DOI:
10.1039/D2RA02537J
(Paper)
RSC Adv., 2022,
12, 16843-16846
As(III) removal through catalytic oxidation and Fe(III) precipitation†
Received
20th April 2022
, Accepted 28th May 2022
First published on 7th June 2022
Abstract
To remove arsenite (As(III)) from wastewater effectively, the catalytic oxidation of As(III) to arsenate (As(V)) and As(V) precipitation with iron ions (Fe(III)) was investigated. The Pt/SiO2 catalyst functioned as a reaction site for As(III) with oxygen in the atmosphere. The combination of the Pt/SiO2 catalyst and Fe(III) precipitant improved the removal of As(III) in the precipitate; Pt/SiO2 worked as both an As(III) oxidation site and precipitation site with Fe(III) precipitant.
1. Introduction
Arsenic which has various oxidation states is highly toxic to humans, and its removal is indispensable for maintaining environmental safety.1–4 As arsenite (AsO2−, As(III)) is difficult to recover because of its high solubility in water, its removal generally requires a pre-oxidation process to form arsenate (AsO3−, As(V)), which is easily recovered.1,5,6 To convert As(III) to As(V) under mild conditions, catalytic oxidation is a promising process, among which photocatalytic oxidation is the mildest method because oxidants in the process are produced by photoreaction without external addition.7–9 Although processes without photocatalytic reactions have been reported, they require strong oxidants such as ozone and chlorine, which pose an environmental risk.10,11 As milder oxidation processes, biological oxidation12,13 and electrochemical removal14,15 have also been reported; however, it is difficult to treat a large amount of wastewater using these processes. Therefore, the development of an alternative pre-oxidation process without strong oxidants is necessary to maintain environmental safety.
We investigated the catalytic oxidation of As(III) in water to As(V) using dissolved oxygen on a heterogeneous catalyst. Oxygen is a mild oxidant without environmental risks, and it can be used to treat a large amount of wastewater. We found that a supported Pt catalyst (Pt/SiO2) can activate oxygen to react with As(III) in water.16 To develop a catalytic oxidative method for effective As removal, we investigated the combined effect of Pt/SiO2 with Fe(III), which is one of the precipitants for As removal.2,17 It is important for a practical utilization to simplify the process by the combination of the precipitant. In this study, the As precipitation behaviour with the Fe(III) precipitant and the catalytic performance of the Pt/SiO2 catalyst at various temperatures were confirmed, and the combination of Pt/SiO2 and Fe(III) for effective As(III) removal was evaluated.
2. Experimental
2.1. Catalyst preparation
According to our previous research,16 a Pt catalyst supported on SiO2 (Pt/SiO2) was prepared using an impregnation method. Silica (CARiACT Q-50, Fuji Silysia Chemical Ltd) was suspended in distilled water containing Pt(NH3)2(NO2)2 (Tanaka Kikinzoku Kogyo K. K.) as a metal precursor. After evaporating water at 80 °C, the obtained powder was reduced with 10% H2/N2 at 350 °C for 2 h. The metal content was 1 wt%, and the specific metallic surface area was 1.3 m2 gcat−1, which was calculated using the CO chemisorption technique (BELCAT-B, MicrotracBEL).
2.2. As precipitation tests with Fe(III)
Arsenic precipitation tests without Pt/SiO2 were performed in test tubes. An arsenic solution with a concentration of 20 mg L−1 was prepared using sodium arsenite (NaAsO2, Aldrich) or sodium arsenate (NaAsO3, Aldrich), and hydrogen chloride (HCl, Aldrich) was added to the solution to keep pH = 3 in all experiments. It is known that protons formed in As(III) oxidation decrease pH of the solution,18,19 and we have confirmed the change in pH (Fig. S1†). The temperature range of precipitation was 30–70 °C, and the amount of Fe(III) precipitant (FeCl3, Kanto Chemical) was in the range of Fe/As = 1–100. After 120 min from the addition of Fe(III), the solution was filtered through a PTFE filter of 0.22 μm pore size PTFE filter to remove the precipitate. The total concentration of arsenic ions (As(III) + As(V)) in the filtered solution was analysed using inductively coupled plasma optical emission spectrometry (ICP-OES; ICPE-9820, Shimadzu). The As(V) concentration was determined using the molybdenum blue method.16,20 The absorbance of the coloured solution at 750 nm and 846 nm was measured using a UV-vis spectrophotometer (UC-2400, Shimadzu). The amount of precipitate was calculated from the difference in the concentration of the solution. In the case of Fe/As = 100, because the ICP peak of As (189 nm) overlapped with that of the Fe satellite peak (192 nm), the total concentration of As ions could not be determined.
2.3. As oxidative removal
Arsenite(III) oxidation tests were performed in a batch reactor with a continuous air supply. An arsenite solution (100 mL) with a concentration of 30 mg L−1 was also prepared with NaAsO2 and HCl. After the temperature was increased to 30–70 °C, air was supplied for 30 min to reach the saturation solubility of oxygen, and then, 100 mg of the catalyst was added to the solution. During the oxidation tests, air was continuously supplied to the solution. The sampled solution was filtered through a PTFE filter and analysed using ICP-OES and UV-vis spectrophotometry. In a combined removal test of As(III) oxidation and precipitation with Fe(III), Fe(III) precipitant with Fe/As = 10 was added to the solution before catalyst addition. After the combined removal test, the residue was observed using scanning electron microscopy (SEM; SU3500, Hitachi).
3. Results and discussion
3.1. As(III) and As(V) precipitation with Fe(III)
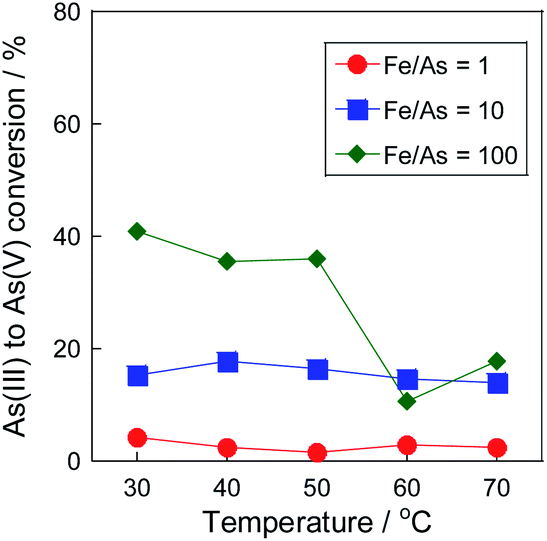
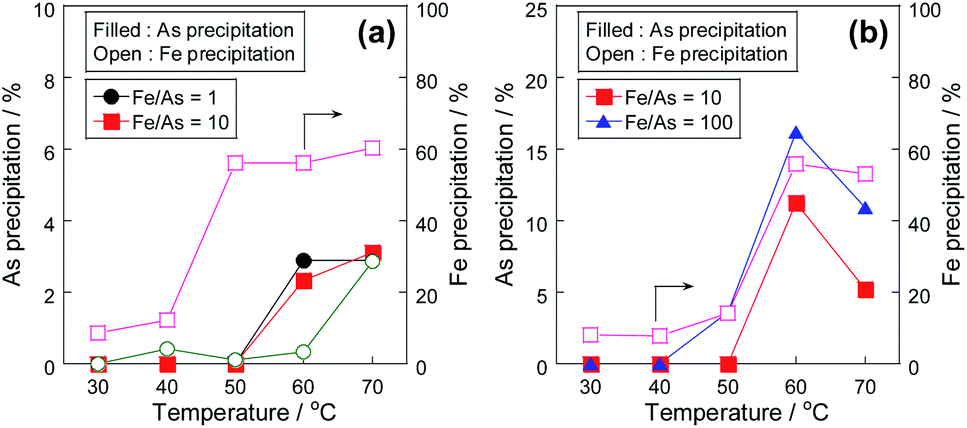
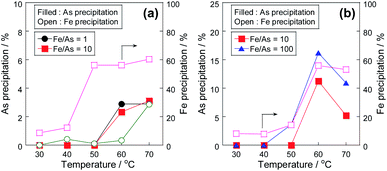
The precipitation tests of arsenite(III) and arsenate(V) without Pt/SiO2 were performed at various temperatures to investigate the precipitation behaviour of arsenic with the Fe(III) precipitant. While the co-precipitation of As(V) with iron precipitates is widely known,17,21–23 in this study, the difference in the precipitation behaviour between As(III) and As(V) was compared. Fig. 1 shows the conversion of As(III) to As(V) with various Fe(III) concentrations, and Fig. 2 shows the As and Fe precipitation ratios in Fig. 1. At low Fe/As molar ratios of 1 and 10, As(III) was oxidized with Fe(III), and the As(III) conversion was almost the same at these temperatures. However, the As(III) conversion decreased at high temperatures in the case of excess Fe(III) concentration of Fe/As = 100. At high temperatures, Fe precipitation was confirmed, as shown in Fig. 2; therefore, Fe(III) did not work as the oxidant because of the formation of its precipitate. The precipitation mechanism of As(III) with Fe(III) has been reported. Fe(III) reacts with water at high temperatures to form iron hydroxide, which then forms a co-precipitate with arsenic ions.2,17 The precipitation behaviour of Fe(III) was similar to that of As, as shown in Fig. 2, and we consider that the same co-precipitation mechanism can be used in this study.
 |
| | Fig. 1 Temperature dependency of As(III) conversion to As(V) in the precipitation test. Initial As concentration: 20 mg L−1 and without stirring. | |
 |
| | Fig. 2 Temperature dependency of the precipitation ratio of (a) As(III) and (b) As(V) with Fe(III) precipitant. Initial As concentration: 20 mg L−1 and without stirring. | |
In the case of the As(III) precipitation, while the precipitation proceeded from 50 °C, the precipitation ratio was only 3% at 70 °C. In contrast to the results of As(III) precipitation, the precipitation ratio in the case of As(V) was over 10% at 60 °C, as shown in Fig. 2(b). Because the precipitation ratio of Fe(III) was almost the same, the formation of co-precipitation with As(V) was superior to that with As(III), as previously reported;2,17 thus, it was confirmed that the development of the conversion system of As(III) to As(V) in the solution was indispensable for environmental safety. However, when Fe(III) was added to the As(III) solution, as shown in Fig. 1, Fe(III) worked as the oxidant at low temperatures but did not work at high temperatures due to the formation of its precipitate. Therefore, to develop a combinative removal of As precipitates, a process that can oxidize As(III) to As(V) effectively at high temperatures is necessary; thus, we investigated the addition of a Pt/SiO2 catalyst to promote As(III) oxidation with air.
3.2. As(III) oxidation on Pt/SiO2 catalyst
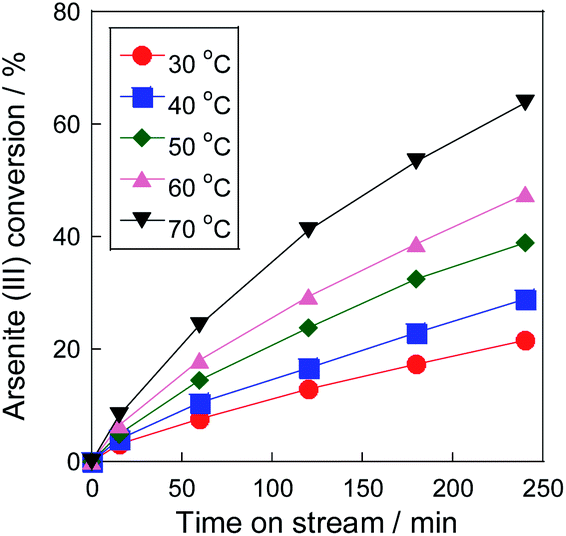
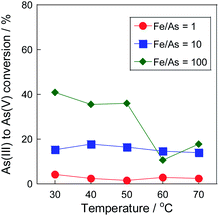
Fig. 3 shows As(III) conversion on the Pt/SiO2 catalyst with air at various temperatures. The conversion increased as the increase of the reaction temperature, and reached over 60% within 240 min at 70 °C. In our previous research,16 a 5 wt% Pt/SiO2 catalyst showed 100% of As(III) conversion within only 20 min at 80 °C with pure O2 flow. From the temperature dependency, the conversion would not reach such the high conversion in the conditions, therefore, O2 concentration was found to be an important factor for high As(III) conversion on the Pt/SiO2 catalyst. Of note, we revealed that Pt/SiO2 can oxidize As(III) even with air, which is the most practical gas for commercial utilizations. Therefore, the catalytic oxidation using Pt/SiO2 is effective method for As(III) oxidation at 70 °C in subsequent to the precipitation step with Fe(III) precipitant.
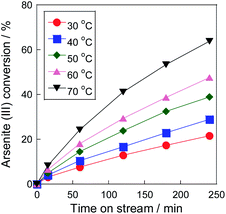 |
| | Fig. 3 Temperature dependency of As(III) oxidation on Pt/SiO2 catalyst. Initial As(III) concentration: 30 mg L−1. | |
From the dependency, the apparent activation energy (Ea) of As(III) oxidation on the Pt/SiO2 catalyst was calculated using the Arrhenius plot with a differential kinetic analysis (Fig. S2†). The apparent activation energy was approximately 30 kJ mol−1, which was similar to that reported in our previous study using a Pt/ZrO2 catalyst.16 Because the apparent activation energies in other removal systems are 60 kJ mol−1 with sulfite,24 46 kJ mol−1 with peroxydisulfate,25 and 53 kJ mol−1 with zero-valent iron and persulfate,26 it was found that the Pt catalyst is effective for As(III) oxidation over a wide temperature range. Please note that the experiment was carried out under dilute conditions, and the temperature dependency of dissolved oxygen was not considered in the calculation. The O2 concentration at 30 °C was almost twice that at 70 °C, which is almost the same concentration as that of arsenite in the solution (30 ppm). Therefore, it was assumed that the actual activation energy was higher than the calculated value of 30 kJ mol−1. Furthermore, the turnover frequency (TOF), calculated using the metallic surface area derived from CO chemisorption was 2.7 h−1 at 30 °C and 8.6 h−1 at 70 °C. Assuming that the arsenic concentration in wastewater is 1 ppm, the liquid hourly space velocity (LHSV) calculated from the TOF at 70 °C is approximately 6000 h−1 in wastewater basis. The throughput in the photocatalytic process was calculated as 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1.27 The removal performance was inferior to these processes in maximum, but our catalytic process was independent of environmental factors, such as weather and climate. Therefore, it can be said that the process has a possibility for the commercial utilization as the pre-oxidation method of As(III) in water with air.
000 h−1.27 The removal performance was inferior to these processes in maximum, but our catalytic process was independent of environmental factors, such as weather and climate. Therefore, it can be said that the process has a possibility for the commercial utilization as the pre-oxidation method of As(III) in water with air.
3.3. Combinative As(III) removal
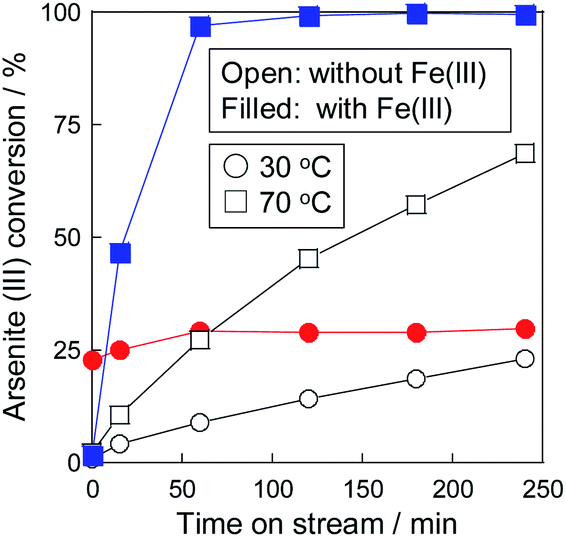
Considering the results of the co-precipitation and catalytic oxidation, a combinative As(III) removal of the oxidation and precipitation was performed. Fig. 4 shows the As(III) conversion at 30 and 70 °C in the combinative removal system. In the test at 30 °C, Fe(III) did not work as the precipitant but worked as the oxidant, as mentioned in the previous section. While As(III) is catalytically oxidized by oxygen in air, as shown in Fig. 3, oxidation on the catalyst could not be observed in the presence of Fe(III). It was considered that the Fe(III) ions in the solution inhibited the adsorption of reactants onto the catalyst.
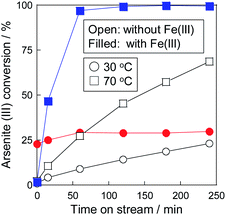 |
| | Fig. 4 Combinative arsenite removal as precipitate via a pre-oxidation on Pt/SiO2 and a precipitation with Fe(III) in one-pot at 30 °C and 70 °C. | |
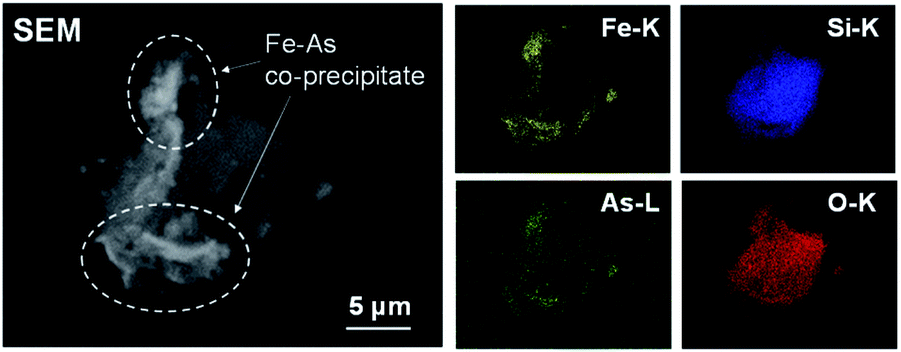
In contrast, the promotion of As(III) conversion and the formation of As precipitate was achieved in the combinative removal test at 70 °C. The As removal performance was higher than the conversion at each step (Fig. 2 and 3), indicating that the synergetic effect between Pt/SiO2 and Fe(III) was confirmed in the combined removal test. It is considered that the reason for the improvement in the removal performance is not the shift of the chemical equilibrium, but that Pt/SiO2 is responsible for not only the reaction site but also the nucleation of the precipitate. Electron microscopic observations of the residue, as shown in Fig. 5, revealed that an Fe–As precipitate was formed on Pt/SiO2. Syam Babu et al. reported an Fe–As precipitation mechanism following formulas:28
| | |
3FeOOH + HAsO42− → (FeO)3AsO4 + H2O + 2OH−
| (1) |
| | |
Fe3+ + H3AsO4 + 2H2O → FeAsO4·2H2O + 3H+
| (2) |
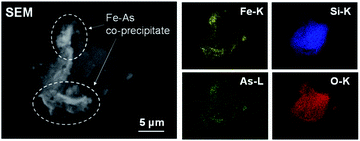 |
| | Fig. 5 SEM image and EDS mapping of precipitate in the sequential removal test at 70 °C. | |
The Fe/As molar ratio of the precipitate determined from the EDS spectrum was approximately 10, which was larger than the stoichiometric ratio from eqn (1) and (2). The result revealed that As ions were not formed the compound such as (FeO)3AsO4 shown in eqn (1), but removed by co-precipitation involved in Fe precipitation.
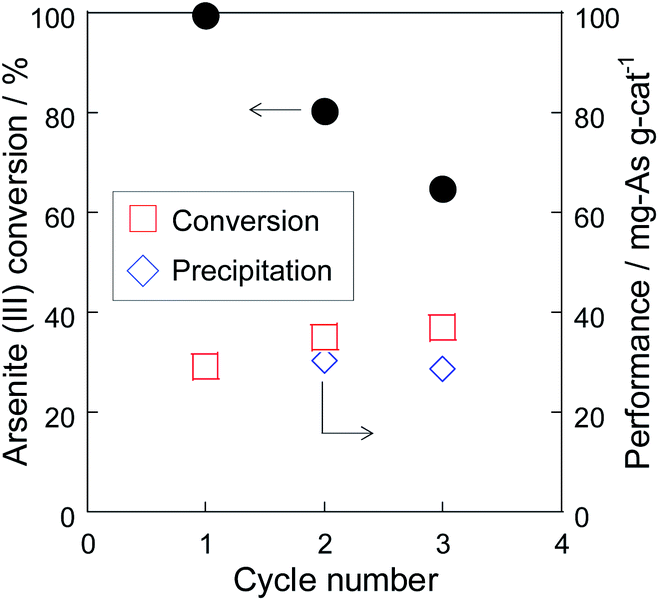
To evaluate the stability of Pt/SiO2, the residue recovered from the co-precipitation test was repeatedly used as the catalyst of a cyclic test (Fig. 6). In the cyclic test, the recovery ratio of the catalyst residue was almost 80% due to the loss in the sampling and the filtration. Although the As(III) conversion decreased due to the decrease of the catalyst weight, the catalytic performance, which meant the amount of the As(III) conversion per catalyst weight [mgAs gcat−1], did not decrease as show in Fig. 6. In addition, the performance of the As precipitation did not decrease in the cyclic test, indicating that the catalyst could be used repeatedly. It was found that Pt/SiO2 was also responsible for the nucleation site of the precipitate, but the precipitate did not remain strongly on Pt/SiO2 and the residue could be used repeatedly as the catalyst. Therefore, the combinative As(III) removal using Pt/SiO2 and Fe(III) significantly promoted the As removal performance due to the synergetic effect of oxidation and precipitation.
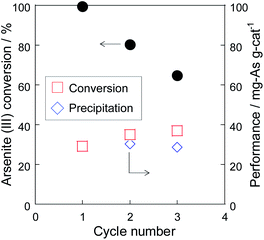 |
| | Fig. 6 Cyclic test of the As removal using the residue of the co-precipitation test at 70 °C. | |
4. Conclusions
The combination of As(III) oxidation and precipitation was investigated to ensure the effective removal of As(III) from wastewater using a Pt/SiO2 catalyst and an Fe(III) precipitant. As precipitation with Fe(III) required a pre-oxidation step to form As(V) and a high temperature of approximately 70 °C to form Fe(III) precipitates. The Fe(III) also functioned as an oxidant for As(III) even at a low temperature of 30 °C, whereas its oxidative performance decreased at a high temperature of 70 °C. The addition of the Pt/SiO2 catalyst promoted As(III) oxidation with oxygen in air, and, a significant improvement in As(III) removal was achieved with the synergetic effect of the Pt/SiO2 catalyst and the Fe(III) precipitant.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (grant number 18H03856).
References
- D. Mohan and C. U. Pittman Jr, J. Hazard. Mater., 2007, 142, 1–53 CrossRef CAS.
- L. Hao, M. Liu, N. Wang and G. Li, RSC Adv., 2018, 8, 39545–39560 RSC.
- B. Liu, K. H. Kim, V. Kumar and S. Kim, J. Hazard. Mater., 2020, 388, 121815 CrossRef CAS PubMed.
- S. Alka, S. Shahir, N. Ibrahim, M. J. Ndejiko, D.-V. N. Vo and F. A. Manan, J. Cleaner Prod., 2021, 278, 123805 CrossRef CAS.
- R. Amen, H. Bashir, I. Bibi, S. M. Shaheen, N. K. Niazi, M. Shahid, M. M. Hussain, V. Antoniadis, M. B. Shakoor, S. G. Al-Solaimani, H. Wang, J. Bundschuh and J. Rinklebe, Chem. Eng. J., 2020, 396, 125195 CrossRef CAS.
- S. Wang and C. N. Mulligan, Environ. Int., 2008, 34, 867–879 CrossRef CAS PubMed.
- F. S. Zhang and H. Itoh, Chemosphere, 2006, 65, 125–131 CrossRef CAS PubMed.
- G.-h. Moon, D.-h. Kim, H.-i. Kim, A. D. Bokare and W. Choi, Environ. Sci. Technol. Lett., 2014, 1, 185–190 CrossRef CAS.
- Q. Yang, Y. Dai, Z. Huang, J. Zhang, M. Zeng and C. Shi, RSC Adv., 2019, 9, 29689–29698 RSC.
- M.-J. Kim and J. Nriagu, Sci. Total Environ., 2000, 247, 71–79 CrossRef CAS.
- S. Sorlini and F. Gialdini, Water Res., 2010, 44, 5653–5659 CrossRef CAS PubMed.
- A. V. Bagade, S. P. Bachate, B. B. Dholakia, A. P. Giri and K. M. Kodam, J. Hazard. Mater., 2016, 318, 742–750 CrossRef CAS PubMed.
- B. Casentini, M. Gallo and F. Baldi, J. Environ. Chem. Eng., 2019, 7, 102908 CrossRef CAS.
- C. S. Fan, S. C. Tseng, K. C. Li and C. H. Hou, J. Hazard. Mater., 2016, 312, 208–215 CrossRef CAS PubMed.
- M. M. Alam, M. A. Rashed, M. M. Rahman, M. M. Mohanmmed, Y. Nagao and M. A. Hasnat, RSC Adv., 2018, 8, 8071–8079 RSC.
- J. Zhao, H. Matsune, S. Takenaka and M. Kishida, Chem. Eng. J., 2017, 325, 270–278 CrossRef CAS.
- V. T. Luong, E. E. Canas Kurz, U. Hellriegel, T. L. Luu, J. Hoinkis and J. Bundschuh, Water Res., 2018, 133, 110–122 CrossRef CAS PubMed.
- D. H. Kim, A. D. Bokare, M. Koo and W. Choi, Environ. Sci. Technol., 2015, 49, 3506–3513 CrossRef CAS.
- C. Wu, H. Mahandra and A. Ghahreman, Ind. Eng. Chem. Res., 2020, 59, 9882–9889 CrossRef CAS.
- R. K. Dhar, Y. Zheng, J. Rubenstone and A. van Geen, Anal. Chim. Acta, 2004, 526, 203–209 CrossRef CAS.
- C. C. Fuller, J. A. Davis and G. A. Waychunas, Geochim. Cosmochim. Acta, 1993, 57, 2271–2282 CrossRef CAS.
- C. M. van Genuchten, S. E. Addy, J. Pena and A. J. Gadgil, Environ. Sci. Technol., 2012, 46, 986–994 CrossRef CAS PubMed.
- A. Ahmad, S. Rutten, M. Eikelboom, L. de Waal, H. Bruning, P. Bhattacharya and A. van der Wal, Sep. Purif. Technol., 2020, 235, 116644 CrossRef.
- W. Ding, X. Huang, W. Zhang, F. Wu and J. Li, Chem. Eng. J., 2019, 359, 1518–1526 CrossRef CAS.
- L. Chai, J. Yang, F. Liao, Q. Li, Q. Wang, H. Liu, Q. Dong and Z. Yin, Mol. Catal., 2017, 438, 113–120 CrossRef CAS.
- I. Hussain, M. Li, Y. Zhang, S. Huang, W. Hayat, Y. Li, X. Du and G. Liu, J. Environ. Chem. Eng., 2017, 5, 3983–3990 CrossRef CAS.
- Z. Wei, Y. Fang, Z. Wang, Y. Liu, Y. Wu, K. Liang, J. Yan, Z. Pan and G. Hu, Chemosphere, 2019, 225, 434–442 CrossRef CAS.
- D. Syam Babu, K. Vijay, P. V. Nidheesh and M. Suresh Kumar, Sustain. Energy Technol. Assessments, 2021, 47, 101476 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Hiromichi Kondo,
Eriko Konishi,
Tsuyoshi Yamamoto,
Yoshifumi Tsuge,
Takayuki Watanabe
,
Hiromichi Kondo,
Eriko Konishi,
Tsuyoshi Yamamoto,
Yoshifumi Tsuge,
Takayuki Watanabe