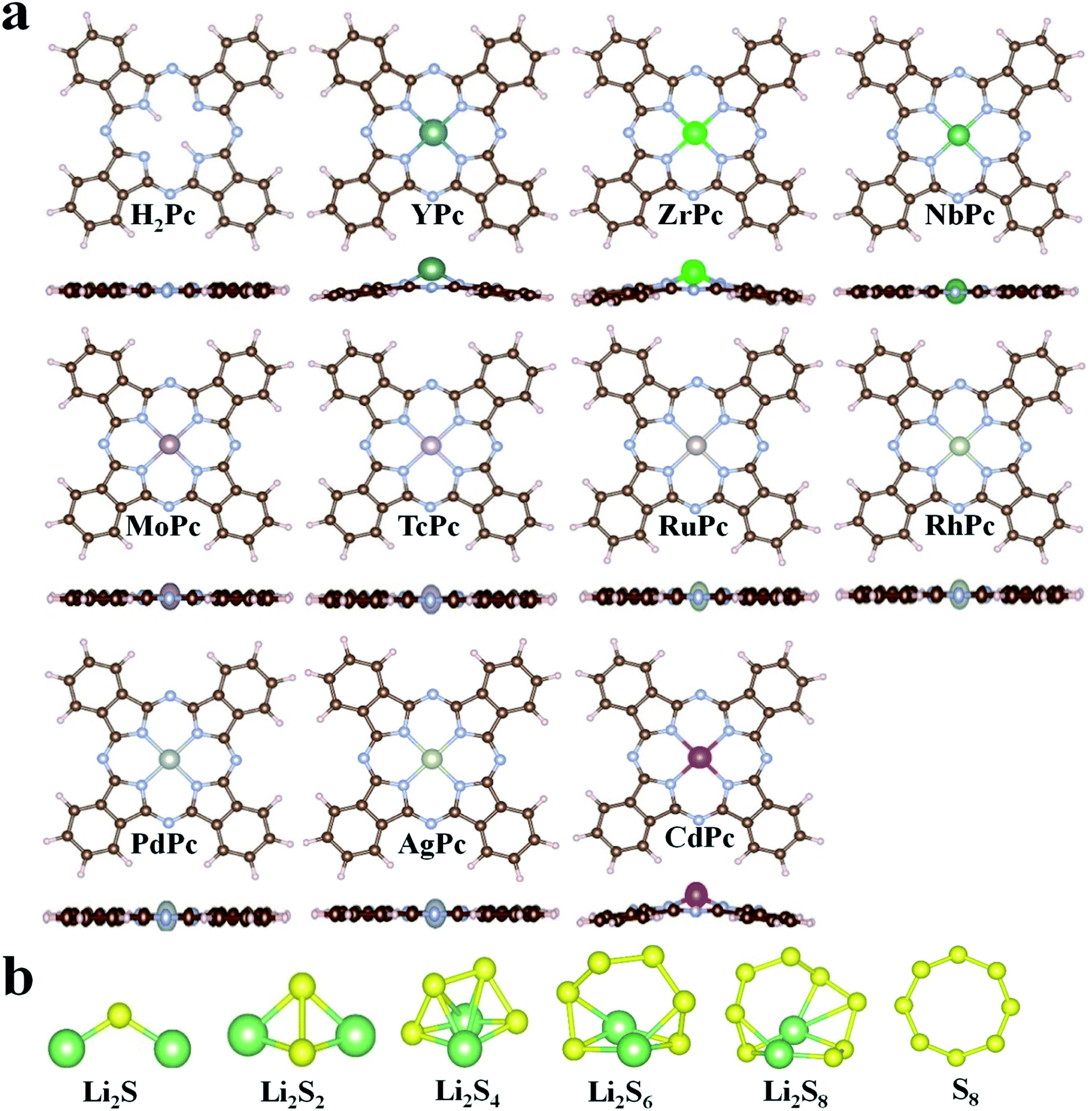
In this study, all 4d transition metal phthalocyanine (TMPc) structures were considered for the performance analysis for anchoring S8/LiPSs. The optimized structures of H2Pc/TMPc (TM = Y, Zr, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Cd) and S8/LiPSs are displayed in Fig. 1a and b. As illustrated in Fig. 1a, the non-metal-doped phthalocyanine (H2Pc) structure composed of four isoindole groups and an inner porphyrazine ring is a large conjugated system with 18 electrons and is a completely artificially synthesized compound. Theoretically, all transition metal (TM) ions can be used to replace the two hydrogen atoms situated in the center of H2Pc structure, thereby forming a stable TMPc structure25 (except AgPc with a positive formation energy and the details are described in Table S1†) for better anchoring S8/LiPSs. Most of the optimized TMPc structures (except YPc, ZrPc, CdPc) can well maintain the initial planar structure of H2Pc. The large radius of metal atoms Y, Zr and Cd result in a non-planar configuration and the introduced metal atoms are above the macrocyclic plane. Fig. 1b exhibits the fully relaxed S8/Li2Sx (x = 1, 2, 4, 6, 8) structures, which are consistent with the previous investigations.44,45
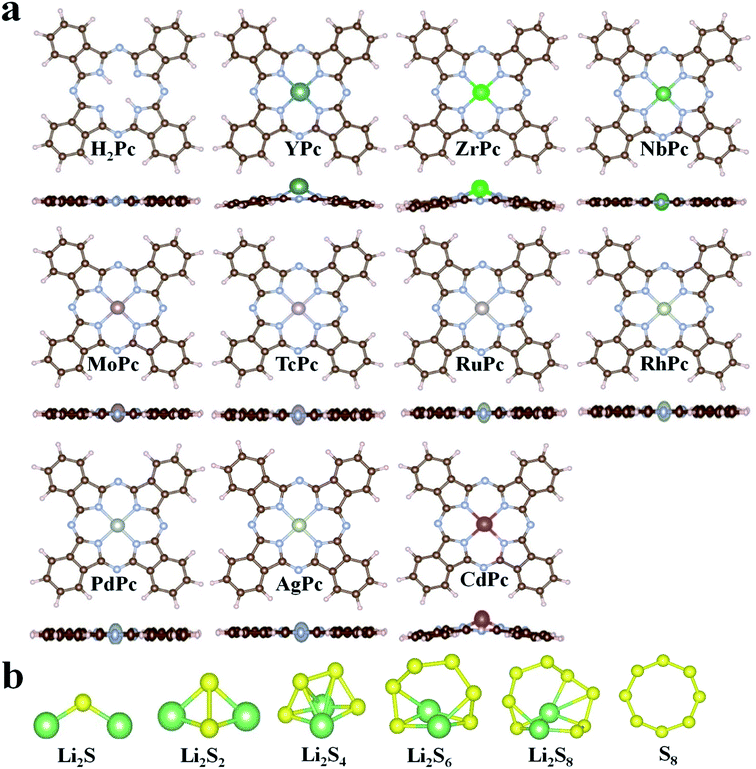 |
| | Fig. 1 (a) The schematic diagram of the optimized H2Pc24 and TMPc structures. (b) The fully relaxed Li2S, Li2S2, Li2S4, Li2S6, Li2S8 and S8 molecules. The N, C, H, S and Li are represented by silver, brown, lavender blush, yellow and green balls, respectively. | |
3.2 Analysis of anchoring performance of S8/LiPSs adsorbed on H2Pc/TMPc substrates
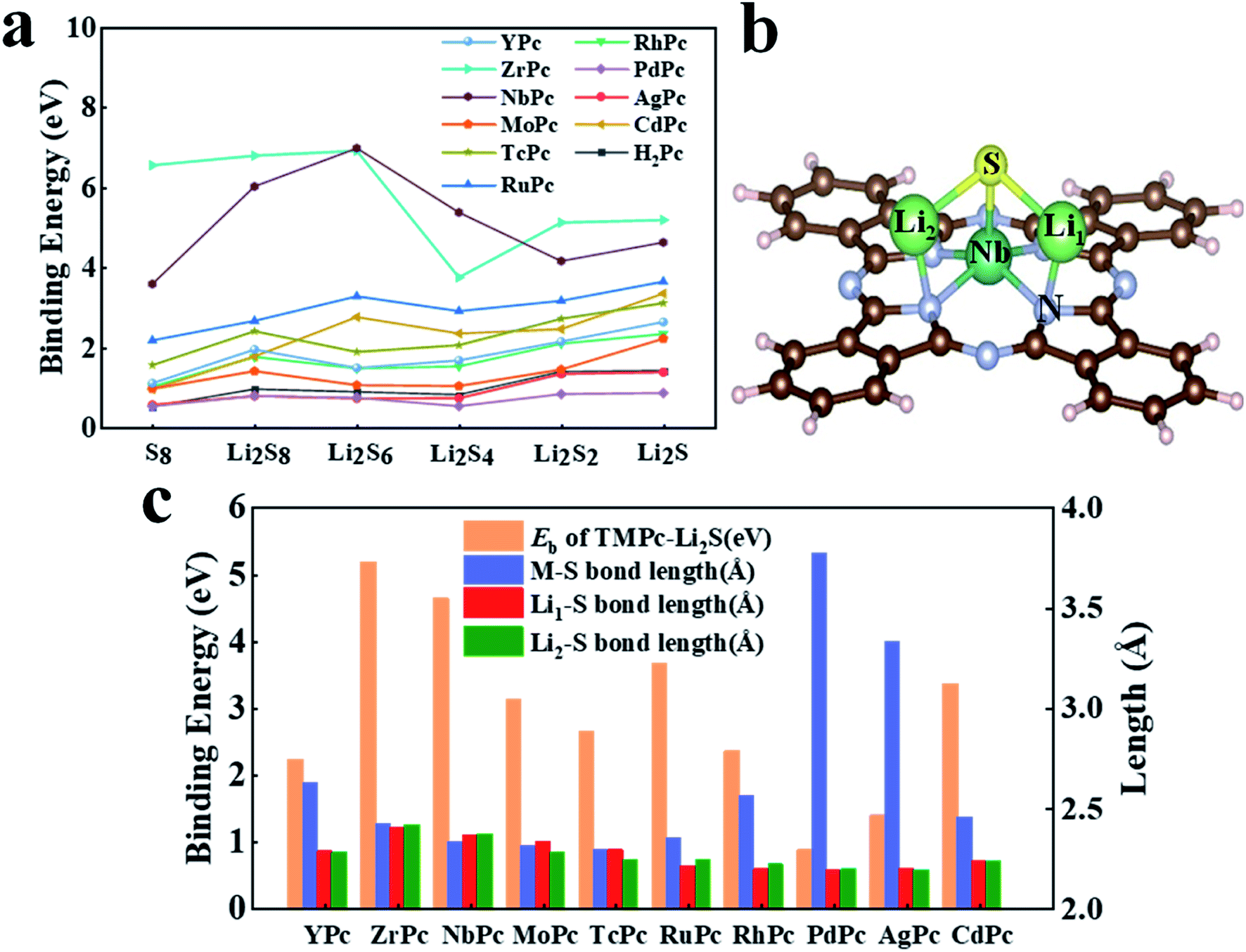
Li–S battery is a rechargeable battery with elemental sulfur as the positive electrode and metal lithium as the negative electrode. Because of its wide range of material sources, high energy density, large theoretical capacity and so on, it has received extensive attention and research. However, the biggest technical obstacle of Li–S battery studies lies in the “shuttle effect” of LiPSs46 (especially Li2S4, Li2S6 and Li2S8), which simultaneously reduces the performance of the cathode and anode. In the research of this article, we fully investigated the interaction between various H2Pc/TMPc substrates with S8/LiPSs clusters. The binding energy results of H2Pc/TMPc adsorbing S8/LiPSs are summarized and displayed in Fig. 2a. It can be intuitively seen that most of TMPc structures, especially ZrPc and NbPc, exhibit much better anchoring performance for S8/LiPSs than the initial phthalocyanine structure (H2Pc), revealing that the shuttle effect of LiPSs can be effectively suppressed by the transition-metal adjusted Pc structure. The binding strength between most of the soluble LiPSs (e.g., Li2S4, Li2S6 and Li2S8) and H2Pc/TMPc substrates is weaker than that of insoluble LiPSs (e.g., Li2S and Li2S2), and the binding energy of S8 is the minimum. The stable structures of S8/LiPSs species adsorbed on all the H2Pc/TMPc substrate materials could be found in Fig. S1 and S2.† Fig. 2b is a schematic diagram of the optimized structure of Li2S molecules adsorbed on the NbPc substrate. It can be seen that lithium-atom (Li1/Li2) are easier to bond with nitrogen-atom forming Li–N bonds, while TM (Nb) atoms tend to combine with sulfur-atom to form TM(Nb)–S bonds. The two newly formed bonds (Li–N and TM–S) are of vital importance in anchoring LiPSs and the interaction of TM–S bonds play a dominating role. In addition, the strength of the interaction between TMPc and Li2S can also be reflected by the bond lengths of TM–S and Li–S bonds. The columns of different colors in Fig. 2c show the results of our statistics. The red, green, blue and orange columns represent the binding energy of TMPc adsorbing Li2S, the bond lengths of TM–S, Li1–S and Li2–S (denoted as Li1/2–S bonds), respectively. As shown in Fig. 2c, there is a correlation between the binding strength of TMPc adsorbing Li2S and the TM–S bond lengths. The binding energies of PdPc, AgPc and YPc to Li2S are relatively small among the TMPc structures, and the TM–S bond lengths after structure optimization is obviously larger than other structures. Among them, the Pd–S bond length of PdPc–Li2S is the longest one (3.77 Å) and its binding energy is also the smallest (0.86 eV). Moreover, the Li1/2–S bond lengths of all TMPc structures after adsorption of Li2S molecules have varying degrees of elongation, and the longer Li1/2–S bond corresponds to a stronger binding energy. Taking the ZrPc–Li2S and PdPc–Li2S systems as examples, the Li1/2–S bond lengths of the Li2S molecules before and after adsorption are both 2.09 Å, while the Li1–S bond length in the two systems increased to 2.39 Å and 2.18 Å, respectively. Furthermore, the bond length of Li2–S increased to 2.41 Å and 2.19 Å, and the corresponding binding energies are 5.18 eV and 0.86 eV. In short, in all the considered TMPc structures tuned by the 4d transition metal ions, ZrPc and NbPc have excellent anchoring properties for S8/LiPSs, which shows that the two structures can effectively suppress the shuttle effect in Li–S batteries.
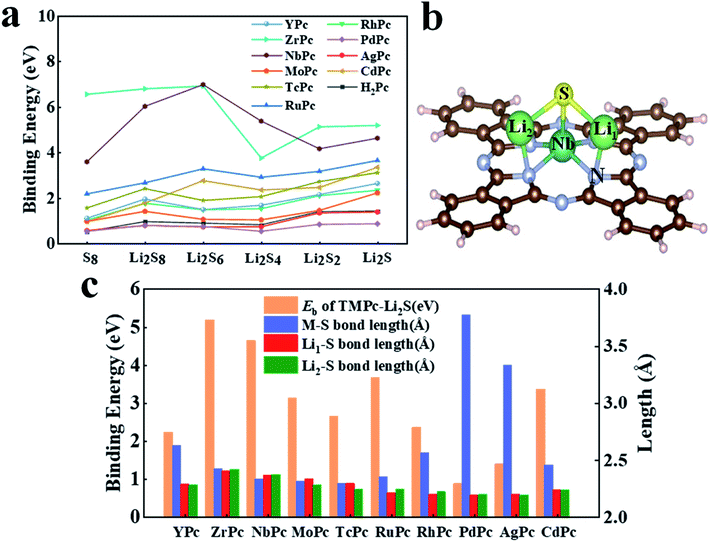 |
| | Fig. 2 (a) Binding energy of S8/Li2Sx (x = 1, 2, 4, 6, 8) adsorbed on different H2Pc24/TMPc surfaces; (b) the schematic diagram of NbPc–Li2S binding system with atomic label (Li1, Li2, S, Nb and N atoms); (c) the binding energy, TM–S bond lengths and Li1/2–S bond lengths histogram of different TMPc–Li2S systems. | |
3.3 Electronic structure analysis of TM (Zr, Nb, Ru, Pd)Pc anchoring S8/LiPSs
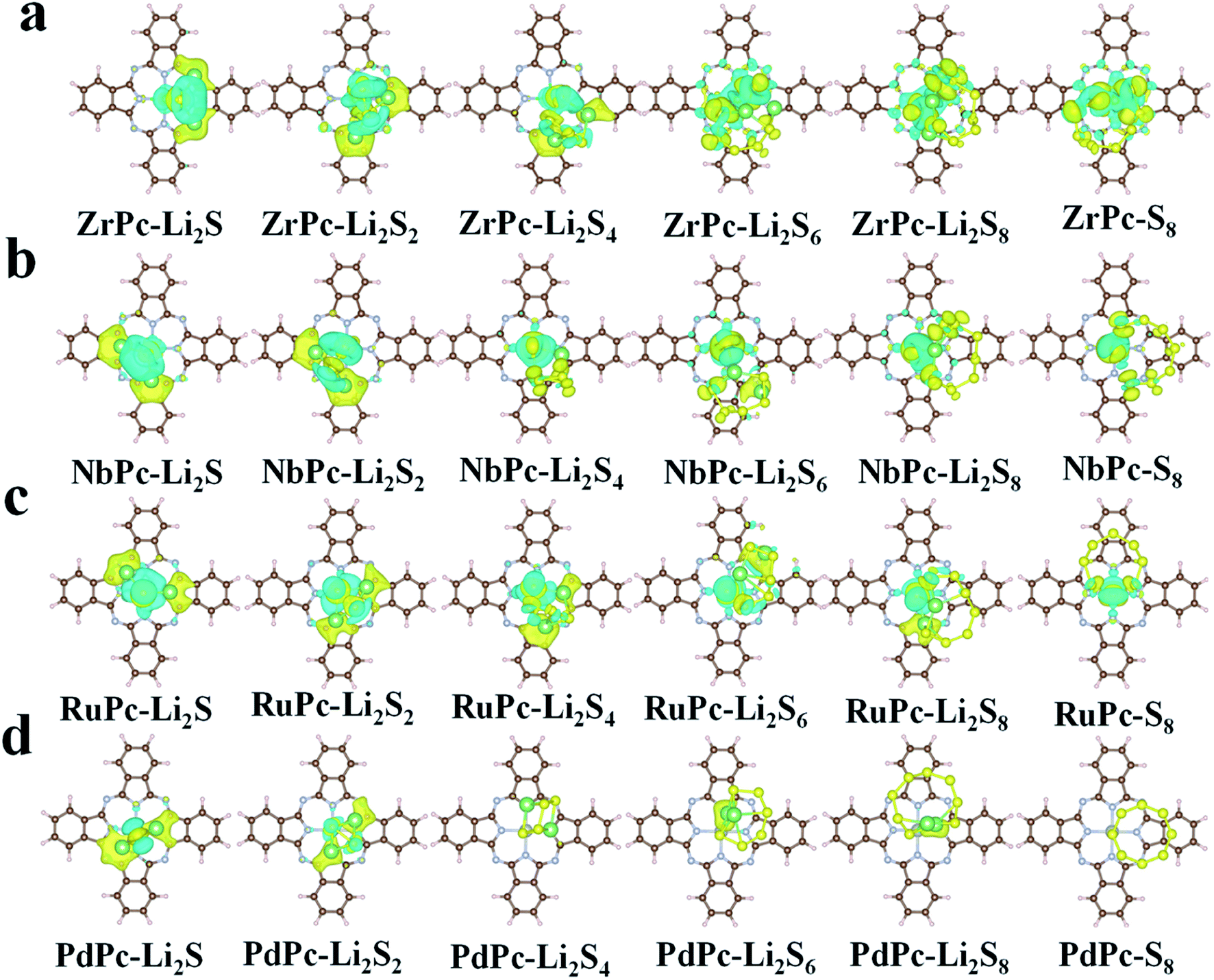
In order to further understand the potential binding mechanism between S8/LiPSs and TMPc substrates, taking ZrPc, NbPc, RuPc and PdPc systems as examples, we studied the charge density difference before and after adsorption. As shown in Fig. 3, the electron accumulation is displayed in the yellow area, while the electron consumption is displayed in the cyan area, respectively. The charge density difference diagrams clearly exhibit the differentiation in charge transfer between each structure. It is worth northing that the charge transfer between PdPc and S8/LiPSs is significantly less than that between Zr(Nb, Ru)Pc and S8/LiPSs, which is highly consistent with the results of binding energy. Through comparative analysis, the charge transfer of ZrPc–Li2S, NbPc–Li2S and RuPc–Li2S systems mainly occurs in the TM(Zr, Nb, Ru)–S bonds and Li–N bonds. However, for the PdPc–Li2S system, the charge mainly accumulates between Li atom and PdPc substrate rather than the Pd/S atoms (Fig. 3d). So far, the above results show that Zr–S, Nb–S and Ru–S bonds are much stronger than the Pd–S bond. In addition, the binding energy of PdPc–Li2S is similar to that of H2Pc–Li2S (Fig. 2a), indicating that the interaction of lithium atoms and nitrogen atoms is not sufficient to anchor S8/LiPSs clusters. In general, a strong TM–S bond is essential to alleviate the shuttle effect of LiPSs.
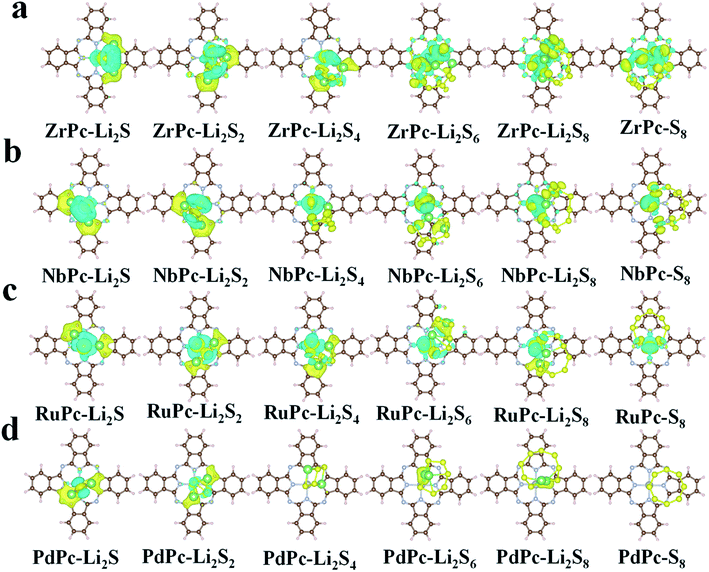 |
| | Fig. 3 Charge density differences of S8/LiPSs adsorbed on (a) ZrPc, (b) PdPc, (c) RuPc and (d) PdPc with an iso-surface value set to 0.002 e Å−3. The yellow area and cyan area in the figure indicate charge accumulation and depletion, respectively. | |
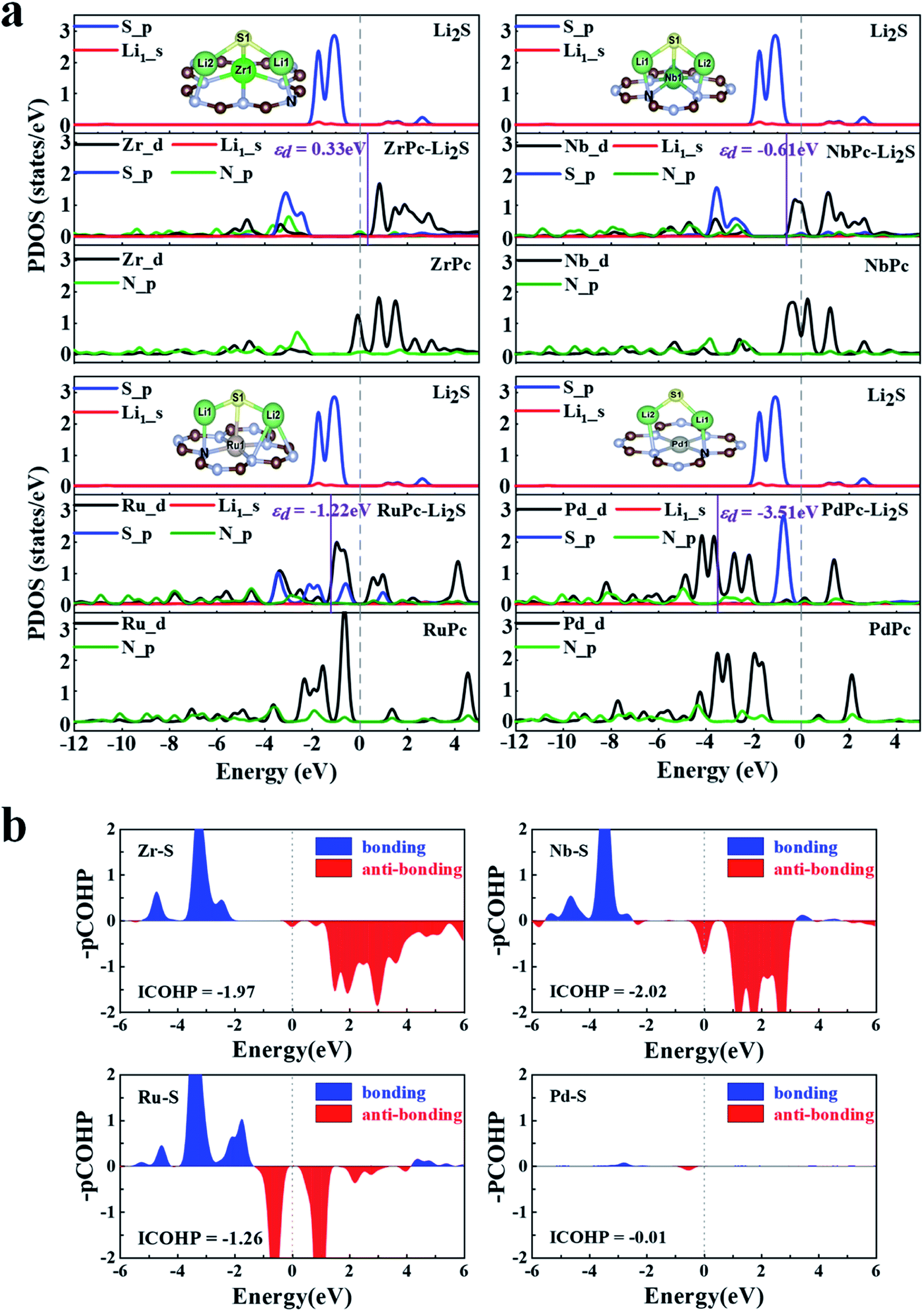
To reveal the mechanisms of TMPc substrates anchoring S8/LiPSs, we calculated the projected density of states (PDOS) of different TM(Zr, Nb, Ru and Pd)Pc before and after the adsorption of Li2S and the corresponding energy of the d-band center (εd), which are presented in Fig. 4. Focusing on the TM–S and Li1–N (Fig. 2b) bonds, the PDOS of the 4d orbitals of Zr, Nb, Ru and Pd (Zr_d, Nb_d, Ru_d and Pd_d) before adsorption and the 2p orbitals of N-atom (N_p) in the Zr(Nb, Ru, Pd)Pc structures are drawn. As shown at the bottom of the PDOS diagrams in Fig. 4a, ZrPc, NbPc and RuPc show a large overlap between the d orbital and p orbital in a wide energy range, which means a stronger bonding between Zr(Nb, Ru) and N atoms. The middle part in Fig. 4a show that the adsorption of Li2S induces a strong hybridization of the 2p orbital (S_p) and the d orbital of Zr/Nb, revealing a stronger interaction between Zr–S and Nb–S. Meanwhile, the PDOSs of S_p and Zr/Nb_d split into two parts, that is the bonding state and the anti-bonding state, and are distributed on both sides of the Fermi level. Compared with PdPc and Li2S alone, the PDOS changes of Pd_d and S_p in PdPc–Li2S system are very small, indicating a much weaker interaction between Pd and S atom. The analysis from the PDOS plots reveals that interactions between the TM_d and S_p orbitals engender the strong TM–S interactions, such as the Zr–S and Nb–S bonds. In addition, the energy of the d-band center can be approximately employed to describe the adsorption energy of the adsorbate. And the closer the value of the d-band center is to the Fermi level, the stronger the adsorption effect on S8/LiPSs.47 Therefore, we calculated the energy of the d-band center in the ZrPc–Li2S, NbPc–Li2S, RuPc–Li2S, and PdPc–Li2S systems. As shown by the purple solid line in Fig. 4, the corresponding energy values of the d-band center (εd) are 0.33 eV (Zr_d), −0.61 eV (Nb_d), −1.22 eV (Ru_d) and −3.51 eV (Pd_d), respectively. Obviously, the energy of the d-band center of the ZrPc–Li2S and NbPc–Li2S systems is relatively closer to the Fermi level, indicating that the effect of Zr/NbPc anchoring LiPSs is indeed much stronger than other TMPc structures.
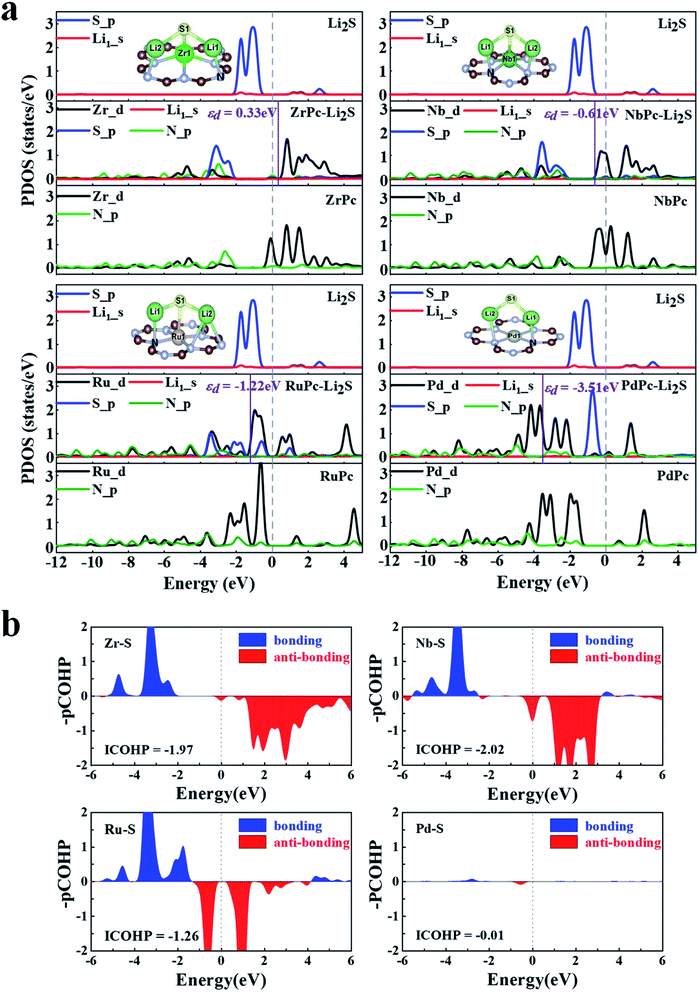 |
| | Fig. 4 (a) The partial density of states (PDOSs) plots and d-band central energies (εd) for ZrPc–Li2S, NbPc–Li2S, RuPc–Li2S and PdPc–Li2S binding systems, where vertical dashed and solid lines represent the Fermi level and the energy of the d-band center, respectively. (b) The projected crystal Hamilton population (pCOHP) and corresponding integrated value (ICOHP) of Zr–S bond in ZrPc–Li2S, Nb–S bond in NbPc–Li2S, Ru–S bond in RuPc–Li2S and Pd–S bond in PdPc–Li2S. | |
The COHP analysis is applied to explain the change of TM–S bonding strength. Hence COHP analysis of the TM–S bonds for ZrPc–Li2S, NbPc–Li2S, RuPc–Li2S and PdPc–Li2S systems are calculated and plotted in Fig. 4b. The Zr/Nb–S bonds have more electronic states near the Fermi level than the Ru/Pd–S bonds, as shown in Fig. 4b. The above findings indicate that the Zr/Nb–S bonds in Zr/NbPc–Li2S systems have a much stronger interaction than the Ru/Pd–S bonds of Ru/PdPc–Li2S systems. In order to obtain a quantitative and in-depth understanding of the interaction between TM–S bonds, the integral value of COHP (ICOHP) is further calculated. A more negative value of the ICOHP indicates that the stronger interaction of the TM–S bonds. The ICOHP results of different TM–S bonds in the four systems are −1.97 (Zr–S), −2.02 (Nb–S), −1.26 (Ru–S) and −0.01 (Pd–S), which is in good agreement with the trend of their binding energies. Among them, the ICOHP values of Zr/Nb–S are very close, which further verifies the conclusion that both ZrPc and NbPc all have excellent anchoring effect on S8/LiPSs.
3.4 Catalytic performance analysis
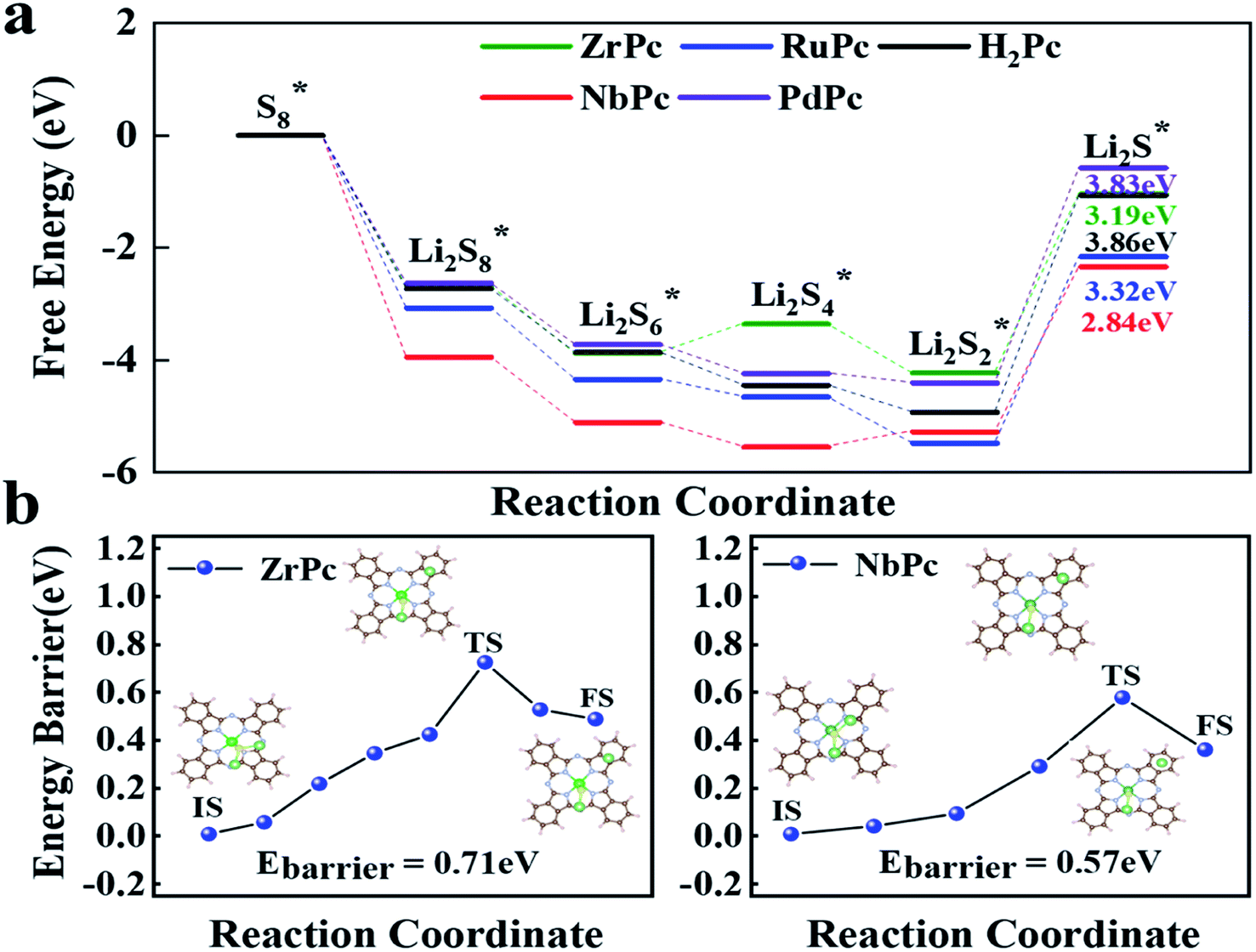
Through the previous analysis of H2Pc/TMPc structures anchoring S8/LiPSs, it can be found that ZrPc and NbPc can well inhibit the shuttle effect, and are promising candidates for SRRs electrocatalysts. In addition, the rapid conversion of LiPSs during charging and discharging of lithium–sulfur batteries is also an important factor affecting its overall performance. The discharge process is mainly the conversion of soluble Li2S4, Li2S6 and Li2S8 to insoluble or insulating Li2S/Li2S2, making it difficult for the reaction to proceed quickly and sustainably. The charging process is a reversible reaction of the discharging process and the first reaction is the dissociation of Li2S (Li2S → LiS + Li+ + e−), which is the key step to produce Li–S batteries with high capacity and high rate. In view of this, the free energy diagram in the discharge reaction process (S8* → Li2S*) and the dissociation energy barrier of Li2S* in the charging reaction process are further simulated and analyzed for estimating catalytic performance of TMPcs.
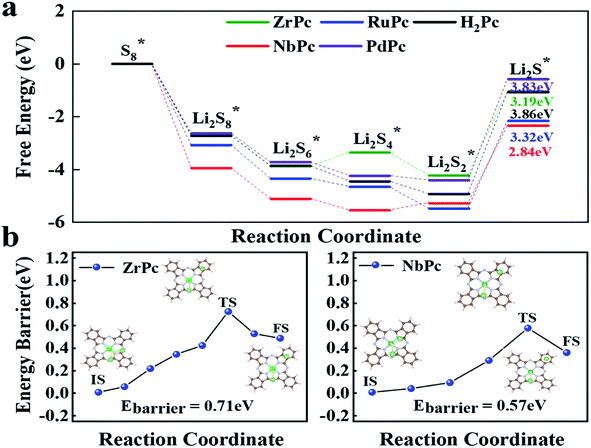
The entire charge and discharge reaction process involve the transfer of 16 electrons. The SRRs from S8* to Li2S* are divided into six basic reactions (eqn (3)–(8)). In the discharge process, S8* is first converted to Li2S8*, and then gradually reduced to Li2S6*, Li2S4*, Li2S2* and Li2S*.48 Fig. 5a presents the free energy diagrams of ZrPc, NbPc, RuPc, PdPc and H2Pc during SRRs. Obviously, the conversion from S8* to Li2S8* has the most negative ΔG, demonstrating that this reaction is the easiest to occur. Only the last step (Li2S2* is reduced to Li2S*) has the most positive Gibbs free energy (ΔG), indicating that the rate-determining step of the entire SRRs process is the last step. Among them, the ΔGs of ZrPc, NbPc, RuPc, PdPc and H2Pc for the rate-determining step are 3.19 eV, 2.94 eV, 3.32 eV, 3.83 eV and 3.86 eV, respectively. Our simulations indicate that ZrPc and NbPc have a smaller rate-limiting step ΔG, which are excellent to promote the kinetics of SRRs.
 |
| | Fig. 5 (a) Relative free energy profiles for the discharging process from S8 to Li2S on the ZrPc, NbPc, RuPc, PdPc and H2Pc surfaces. (b) The decomposition free energy barriers of the Li2S on ZrPc and NbPc surfaces, and the corresponding initial, transition, and final structures are shown in the illustration. | |
In addition, Fig. 5b exhibits the results of Li2S dissociated on the surface of ZrPc and NbPc. The ZrPc/NbPc–Li2S systems have a close (0.71 eV and 0.57 eV) and low Li2S decomposition barrier (initial, transient and final state are plotted in the insets of Fig. 5b). On the contrary, the PdPc/H2Pc–Li2S systems cannot obtain the stable configuration after the dissociation of Li2S* (as exhibited in Fig. S3†). The LiS and Li radicals in the dissociated state are fully optimized and then recombined to form Li2S, indicating that they have almost no catalytic activity for the decomposition of Li2S. It is worth mentioning that the dissociation performance of the base material for Li2S has a certain correlation with the strength of anchoring Li2S*. This is due to the fact that the Li–S bonds in the H2Pc/TMPc–Li2S system are elongated and weakened to different degrees. The ZrPc/NbPc–Li2S systems have a strong Zr/Nb–S bonds and hence possess a lower Li2S* dissociation energy barrier attributed to the strong interaction of the Zr/Nb–S bonds weakening the Li1/2–S bonds (according to Table S2,† the bond lengths of Li–S in pristine Li2S, ZrPc–Li2S, NbPc–Li2S, PdPc–Li2S and H2Pc–Li2S are ∼2.09 Å, ∼2.41 Å, ∼2.37 Å, ∼2.19 Å, ∼2.19 Å, respectively), so as to better promote the dissociation of Li2S. Consequently, ZrPc and NbPc also exhibit excellent catalytic performance during the charging reaction.
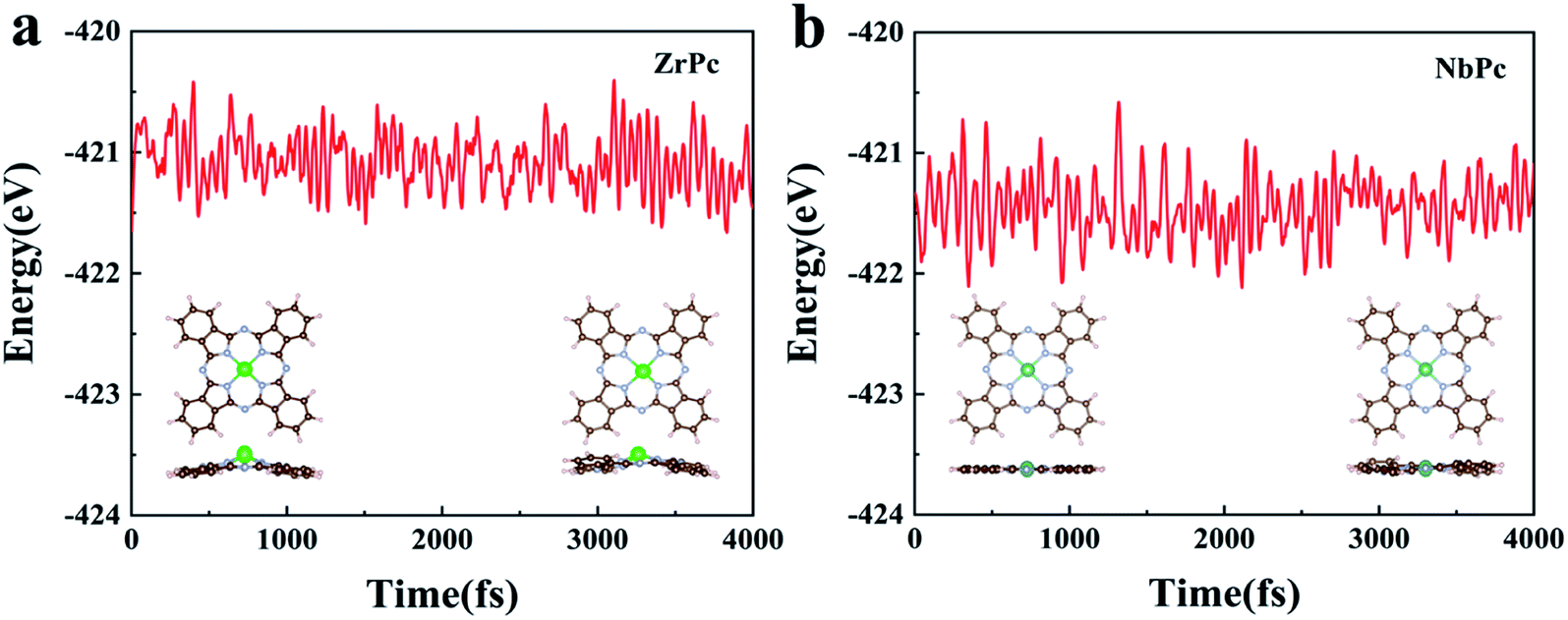
For practical usage of TMPc catalysts, the thermodynamic stability of ZrPc and NbPc are evaluated using AIMD simulations at 400 K for 4 ps with 1 fs interval. The results of final structures and the total energy change are shown in Fig. 6. The AIMD simulation results of the remaining TMPc (TM = Y, Mo, Tc, Ru, Rh, Pd, Ag, Cd) structures are given in Fig. S4.† Most of TMPc structures are thermodynamically stable, which is basically consistent with the results of their formation energy. For ZrPc and NbPc, our results show that the total energy oscillate within a small range, and the geometric morphology are also preserved well without significant distortion and bond breaking, guaranteeing their relatively high stability. Based on this, it is of great interest that ZrPc and NbPc can serve as sulfur host materials in LiPSs with high experimental feasibility under different reaction environments.
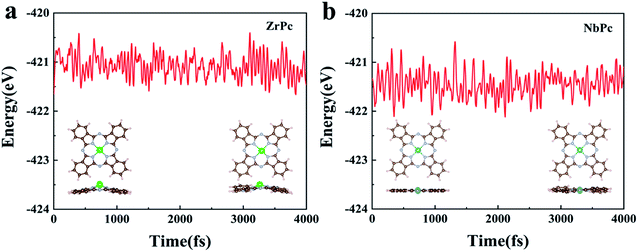 |
| | Fig. 6 (a) and (b) are the results of AIMD simulations of ZrPc and NbPc structures, respectively. | |
4. Conclusions
In this work, density functional theory (DFT) calculations were used to explore the anchoring and catalytic performance of H2Pc and TMPc-based electrocatalysts for adsorbing S8/LiPSs clusters. The results ensure higher adsorption ability of TMPc (especially ZrPc and NbPc) for anchoring the S8/LiPSs clusters than H2Pc, as well as two bonding patterns (TM–S and Li–N bonding). Moreover, TM–S bonds are more vital to anchor LiPSs than Li–N bonds, due to the strong interaction between the 4d orbital of TM and the 2p orbital of S. The small decomposition barriers and low rate-limiting step ΔG indicate that ZrPc and NbPc have excellent anchoring properties for S8/LiPSs and can better inhibit the “shuttle effect” when used as the sulfur hosts in Li–S batteries. The AIMD simulation results also further predict the experimental feasibility of the TMPc substrate materials. Our research gave atomic insights for explaining the interaction between H2Pc/TMPc substrates and S8/LiPSs molecules, and provided a feasible strategy for the rational design of better sulfur cathode hosts.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 22168036), Department of Education of Tibet Autonomous Region ([2021]1-21 Theoretical research and design of two dimensional electrocatalytic materials).
Notes and references
- M. Zhao, B. Q. Li, H. J. Peng, H. Yuan, J. Y. Wei and J. Q. Huang, Angew. Chem., Int. Ed., 2020, 59, 12636–12652 CrossRef CAS PubMed.
- J. X. Zhao, Y. G. Yang, R. S. Katiyar and Z. F. Chen, J. Mater. Chem. A, 2016, 4, 6124–6130 RSC.
- H. Yuan, H. J. Peng, J. Q. Huang and Q. Zhang, Adv. Mater. Interfaces, 2019, 6, 1802046 CrossRef CAS.
- T. Li, X. Bai, U. Gulzar, Y. J. Bai, C. Capiglia, W. Deng, X. F. Zhou, Z. P. Liu, Z. F. Feng and R. P. Zaccaria, Adv. Funct. Mater., 2019, 29, 1901730 CrossRef.
- Y. Hu, W. Chen, T. Y. Lei, Y. Jiao, J. W. Huang, A. J. Hu, C. H. Gong, C. Y. Yan, X. F. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 2000082 CrossRef CAS.
- M. Zhang, W. Chen, L. X. Xue, Y. Jiao, T. Y. Lei, J. W. Chu, J. W. Huang, C. H. Gong, C. Y. Yan, Y. C. Yan, Y. Hu, X. F. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 1903008 CrossRef CAS.
- A. A. Razzaq, Y. Z. Yao, R. Shah, P. W. Qi, L. X. Miao, M. Z. Chen, X. H. Zhao, Y. Peng and Z. Deng, Energy Storage Mater., 2019, 16, 194–202 CrossRef.
- P. C. Shi, Y. Wang, X. Liang, Y. Sun, S. Cheng, C. H. Chen and H. F. Xiang, ACS Sustainable Chem. Eng., 2018, 6, 9661–9670 CrossRef CAS.
- Y. Yang, G. Y. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018–3032 RSC.
- Z. Q. Ye, Y. Jiang, J. Qian, W. L. Li, T. Feng, L. Li, F. Wu and R. J. Chen, Nano Energy, 2019, 64, 103965 CrossRef CAS.
- R. Jayan and M. M. Islam, J. Phys. Chem. C, 2020, 124, 27323–27332 CrossRef CAS.
- J. Lei, T. Liu, J. J. Chen, M. S. Zheng, Q. Zhang, B. W. Mao and Q. F. Dong, Chem, 2020, 6, 2533–2557 CAS.
- J. F. Wu, S. Y. Ding, S. H. Ye and C. Lai, J. Energy Chem., 2020, 42, 27–33 CrossRef.
- W. X. Wu, Y. M. Zhang, Y. H. Guo, J. X. Bai, C. H. Zhang, Z. F. Chen, Y. X. Liu and B. B. Xiao, Appl. Surf. Sci., 2020, 526, 146717 CrossRef CAS.
- Y. P. Zheng, H. H. Li, H. Y. Yuan, H. H. Fan, W. L. Li and J. P. Zhang, Appl. Surf. Sci., 2018, 434, 596–603 CrossRef CAS.
- D. Gounden, N. Nombona and W. E. van Zyl, Coord. Chem. Rev., 2020, 420, 213359 CrossRef CAS.
- I. E. Brumboiu, S. Haldar, J. Luder, O. Eriksson, H. C. Herper, B. Brena and B. Sanyal, J. Chem. Theory Comput., 2016, 12, 1772–1785 CrossRef CAS PubMed.
- Y. N. Zhou, G. P. Gao, W. Chu and L. W. Wang, Nanoscale Adv., 2020, 2, 710–716 RSC.
- S. Q. Liu, Y. W. Liu, X. P. Gao, Y. J. Tan, Z. M. Shen and M. H. Fan, Appl. Surf. Sci., 2020, 500, 144032 CrossRef CAS.
- X. H. Wan, Z. F. Zhang, H. Niu, Y. H. Yin, C. G. Kuai, J. Wang, C. Shao and Y. Z. Guo, J. Phys. Chem. Lett., 2021, 12, 6111–6118 CrossRef CAS PubMed.
- M. Cheviri and S. Lakshmipathi, Chem. Phys. Lett., 2020, 739, 136942 CrossRef CAS.
- J. Kim, H. Shin, D. J. Yoo, S. Kang, S. Y. Chung, K. Char and J. W. Choi, Adv. Funct. Mater., 2021, 11, 2106679 CrossRef.
- J. S. Ma, M. P. Yu, J. W. Zhu, W. T. Li, W. Gong and H. Qiu, Ionics, 2021, 27, 3007–3016 CrossRef CAS.
- B. Yu, Q. He and Y. Zhao, Appl. Surf. Sci., 2021, 558, 149928 CrossRef CAS.
- M. S. Liao and S. Scheiner, J. Chem. Phys., 2001, 114, 9780–9791 CrossRef CAS.
- R. Zeis, T. Siegrist and C. Kloc, Appl. Phys. Lett., 2005, 86, 022103 CrossRef.
- R. B. dos Santos, R. Rivelino, F. de Brito Mota, G. K. Gueorguiev and A. Kakanakova-Georgieva, J. Phys. D: Appl. Phys., 2015, 48, 295104 CrossRef.
- A. Kakanakova-Georgieva, G. K. Gueorguiev, R. Yakimova and E. Janzén, J. Appl. Phys., 2004, 96, 5293–5297 CrossRef CAS.
- R. B. Dos Santos, R. Rivelino, B. Mota Fde, A. Kakanakova-Georgieva and G. K. Gueorguiev, Dalton Trans., 2015, 44, 3356–3366 RSC.
- B. Hammer, L. B. Hansen and J. K. Norskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- Y. M. Juan, E. Kaxiras and R. G. Gordon, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 9521–9525 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- V. L. Deringer, A. L. Tchougreeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
- R. Dronskowski and P. E. Blochl, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- S. Maintz, V. L. Deringer, A. L. Tchougreeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- Y. M. Zhao and J. X. Zhao, Appl. Surf. Sci., 2017, 412, 591–598 CrossRef CAS.
- L. H. Song, M. G. Zhang and J. Guo, Chem. Phys. Lett., 2021, 777, 138711 CrossRef CAS.
- Z. L. Yi, F. Y. Su, L. Huo, G. Y. Cui, C. L. Zhang, P. D. Han, N. Dong and C. M. Chen, Appl. Surf. Sci., 2020, 503, 144446 CrossRef CAS.
- Z. Y. Gao, H. Y. Huang, S. P. Xu, L. L. Li, G. Yan, M. L. Zhao, W. J. Yang and X. J. Zhao, Mol. Catal., 2020, 493, 111091 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- D. G. Sangiovanni, G. K. Gueorguiev and A. Kakanakova-Georgieva, Phys. Chem. Chem. Phys., 2018, 20, 17751–17761 RSC.
- L. J. Wang, T. R. Zhang, S. Q. Yang, F. Y. Cheng, J. Liang and J. Chen, J. Energy Chem., 2013, 22, 72–77 CrossRef CAS.
- Y. M. Zhao, L. Yang, J. X. Zhao, Q. H. Cai and P. Jin, Phys. Chem. Chem. Phys., 2017, 19, 18208–18216 RSC.
- H. Ci, M. Wang, Z. Sun, C. Wei, J. Cai, C. Lu, G. Cui, Z. Liu and J. Sun, J. Energy Chem., 2021, 474–482 Search PubMed.
- Y. Zhou, Z. Z. Zhou, R. X. Shen, R. G. Ma, Q. Liu, G. Z. Cao and J. C. Wang, Energy Storage Mater., 2018, 13, 189–198 CrossRef.
- R. S. Assary, L. A. Curtiss and J. S. Moore, J. Phys. Chem. C, 2014, 118, 11545–11558 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02049a |
| ‡ Both authors are co-first authors and contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence