DOI:
10.1039/D2RA03424G
(Paper)
RSC Adv., 2022,
12, 23937-23945
The adsorption behaviors of N2O on penta-graphene and Ni-doped penta-graphene
Received
2nd June 2022
, Accepted 9th August 2022
First published on 24th August 2022
Abstract
In order to develop the adsorption application of penta-graphene (PG) to N2O gas molecule, we calculated the sensing properties of PG and Ni-doped PG to N2O molecule via first-principles calculations. Based on the calculated adsorption energy, charge transfer, band gap, density of states and partial density of states, we observed that this gas molecule was weakly physically adsorbed on the surface of intrinsic PG, while the adsorption behaviors on the surface of Ni-doped PG were greatly influenced by the doping sites and adsorption orientations. With the Ni atom doped at the sp2 hybridized carbon site, strong chemical adsorption between the gas molecule and the substrate was induced. The adsorption structure of the N2O molecule with its N atom close to the substrate exhibited better stability. Moreover, an external perpendicular electric field could enhance the adsorption performance of the N2O molecule and adjust the charge transfer between the molecule and substrate. Our results broaden the adsorption applications of PG and indicate that Ni-doped PG is a potential candidate for N2O gas sensors.
1. Introduction
Penta-graphene (PG) is a new two-dimensional material with a metastable carbon pentagonal structure. Compared with conventional hexagonal graphene, which has robust sp2 binding among carbon atoms, PG contains both sp2 and sp3 hybridized carbon atoms.1 The unique hybridized bonds enable PG exceptional mechanical properties,2 low thermal conductivity,3 compelling optical property,4 ultrahigh carrier migration rate,5 and energetic and dynamic stabilities.1 Because of its excellent features, PG is anticipated to develop into a potential anode candidate for Li/Na-ion batteries,6 a competitive and metal-free catalyst for CO oxidation at a low temperature,7 and a potential gas sensor for some harmful gases.8–10 The band gap of PG makes it a semiconductor, and the tetrahedral character of the sp3-hybridized carbon gives PG a buckle shape, which is not ideally planar but rather oscillates out-of-plane in a periodically corrugated manner. Therefore, PG can adsorb gas molecules in rich configurations, and it is considered a potential candidate for developing gas sensors.
Recently, the identification and elimination of harmful gases from carbon-based structures have attracted considerable attention. Many researchers have shifted their main focus from traditional 2D materials, such as graphene,11 MoS2 (ref. 12) and BN,13 to other graphene-like 2D materials14–16 and carbon-based materials, such as graphyne17 and PG. It has been reported that the CO, H2O, H2S, NH3 and SO2 gas molecules are physically adsorbed on intrinsic PG,18 whereas the NO and NO2 molecules on PG belong to chemisorption. Moreover, doping can improve the adsorption ability of gas molecules. It has also been reported that transition-metal-doped PG is a potential hydrogen-storage material19 and Zhang20 et al. observed that transition-metal-doped PG can significantly enhance the adsorption performance of CO and CO2 gas molecules. However, to the best of our knowledge, no previous reports have focused on the adsorption behaviors of N2O gas molecules on the intrinsic and doped PG, which is highly desirable to be explored. N2O gas molecules mainly arise from the selective catalytic reduction of NOx and the burning of fossil fuels and biomass.21,22 Since the excessive emission of N2O accelerates the exhaustion of ozone in the stratosphere,23 it is necessary to lower the concentration level of N2O gas in chemical processes using highly sensitive materials with efficient adsorption performance.
Among the transition metal atoms, we mainly focus on the last three successive groups because they have filled and near filled d shell electrons. Moreover, for the sake of electrochemical stability and economic cost, we chose a cheap and promising metal, namely nickel, as the dopant in our study, which improved the admirable catalytic behavior24 and outstanding gas adsorption application25,26 in previous studies. Although the Ni-doped penta-graphene has not been synthesized experimentally, the Ni-doped graphene and intrinsic penta-graphene have been synthesized experimentally.27,28 We believe that the synthesis of transition metal doped penta-graphene can be achieved in the near future. In our study, the sensing properties of PG and Ni-doped PG to the small gas molecule N2O were investigated via first-principles calculations. Based on the unique structure of PG, different doping sites and adsorption orientations were considered before adsorption. The adsorption energy, charge transfer, band gap, density of states, and partial density of states were calculated to analyze the different adsorption behaviors. Moreover, in order to enhance the adsorption performance of N2O on the substrate, an external perpendicular electric field was employed to tune its adsorbability.
2. Methods
The projected-augmented wave (PAW) and plane wave basis method of the Vienna ab initio simulation package (VASP)29,30 were utilized to calculate the adsorption of N2O molecule via density functional theory (DFT).31 For the exchange-correlation potential, the generalized gradient approximation (GGA)32 of the Perdew–Burke–Ernzerhof (PBE)33 function was applied. Since PBE function fails to precisely convey van der Waals interactions caused by the undulating movement of electric charge, the DFT-D2 method was used to solve this problem.34 Spin polarization must be considered in the structural relaxation process. We constructed a 3 × 3 × 1 supercell PG containing 54 atoms. In the structural optimization process, the truncated energy was set to 520 eV, the k-point grid centered on gamma in the Brillouin zone was set to 3 × 3 × 1, and the convergence condition was set to 0.1 meV. In order to obtain an accurate density of states, the mesh point of k space was increased to 9 × 9 × 1. To eliminate the influence of the interaction between the adjoining PG layers, the height of the vacuum layer was set to 15 Å. The adsorption energy Ead was calculated using the following formula:| | |
Ead = Egas/substrate − Egas − Esubstrate,
| (1) |
where Egas/substrate, Egas and Esubstrate represent the total energies of N2O adsorbed on the substrate, isolated N2O molecule, and isolated substrate, respectively.
3. Results and discussion
3.1. The intrinsic PG
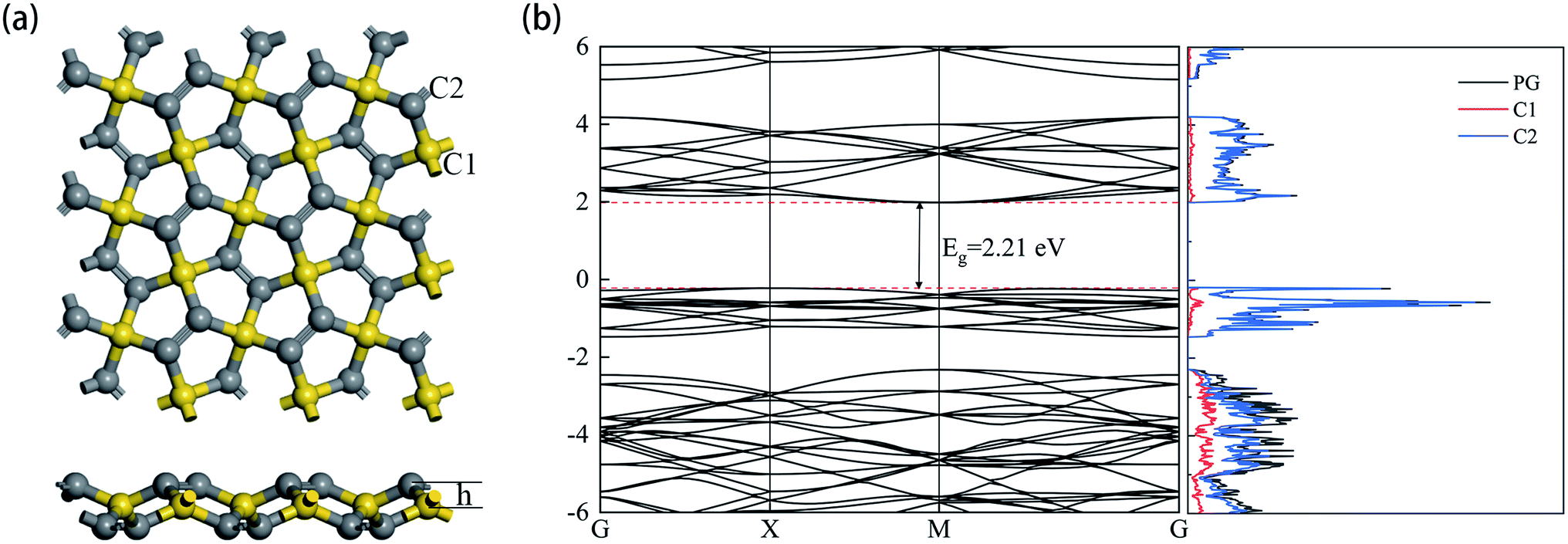
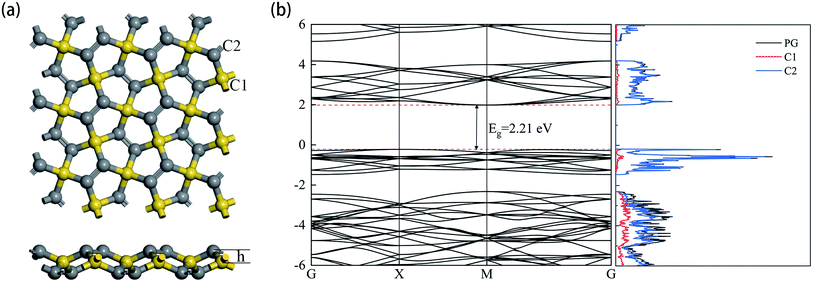
The optimized structure of the 3 × 3 × 1 supercell PG model is shown in Fig. 1a. The sp3 and sp2 hybridized carbon atoms are marked C1 and C2 in yellow and gray colors, respectively. The detailed parameters related to the PG sheet are shown in Table 1, which are consistent with previous studies.35 The plane composed of C1 atoms was 0.604 Å away from the coplanar structure consisting of C2 atoms. The band structure and projected density of states (DOS) are shown in Fig. 1b. The intrinsic PG exhibits a band gap Eg = 2.21 eV, and the primary contribution to the total DOS is derived from C2 atoms.
 |
| | Fig. 1 (a) Top and side views (b) the electronic band structure and density of the states of the relaxed PG sheet. | |
Table 1 Calculated lattice constant, bond length and bond angle between adjacent C1 and C2 atoms of the optimized 3 × 3 × 1 PG sheet
| Lattice constant (Å) |
Bond length (Å) |
Bond angle |
| a = b = 10.920 |
C1–C2 = 1.550 |
C2–C1–C2 = 134.386° |
| c = 15.00 |
C2–C2 = 1.339 |
C1–C2–C1 = 112.446° |
| C2–C2–C1 = 113.498° |
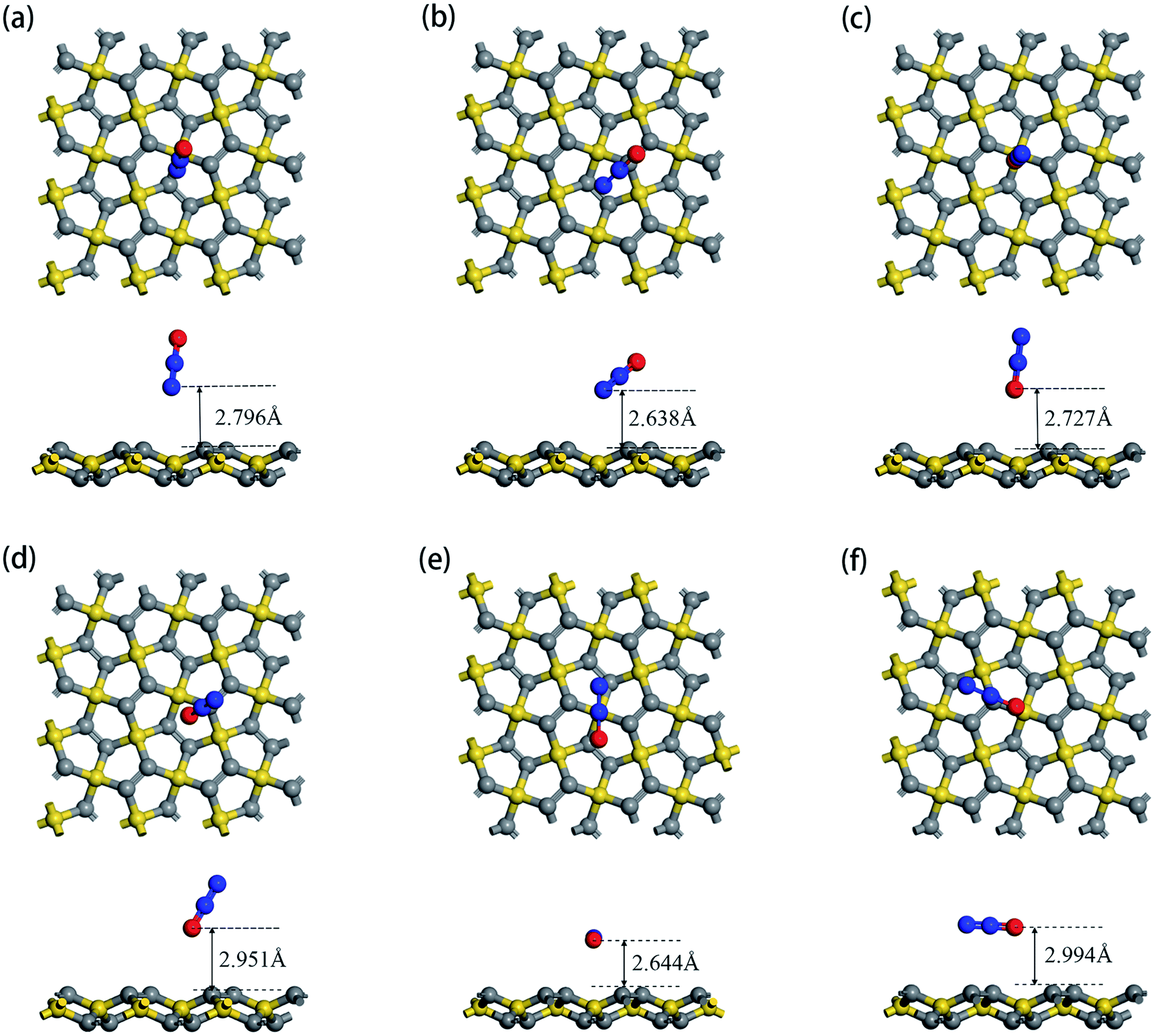
According to the structure of N2O gas molecules, three orientations were considered before the adsorption calculation: N2O molecule is vertical to the substrate end with O atom (O end), N2O molecule is vertical to the substrate end with N atom (N end), and N2O molecule is horizontal to the substrate. Because PG has two adsorption sites, C1 and C2, N2O has six adsorption orientations, as shown in Fig. 2. The corresponding characteristic parameters (including adsorption distance, adsorption energy, band gap and charge transfer) are listed in Table 2. The results show that N2O gas molecules with different adsorption orientations are far away from the PG sheet with adsorption distances from 2.638 Å to 2.994 Å, and the adsorption energies are quite small, ranging from −0.203 eV to −0.091 eV. Simultaneously, the N2O molecules act as electron acceptors during the adsorption process, obtaining trace amounts of electrons from the PG sheet, which are around 0.01e. With N2O adsorption, the band gaps of PG slightly decreased by around 0.01 eV, which is almost negligible. Combined with relatively small amounts of charge transfer, weak adsorption energies and large substrate-molecule adsorption distances, we believe that N2O molecules are physically adsorbed on the PG monolayer via van der Waals forces, and primitive PG is not a good choice for adsorbing and detecting the N2O gas molecule.
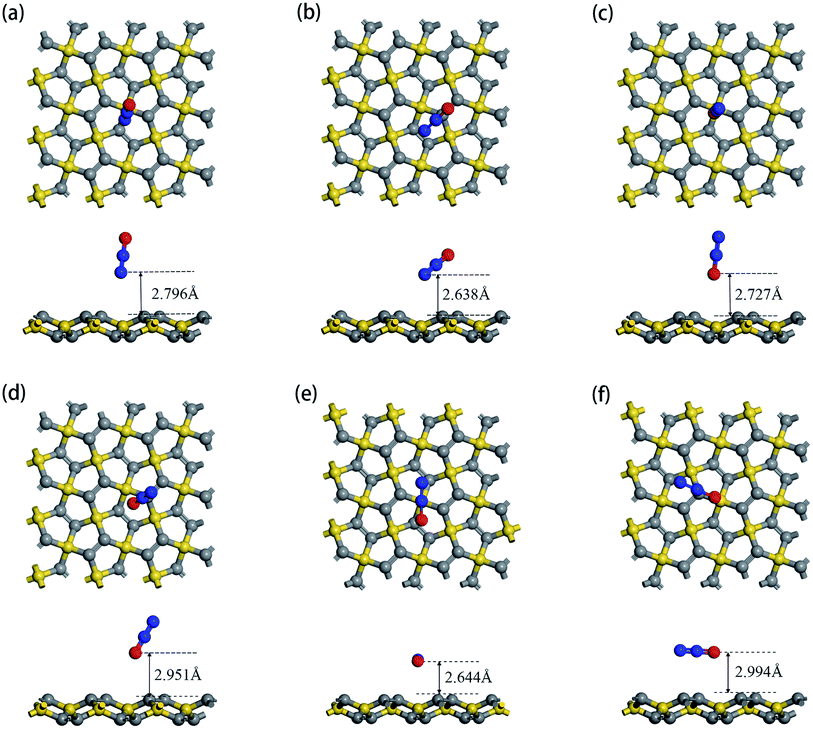 |
| | Fig. 2 Top view and side view of relaxed geometry structure of N2O molecule adsorpted on primitive PG sheet by different forms: N2O is vertical to PG with its O end upward at C1 site (a) and C2 site (b); N2O is vertical to PG with its O end downward at C1 site (c) and C2 site (d); N2O is horizontal to PG at C1 site (e) and C2 site (f). | |
Table 2 The adsorption energy Ead, charge transfer Q, and band gap Eg of N2O adsorbed on the PG sheet
| Orientation |
Adsorption site |
Ead (eV) |
Eg (eV) |
Q (e) |
| N end |
C1 |
−0.114 |
2.195 |
0.010 |
| N end |
C2 |
−0.152 |
2.193 |
0.010 |
| O end |
C1 |
−0.091 |
2.197 |
0.011 |
| O end |
C2 |
−0.102 |
2.198 |
0.009 |
| Horizontal |
C1 |
−0.203 |
2.197 |
0.015 |
| Horizontal |
C2 |
−0.156 |
2.189 |
0.018 |
3.2. The Ni-doped PG
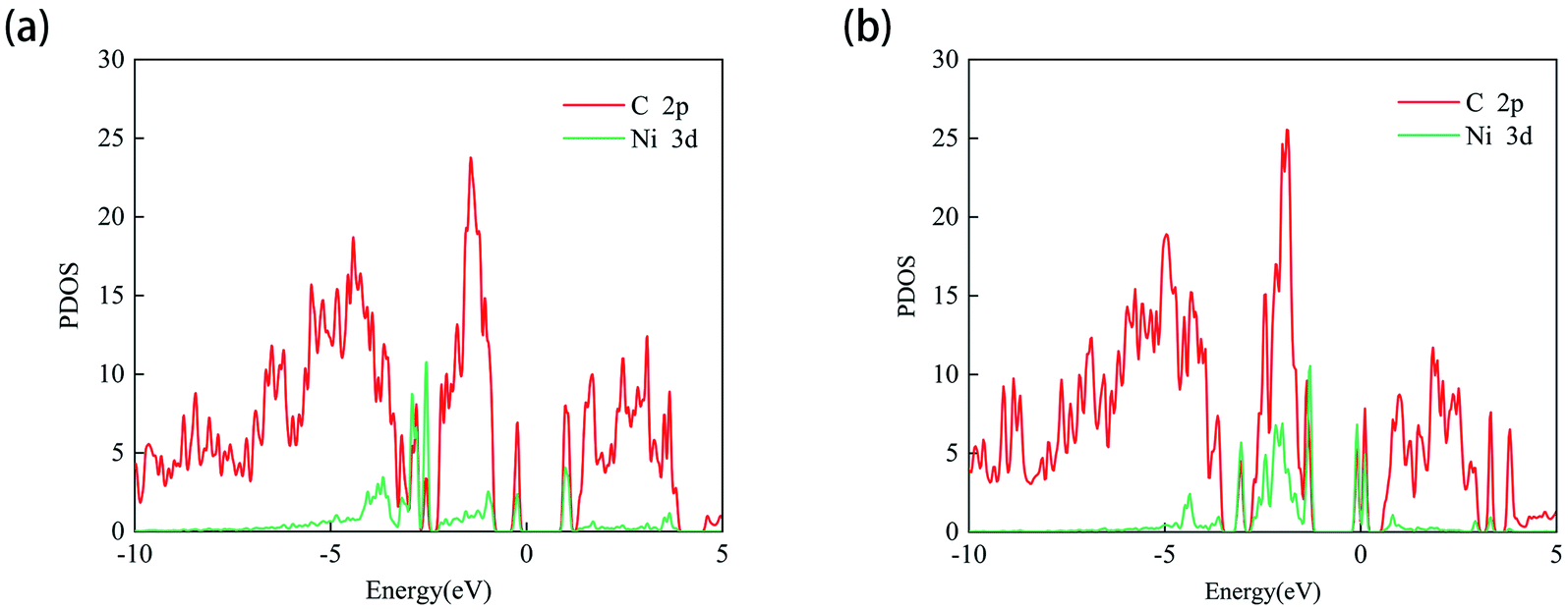
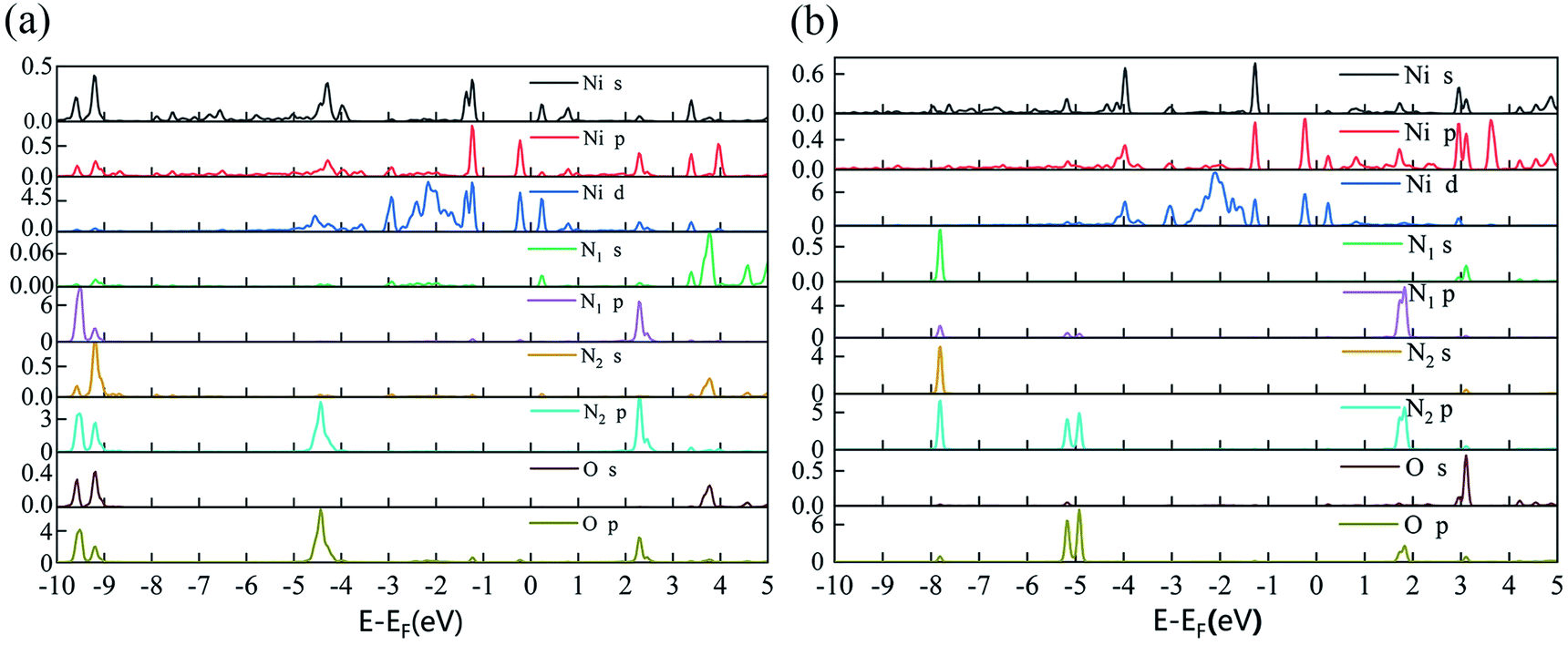
Since the adsorption of N2O gas molecules on the intrinsic PG is weak physical adsorption, the doping of transition metal Ni on PG is considered to modify the electronic properties of PG and enhance the adsorption of N2O on PG. Two different doping sites, C1 and C2, are used for Ni atoms to decorate after the formation of C1 and C2 vacancies, and these two systems are defined as Ni-1-PG and Ni-2-PG, respectively. The binding energy, band gap and charge transfer of Ni-doped PG are listed in Table 3. The binding energies of Ni-1-PG and Ni-2-PG are −6.742 eV and −5.037 eV, respectively; the charge transfer from the Ni atom to its adjacent C atoms is 0.643e and 0.560e, which helped in the stabilization of the structures.24 The calculated PDOS results are shown in Fig. 3, and strong overlapping peaks can be observed below and above the Fermi level. This reveals that the Ni atom can use its valence electrons to saturate the dangling-bond states around the vacancy site, and such strong coupling between the C-2p and Ni-3d states can maintain the Ni atom stability of the defect site. Moreover, the doping of the Ni atom significantly reduces the band gap of the structure. For Ni-1-PG, the value of the band gap decreases from 2.21 eV to 1.18 eV because new bands appear in the valence band in the band gap. While for Ni-2-PG, it decreases to 0.159 eV since new bands appear in both the valence and conduction bands of the gap. These consequences are due to the fact that donor atom Ni spreads its excess electrons to its surroundings, losing 0.643e and 0.560e at the C1 and C2 sites, respectively.
Table 3 The binding energy Eb, band gap Eg and charge transfer Q of Ni-1-PG and Ni-2-PG
| System |
Eb (eV) |
Eg (eV) |
Q (e) |
| Ni-1-PG |
−6.742 |
1.180 |
−0.643 |
| Ni-2-PG |
−5.037 |
0.159 |
−0.560 |
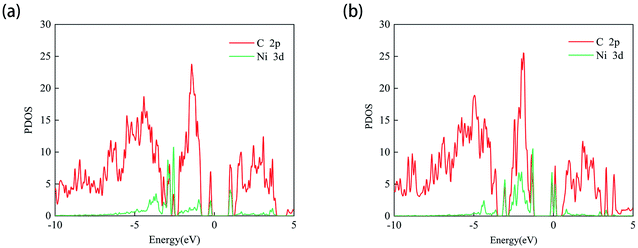 |
| | Fig. 3 PDOS of the Ni-1-PG (a) and Ni-2-PG (b) systems. The Fermi level is set to zero. | |
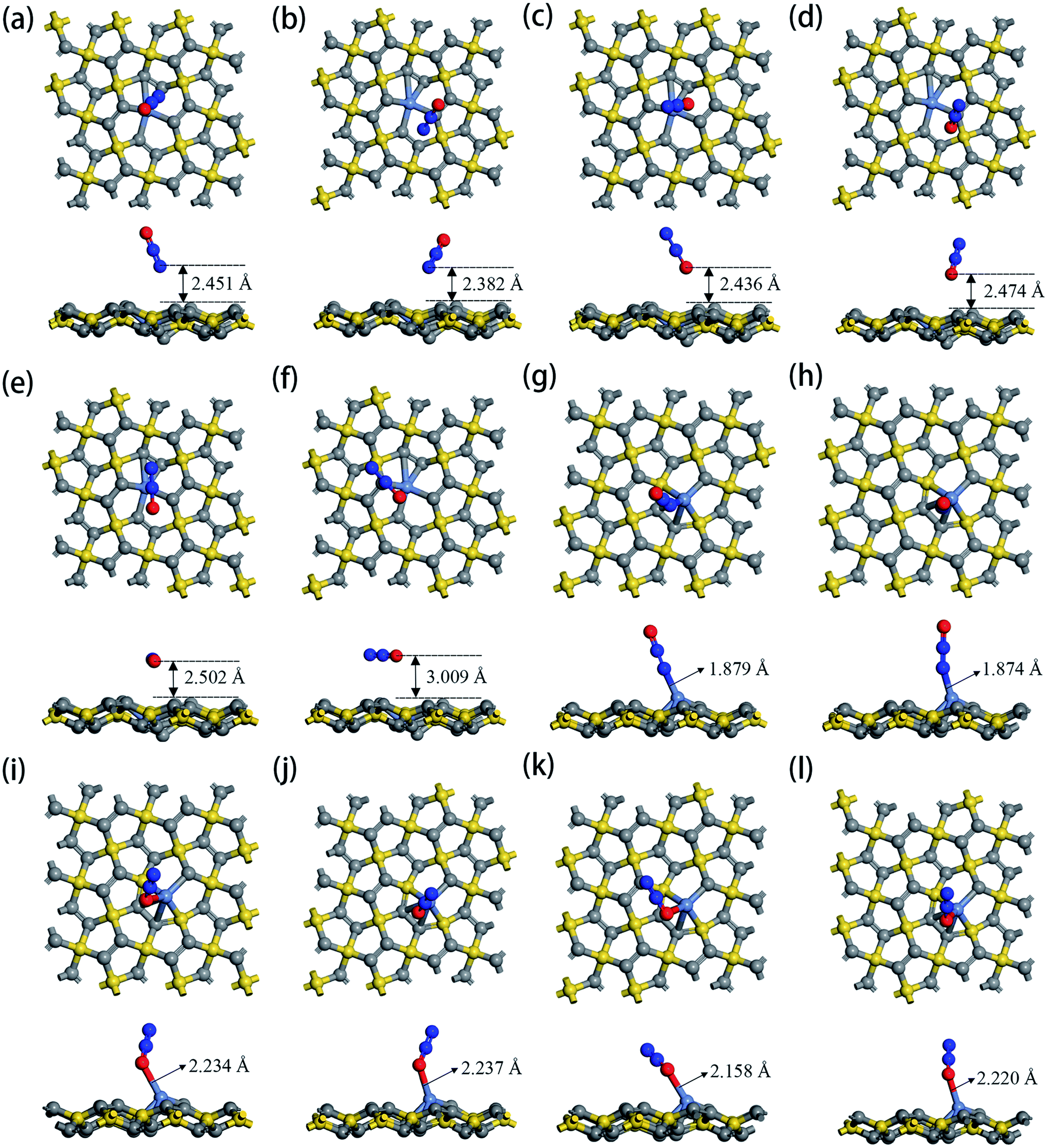
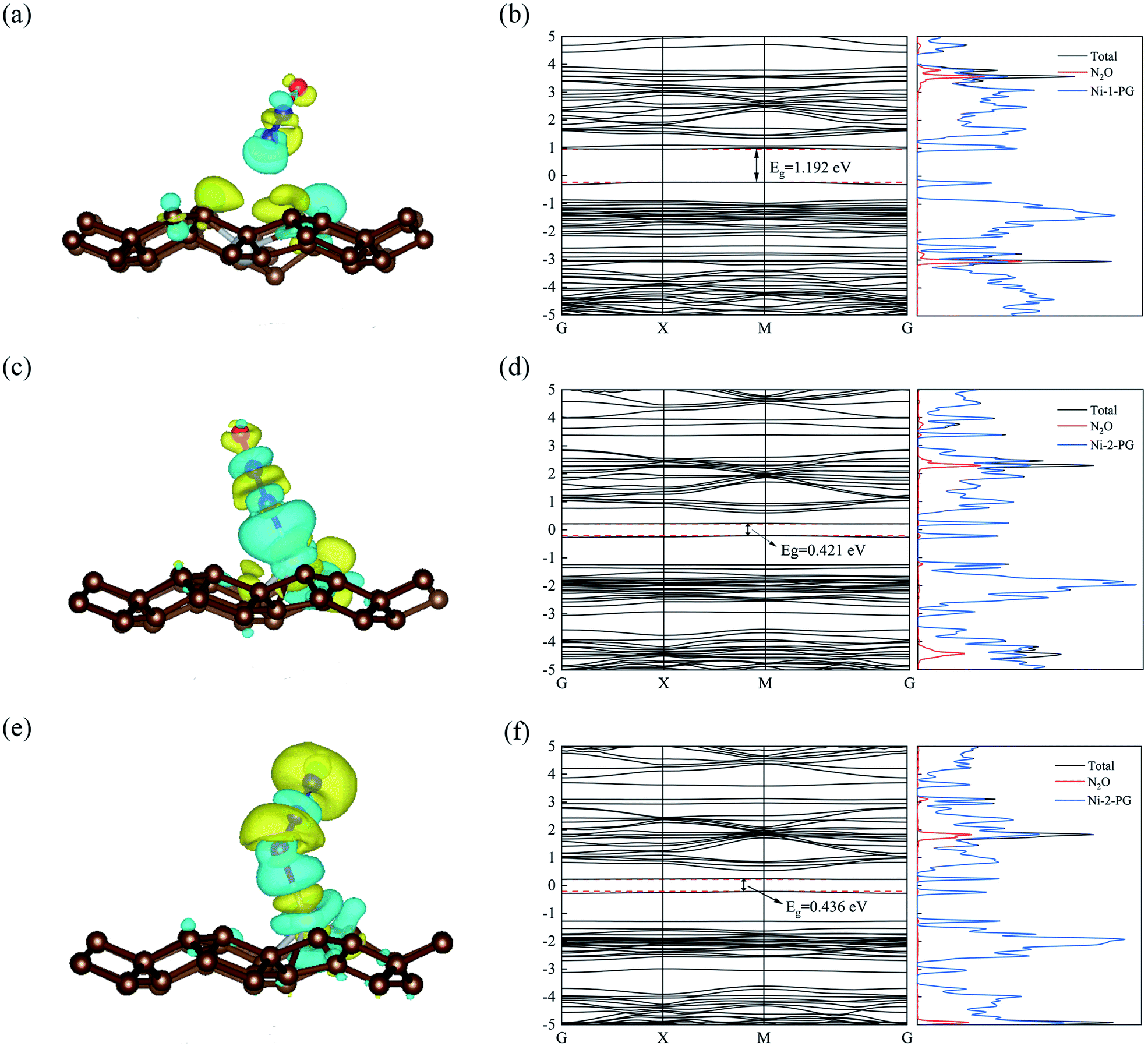
The optimized geometric configurations of N2O absorbed on Ni-1-PG and Ni-2-PG by pointing its N or O atom to the adsorption positions are shown in Fig. 4. The values of adsorption energy, adsorption distance, band gap and charge transfer of these adsorption configurations are summarized in Table 4. For N2O absorbed on Ni-1-PG systems, the adsorption energies are quite small, which are −0.077 eV, −0.107 eV, −0.056 eV, −0.072 eV, −0.178 eV and −0.089 eV. Furthermore, the adsorption distances are rather large, which are 2.451 Å, 2.382 Å, 2.436 Å, 2.474 Å, 2.502 Å and 3.009 Å. Moreover, after N2O adsorption, the band gap Eg slightly increases and the N2O gas molecule only gets less than 0.02 electrons for these six configurations. We take the configuration N2O1V2 as an example to plot the differential charge density, band structure and DOS, as shown in Fig. 5a and b. The band gap slightly increased from 1.180 eV to 1.192 eV after adsorption, and there is not any hybrid coupling in the band gap from the DOS of N2O molecules and Ni-1-PG, whose corresponding orbital contributions to the adsorption systems occurred at a position far away from the Fermi level. In addition, the charge accumulation and charge depletion regions are around the gas molecule and substrate, and few overlapping areas can be observed between the electron cloud of N2O and Ni-1-PG in Fig. 5a. This means that compared with the properties of N2O on primitive PG, the adsorption between the N2O molecule and Ni-1-PG sheet is improved but is still weak physical adsorption. Therefore, Ni-1-PG is not sensitive to N2O molecules and is inadvisable for detecting or eliminating N2O molecules in general.
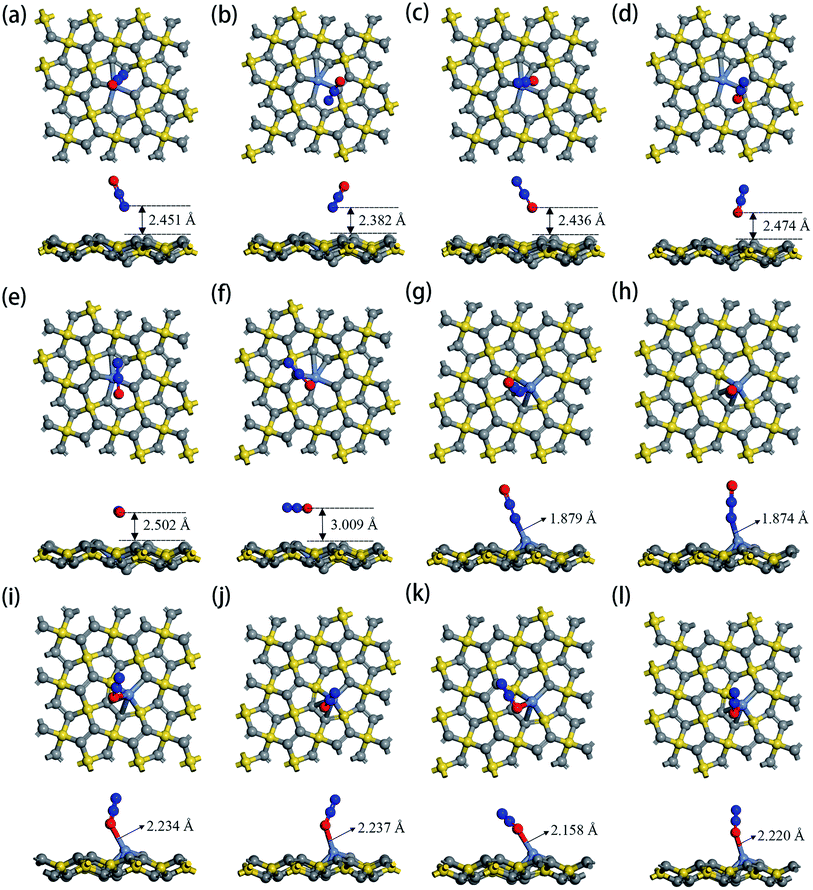 |
| | Fig. 4 Top and side views of optimized configurations of N2O absorbed on Ni-1-PG and Ni-2-PG: (a) N2O1V1, (b) N2O1V2, (c) N2O1V3, (d) N2O1V4, (e) N2O1H1, (f) N2O1H2, (g) N2O2V1, (h) N2O2V2, (i) N2O2V3, (j) N2O2V4, (k) N2O2H1, and (l) N2O2H2. | |
Table 4 The calculated adsorption energy Ead, adsorption distance d, band gap Eg and charge transfer Q of different orientations of N2O adsorbed on Ni-1-PG and Ni-2-PG
| System |
Orientation |
Ead (eV) |
d (Å) |
Eg (eV) |
Q (e) |
| N2O on Ni-1-PG |
Vertical to C1 and O is upward(N2O1V1) |
−0.077 |
2.451 |
1.201 |
0.012 |
| Vertical to C2 and O is upward (N2O1V2) |
−0.107 |
2.382 |
1.192 |
0.011 |
| Vertical to C1 and O is downward(N2O1V3) |
−0.056 |
2.436 |
1.192 |
0.013 |
| Vertical to C2 and O is downward(N2O1V4) |
−0.072 |
2.474 |
1.182 |
0.011 |
| Horizontal to C1(N2O1H1) |
−0.178 |
2.502 |
1.211 |
0.019 |
| Horizontal to C2(N2O1H2) |
−0.089 |
3.009 |
1.171 |
0.018 |
| N2O on Ni-2-PG |
Vertical to C1 and O is upward(N2O2V1) |
−0.944 |
1.690 |
0.421 |
0.125 |
| Vertical to C2 and O is upward (N2O2V2) |
−0.935 |
1.749 |
0.429 |
0.128 |
| Vertical to C1 and O is downward(N2O2V3) |
−0.346 |
1.956 |
0.430 |
0.007 |
| Vertical to C2 and O is downward(N2O2V4) |
−0.344 |
2.046 |
0.436 |
0.006 |
| Horizontal to C1(N2O2H1) |
−0.453 |
1.798 |
0.305 |
0.002 |
| Horizontal to C2(N2O2H2) |
−0.358 |
2.035 |
0.452 |
0.006 |
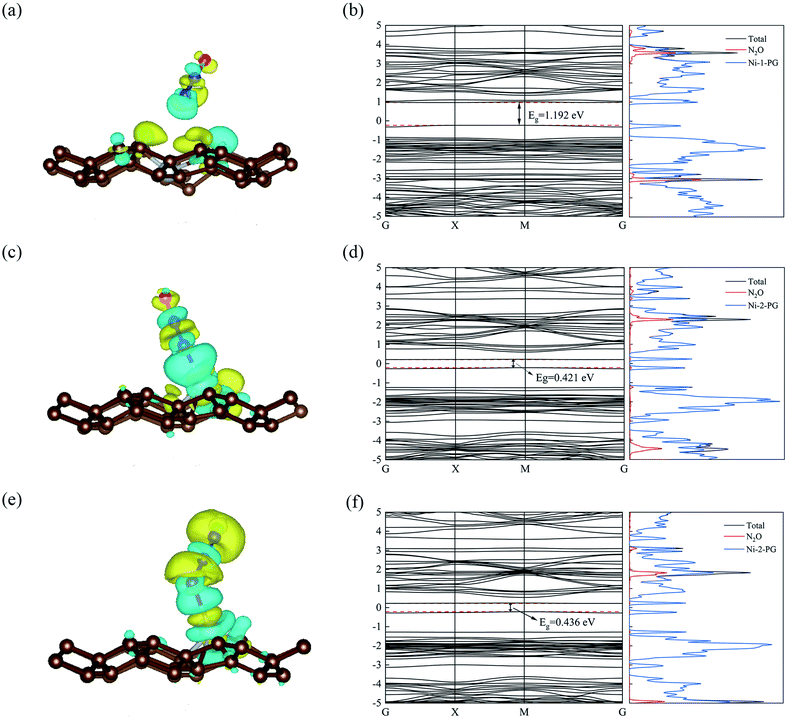 |
| | Fig. 5 The differential charge densities of the relaxed configuration: (a) N2O1V2 (c) N2O2V1 and (e) N2O2V4; the isovalues are 0.0002 a.u., 0.0009 a.u. and 0.0004 a.u., respectively; the blue and yellow represent the gain and loss of electrons, respectively. (b), (d) and (f) denote the band structure and DOS of N2O1V2, N2O2V1 and N2O2V4. The Fermi level is set to zero. | |
In contrast, Ni-2-PG exhibits excellent adsorption performance for N2O, which is affected by the adsorption orientations. When the O atom is upward, the adsorption energies are quite large and the adsorption distances are very small. N2O2V1 has better adsorption stability, with an adsorption energy of −0.944 eV and a shorter adsorption distance of 1.690 Å. This adsorption energy is larger than that of N2O adsorbed on Al-doped graphene (Ead = −0.57 eV),36 Fe-doped MoSe2 (Ead = −0.40 eV),37 and B-doped C3N nanosheet (Ead = −0.80 eV).38 When the O atom is downward, or N2O is horizontal, the adsorption energies are smaller and the adsorption distances are longer compared with those of N2O2V1 and N2O2V2. For example, N2O2V4 has less adsorption stability with an adsorption energy of −0.344 eV and a long adsorption distance of 2.046 Å. The differential charge density, band structure and DOS of N2O2V1 and N2O2V4 are plotted in Fig. 5c–f. For N2O2V1, N2O acts as an electron acceptor and has a large amount of charge (0.125e) from Ni-2-PG during the adsorption process. The charge interaction between N2O and substrate is quite obvious (Fig. 5c). The band gap increases after the adsorption process, but the orbital of the N2O molecules exhibits hybrid coupling with that of Ni-2-PG around the Fermi level (Fig. 5d), suggesting that the interaction between the N2O gas molecule and Ni-2-PG is strong and the N2O molecule is chemically adsorbed on Ni-2-PG. For N2O2V4, the band gap was further increased, and the corresponding orbital contribution of the adsorption system occurred at a position far away from the Fermi level, so the N2O molecule could only obtain a small amount of charge from Ni-2-PG. However, Fig. 5e shows an overlapped charge interaction region between the N2O molecule and substrate, which indicates that the configuration N2O2V4 is still chemical adsorption. In general, the adsorption strength of N2O is determined by the adsorption orientation: when the O atom is downward, the adsorption strength is relatively weaker, which indicates that the N2O molecule is likely to be desorbed from Ni-2-PG within these adsorption configurations; when the O atom is upward, the adsorption behavior is much stronger, and N2O molecule can be stably adsorbed on Ni-2-PG substrate.
Furthermore, the partial density of states (PDOS) of N2O2V1 and N2O2V4 are plotted to further investigate the adsorption effect of N2O on Ni-2-PG, and the results are shown in Fig. 6. For configuration N2O2V1, obvious orbital mixing exists between the N2O and substrate at the position very close to the Fermi level: the p orbital of N1 and O atoms in N2O overlap with the p and d orbitals of Ni on the surface of Ni-2-PG, at about −0.225 eV; the s and p orbitals of N1, p orbital of N2 and O atom overlap with the s, p and d orbitals of Ni, at about −1.241 eV. Furthermore, the p orbitals of N2 and O atoms in N2O have large overlapping peaks with the s, p and d orbitals of the Ni atom at about −4.437 eV, a little far away from the Fermi level. These results indicate that N2O2V1 has stable and strong chemical adsorption performance. While for configuration N2O2V4, the relatively overlapping area of the formant is far away from the Fermi level: the p orbitals of N1, N2 and O atoms in N2O overlap the s, p and d orbitals of Ni atom at about −4.923 eV and −5.178 eV. This means that a chemical bond is formed between the N2O gas molecule and the Ni-2-PG substrate, but the bond is weaker than that of N2O2V1 due to its relatively far and tiny overlapping area. The analysis discussed above demonstrates that Ni-2-PG has a greater adsorption performance, and the N2O molecule can be chemisorbed on it regardless of the adsorption orientation.
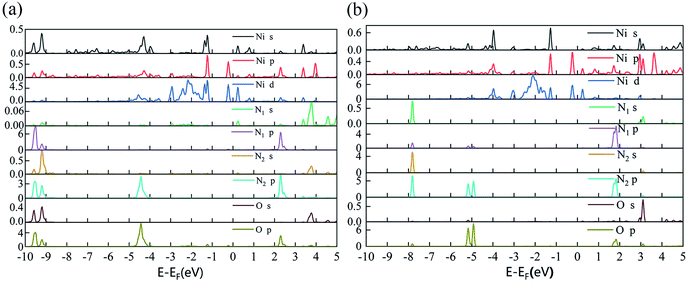 |
| | Fig. 6 The partial density of states of configuration: (a) N2O2V1 and (b) N2O2V4. | |
3.3. Applied electric field
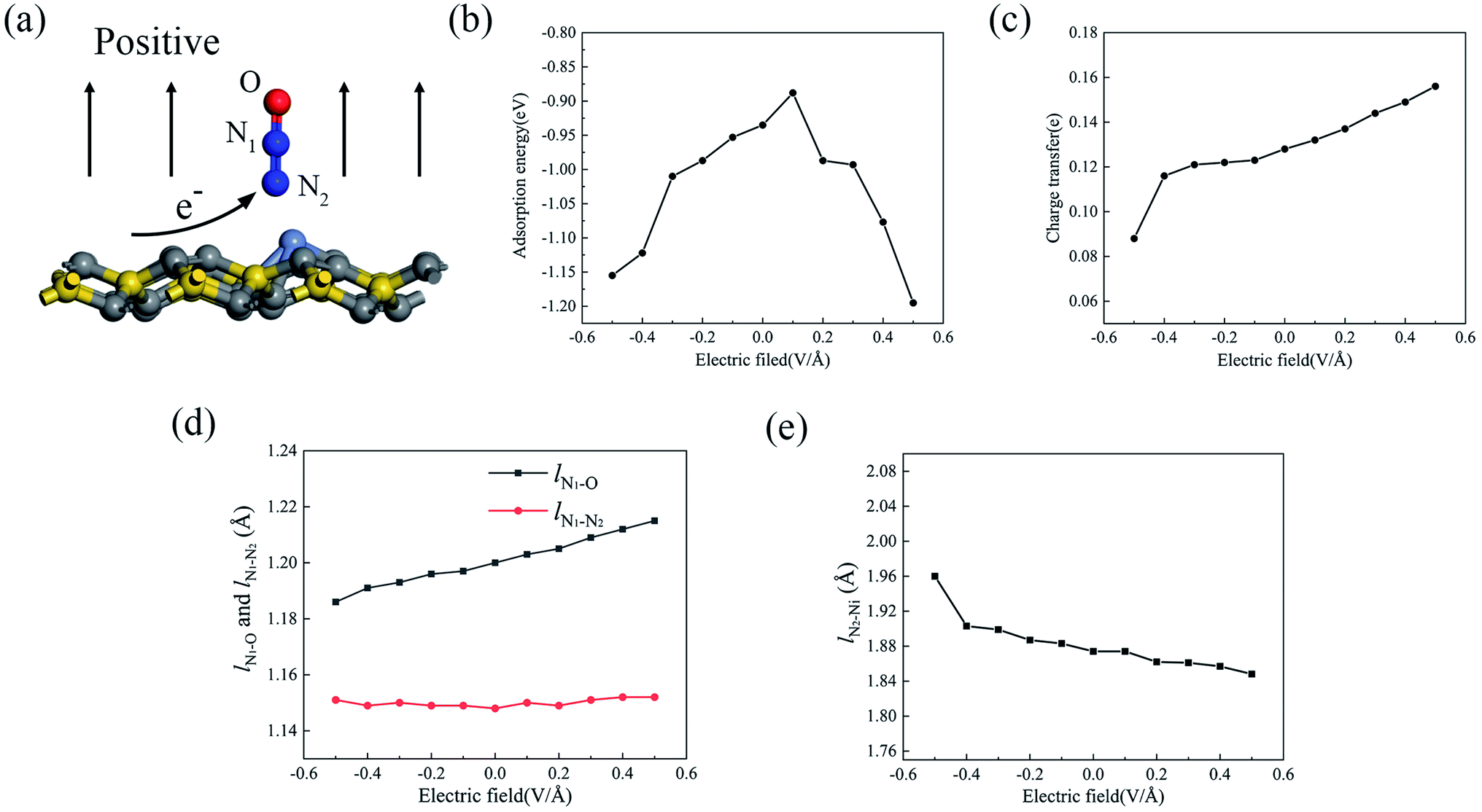
Since the external electric field can influence the adsorption property by adjusting the internal charge state,39–41 we applied an external perpendicular electric field to the adsorption configuration, and N2O2V2 was chosen as a candidate. As shown in Fig. 7a, the direction from the substrate toward the gas molecule (upward) is positive, whereas the opposite direction (downward) is negative. Fig. 7b displays the change in the adsorption energy with respect to the electric field from −0.5 V Å−1 to +0.5 V Å−1. It can be seen that the adsorption energy of N2O shows a significant increase with the increase in the electric field strength when either a positive or negative electric field is applied, except for the positive electric field value of +0.1 V Å−1, whose adsorption energy decreases to the minimum. Under the electric field value of +0.5 V Å−1, the adsorption energy increases to −1.195 eV, which is 28% higher than that with no extra electric field. As mentioned above, the property of adsorption energy under the electric field varies because of the adjustment of the atomic charge state between the absorbed gas molecule and the substrate, which may be caused by charge transfer, adsorption structure, and adsorption distance. Fig. 7c–e show the change curves of the charge transfer, the bond lengths of the N2O molecule (lN1–O and lN1–N2) and the formed chemical bond (lN2–Ni) as a function of the electric field. The positive electric field is capable of promoting the charge transfer process from the substrate to the absorbed molecule, whereas the negative electric field hinders the charge transfer induced by N2O adsorption. It was observed that the charge transfer from the Ni-2-PG substrate to the N2O gas molecule increased linearly when the electric field changed from −0.1 V Å−1 to +0.5 V Å−1. When the electric field reached +0.5 V Å−1, N2O achieves additional 0.028 electrons arising from the Ni-2-PG substrate. This alteration of charge transfer may be relevant to the dipole moment emerging between the absorbed gas molecule and substrate,42 which shows that the same directions of electric field and dipole moment enhance charge transfer while opposite directions weaken it. Furthermore, the dipole moment and charge transfer between the substrate and N2O further enhance the charge redistribution on the surface of Ni-2-PG, which is why the bond length lN2–Ni decreases with an increase in the electric field. This means that the adsorption distance decreases when the electric field is positive and increases when the electric field is negative. Moreover, the variation curves of the bond length lN1–O and lN1–N2 of the N2O gas molecule are plotted in Fig. 7d. With an increase in the applied electric field from negative to positive, the bond length between N1 and O atoms significantly increases, whereas the distance between N1 and N2 atoms remains almost constant at about 1.50 Å. Because the p orbital of the O atom plays an important role in the formation of chemical bonds in the adsorption process, when a negative electric field is applied, the bond length lN1–O decreases, causing a large adsorption energy and more stable adsorption.
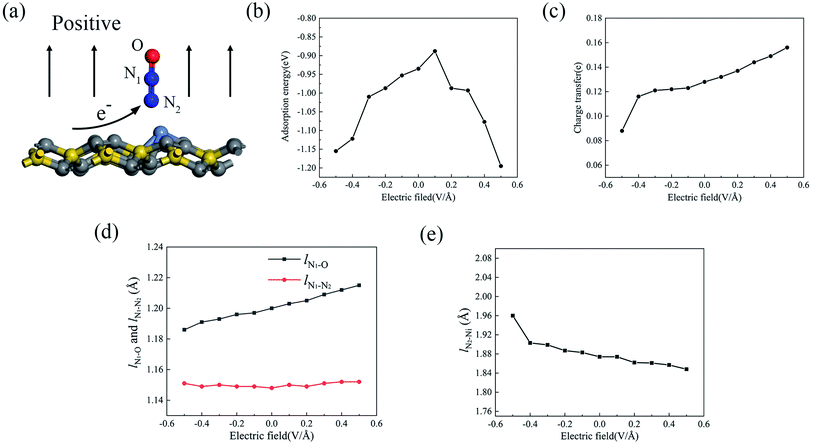 |
| | Fig. 7 (a) The diagram of the N2O2V2 under a positive external perpendicular electric field. The variation curves of (b) adsorption energy, (c) charge transfer, (d) lN1–O and lN1–N2, and (e) lN2–Ni of N2O2V2 as a function of the electric fields. | |
In most cases, an external perpendicular electric field can remarkably enhance the adsorption energy and adsorption performance of the N2O molecule, which is not affected by the direction of the electric field when the value is greater than 0.1 V Å−1. Based on this result, we suggest that applying an electric field is an essential and favorable method for improving the sensitivity of the Ni-2-PG sensors to N2O gas molecules. This result is in good agreement with previous studies, which inferred that an external perpendicular electric field would enhance the adsorption behaviors both theoretically43,44 and experimentally.45
4. Conclusion
In this study, the adsorption behaviors of N2O on PG and Ni-doped PG were theoretically investigated using first-principles calculations. By comparing and analyzing the results of adsorption energy, charge transfer, band gap, DOS and PDOS, we observed that the N2O molecule was physically adsorbed via van der Waals forces on the intrinsic PG and Ni-1-PG, whereas N2O was chemically adsorbed on Ni-2-PG, forming chemical bonds between the gas molecule and substrate. Therefore, the N2O molecule can be stably adsorbed on the Ni-doped PG only when the Ni atom is doped at the sp2 hybridized carbon site (C2). Additionally, the adsorption behaviors were affected by the adsorption orientations, in which the N2O molecule with its N atom close to the substrate exhibited better adsorption stability. Moreover, the application of a perpendicular electric field can improve the sensitivity of Ni-2-PG to N2O by enhancing the adsorption ability and adjusting the charge distribution between the gas molecule and substrate. Therefore, the adsorption application of PG could be broadened to N2O, and Ni-2-PG could be a promising candidate for the adsorption or detection of N2O gas molecules.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The first-principles calculations in this work are based on the VASP package, which is provided by Prof. Gang Zhou in Hubei University of Technology. This work is also supported by the National Natural Science Foundation of China under Grants No. 11404119, by the Doctoral Scientific Research Foundation of Hubei University of Technology (Grant No. BSQD2020106).
References
- S. Zhang, J. Zhou, Q. Wang, X. Chen, Y. Kawazoe and P. Jena, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2372–2377 CrossRef CAS PubMed.
- H. Sun, S. Mukherjee and C. V. Singh, Phys. Chem. Chem. Phys., 2016, 18, 26736–26742 RSC.
- F. Q. Wang, J. Yu, Q. Wang, Y. Kawazoe and P. Jena, Carbon, 2016, 105, 424–429 CrossRef CAS.
- Z. Wang, F. Dong, B. Shen, R. Zhang, Y. Zheng, L. Chen, S. Wang, C. Wang, K. Ho and Y.-J. Fan, Carbon, 2016, 101, 77–85 CrossRef CAS.
- J. Deb, N. Seriani and U. Sarkar, Phys. E, 2021, 127, 114507 CrossRef CAS.
- B. Xiao, Y.-c. Li, X.-f. Yu and J.-b. Cheng, ACS Appl. Mater. Interfaces, 2016, 8, 35342–35352 CrossRef CAS PubMed.
- R. Krishnan, W.-S. Su and H.-T. Chen, Carbon, 2017, 114, 465–472 CrossRef CAS.
- M.-Q. Cheng, Q. Chen, K. Yang, W.-Q. Huang, W.-Y. Hu and G.-F. Huang, Nanoscale Res. Lett., 2019, 14, 1–8 CrossRef CAS PubMed.
- T. Y. Mi, D. M. Triet and N. T. Tien, Physics Open, 2020, 2, 100014 CrossRef.
- C. Feng, X.-H. Luan, P. Zhang, J. Xiao, D.-G. Yang and H.-B. Qin, A first-principle study of the adsorption behavior of NO gas molecules on pristine and Al-doped penta-graphene, 2017 18th International Conference on Electronic Packaging Technology (ICEPT), 2017, pp. 1138–1142, DOI:10.1109/ICEPT.2017.8046642.
- Y. Ni, J. Li, W. Tao, H. Ding and R.-X. Li, Phys. Chem. Chem. Phys., 2021, 23, 2753–2761 RSC.
- H. Li, Q. Zhang, C. C. R. Yap, B. K. Tay, T. H. T. Edwin, A. Olivier and D. Baillargeat, Adv. Funct. Mater., 2012, 22, 1385–1390 CrossRef CAS.
- E. Bengu and L. Marks, Phys. Rev. Lett., 2001, 86, 2385 CrossRef CAS PubMed.
- S.-C. Zhu, C.-T. Yip, S.-J. Peng, K.-M. Wu, K.-L. Yao, C.-L. Mak and C.-H. Lam, Phys. Chem. Chem. Phys., 2018, 20, 7635–7642 RSC.
- N. Liu, J. Liu, S. Wang and K. Yao, Phys. Lett. A, 2020, 384, 126127 CrossRef CAS.
- Y. Feng, X. Wu, J. Han and G. Gao, J. Mater. Chem. C, 2018, 6, 4087–4094 RSC.
- Y. Ni, H. Hua, J. Li and N. Hu, Nanoscale, 2022, 14, 3818–3825 RSC.
- H. Qin, C. Feng, X. Luan and D. Yang, Nanoscale Res. Lett., 2018, 13, 1–7 CrossRef CAS PubMed.
- J. I. G. Enriquez and C. V. Al Rey, Int. J. Hydrogen Energy, 2016, 41, 12157–12166 CrossRef CAS.
- C.-P. Zhang, B. Li and Z.-G. Shao, Appl. Surf. Sci., 2019, 469, 641–646 CrossRef CAS.
- X. Zhang, X. Wang, X. Zhao, Y. Xu, H. Gao and F. Zhang, Chem. Eng. J., 2014, 252, 288–297 CrossRef CAS.
- J. M. Otte, N. Blackwell, R. Ruser, A. Kappler, S. Kleindienst and C. Schmidt, Sci. Rep., 2019, 9, 1–12 CrossRef PubMed.
- M. Cain, J. Lynch, M. R. Allen, J. S. Fuglestvedt, D. J. Frame and A. H. Macey, npj Clim. Atmos. Sci., 2019, 2, 1–7 CrossRef CAS.
- M. D. Esrafili and N. Saeidi, New J. Chem., 2017, 41, 13149–13155 RSC.
- H. Wei, Y. Gui, J. Kang, W. Wang and C. Tang, Nanomaterials, 2018, 8, 646 CrossRef PubMed.
- H. Cui, X. Zhang, Y. Li, D. Chen and Y. Zhang, Appl. Surf. Sci., 2019, 494, 859–866 CrossRef CAS.
- H. Wang, Q. Wang, Y. Cheng, K. Li, Y. Yao, Q. Zhang, C. Dong, P. Wang, U. Schwingenschlögl and W. Yang, Nano Lett., 2012, 12, 141–144 CrossRef CAS PubMed.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- K. Yang, J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2010, 132, 164117 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- B. Rajbanshi, S. Sarkar, B. Mandal and P. Sarkar, Carbon, 2016, 100, 118–125 CrossRef CAS.
- Y.-a. Lv, G.-l. Zhuang, J.-g. Wang, Y.-b. Jia and Q. Xie, Phys. Chem. Chem. Phys., 2011, 13, 12472–12477 RSC.
- N. Mishra, B. P. Pandey, B. Kumar and S. Kumar, Superlattices Microstruct., 2021, 160, 107083 CrossRef CAS.
- M. D. Esrafili and S. Heidari, Chem. Phys. Lett., 2019, 725, 52–58 CrossRef CAS.
- X. F. Yu, Y. C. Li, J. B. Cheng, Z. B. Liu, Q. Z. Li, W. Z. Li, X. Yang and B. Xiao, ACS Appl. Mater. Interfaces, 2015, 7, 13707–13713 CrossRef CAS PubMed.
- S. Ma, D. Yuan, Z. Jiao, T. Wang and X. Dai, J. Phys. Chem. C, 2017, 121, 24077–24084 CrossRef CAS.
- X.-Y. Liang, N. Ding, S.-P. Ng and C.-M. L. Wu, Appl. Surf. Sci., 2017, 411, 11–17 CrossRef CAS.
- S. Tang and Z. Cao, J. Chem. Phys., 2011, 134, 044710 CrossRef PubMed.
- N. Sathishkumar, S.-Y. Wu and H.-T. Chen, Chem. Eng. J., 2020, 391, 123577 CrossRef CAS.
- N. Sathishkumar, S.-Y. Wu and H.-T. Chen, Chem. Eng. J., 2021, 407, 127194 CrossRef CAS.
- T. E. Benavidez, D. Torrente, M. Marucho and C. D. Garcia, Langmuir, 2015, 31, 2455–2462 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab