
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic N-methyl amidation of carboxylic acids under cooperative conditions†
Li Yingxian‡
a,
Chen Wei‡a,
Zhao Linchuna,
Zhang Ji-Quana,
Zhao Yonglong a,
Li Chuna,
Guo Bingb,
Tang Lei*a and
Yang Yuan-Yong*a
a,
Li Chuna,
Guo Bingb,
Tang Lei*a and
Yang Yuan-Yong*a
aState Key Laboratory of Functions and Applications of Medicinal Plants, School of Pharmacy, Guizhou Provincial Engineering Technology Research Center for Chemical Drug R&D, Guizhou Medical University, 550014 Guiyang, P. R. China. E-mail: yangyuanyong@gmc.edu.cn; 2317972657@qq.com
bGuizhou Provincial Key Laboratory of Pathogenesis and Drug Research on Common Chronic Diseases, Guizhou Medical University, 550004 Guiyang, P. R. China
First published on 15th July 2022
Abstract
Amide is a fundamental group that is present in molecular structures of all domains of organic chemistry and the construction of this motif with high atom economy is the focus of the current research. Specifically, N-methyl amides are valuable building blocks in natural products and pharmaceutical science. Due to the volatile nature of methyl amine, the generation of N-methyl amides using simple acids with high atom economy is rare. Herein, we disclose an atom economic protocol to prepare this valuable motif under DABCO/Fe3O4 cooperative catalysis. This protocol is operationally simple and compatible with a range of aliphatic and (hetero)aromatic acids with very good yields (60–99%). Moreover, the Fe3O4 can be easily recovered and high efficiency is maintained for up to ten cycles.
Amide is a fundamental group that is present in molecular structures of all domains of organic chemistry.1 It is widely distributed in natural products, synthetic drugs and functional polymers, and is also the key chemical connection in proteins.2 It has been shown that amide bond formation alone accounts for 65% of all preliminary screening reactions in the pharmaceutical industry.3 This means the generation of amide bonds with high atom efficiency is of high practical importance. And not surprisingly, ‘amide formation avoiding poor atom economy reagents’ was voted as the top challenge for organic chemistry by the ACS Green Chemistry Institute in 2007.3
From synthetic point of view, the ideal way to produce amide bonds would be the direct coupling of readily available carboxylic acids and amines, but this process is thermodynamically unfavourable due to the formation of the corresponding carboxylate-ammonium salt,4 therefore, stoichiometric amount of coupling reagents, such as DCC, DIC, EDCI, HATU, HBTU, HCTU, SOCl2, BOP, acid chloride etc, are generally required to sidestep thermal conditions for amide bond formation.5 These reagents are highly successful, but the process generally suffers from poor atom economy and side products removal issue especially in the large-scale applications.5 To overcome these drawbacks, “nonclassical” amide bonds formation routes were investigated.6 In these processes, the catalyst takes the role of a coupling reagent in generating an active ester suitable for amidation in a waste-free manner. However, these processes have not been applied in the preparation of N-methyl amides, probably because the methyl amine was delivered in its hydrochloride salt, alcoholic or aqueous form due to its volatile nature.
On a different note, N-methyl amides are extensively presented in numerous natural products and pharmaceutical molecules, as shown in Fig. 1,7 and the methylation of amides is a promising way to improve the pharmacological property of molecules.8 However, the synthesis of N-methyl amides compounds relies heavily on non-catalytic approaches.5,9 Catalytic approaches were also investigated by Hisaeda,10 Kundu,11 Li,12 Guo,13 Yu,14 Maruoka,15 Wang,16 Chen,17 Lamaty18 and their co-workers starting from nitriles, primiary amides, aldoximes, aldehydes, lignin, carbamoylsilane and alcohols. Until recently, Thakur,19 Marce,20 Sadeghzadeh21 and their co-workers developed elegant N-methyl amidation approach starting from carboxylic acids under nano-MgO, diatomite Earth@IL/ZrCl4 and Mg(NO3)2·6H2O catalysis respectively, while limitations like poor substrate scope or sophisticated tailored catalyst still persist. Mindful of all the above issues, developing an N-methyl amidation process of simple carboxylic acids, which is still of great challenge in synthesis, and establishing a broad (hetero)aryl scope with high atom economy from commercial available reagents and catalysts were critical considerations in this study. Moreover, the significance of N-methyl amides combined with our interests in the development of green synthetic approaches motivated us to explore the direct coupling of the carboxylic acids and isothiocyanates. To the best of our knowledge, this is the first successful work using isothiocyanatomethane to prepare N-methyl amides.
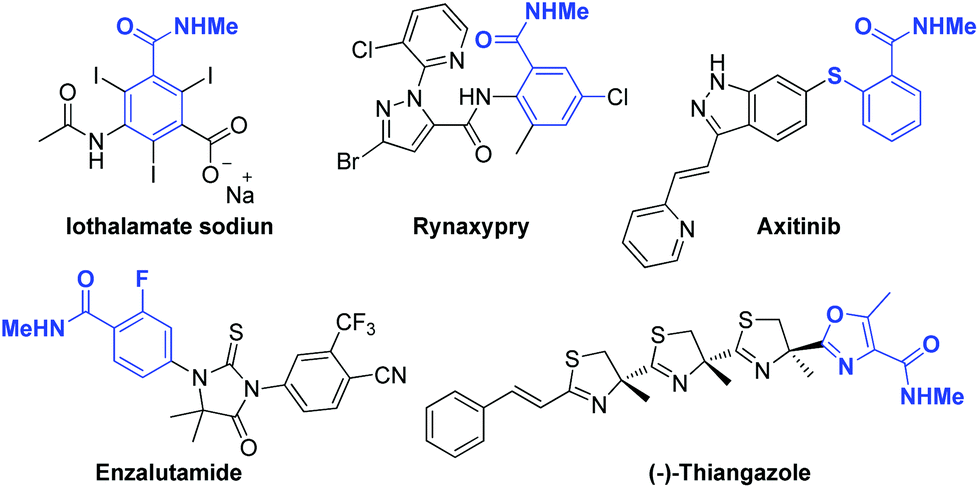 | ||
| Fig. 1 Marketed drugs bearing N-methyl amide group. | ||
Our initial investigation begins with phenylacetic acid and isothiocyanatomethane as model substrate for condition optimization. Using acetonitrile as solvent, only trace amount of product was detected under catalyst free or p-toluenesulfonic acid (PTSA) catalysis conditions (Table 1, entry 1 and 2). Then, triethylamine was applied to increase the nucleophilicity of the acid and 17% yield was obtained after 48 hours (Table 1, entry 3). Consequently, different nucleophilic bases that can activate the isothiocyanate were screened and the chemical yield was increased to 65% under DABCO catalysis (Table 1, entry 3–7). Later on, in hoping of better reaction efficiency, Lewis acids that capable of lower the LUMO of the electrophile were screened. Delightfully, the chemical yield could be increased to 71% with mild Lewis acid lithium bromide and further increased to 75% with Mn(OAc)2 (Table 1, entry 8 and 9). Because the acetate from the Mn(OAc)2 may consume the isothiocyanatomethane, MnO was used to replace Mn(OAc)2 and better yield was achieved (Table 1, entry 10). With these encouraging results, further endeavor were devoted to the identification of efficient metal oxides and eventually the cheap and abundant Fe3O4 was found to be optimal (Table 1, entry 11–13). More importantly, the benefit of Fe3O4 was twofold. Firstly, due to the magnetic nature of Fe3O4, it could be easily recovered under magnetic conditions. Secondly, iron is not a toxic element comparing with other transition metals, therefore the trace amount of Fe3O4 residue will not be a problem even in the industry production. Further interrogation of reaction time and temperature reveals that shorter reaction time and lower reaction temperature was detrimental (Table 1, entry 14 and 15). Therefore, 10 mol% DABCO and 10 mol% Fe3O4 at 85 °C in MeCN was established as the optimal reaction conditions for substrate screening.
|
||||
|---|---|---|---|---|
| Entry | Additive | Time (h) | Catalyst | Yield (%) |
| a Reactions were run on 1 mmol 1a and 1.1 mmol 2a with 10 mol% catalyst and 10 mol% additive in 1 mL of MeCN at 85 °C for 48 hours unless otherwise noted.b Reaction was conducted at 60 °C. | ||||
| 1 | — | 24 | — | 5 |
| 2 | — | 24 | PTSA | — |
| 3 | — | 48 | TEA | 17 |
| 4 | — | 48 | DBU | 45 |
| 5 | — | 48 | DMAP | 43 |
| 6 | — | 48 | DBN | 51 |
| 7 | — | 48 | DABCO | 65 |
| 8 | LiBr | 48 | DABCO | 71 |
| 9 | Mn(OAc)2 | 48 | DABCO | 75 |
| 10 | MnO | 48 | DABCO | 79 |
| 11 | MgO | 48 | DABCO | 88 |
| 12 | Al2O3 | 48 | DABCO | 85 |
| 13 | Fe3O4 | 48 | DABCO | 98 |
| 14 | Fe3O4 | 24 | DABCO | 75 |
| 15b | Fe3O4 | 48 | DABCO | 80 |
Firstly, different acids were employed to react with isothiocyanatomethane and the results were summarized in Table 2. Various aliphatic acids are compatible with our optimized reaction conditions, good to excellent yields were realized (Table 2, entry 3a–3y). Generally, electro-rich substrates give better yields compared with electro-deficient substrates (Table 2, entry 3a–3z). Moreover, functional groups like hydroxyl (3i), alkynyl (3p), alkenyl (3o), ether (3r) and amide (3s) were tolerated with our reaction conditions. Later on, steric groups were installed onto the acid and isothiocyanate partner to study the steric effect on the substrates (Table 2, entry 3q and 3w). The results reveal that the steric effect pose negligible effect on the acid so that different marketed drugs like duplosan (3r), indomethacin (3s), ketoprofen (3t) and naproxen(3u) were transformed into the corresponding methyl amide under current reaction conditions with very good yields (Table 2, entry 3r–3u). Finally, different isothiocyanates were tested and found to be compatible with current reaction conditions, good yields were achieved for both aliphatic and aromatic isothiocyanates (Table 2, entry 3v–3y).
| a Reactions were run on 1 mmol 1 and 1.1 mmol 2 with 10 mol% DABCO and 10 mol% Fe3O4 in 1 mL of MeCN for 48 hours at 85 °C unless otherwise noted. |
|---|
 |
Subsequently, aromatic and heteroaromatic acids were tested for their compatibility with current reaction conditions and the results were summarized in Table 3. The results reveal our reaction conditions was compatible with various aromatic and heteroaromatic acids and good to excellent yields were obtained (Table 3, entry 3aa–3ar). Generally, electron-withdrawing groups take longer reaction time to get satisfied yield (Table 3, entry 3af and 3ag), while dinitro substrate give moderate yield even under prolonged reaction time (Table 3, entry 3ah). Interestingly, when 2-mercaptobenzoic acid was used as substrate, the cyclic product 3ak was obtained as the sole product in excellent yield. More importantly, methyl amides derived from aromatic and heteroaromatic acids are of medicinal significance. For example, compound 3aj, 3ak and 3ap are key fragment of enzalutamide, axitinib and (−)-thiangazole (as shown in Fig. 1) respectively, and all could be convenient prepared using current procedure with excellent yields.
| a Reactions were run on 1 mmol 1 and 1.1 mmol 2 with 10 mol% DABCO and 10 mol% Fe3O4 in 1 mL of MeCN for 48 hours at 85 °C unless otherwise noted. |
|---|
 |
Furthermore, to demonstrate the synthetic utilization of our methodology, the preparation of bioactive compounds was demonstrated Scheme 1. Compounds 3as is a patent HDAC4 inhibitor.22 Under the standard reaction conditions, 3as could be obtained from commercial available 1as in 92% yield. Our procedure is much more atom economy as it excluded the usage of activating reagent and excess amount of base. In a recent report, Yang group reported their pilot-scale synthesis of substituted phenylacetamides to tetrahydroisoquinoline-2-ones.23 In their practice, corrosive thionyl chloride was applied as activating reagent and large excess amount of methyl amine was required, however, moderate yield of 3a was obtained, while our method can achieve better yield along with the exclusion of corrosive thionyl chloride. Following this report, tetrahydroisoquinoline-2-one 4a could be obtained in 82% yield, which could be used in the preparation of various bioactive 4-aryl-tetrahydroisoquinolines 5a with known procedure.24
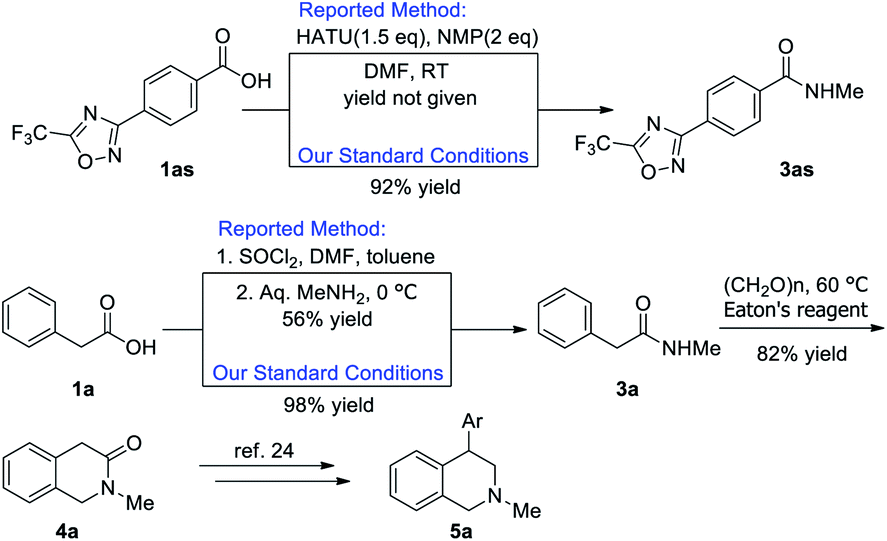 | ||
| Scheme 1 Application of N-methyl amide. | ||
Finally, owing to the magnetic nature of Fe3O4, we try to recover the Fe3O4 from the reaction system and test its efficiency. As the Fe3O4 is always stick to the magnetic stir bar, after the termination of the reaction, the reaction solution was pour out and the tub along with the magnetic stir bar was rinsed with MeCN three times, oven dried and used for the next cycle. The results shown that the Fe3O4 could be used 10 times and still maintained very good efficiency (Fig. 2).
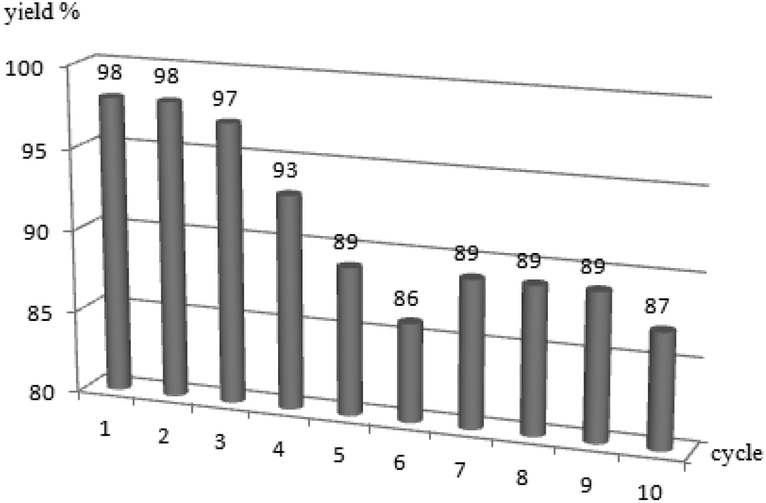 | ||
| Fig. 2 The efficiency of recovered Fe3O4. | ||
Combined with the literature reports and experimantal observation,25 a plausable mechanism was proposed in Scheme 2. Firstly, the carboxylic acid reacts with the Fe3O4 to get iron (II and III) carboxylate A, which will coordinate to the intermediate B generated from DABCO and isothiocyanate to get intermediate C. Then, one of the carboxylate attack intermediate B to release DABCO and generates intermediate D. Intermediate D go through an intramolecular addition to generate intermediate E, which go through a rearrangement reaction to get intermediate F with the release of carbonyl sulfide. Finally, the protonation of F with carboxylic acid to get the final product and regenerate the iron (II and III) carboxylate A.
 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Conclusions
To summarize, we have developed a catalytic atom economy amidation process of simple acids with isothiocyanates using cheap and abundant DABCO and Fe3O4. Our method is operational simple, exclusion of air or moisture is not required, and compatible with a range of acids and isothiocyanates. Moreover, the Fe3O4 could be easily recovered under magnetic conditions for up to 10 times with high efficiency. Particularly, considering the volatile nature of methyl amine, our method would be a valuable supplement of existing methyl amides preparation mythology, which will found wide application in pharmaceutical relevant compounds preparation and natural products synthesis.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for financial support from the National Natural Science Foundation of China (22061012), Department of Science & Technology of Guizhou Province ([2020]4Y208), Department of education of Guizhou Province ([2021]047), National-Local Joint Engineering Research Center for Innovative & Generic Chemical Drug, and Guizhou High-level Innovative Talents Supporting Program (2016-4015).Notes and references
- (a) A. Greenberg, C. M. Breneman and J. F. Liebman, The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, Wiley, 2000 Search PubMed; (b) V. Pattabiraman and J. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed.
- (a) A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55 CrossRef CAS PubMed; (b) T. Wieland, and M. Bodanszky, The world of peptides: a brief history of peptide chemistry, Springer, 1991 CrossRef.
- D. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- O. Mahe, J. Desroches and J. F. Paquin, Eur. J. Org. Chem., 2013, 20, 4325 CrossRef.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140 CrossRef CAS.
- (a) R. M. Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029 CrossRef PubMed; (b) J.-Y. Chen, H.-Y. Wu, Q.-W. Gui, X.-R. Han, Y. Wu, K. Du, Z. Cao, Y.-W. Lin and W.-M. He, Org. Lett., 2020, 22, 2206 CrossRef CAS PubMed; (c) W.-H. Bao, Z. Wang, X. Tang, Y.-F. Zhang, J.-X. Tan, Q. Zhu, Z. Cao, Y.-W. Lin and W.-M. He, Chin. Chem. Lett., 2019, 30, 2259 CrossRef CAS; (d) R. M. Lanigan, P. Starkov and T. D. Sheppard, J. Org. Chem., 2013, 78, 4512 CrossRef CAS PubMed; (e) T. M. E. Dine, W. Erb, Y. Berhault, J. Rouden and J. Blanchet, J. Org. Chem., 2015, 80, 4532 CrossRef PubMed.
- (a) E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215 CrossRef CAS PubMed; (b) J. Chatterjee, C. Gilon, A. Hoffman and H. Kessler, Acc. Chem. Res., 2008, 41, 1331 CrossRef CAS PubMed.
- J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed., 2013, 52, 254 CrossRef CAS PubMed.
- (a) Q. Yang, L. G. Ulysse, M. D. McLaws, D. K. Keefe, P. R. Guzzo and B. P. Haney, Org. Synth., 2012, 89, 44 CrossRef CAS; (b) L. G. Ulysse, L. Q. Yang, M. D. McLaws, D. K. Keefe, P. R. Guzzo and B. P. Haney, Org. Process Res. Dev., 2010, 14, 225 CrossRef CAS; (c) S. J. Ferrara and T. S. Scanlan, J. Med. Chem., 2020, 63, 9742 CrossRef CAS PubMed; (d) A. Khalafi-Nezhad, A. Parhami, M. N. S. Rad and A. Zarea, Tetrahedron Lett., 2005, 46, 6879 CrossRef CAS.
- H. Shimakoshi and Y. Hisaeda, Angew. Chem., Int. Ed., 2015, 54, 15439 CrossRef CAS PubMed.
- (a) B. Paul, M. Maji and S. Kundu, ACS Catal., 2019, 9, 10469 CrossRef CAS; (b) B. Paul, M. Maji, D. Panja and S. Kundu, Adv. Synth. Catal., 2019, 361, 5357 CrossRef CAS; (c) B. Paul, D. Panja and S. Kundu, Org. Lett., 2019, 21, 5843 CrossRef CAS PubMed.
- C. Bai, X. Yao and Y. Li, ACS Catal., 2015, 5, 884 CrossRef CAS.
- M.-Z. Zhang, Q.-H. Guo, W.-B. Sheng and C.-C. g Guo, Adv. Synth. Catal., 2015, 357, 2855 CrossRef CAS.
- Y. Wang, Z. Wu, Q. Li, B. Zhu and L. Yu, Catal. Sci. Technol., 2017, 7, 3747 RSC.
- R. Sakamoto, S. Sakurai and K. Maruoka, Chem. – Eur. J., 2017, 23, 9030 CrossRef CAS PubMed.
- H. Li, M. Liu, H. Liu, N. Luo, C. Zhang and F. Wang, ChemSusChem, 2020, 13, 4660 CrossRef CAS PubMed.
- W. Tong, P. Cao, Y. Liu and J. Chen, J. Org. Chem., 2017, 82, 11603 CrossRef CAS PubMed.
- X. Bantreil, N. Kanfar, N. Gehin, E. Golliard, P. Ohlmann, J. Martinez and F. Lamaty, Tetrahedron, 2014, 70, 5093 CrossRef CAS.
- V. K. Das, R. R. Devi and A. J. Thakur, Appl. Catal., A, 2013, 456, 118 CrossRef CAS.
- A. R. Chhatwal, H. V. Lomax, A. J. Blacker, J. Williams and P. Marcé, Chem. Sci., 2020, 11, 5808 RSC.
- M. Ahmadi, L. Moradi and M. Sadeghzadeh, Res. Chem. Intermed., 2018, 44, 7873 CrossRef CAS.
- C. Hebach, E. Joly, J. Kallen, J. G. Ternois, and M. Tintelnot-Blomley, US Pat., 9056843, 2015.
- L. G. Ulysse, L. Q. Yang, M. D. McLaws, D. K. Keefe, P. R. Guzzo and B. P. Haney, Org. Process Res. Dev., 2010, 14, 225 CrossRef CAS.
- S. Liu, C. Zha, K. Nacro, M. Hu, W. Cui, Y. L. Yang, U. Bhatt, A. Sambandam, M. Isherwood, L. Yet, M. T. Herr, S. Ebel-toft, C. Hassler, L. Fleming, A. D. Pechulis, A. Payen-Fornicola, N. Holman, D. Milanowski, I. Cotterill, V. Mozhaev, Y. Khmelnitsky, P. R. Guzzo, B. J. argent, B. F. Molino, R. Olson, D. King, S. Lelas, Y. W. Li, K. Johnson, K. T. Molski, A. Orie, A. Ng, R. Haskell, W. Clarke, R. Bertekap, J. O'Connell, N. Lodge, M. Sinz, S. Adams, R. Zaczek and J. E. Macor, ACS Med. Chem. Lett., 2014, 5, 760 CrossRef CAS PubMed.
- J. Tan, Y. Guo, F. Zeng, G. Chen and W. He, Chinese J. Org. Chem., 2018, 38, 1740 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03255d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |