
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative glycoproteomics of high-density lipoproteins†
Xinyu Tang a,
Maurice Wongb,
Jennyfer Tenab,
Chenghao Zhua,
Christopher Rhodesa,
Qingwen Zhoub,
Anita Vinjamurib,
Armin Oloumib,
Sucharita Bodduc,
Guillaume Luxardic,
Emanual Maverakisc,
Carlito B. Lebrillab and
Angela M. Zivkovic*a
a,
Maurice Wongb,
Jennyfer Tenab,
Chenghao Zhua,
Christopher Rhodesa,
Qingwen Zhoub,
Anita Vinjamurib,
Armin Oloumib,
Sucharita Bodduc,
Guillaume Luxardic,
Emanual Maverakisc,
Carlito B. Lebrillab and
Angela M. Zivkovic*a
aDepartment of Nutrition, University of California, Davis, One Shields Ave., Davis, CA 9516, USA. E-mail: amzivkovic@ucdavis.edu
bDepartment of Chemistry, University of California, Davis, One Shields Ave., Davis, CA 95616, USA
cDepartment of Dermatology, University of California, Davis School of Medicine, 3301 C Street, Suite 1400, Sacramento, CA 95816, USA
First published on 23rd June 2022
Abstract
In this work, we developed a targeted glycoproteomic method to monitor the site-specific glycoprofiles and quantities of the most abundant HDL-associated proteins using Orbitrap LC-MS for (glyco)peptide target discovery and QqQ LC-MS for quantitative analysis. We conducted a pilot study using the workflow to determine whether HDL protein glycoprofiles are altered in healthy human participants in response to dietary glycan supplementation.
1 Introduction
High-density lipoproteins (HDL) range in size from 8 to 12 nm in diameter and have a density between 1.063 and 1.21 g mL−1. HDL are most known for their critical role in regulating cholesterol concentrations, but they perform other protective functions, including immunomodulatory, antioxidant, and antiproteolytic, among other functions.2 The HDL proteome is complex and dynamic. HDL-associated proteins assist in stabilizing particle structure and solubilizing the lipid component, participate in signaling and interactions with lipoprotein receptors, and serve as cofactors for enzymes of lipid metabolism.1 Apolipoproteins and other HDL-associated proteins such as Apo-E, ApoA-IV, ApoA-V, Apo-M, and serum amyloid A (SAA) are also involved in immune responses to infection and inflammation.3–5 Many of these proteins are glycosylated with N- and O-glycans,6,7 and they typically exhibit glycan structural heterogeneity at each glycosite. Glycosylation can modulate HDL protein function, affect lipid- and protein-binding affinities, and is correlated with the functional capacity of HDL.8–10 The effects of glycosylation of most HDL proteins are still incompletely understood. There is a growing interest in the quantification of proteins and site-specific glycoforms of HDL particles.11Previous work from our group has established the applicability and reliability of the multiple reaction monitoring (MRM) method in the quantitative analysis of peptides and glycopeptides from serum12,13 and purified HDL.6,7 We expanded upon the previous methods to craft a workflow tailor-made for HDL by employing LC-MS with an Orbitrap analyzer to discover and identify new HDL-associated tryptic (glyco)peptides. We also optimized the tryptic digestion procedure to ensure reproducibility and sensitivity toward target glycopeptides. In the previous method we monitored 8 proteins, including 23 glycopeptides.7 The new method includes 339 transitions, spanning 47 peptides and 170 glycopeptides from 33 proteins. Absolute quantitation was achieved for the proteins ApoA-I, ApoC-I, Apo-D, Apo-E, and clusterin (Clus) by calibration with commercially available protein standards (Table S1†).
We then used the new panel to determine whether dietary glycan supplementation affects HDL glycoprofiles. Twenty-two healthy adult men/women (age 18–45) with a Body Mass Index (BMI) range of 18.5–25 were randomized to four treatment groups: placebo (n = 4), N-acetylglucosamine (n = 6), Spirulina (n = 6), and galactose (n = 6). Blood samples were collected before and after four weeks of supplementation. HDL was isolated using an optimized, validated method,14 and peptides and glycopeptides were quantified using the new workflow.
2 Results and discussion
2.1 Identification of (glyco)peptides on HDL
Target discovery of HDL-associated proteins was accomplished through bottom-up glycoproteomics using Orbitrap MS on a total of 80 samples from young children.15 Proteomics of HDL and other nanoparticles have been performed previously.5,11,16–18 We considered both N- and O-linked glycosylation in searching for potential glycopeptides. Site-specific identification of O-linked glycopeptides is more challenging because tryptic digestion often yields peptides with multiple possible O-glycosylation sites, compounding the microheterogeneity at each site. Several issues can arise: ambiguity in assigning glycan compositions and positions of glycosylation in a given peptide and the multiplicity of glycoforms which further divides the glycopeptide signal. We indicated the ambiguity in position and/or glycan composition when appropriate. In Fig. 1 we show some examples of peptide and glycopeptide identifications from MS/MS spectra.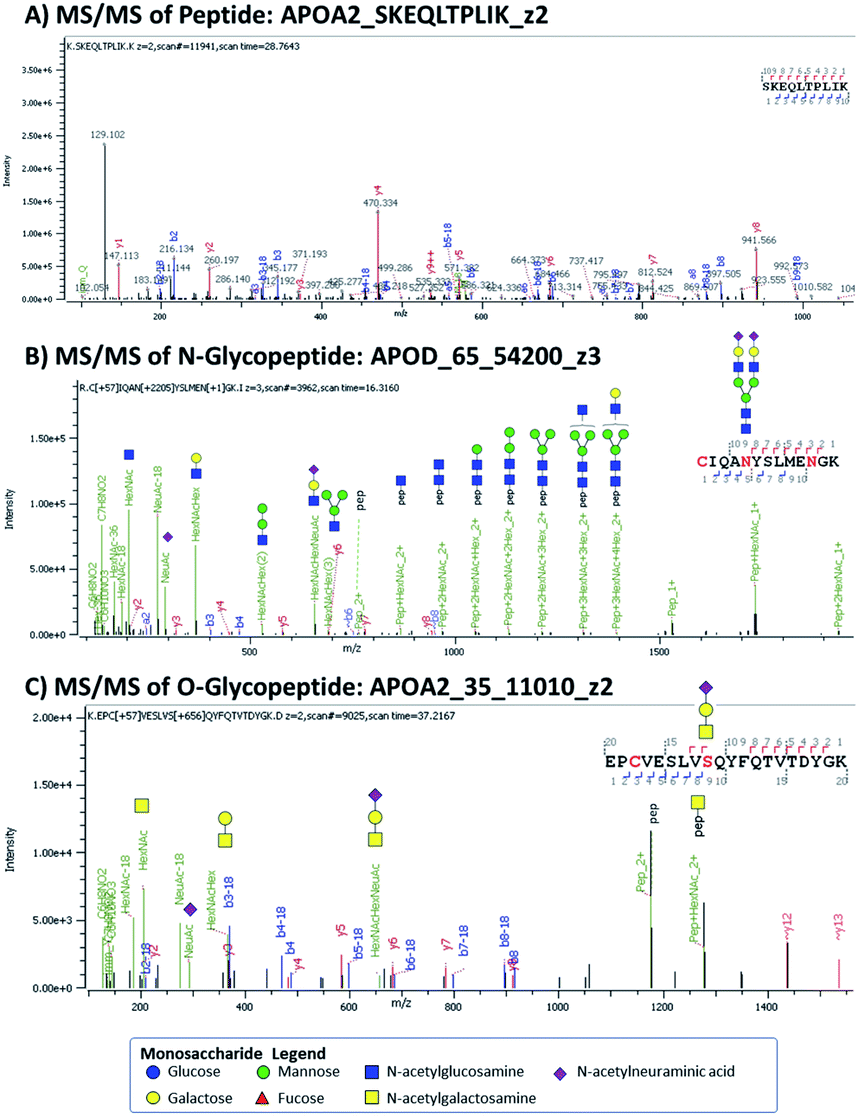 | ||
| Fig. 1 MS/MS spectra of (A) APOA2 peptide, (B) an N-glycopeptide from ApoD, and (C) an O-glycopeptide from ApoA2. The peptides are labeled as Protein_Sequence_ChargeState, while the glycopeptides are labeled as Protein_Position_GlycanComposition _ChargeState. Glycan compositions are written as 4- or 5-digit numbers denoting the number of hexose, HexNAc, fucose, NeuAc, and sulfate respectively. | ||
For each HDL protein, we chose the top-ranked peptides with the highest abundances and good sequence coverage for possible MRM-based detection. Some of the MRM transitions were obtained from previous work6,9,12 and validated in the current workflow. Here we describe some of the proteins and their glycoforms that were analyzed in the MRM method.
ApoA-I is the predominant protein constituent in HDL and an important structural component.1 It is known to play a role in cholesterol efflux and to act as a cofactor for lecithin cholesterol acyltransferase (LCAT).1 Transitions from 2 ApoA-I peptides were included; absolute quantitation was based on the peptide with sequence LAEYHAK, which showed better linearity as seen in Fig. 2. ApoA-II, ApoA-IV, and ApoA-V are minor protein constituents that can affect lipoprotein metabolism. Three possible O-glycosylation sites in ApoA-II featuring sialylated Core 1 glycans were found. ApoC-I, ApoC-II, ApoC-III, and ApoC-IV have relatively low molecular weights of just around 10–15 kDa. ApoC-I is an inhibitor of cholesteryl ester transfer protein (CETP) and reduces the esterification of free fatty acids. ApoC-II is an activator of lipoprotein lipase.19 ApoC-III is the most abundant C apolipoprotein in human plasma and is O-glycosylated at the threonine residue in position 94 (position 74 after signal peptide cleavage). In our transition list, we included 10 different glycan structures on this site, as well as some variants in the peptide sequence.
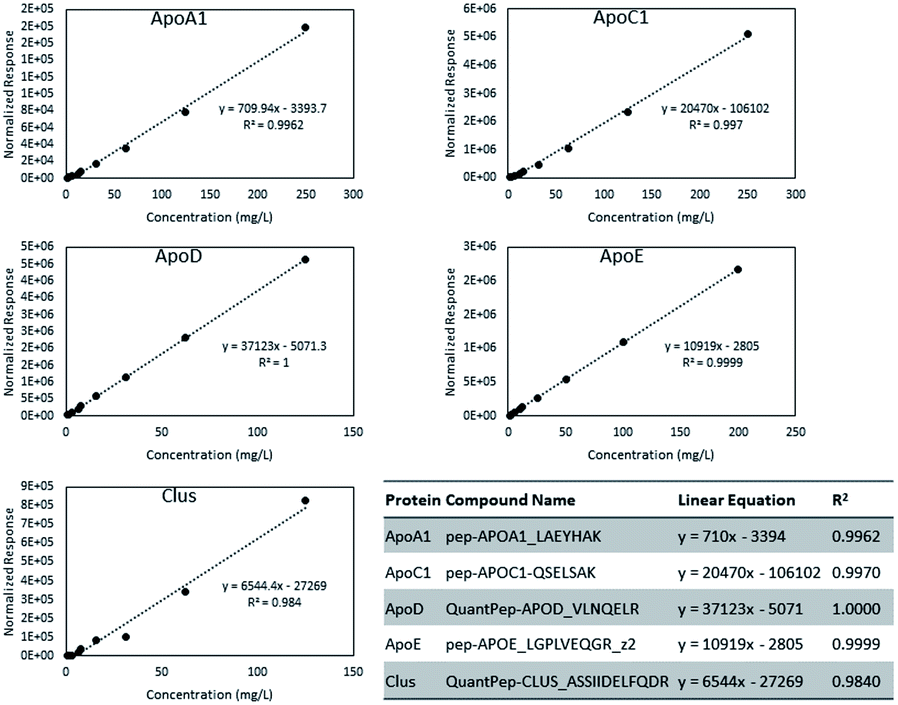 | ||
| Fig. 2 Calibration curves for Apo A-I, Apo C-I, Apo D, Apo E, and Clusterin/Apo J. | ||
Apo-D is closely associated with LCAT and can act as a multi-functional transporter. In our study, we included two N-glycosylation sites in positions 65 and 98, featuring mostly complex-type glycoforms.
Apo-E has been heavily studied because of its links to Alzheimer's disease and cardiovascular disease.20,21 In this sample set, two O-glycosylation sites were found at positions 215 and 307/308, both with sialylated Core 1 structures. Positions 307 and 308 feature a threonine and a serine, respectively; the exact site of attachment could not be disambiguated based on MS/MS data. Clusterin (Clus, also known as Apo-J) is a ubiquitous glycoprotein that acts as a versatile chaperone. Its expression is increased in Alzheimer's disease, and it is involved in the clearance of amyloid-β peptides.22 Different kinds of tumors have also been found to overexpress the protein or exhibit aberrant Clus glycosylation.23 Changes in the glycosylation of Clus have yielded possible biomarkers for Alzheimer's disease22 and breast cancer.23 In this study, N-glycosylation with complex-type structures was monitored at positions 86, 291, and 374.
Apo-M binds a variety of lipids and is involved in lipid transport. We found one N-glycosylation site at position 135, for which 8 different glycan compositions were observed. SAA proteins are major acute-phase reactants that rapidly increase in concentration during acute inflammation. SAA proteins are polymorphic, and they can present various isoforms.24 In this study, we included peptides from three variants: SAA1, SAA2, and SAA4. Seven N-glycopeptides from a variant of SAA4 (Uniprot Accession Code B2R5G8) that has an N-glycosylated site in position 94 were also monitored. This glycosite does not exist in the more common SAA4 variant. We further included several other minor protein constituents in HDL for a total of 34 unique proteins. The complete list of proteins and their corresponding Uniprot accession codes is provided in Table S2.†
2.2 Optimization of digestion protocol and method parameters
The HDL sample preparation and digestion workflow were optimized to minimize sample dilution while maintaining the efficacy of tryptic digestion and achieving adequate detection and good linearity for both peptides and glycopeptides. The optimized method featured a final dilution factor of 5 and a final protein concentration of around 0.5 μg μL−1. This allowed us to inject 5 μL of the digested HDL sample to meet the target peptide level. We avoided cleanup steps such as solid-phase extraction to avoid further perturbations on the sample. The high lipid content of the HDL sample and the relatively low dilution factor necessitated additional conditioning and washing of the HPLC C18 column. A complete list of the MRM transitions, including the precursor and product ion m/z, retention times, and collision energies, are listed in Table S3.†A synthetic peptide serving as an internal standard (ISTD) was added to each sample with a final concentration of 1 μg mL−1 to monitor the signal stability of the batch analysis. The ISTD signals from the 80 HDL samples had a coefficient of variation (CV) of 10.5%, demonstrating the stability of the instrument response throughout the batch analysis. The ISTD signal was also used to normalize all the peptide and glycopeptide signals for more consistent quantitation. Normalization was done by dividing the response of each analyte by that of the ISTD from the same sample run, then multiplying by the average ISTD response. The normalized response was used for all subsequent data processing.
Absolute quantification was determined for five proteins: ApoA-I, ApoC-I, Apo-D, Apo-E, and Clus. Fig. 2 shows the calibration curves for the five proteins. From the mass per volume concentration of the proteins, we also determined the number of protein units per volume of purified HDL. The molecular weight of the protein without its signal peptide was used for this calculation. Calibration curve plots based on molar concentrations are shown in Fig. S1.† Because all signals were normalized to that of the ISTD, we compared the relative molar response factors based on the slope of the linear equations. In Fig. S2,† we present the distribution of protein concentrations from 80 individuals in terms of both weights per volume and molar concentration. The median molar abundance of ApoA-I in HDL was 80 times greater than that of ApoC-I. The CVs of the molar abundances ranged between 34% to 43% for ApoA-I, ApoC-I, and Apo-D. Among the five proteins, Apo-E displayed the most variation at 76%, while the CV of Clus was surprisingly low at just 4.0%.
2.3 Batch reproducibility
Pooled serum QC samples were run after every 10 sample injections to monitor the performance of the instrument. A total of 14 QC samples were run throughout the batch. For the five quantified proteins, their CVs were 4.4%, 3.2%, 4.4%, 5.5%, and 6.2% for the absolute quantification of ApoA-I, ApoC-I, Apo-D, Apo-E, and Clus respectively.Among the 47 peptides monitored, 34 had CVs of less than 15%, while an additional 4 had CVs between 15 and 30%. Among the 9 peptides that have high CVs > 30%, 4 can be considered redundant because they were from proteins with better scoring peptides, while 4 more are from extremely low abundance proteins (Apo-F, Apo-(A), ApoC-IV, ApoA-V). The signal for the peptide from serum paraoxonase/arylesterase 1 protein (PON1) showed higher variability and lower signal compared to previous results.12 We believe this result was an unintended effect of the lower sample volume during tryptic digestion, leading to lower cleavage efficiency. PON1 is known to bind strongly to phospholipids and might not be fully dissociated and/or denatured in the less dilute digestion conditions that we employed. Of the 168 glycopeptides monitored, 69 had CVs of less than 30%, while a further 24 had CVs between 30-50%. About 60 of the glycopeptides were not present in appreciable amounts in the QC standards and had mean signals lower than 1000 ion counts, and so had high CVs. Heatmaps of the peptide and glycopeptide ISTD-normalized responses from all quality control runs are shown in Fig. S3.† Overall, the method showed good reproducibility throughout the digestion and instrument analysis for the target proteins; the limitation was mostly due to the naturally low abundance of certain analytes.
2.4 Relative glycoprotein quantitation
Relative quantitation can be obtained for the other 33 proteins for which there were no standards. The ISTD-normalized responses of the protein peptides are shown in Fig. 3. The normalized responses can be compared relative to the same peptide across all samples; the variability across samples is demonstrated by the heatmap. However, we cannot directly compare different peptides to each other or infer their actual concentrations based solely on their normalized responses because their molar response factors can vary by several orders of magnitude.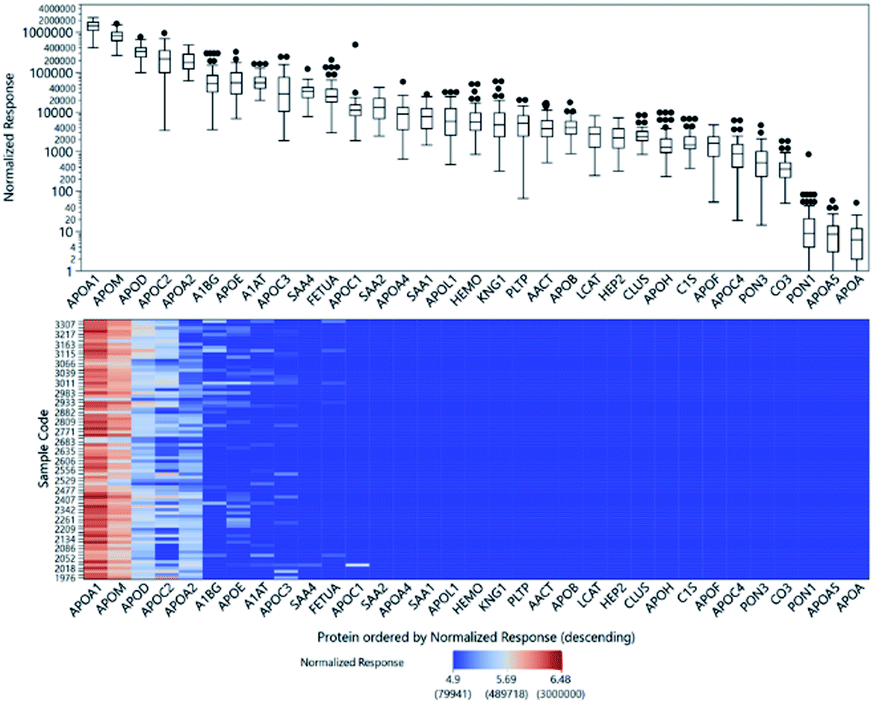 | ||
| Fig. 3 Top: Box plot of the ISTD-normalized responses of protein peptides showing the distribution of relative abundances from 80 individuals. The y-axis is plotted in the log 10 scale. Bottom: Heatmap of responses of protein peptides showing the ISTD-normalized response for each of the 80 samples. The color scaling is also done in the log 10 scale. | ||
Relative glycopeptide comparisons were performed by normalizing the glycopeptide responses to the peptide response from the same protein to ensure that the changes observed for the specific glycoforms would not be affected by changes in protein abundance. In Fig. S4,† we illustrate the variation in glycan expression among the 80 samples.
To our knowledge, this is the first method to comprehensively quantify both the proteins and their glycosylation alterations of isolated HDL particles. As a proof of principle, we applied the method to determine compositional alterations of the proteins and their glycoprofiles in HDL isolated from participants before vs. after 4 weeks of supplementation with different monosaccharides.
2.5 Effects of dietary supplementations on HDL proteins and glycoprofiles
The baseline characteristics of the participants in each experimental arm are summarized in Table S4.† The age, BMI, systolic, and diastolic blood pressure of subjects were not significantly different among experimental arms (ANOVA p value > 0.05 for all characteristics). The significantly altered glycans are shown in Fig. 4. The mono-fucosylated fraction of Alpha-1 Antitrypsin (A1AT) differed across treatment groups and significantly decreased after Spirulina treatment compared to placebo (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2FC = 0.68 ± 0.77 vs. −0.91 ± 0.70 after taking the placebo vs. Spirulina for four weeks, Fig. 4A). The abundance of disialylated Clus increased after four weeks of galactose supplementation (log2FC = −0.23 ± 0.88 vs. 1.09 ± 1.12 after taking the placebo vs. galactose for four weeks, Fig. 4B), while the sialylation fractions of other glycopeptides remain unchanged across all supplement groups. Mono-fucosylated ApoE decreased after galactose treatment compared to control (log2FC = 0.93 ± 1.30 vs. −0.14 ± 0.86 after taking the placebo vs. galactose for four weeks), and an N-glycan on A1AT, A1AT_107_6513, was decreased after the Spirulina supplement (log2FC = 0.61 ± 1.30 vs. −0.98 ± 1.35 after taking the placebo vs. Spirulina for four weeks), although the differences of both glycopeptides between treatment group means did not reach statistical significance (Fig. 4C and D). Table S5† lists all glycopeptides with differential fold changes comparing supplements to placebo.
2FC = 0.68 ± 0.77 vs. −0.91 ± 0.70 after taking the placebo vs. Spirulina for four weeks, Fig. 4A). The abundance of disialylated Clus increased after four weeks of galactose supplementation (log2FC = −0.23 ± 0.88 vs. 1.09 ± 1.12 after taking the placebo vs. galactose for four weeks, Fig. 4B), while the sialylation fractions of other glycopeptides remain unchanged across all supplement groups. Mono-fucosylated ApoE decreased after galactose treatment compared to control (log2FC = 0.93 ± 1.30 vs. −0.14 ± 0.86 after taking the placebo vs. galactose for four weeks), and an N-glycan on A1AT, A1AT_107_6513, was decreased after the Spirulina supplement (log2FC = 0.61 ± 1.30 vs. −0.98 ± 1.35 after taking the placebo vs. Spirulina for four weeks), although the differences of both glycopeptides between treatment group means did not reach statistical significance (Fig. 4C and D). Table S5† lists all glycopeptides with differential fold changes comparing supplements to placebo.
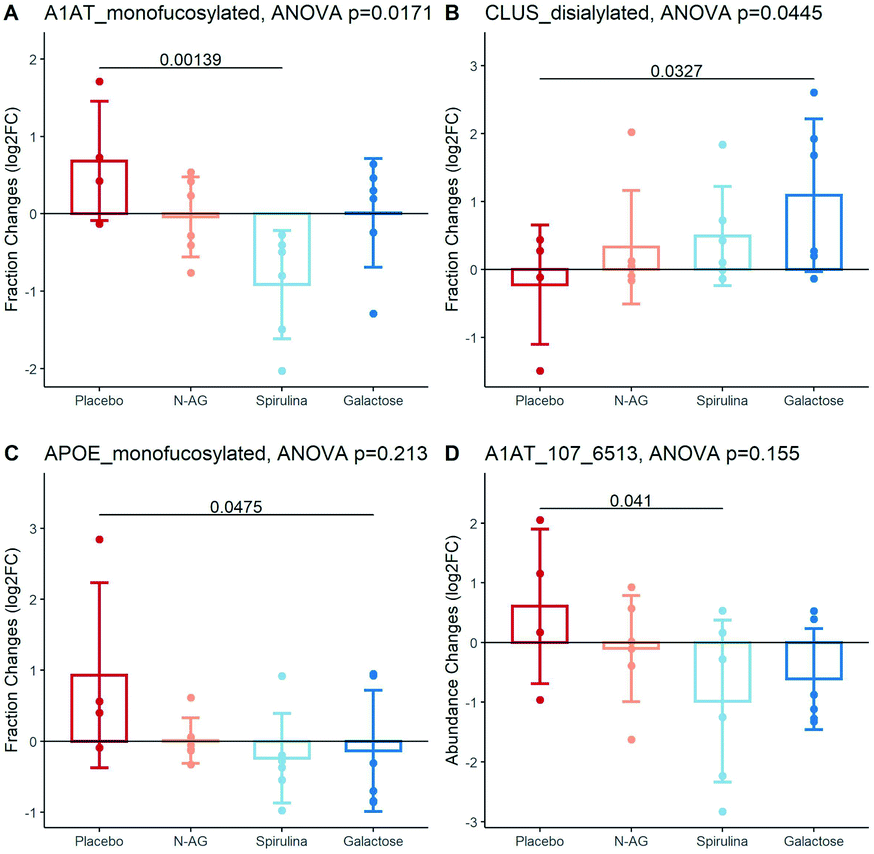 | ||
| Fig. 4 HDL glycan composition changes in response to supplements. (A) Fucosylation fraction changes on A1AT after treatment. (B) Sialylation fraction changes on CLUS after treatment. (C) Fucosylation fraction changes on APOE after treatment. (D) HDL glycan, A1AT_107_6513, changes after treatment. Unadjusted p-values were labeled. | ||
As expected, the relative quantities of the proteins themselves were not affected by any supplement treatment. These results show for the first time that dietary glycan composition can affect HDL protein glycoprofiles without altering HDL protein concentrations, including changes in important functional proteins (i.e., Clus, Apo-E, A1AT) known to be involved in a number of disease conditions, including Alzheimer's disease, metabolic disorders, and other chronic inflammatory diseases.22,25
3 Conclusions
We have demonstrated the applicability of the developed HDL sample preparation and MRM MS workflow for the quantitation of proteins and glycopeptides in large clinical batches. Further developments will aim at the absolute quantitation of more target proteins for which purified standards can be obtained, as well as better sensitivity for glycopeptides from a variety of age groups and disease conditions. This method is sensitive enough to detect subtle glycosylation composition changes on HDL even in a pilot study with 4–6 subjects in each group. The pilot study demonstrated the potential of dietary modification to modulate HDL glycoprofiles. These results demonstrate the potential of the MRM MS method for glycoprofiling HDL particles and other lipoproteins for the development of biomarkers as well as to determine the effects of various interventions (e.g., diet, lifestyle, medications).4 Experimental
4.1 Sample preparation for untargeted glycoproteomics analysis
For untargeted glycoproteomics analysis, blood plasma samples collected from a previously conducted study in 18 month old children in Ghana.26 Purified HDL particles were isolated through a two-step HDL isolation method which isolates HDL particles first by density using sequential flotation ultracentrifugation followed by size exclusion chromatography, as described previously.14 Briefly, 500 μL of plasma was underlaid under KBr solution at a density of 1.0060 g mL−1 to remove triglyceride-rich, low density (<1.006 g mL−1) particles, including chylomicrons and VLDL, and submitted to ultracentrifugation in an Optima MAX-TL Ultracentrifuge with (Beckman-Coulter) fixed angle rotor at 110000 RPM and 14 °C for 30 minutes. After centrifugation, the supernatant was removed by aspiration, and the remaining fraction containing HDL, LDL, albumin, and plasma proteins was adjusted to a density of 1.210 g mL−1 with 1.340 g mL−1 KBr solution and underlaid under clean 1.210 g mL−1 density solution, then submitted to ultracentrifugation at 110000 RPM and 14 °C for 3 hours and 30 minutes. The supernatant was removed by aspiration and dialyzed using an Amicon Ultra-4 50 kDa centrifugal filter (Millipore) by centrifugation at 4500 RPM for 8 minutes. A final volume of 250 μL was then transferred to an amber vial for FPLC analysis using a single Superdex 200 Increase 10/300 GL agarose-crosslinked column (GE Healthcare) on an AKTA P-920 FPLC (Amersham Biosciences). Four 1 mL fractions of HDL were pooled together and dialyzed to 100 μL, of which one 25 μL aliquot was used for glycoproteomic analysis.
For sample digestion, the HDL fractions were diluted in a total of 100 μL with 50 mM ammonium bicarbonate buffer at pH 7.5. All reagents used for sample preparation were freshly prepared in a buffer of 50 mM ammonium bicarbonate. Proteins were then denatured with 2 μL of 550 mM dithiothreitol (Promega, Madison, WI) for 1 h at 65 °C and alkylated with 4 μL of 450 mM iodoacetamide (Sigma-Aldrich, St. Louis, MO) for 30 min at room temperature away from light. Proteins were digested with 2 μg sequencing grade trypsin (Promega) for 18 h at 37 °C. Samples were purified through Bond Elut C18 Solid phase extraction (Agilent, Santa Clara, CA), dried in a vacuum concentrator and reconstituted in 50 μL LC-MS grade water.
4.2 Untargeted glycoproteomics analysis
Samples were run on an Agilent 1290 Infinity II High Performance Liquid Chromatography (HPLC) coupled to a Fusion Lumos MS/MS Orbitrap (Thermo Fisher Scientific). The HPLC was equipped with a 150 mm Agilent Zorbax Eclipse Plus C18 column with 1.8 μm particle size. Peptides and glycopeptides were eluted with a binary gradient of (A) 3% acetonitrile with 0.1% formic acid in water, and (B) 90% acetonitrile with 0.1% formic acid in water. The HPLC was set at a flow rate of 0.3 mL min−1 and programmed to ramp from 0% to 20% B in 20 min, 30% at 40 min, 44% at 47 min, and 100% at 48 min followed by a flushing and equilibration cycle. The electrospray ionization (ESI) voltage was set to 3500 V in the positive mode.The Orbitrap was operated in positive mode with a precursor scan resolution of 60000 and a range of 350–2000 m/z. Fragmentation was accomplished by stepped High-energy Collisional Dissociation (HCD). The collision energy was set at 30% and stepped at 10%. Ions for fragmentation were filtered to include a precursor mass range of 700–2000 m/z and charge states between 2 and 6.
Peptides and glycopeptides were identified with Byonic software (Protein Metrics Inc). For glycopeptide identification, a database of protein sequences and a library of glycan compositions are required as inputs. The human proteome database was downloaded from http://Uniprot.org. We used in-house libraries for N-glycan and O-glycan compositions. (Glyco)peptides were identified based on the accurate mass of the precursor ions with tolerance set at 10 ppm and by matching MS/MS fragmentation with theoretical MS/MS spectra generated from in silico digestion of the provided protein database.
4.3 Sample preparation for targeted quantitative glycoproteomics analysis
Tryptic digestion of the HDL samples was done in a 96-well format to facilitate batch processing. Samples were randomized prior to plating. All reagents were freshly prepared in a buffer of 50 mM ammonium bicarbonate. A sample volume of 10 μL purified HDL was used for tryptic digestion. After every 20 samples, 10 μL of commercially available human serum (Sigma-Aldrich) was also digested to serve as sample preparation controls. Protein standards (APOA1, APOC1 APOD, APOE, Clusterin; all from Sigma-Aldrich) were mixed in known amounts (250, 250, 125, 200, and 125 μg mL−1 respectively) and digested with the batch to serve as calibration standards. Serial dilution of the digested protein mixture provided the calibration curve for absolute quantitation of the 5 proteins. The digested mixture was diluted by factors of 160, 80, 40, 20, 16, 8, 4, 2, and 1 to obtain 9 calibration standards, from which calibration curves spanning 4 orders of magnitude were calculated.After pipetting the samples, controls, and standards onto the 96-well plate, 10 μL of 100 mM dithiothreitol was added to each well to reduce the protein disulfide bonds. Protein denaturation was continued by heating in a water bath for 1 h at 65 °C. Samples were then alkylated with 5 μL of 360 mM iodoacetamide for 30 min at room temperature away from light. Excess iodoacetamide was quenched with 5 μL of 100 mM dithiothreitol. Proteins were digested with 10 μL of 200 μg mL−1 sequencing grade trypsin for 18 h at 37 °C. The digestion was stopped by acidifying the solution with 5 μL 10% (v/v) formic acid (Fluka). To account for batch variability and possible run-order effects, 5 μL of a 1 μg mL−1 synthetic peptide with sequence RPAIAINNPYVPR (Bionexus, Oakland, CA) was added as an internal standard. The final volume of the HDL digest is 50 μL, a 5-fold dilution of the purified HDL solution. The samples were injected into the LC-MS instrument without further cleanup.
4.4 Targeted glycoproteomics analysis using QqQ
(Glyco)peptides were quantified on an Agilent 1290 Infinity II LC system coupled to an Agilent 6495B Triple Quadrupole MS. Injection volumes were set at 5 μL for protein standards and HDL samples, and 1 μL for serum digests. A pooled sample of the digested serum was run after every 10 HDL samples to serve as quality control (QC). They were used to monitor the stability of the instrument and the reproducibility of the batch analysis.The HPLC was equipped with a 150 mm Agilent Zorbax Eclipse Plus C18 column with 1.8 μm particle size. A C18 column guard was used to protect the column from buildup of lipids and other hydrophobic substances in the sample. A binary gradient of (A) 3% acetonitrile with 0.1% formic acid in water, and (B) 90% acetonitrile with 0.1% formic acid in water was set at a flow rate of 0.5 mL min−1. The HPLC pump parameters were programmed to ramp from 0% to 20% B in 20 min, 30% at 40 min, 44% at 47 min, and 100% at 48 min followed by 12 min column flushing cycle with 100% B and 7 min equilibration at 100% A. The Electrospray Ionization (ESI) voltage was set to 3500 V in the positive mode.
A transition list for target analytes was created by combining previously reported transitions6,12 with new transitions selected from untargeted glycoproteomics analysis. The instrument was run on Dynamic Multiple Reaction Monitoring (DMRM) mode to minimize the number of transitions being monitored at each scan cycle. For peptides, at least 2 product ions were selected for monitoring. Quantitation was based on the area of the more abundant product ion while the others are for qualitative identification. Product ions for glycopeptides were based on diagnostic glycan fragments. Glycans yield characteristic oxonium ions after Collision Induced Dissociation (CID) with mass-to-charge ratios (m/z) of 204.08, 274.09, and 366.14 for N-acetylhexosamine (HexNAc), N-acetylneuraminic acid (NeuAc) with loss of H2O, and hexose + HexNAc (Hex1HexNAc1) respectively.
4.5 Participants and study design of dietary supplementation pilot study
The study was a four-week, double-blinded, randomized, placebo-controlled study. Twenty-two healthy adults (eleven males and ten females, age 18–45) with a Body Mass Index (BMI) range of 18.5–25 were recruited to the study and if they passed screening for inclusion and exclusion criteria were consented and randomized to four treatment groups: placebo (4), N-acetylglucosamine (6), Spirulina (6), and galactose (6). Exclusion criteria were prescription medications, supplements, smoking, drinking, special diet, intense exercise pattern, special diet features, weight changes, medical conditions, chronic disease, infection, autoimmune conditions, glycosylation-related diseases, low blood hemoglobin, and severe food or drug allergic reactions. Women were further excluded if they had a pregnancy within the last year or used hormonal contraception. Women were also excluded if they were perimenopausal or postmenopausal. The participants were randomized to take a dietary supplement daily for 4 weeks, either placebo, 2.8 grams N-AG, 3000 milligrams Spirulina, or 25 grams galactose. The placebo contained one teaspoon cellulose powder. These doses were determined by a primary in vitro study which showed effects on cell surface glycosylation at these doses of monosaccharide in the culture medium. All participants came in to the study center for a morning blood draw at baseline and after four weeks. Blood samples were processed within 1 hour of the blood draw and plasma was aliquoted and stored immediately at −80 °C until analysis. HDL particles were isolated from plasma as described above. The study was approved by the University of California Davis Institutional Review board and registered at http://clinicaltrials.gov (NCT05040204).4.6 Statistics
Protein concentrations of APOC1, APOA1, APOD, APOE, and CLUS were calculated based on the calibration curves. The relative abundance of the rest of the proteins was determined from the ion counts of the indicator peptide for each protein, as described previously.13 The relative abundance of each glycopeptide was calculated as the ratio of the glycopeptide ion abundance to the ion abundance of the indicator peptide for the parent protein, as described previously.13 The coefficient of variation (CV) was calculated for all (glyco)peptides based on pooled samples.All statistical analyses were conducted in R version 4.1.0. Glycopeptides undetected in ≥ 5 samples or with CVs ≥ 30% across all samples were excluded from further analysis. Any remaining unobserved values were imputed as the minimum observed values of the specific glycopeptide. The fucosylated and sialylated peptide fractions were calculated as the sum of relative glycopeptide abundance of non-, mono-, di-, or poly-glycosylated peptides relative to that of the total peptides to evaluate the change in overall fucosylation and sialylation of each protein.
The effects of placebo/supplements on HDL-related (glyco)peptides and their fucosylation/sialylation status were quantified as the fold changes of relative abundance/fractions comparing post-treatment to baseline. The differences in fold changes of abundances between the four groups were analyzed using a one-way ANOVA test. A pair-wise differential abundance test was performed to identify (glyco)peptides differentially altered between placebo and each supplement using a linear model with the limma package.27 The resulting p-values were adjusted with multiple testing corrections using the Benjamini–Hochberg method. The fold change data were log2 transformed before linear model analysis and the Shapiro–Wilk test was performed to confirm the normality. The same data transformations and linear model were used for analysis of APOC1, APOA1, APOD, APOE, and CLUS using the standard curve derived concentrations, but no multiple testing corrections were applied.
Author contributions
A. M. Z., C. B. L., and E. M. conceived the project, M. W., J. T., C. Z., C. R., Q. Z., A. V., A. O., S. B., G. L. conducted the research, X. T. and M. W. analysed and visualized the data, X. T., M. W., J. T., and A. Z. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institute of Health (R01GM049077 and R01AG062240) and National Institute of Food and Agriculture (CA-D-NUT-2242-H).Notes and references
- R. W. Mahley, T. L. Innerarity, S. C. Rall and K. H. Weisgraber, J. Lipid Res., 1984, 25, 1277–1294 CrossRef CAS.
- B. J. Ansell, K. E. Watson, A. M. Fogelman, M. Navab and G. C. Fonarow, J. Am. Coll. Cardiol., 2005, 46, 1792–1798 CrossRef CAS PubMed.
- W. Khovidhunkit, P. N. Duchateau, K. F. Medzihradszky, A. H. Moser, J. Naya-Vigne, J. K. Shigenaga, J. P. Kane, C. Grunfeld and K. R. Feingold, Atherosclerosis, 2004, 176, 37–44 CrossRef CAS PubMed.
- D. T. Laskowitz, D. M. Lee, D. Schmechel and H. F. Staats, J. Lipid Res., 2000, 41, 613–620 CrossRef CAS.
- T. Vaisar, S. Pennathur, P. S. Green, S. A. Gharib, A. N. Hoofnagle, M. C. Cheung, J. Byun, S. Vuletic, S. Kassim, P. Singh, H. Chea, R. H. Knopp, J. Brunzell, R. Geary, A. Chait, X. Q. Zhao, K. Elkon, S. Marcovina, P. Ridker, J. F. Oram and J. W. Heinecke, J. Clin. Invest., 2007, 117, 746–756 CrossRef CAS PubMed.
- M. J. Kailemia, W. Wei, K. Nguyen, E. Beals, L. Sawrey-Kubicek, C. Rhodes, C. Zhu, R. Sacchi, A. M. Zivkovic and C. B. Lebrilla, J. Proteome Res., 2018, 17, 834–845 CrossRef CAS PubMed.
- J. Huang, H. Lee, A. M. Zivkovic, J. T. Smilowitz, N. Rivera, J. B. German and C. B. Lebrilla, J. Proteome Res., 2014, 13, 681–691 CrossRef CAS PubMed.
- P. A. Schindler, C. A. Settineri, X. Collet, C. J. Fielding and A. L. Burlingame, Protein Sci., 1995, 4, 791–803 CrossRef CAS PubMed.
- C. Zhu, M. Wong, Q. Li, L. Sawrey-Kubicek, E. Beals, C. H. Rhodes, R. Sacchi, C. B. Lebrilla and A. M. Zivkovic, J. Proteome Res., 2019, 18, 3977–3984 CrossRef CAS PubMed.
- A. T. Remaley, A. W. Wong, U. K. Schumacher, M. S. Meng, H. B. Brewer and J. M. Hoeg, J. Biol. Chem., 1993, 268, 6785–6790 CrossRef CAS PubMed.
- T. Vaisar, Curr. Vasc. Pharmacol., 2012, 10, 410–421 CrossRef CAS PubMed.
- Q. Li, M. J. Kailemia, A. A. Merleev, G. Xu, D. Serie, L. M. Danan, F. G. Haj, E. Maverakis and C. B. Lebrilla, Anal. Chem., 2019, 91, 5433–5445 CrossRef CAS PubMed.
- Q. Hong, L. R. Ruhaak, C. Stroble, E. Parker, J. Huang, E. Maverakis and C. B. Lebrilla, J. Proteome Res., 2015, 14, 5179–5192 CrossRef CAS PubMed.
- J. J. Zheng, J. K. Agus, B. V. Hong, X. Tang, C. H. Rhodes, H. E. Houts, C. Zhu, J. W. Kang, M. Wong, Y. Xie, C. B. Lebrilla, E. Mallick, K. W. Witwer and A. M. Zivkovic, Sci. Rep., 2021, 11(1), 16086 CrossRef CAS PubMed.
- B. V. Hong, C. Zhu, M. Wong, R. Sacchi, C. H. Rhodes, J. W. Kang, C. D. Arnold, S. Adu-Afarwuah, A. Lartey, B. M. Oaks, C. B. Lebrilla, K. G. Dewey and A. M. Zivkovic, ACS Omega, 2021, 6, 32022–32031 CrossRef CAS PubMed.
- H. N. Abdelhamid and H. F. Wu, TrAC, Trends Anal. Chem., 2015, 65, 30–46 CrossRef CAS.
- H. F. Wu, J. Gopal, H. N. Abdelhamid and N. Hasan, Proteomics, 2012, 12, 2949–2961 CrossRef CAS PubMed.
- Z. Y. Chen, H. N. Abdelhamid and H. F. Wu, Rapid Commun. Mass Spectrom., 2016, 30, 1403–1412 CrossRef CAS PubMed.
- M. C. Jong, M. H. Hofker and L. M. Havekes, Arterioscler., Thromb., Vasc. Biol., 1999, 19, 472–484 CrossRef CAS PubMed.
- Y. Yamazaki, N. Zhao, T. R. Caulfield, C. C. Liu and G. Bu, Nat. Rev. Neurol., 2019, 15, 501–518 CrossRef CAS PubMed.
- A. D. Marais, Pathology, 2019, 51, 165–176 CrossRef CAS PubMed.
- X. Li, Y. Ma, X. Wei, Y. Li, H. Wu, J. Zhuang and Z. Zhao, Neurosci. Bull., 2014, 30, 162–168 CrossRef CAS PubMed.
- A. Merlotti, A. L. Malizia, P. Michea, P. E. Bonte, C. Goudot, M. S. Carregal, N. Nuñez, C. Sedlik, A. Ceballos, V. Soumelis, S. Amigorena, J. Geffner, E. Piaggio and J. Sabatte, Oncoimmunology, 2019, 8(9), e1629257 CrossRef PubMed.
- B. Kluve-Beckerman, G. L. Long and M. D. Benson, Biochem. Genet., 1986, 24, 795–803 CrossRef CAS PubMed.
- F. de Serres and I. Blanco, J. Intern. Med., 2014, 276, 311–335 CrossRef CAS PubMed.
- S. Adu-Afarwuah, A. Lartey, H. Okronipa, P. Ashorn, J. M. Peerson, M. Arimond, U. Ashorn, M. Zeilani, S. Vosti and K. G. Dewey, Am. J. Clin. Nutr., 2016, 104, 797–808 CrossRef CAS PubMed.
- M. E. Ritchie, B. Phipson, D. Wu, Y. Hu, C. W. Law, W. Shi and G. K. Smyth, Nucleic Acids Res., 2015, 43, e47 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02294j |
| This journal is © The Royal Society of Chemistry 2022 |